

Abstract
Exosomes are small extracellular vesicles released by cells and play important roles in intercellular communication and pathogen transfer. Exosomes have been implicated in several neurodegenerative diseases, including prion disease and Alzheimer disease. Prion disease arises upon misfolding of the normal cellular prion protein, PrPC, into the disease-associated isoform, PrPSc. The disease has a unique transmissible etiology, and exosomes represent a novel and efficient method for prion transmission. The precise mechanism by which prions are transmitted from cell to cell remains to be fully elucidated, although three hypotheses have been proposed: direct cell-cell contact, tunneling nanotubes, and exosomes. Given the reported presence of exosomes in biological fluids and in the lipid and nucleic acid contents of exosomes, these vesicles represent an ideal mechanism for encapsulating prions and potential cofactors to facilitate prion transmission. This study investigates the relationship between exosome release and intercellular prion dissemination. Stimulation of exosome release through treatment with an ionophore, monensin, revealed a corresponding increase in intercellular transfer of prion infectivity. Conversely, inhibition of exosome release using GW4869 to target the neutral sphingomyelinase pathway induced a decrease in intercellular prion transmission. Further examination of the effect of monensin on PrP conversion revealed that monensin also alters the conformational stability of PrPC, leading to increased generation of proteinase K-resistant prion protein. The findings presented here provide support for a positive relationship between exosome release and intercellular transfer of prion infectivity, highlighting an integral role for exosomes in facilitating the unique transmissible nature of prions.
Keywords: cell biology, exosome (vesicle), extracellular vesicles, prion, prion disease, exosomes, monensin, neutral sphingomy, neutral sphingomyelinase, transwell
Introduction
Exosomes are small membrane-bound extracellular vesicles (80–160 nm) released from cells and have been implicated in many diseases, including cancer, viral infection, and neurodegenerative diseases (1–3). Exosomes can be isolated from biological fluids such as blood and urine and hold diagnostic potential (4, 5). Exosome biogenesis occurs within the endosomal system, with invagination of endosomal membranes to form intraluminal vesicles within multivesicular bodies (6). The intraluminal vesicles are then either delivered to lysosomes for degradation or released from cells as exosomes upon fusion of the multivesicular body with the plasma membrane.
Exosomes play important roles in intercellular communication, including regulation of immune responses following bacterial invasion, presentation of antigens for activation of T cells, and, more recently, a novel mechanism of intercellular transfer of mRNA and microRNA, resulting in an additional level of regulation (6–9). Exosomes have also been implicated in the pathogenesis of several neurodegenerative diseases, including prion disease, Alzheimer and Parkinson diseases, and amyotrophic lateral sclerosis (10–15). Processed forms of the specific neurodegenerative disease-associated proteins have been identified in exosomes, leading to the suggestion that exosomes could facilitate their intercellular spread. An example is the role of exosomes in transmission of infectious prions arising from misfolding of the prion protein (10, 11, 16).
Prion diseases are fatal neurodegenerative diseases affecting animals and humans, such as scrapie in sheep, bovine spongiform encephalopathy in cattle, and Creutzfeldt-Jakob disease in humans (17). The disease is characterized by progressive spongiform vacuolation of the brain and neuronal loss and arises upon misfolding of the cellular prion protein (PrPC)4 into the disease-associated isoform, PrPSc. Studies have shown that not only is PrPSc released from cells in exosomes, but the exosomes are also capable of transmitting infection between homologous and heterologous cell types and to mice (10, 11). This supports the hypothesis that exosomes could contribute significantly toward transmission of infection from the periphery to the central nervous system (11).
The precise mechanism by which prions are transmitted between cells remains to be resolved. Although alternative mechanisms of intercellular transmission have also been proposed, such as direct cell-cell contact (18) and tunneling nanotubes (19), the presence of exosomes in biological fluids suggest a prominent role for exosomes in transmission between different parts of the body. In addition, with the lipid and nucleic acid cargo, exosomes represent an ideal mechanism for encapsulating potential cofactors for PrP conversion to facilitate efficient intercellular transmission. Evidence has been gathered to suggest that cofactors are required for prion conversion either by assisting the conformational change or by exerting an effect on the cell to increase susceptibility to infection (20–23). In particular, lipids, such as phosphatidylethanolamine, and RNA have been identified as essential cofactors required for the efficient generation of infectivity de novo (21, 23). Given the distinguishing transmissible nature of prion diseases and that exosomes have been implicated to play a role in intercellular prion transmission, we aimed to further investigate the relationship between exosome release and intercellular transmission of prions.
Experimental Procedures
Reagents and Antibodies
Unless specified otherwise, all reagents were from Sigma-Aldrich. Antibodies used and their working dilutions were as follows: α-PrP antibodies 03R19 (1:25,000, in-house) (24) and L3 (1:5000, in-house) (25), α-tubulin (1:25,000), and α-mouse HRP (1:25,000) and α-rabbit HRP (1:25,000) from GE Healthcare (Sydney, NSW, Australia).
Maintenance and Infection of Cultured Cell Lines with Prions and Cell Blot Assay
MoRK13 and GT1-7 cells were cultured in Opti-MEM (Life Technologies) and infected with a mouse-adapted strain of human prions (M1000) as described previously (25, 26). At six passages after infection, stable infection of cells was assessed using a cell blot assay as described previously (25).
Toxicity Assay
Toxicity assays were carried out usingthe Live/Dead® viability/cytotoxicity kit (Life Technologies) according to the instructions of the manufacturer as described previously (25).
Transwell Assay
Recipient A2 MoRK13 cells were seeded into 6-well tissue culture plates (Corning) at a density of 250,000 cells/well and allowed to attach overnight. Cells were washed once with warm PBS, and 1 ml of complete Opti-MEM was added to each well. Use of puromycin for standard culturing of MoRK13 cells was excluded during the transwell assay (26). Using tweezers sterilized by soaking with 80% (v/v) ethanol, Costar 24-mm Transwell® permeable support inserts with 0.4-μm polyester membranes (Corning) were placed on top of each well. Prion-infected MoRK13 or GT1-7 donor cells were seeded into the inserts at a density of 200,000 cells/insert and made up to 1.5 ml final volume using complete Opti-MEM. For MON or GW4869 treatment, the compounds and vehicle-only controls were diluted appropriately in complete Opti-MEM and added to the culture medium in the inserts. The plates were incubated for 48 h at 37 °C, 5% (v/v) CO2. Culture medium in the inserts were then aspirated, and the insert was removed using sterilized tweezers. The culture medium in the wells were aspirated and replaced with fresh Opti-MEM containing 2.5 μg/ml puromycin. Cells were then passaged under standard conditions with an initial 1:3 split into a new 6-well tissue culture plate (Nunc/Thermo Scientific, Roskilde, Denmark), followed by a 1:5 split and then 1:10 splits for all subsequent passages. At passage 8 after infection, cells were seeded onto sterile plastic coverslips in 6-well tissue culture plates at a density of 450,000 cells/well and allowed to attach and recover for 48 h. Cells were then analyzed using the cell blot assay as described previously (25).
Exosome Isolation and Acetylcholinesterase (AChE) Assay
For exosome collection, 175-cm2 flasks (Nunc) were seeded with 7 × 106 cells and allowed to recover for 24 h. For GW4869 and MON treatment, the culture medium were replaced with exosome-free medium containing appropriate concentrations of GW4869, MON, or vehicle-only controls and cultured for 2 days as described previously (25). Exosomes were isolated from the culture supernatant using differential ultracentrifugation, and the freshly collected exosomes were used for the AChE activity assay as described previously (25–29).
Protease Digestion, SDS-PAGE, and Immunoblotting
Cells were lysed as described previously (25). Total protein concentrations were determined using the BCA protein assay (Pierce, Thermo Scientific) according to the protocol of the manufacturer. 100 μg of protein was treated with proteinase K (PK, 25 μg/ml) as described previously (25, 26). 40 μg of protein was incubated on ice for non-PK-treated samples. Proteins were precipitated using ice-cold methanol and prepared for SDS-PAGE, followed by immunoblotting, as described previously (25, 26). On immunoblots, PrPC refers to the total levels of PrP detected in non-infected cells, and PrPTot refers to the total level of PrP detected in a prion-infected cell. PrPSc refers to the total level of PrP detected in a prion-infected cell after treatment with PK.
Protein Misfolding Cyclic Amplification (PMCA) Assay
The PMCA assay was performed as described previously with varying dilutions of MON and vehicle-only control included in the reaction (26). Non-PMCA controls were stored at −80 °C for the duration of the reaction to define baseline protease-resistant PrPSc levels. PMCA and non-PMCA controls were analyzed by immunoblotting as described previously (26).
Confocal Microscopy
Cells were prepared for confocal microscopy as described previously with modifications (30). Cells were seeded into 8-well μSlide chamber slides (iBidi GmbH, Martinsried, Germany) and allowed to attach overnight. Cells were fixed with 4% (w/v) paraformaldehyde and permeabilized with 0.5% (v/v) Triton X-100. Cells were blocked with 2% (w/v) bovine serum albumin in PBS and incubated with primary antibodies for 2 h followed by secondary antibodies for 2 h. The primary antibodies and dilutions were as follows: 03R19 (1:200), SAF32 (1:150), and α-GM130 (1:150) (BD Biosciences). The secondary antibodies and dilutions were as follows: DAPI (1:1000), Alexa Fluor 568-conjugated anti-rabbit (1:450), and Alexa Fluor 647-conjugated anti-mouse (1:450) (Life Technologies). Cells were imaged on a Leica DMIRE2 confocal microscope with SP2 control software. Images were stacked to ensure equal adjustments to all images.
Prion-infected Cell Assay (PICA)
The PICA was performed using a subclone of the MoRK13 cell line A2 to provide a quantitative analysis of infectivity as described previously (26). Lysates from prion-infected GT1-7 cells that were treated with either vehicle or MON for 24 h and then cultured under standard conditions for three further passages were used to prepare serial titrations of inoculum for infecting the A2 cells (26). A2 cells were co-incubated with the inoculum for 64 h and the inoculum were removed by aspiration, and then the cells were washed once with warm PBS and twice with complete Opti-MEM, refreshed with complete Opti-MEM containing 2.5 μg/ml puromycin, and grown to confluency. Cells were passaged with a 1:3 split into a new plate, followed by a 1:5 split and then 1:10 splits for all subsequent passages. At passage 8 after inoculation, the A2 cells were analyzed using a PICA with the α-PrP L3 antibody to measure de novo synthesis of PrPSc in the cells, as described previously (26). The results were normalized to background signal from control A2 cells that were not inoculated and then plotted as a dose-response curve.
Image Processing and Statistical Analysis
Densitometric analysis was carried out using ImageJ (31). Statistical analyses were performed using GraphPad Prism 6 (GraphPad, San Diego, CA). Data were analyzed as follows. Toxicity assay data are shown as mean ± S.E. of four experiments, and statistical significance tests were performed with two-sample t tests. AChE and densitometric analyses data are shown as mean change ± S.E. in comparison with three control experiments, and statistical significance tests were performed with one-sample t tests to compare each value against a standardized control of 100%. Advice was sought from the University of Melbourne Statistical Consulting Centre. Statistical significance was defined as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Results
Exosomes Facilitate Intercellular Transmission of Prions across Transwells
To determine the relationship between exosome release and prion transmission, transwell assays were used in conjunction with two cell lines: a stably transfected rabbit kidney epithelial cell line overexpressing mouse PrP (MoRK13) and a mouse hypothalamic neuronal cell line expressing endogenous PrP (GT1-7) (11, 32, 33). RK13 cells are permissive to infection with murine prions when expressing a murine PrP transgene. We have demonstrated previously that both MoRK13 and GT1-7 cells release exosomes containing prion infectivity and that these vesicles can transmit prion infectivity to the same and different cell lines in addition to inducing clinical prion disease in susceptible mice (11). In the transwell assay, two populations of MoRK13 cells, a prion-infected (donor cells) and a non-infected population (recipient cells), were co-cultured but prevented from direct cell-cell contact by a semipermeable membrane (Fig. 1A). Exosomes are smaller than the pore size and, therefore, are able to diffuse across the membrane. Cell blot analysis of the recipient cells following the transwell assay showed that no PrPSc was detected in recipient cells that had been co-cultured with non-infected donor cells, indicating that the recipient cells were not infected (Fig. 1B). In contrast, PrPSc was detected in recipient cells that had been co-cultured with prion-infected donor cells, indicating that successful transfer of prions occurred across the transwell membrane in the absence of direct cell-cell contact, most likely mediated by exosomes.
FIGURE 1.

Successful intercellular transmission of prions across transwell membranes. A, schematic depicting the setup for a transwell assay with a non-infected population of cells (recipient, blue) separated from a prion-infected population (donor, red) by a membrane with 0.4-μm pores. B, cell blot assay confirming the presence of PrPSc and transmission of prion infection to recipient cells via the transwell assay.
Monensin-stimulated Exosome Release Correlates with Increased Intercellular Prion Transmission
To further investigate the relationship between exosomes and intercellular transmission of prions across the transwell membrane, the effect of increasing exosome release on prion transmission across transwells was examined. To achieve this, MoRK13 cells were treated with the ionophore monensin (MON), which has been shown previously to increase exosome release from K562 cells (34). Quantification of exosomes using the AChE assay indicates that treatment with an increasing amount of MON induces a corresponding increase of AChE activity, and, therefore, MON stimulates exosome release from MoRK13 cells in a dose-dependent manner (Fig. 2A). When used in combination with the transwell assay, treatment of the prion-infected MoRK13 donor cells with increasing amounts of MON results in a corresponding increase in the amount of PrPSc detected in the recipient cells (Fig. 2B). This indicates that there is a linear relationship between exosome release and intercellular transfer of prions across the transwell membrane, suggesting that exosomes facilitate prion transmission across the transwell.
FIGURE 2.

Monensin stimulates exosome release, which is associated with a corresponding increase in intercellular prion transmission. A, AChE quantitation of isolated exosomes, indicating increasing exosome release with MON treatment. B, cell blot analysis revealing increasing levels of PrPSc and increasing transfer of prion infectivity in recipient cells after exposure to donor cells treated with increasing amounts of MON in a transwell assay. Data are presented as mean ± S.E. (n = 3). *, p < 0.05; **, p < 0.01.
This finding was further validated in the mouse hypothalamic neuronal GT1-7 cell line. Because of the increased sensitivity of GT1-7 cells to MON treatment, the ionophore was further diluted and tested for toxicity (Fig. 3A). Exosomes released from monensin-treated, prion-infected GT1-7 cells were analyzed by Western blotting to determine whether PrPSc was present within the exosomes (Fig. 3B). Indeed, PrPSc was present in monensin-treated prion-infected GT1-7 cells and exosomes, indicating that the monensin-induced increase in intercellular transfer of prion infectivity is mediated by transfer of PrPSc in exosomes. In agreement with the MoRK13 cells, treatment of GT1-7 cells with MON similarly stimulated exosome release from the neuronal cells (Fig. 3C). Having confirmed the effect of MON on exosome release in GT1-7 cells, the transwell assay was repeated using prion-infected GT1-7 cells as donor cells. A cell blot assay of the recipient cells revealed a similar result, with more PrPSc detected in recipient cells that had been exposed to MON-treated donor cells, indicating that increasing exosome release from GT1-7 cells is also associated with a corresponding increase of intercellular prion transfer. This finding further supports a role for exosomes in prion transmission across the transwell and also validates the results obtained using prion-infected MoRK13 cells as donor cells. Moreover, this result demonstrates the ability of exosomes to facilitate prion transfer between two cell lines of distinct origins and species.
FIGURE 3.

Monensin similarly stimulates exosome release and prion transmission in neuronal cells. A, increasing concentrations of MON were tested for toxicity to GT1-7 cells, and no toxicity was observed at all concentrations tested. B, Western blotting analysis for the presence of PrPTot and PrPSc in control- and monensin-treated, prion-infected GT1-7 cell lysates and exosomes (Exo) released from prion-infected GT1-7 cells. The blot confirms that exosomes released from monensin-treated cells contain PrPSc. C, AChE assay of isolated exosomes indicating that MON also stimulates exosome release from GT1-7 cells. Cell blot analysis of recipient cells reveals increased levels of PrPSc and prion transmission following exposure to prion-infected GT1-7 donor cells in the presence of MON. D, AChE assay of isolated exosomes indicating that treatment with GW4869 decreases exosome release from GT1-7 cells. Cell blot analysis of recipient cells exposed to prion-infected GT1-7 cells in the presence of GW4869 reveals a corresponding decrease in PrPSc and prion transmission. Data are presented as mean ± S.E. (n = 3). **, p < 0.01; ***, p < 0.001. DMSO, dimethyl sulfoxide.
We have demonstrated previously that disruption of the neutral sphingomyelinase pathway in GT1-7 cells hinders exosome release (25). To further validate these findings, GT1-7 cells were treated with GW4869, a neutral sphingomyelinase 2 inhibitor, and a decrease in exosome release was observed (Fig. 3D) (35). When used in combination with the transwell assay, recipient cells that had been exposed to donor cells in the presence of GW4869 showed a decrease in PrPSc levels, indicating that decreasing exosome release is associated with a corresponding decrease in intercellular prion transmission. Together, these results illustrate a linear relationship between exosome release and intercellular prion transmission, further highlighting and implicating a role for exosomes in aiding prion dissemination.
Monensin Does Not Alter the Conversion of PrP in an in Vitro Conversion Assay
In addition to increasing prion transmission via increased exosome release, the effect of the ionophore on PrP conversion was investigated further. The direct effect of MON on PrP conversion was examined using the PMCA, which allows in vitro amplification and generation of de novo PrPSc from a reservoir of PrPC (the substrate) in the presence of a minute amount of PrPSc template (the seed) (36). Varying concentrations of MON were included in the PMCA reactions to determine whether MON alters the propensity of PrPSc to convert PrPC. Following the assay, both the samples that had been subjected to PMCA and those that had been stored at −80 °C for the duration of the PMCA as a control were analyzed by immunoblotting for the presence of PrPSc after PK digestion (Fig. 4A). As expected, no PrPSc was detected in the −80 °C control samples. In contrast, PrPSc was detected in the PMCA samples, indicating successful conversion of the substrates by the seed during the assay. However, no changes were observed in the amount of PrPSc generated, suggesting that MON does not affect the propensity of PrPSc to convert PrPC in vitro.
FIGURE 4.
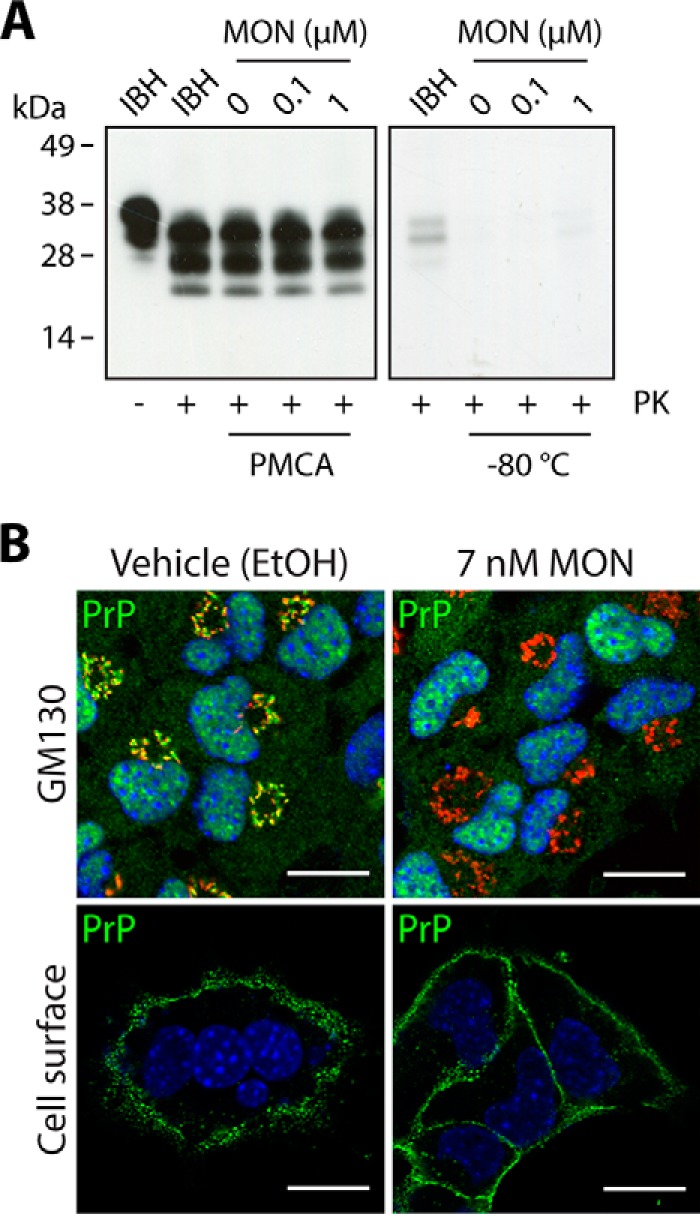
Monensin does not induce de novo generation of PrPSc but impairs Golgi trafficking of PrP. A, immunoblot analysis of PMCA and −80 °C controls indicating no change in de novo generation of PrPSc in the presence of MON. Infected brain homogenates (IBH) are included as a reference point. B, confocal microscopy analysis of PrPC (green) in non-infected vehicle- and MON-treated GT1-7 cells. Treatment with MON abolishes colocalization of PrPC with GM130-positive compartments (red), indicating impaired Golgi trafficking. Visualization of cell surface PrPC revealed unhindered trafficking of PrPC to the plasma membrane. Scale bars = 20 μm.
Monensin Increases the Susceptibility of PrPC Conversion to PrPSc in Prion-infected cells
An alternative way in which MON could affect PrP conversion is by altering the subcellular localization or trafficking of PrPC. MON has been used as a biochemical tool for inhibiting trans Golgi function, and because PrPC traverses the Golgi en route to the cell surface, the effect of MON on posttranslational glycosylation of PrPC was examined (37).
PrP colocalization using immunofluorescent analyses of GT1-7 cells showed that MON treatment abolishes PrPC colocalization with GM130-positive compartments, indicating impaired trafficking of PrPC through the Golgi (Fig. 4B). As a consequence, PrPC secretion to the plasma membrane could be blocked. To evaluate this possibility, non-permeabilized cells were stained to visualize cell surface PrPC (Fig. 4B). Comparison of vehicle- and MON-treated cells revealed no differences, indicating that PrPC is able to bypass the blockage and reach the cell surface, a phenomenon that has been reported for other proteins (38–40). Taken together, these results show that MON modifies the intracellular trafficking of PrPC and blocks trafficking through the Golgi.
Glycosylation plays an important role in promoting correct folding of PrPC, and differences in glycosylation patterns contribute toward prion strain specificity (41–43). Moreover, the glycosylation status of PrPC has also been found to influence transmission of prions from the periphery to the CNS (44). MON-induced aberrant glycosylation of PrPC has been described previously (45) and could therefore alter the susceptibility of PrPC to prion conversion. To determine whether this is the case, vehicle- and MON-treated prion-infected GT1-7 cells were analyzed by immunoblotting to compare the levels of PrPTot and PrPSc present. Two different time points were analyzed: immediately after a 24-h treatment and three passages after a 24-h treatment. Analysis of the cell lysates immediately after treatment showed no changes in the levels of PrPTot or PrPSc (Fig. 5, A and B). However, analysis of the long-term effect of MON treatment revealed a significant increase in PrPSc levels (Fig. 5, C and D). A similar increase was also observed for PrPTot, which can be attributed to the increase of PrPSc. These findings therefore indicate that treatment of prion-infected cells with MON increases the susceptibility of PrPC to conversion, resulting in an increase of PrPSc levels over time.
FIGURE 5.

Monensin induces long-term accumulation of PrPSc. A, immunoblot analysis of PrP in prion-infected GT1-7 cells immediately after vehicle or MON treatment. B, densitometric analysis showed no significant differences in PrPTot and PrPSc levels. C, immunoblot analysis of PrP in prion-infected GT1-7 cells three passages after vehicle or MON treatment. D, densitometric analysis revealed a significant increase in PrPTot and PrPSc levels. E, the levels of prion infectivity in vehicle- and MON-treated GT1-7 cells were quantitated using PICA. A dose-response curve of the relative fluorescence units (RFU) of PrPSc, as a percentage of the background levels in controls cells, was plotted against the log of inoculum used. No significant differences were observed between the infectious responses associated with vehicle- or MON-treated lysates. Data are presented as mean ± S.E. (n = 3). *, p < 0.05; ns, not significant.
The finding that MON treatment increases PrPSc levels suggests that MON could also enhance prion transmission by stimulating PrP conversion in addition to increasing exosome release. This possibility was investigated using the PICA to quantitate the relative levels of infectivity for vehicle- and MON-treated prion-infected GT1-7 cell lysates. Eight passages after inoculation, the recipient cells were analyzed using the PICA to measure de novo synthesis of PrPSc, and the results were plotted as a dose-response curve. If the MON-treated lysates produce higher levels of prion infectivity than vehicle-treated lysates, then the dose-response curve for MON is expected to shift to the left of the curve for vehicle, indicating higher levels of infectivity associated with lower amounts of inoculum. Conversely, if the MON-treated lysates produce lower levels of prion infectivity, then the curve for MON is expected to shift to the right of the curve for vehicle. As shown in Fig. 5E, no differences were detected between the curves, indicating that MON-treated lysates elicit the same infectious response as vehicle-treated lysates. Therefore, despite the increase of PrPSc in response to MON treatment, the level of infectivity associated with the lysates remain unchanged, suggesting that the additional PrPSc produced is PK-resistant but not associated with higher levels of infectivity.
Discussion
A unique feature of prion disease that distinguishes it from other neurodegenerative diseases is the transmissible nature of the disease. Exosomes have been proposed as a mechanism that assists with the intercellular transmission of prions (10, 11). We aimed to directly address the relationship between exosomes and intercellular prion transmission. Using transwell assays, we showed that successful intercellular transmission of prions can occur across the transwell membrane, facilitated by exosomes. When the assays were used in conjunction with compounds that up- or down-regulate exosome release, a linear relationship was observed, with a corresponding increase and decrease in intercellular prion transmission, respectively. Furthermore, we showed that the ionophore MON not only facilitates prion transmission by increasing exosome release but that it also alters the conformational stability of PrPC by hindering posttranslational glycosylation, resulting in the conversion of PrPC into a PK-resistant but non-infectious isoform (PrPres).
The finding that successful intercellular transmission of prions can be achieved across the transwell membrane suggests that exosomes are the likely candidate responsible for mediating prion transmission between the cells because both direct cell-cell contact and formation of tunneling nanotubes are prevented by the transwell membrane (18, 19). In direct contrast to this, it was has been reported previously that intercellular prion transfer is inhibited in the presence of transwell membranes (19). In this study, the same experimental setup was utilized for the transwell assay, with bone marrow-derived dendritic cells (BMDCs) as donor cells and MoRK13 cells as recipient cells. It was shown that co-culturing of the cells in the presence of a transwell membrane failed to result in transmission of prions to MoRK13 cells, whereas co-culturing in the absence of the transwell resulted in successful transmission. Although the same MoRK13 cell line was used, there is a crucial difference between the donor cell lines that were used, which could explain the marked differences observed for prion transmission. Although the prion-infected MoRK13 donor cells produced endogenous PrPSc, the prion-infected donor BMDCs contained exogenous PrPSc, which were loaded into the BMDCs by incubation with infected mouse brain homogenate. It can be speculated that the intracellular trafficking of endogenous and exogenous PrPSc could differ and that the exogenous PrPSc might not undergo packaging into exosomes in BMDCs. Consequently, prion transmission from the loaded BMDCs would therefore only be possible via direct cell-cell contact or tunneling nanotubes but not via exosomes. The discrepancy observed for prion transmission across the transwell membrane could therefore be attributed to the difference in the origin of PrPSc in the infected donor cells. Nevertheless, it is important to keep in mind that more than one mechanism of prion transmission is likely to exist in vivo. Depending on the cell type, tissue, or stage of infection, different mechanisms of prion transmission, or combinations thereof, could occur. As a result, all three proposed mechanisms of transmission require further investigation in vivo to evaluate the relative contributions of each pathway toward disease pathogenesis.
Further support for exosome-mediated dissemination of prions across the transwell membrane was obtained from the use of two chemical compounds, GW4869 and monensin, to decrease and increase exosome release, respectively. Using these compounds, a linear relationship between exosome release and intercellular prion transmission was revealed: decreasing exosome release decreases prion transmission, and increasing exosome release increases prion transmission. Although it cannot be ruled out that small aggregates of PrPSc could also diffuse across the transwell membrane and, thereby, contribute toward prion transmission, taken together, the results obtained using GW4869 and monensin suggest a prominent role for exosomes in mediating the transmission of prions. In support of this, a similar finding has been reported in a study in which the transwell assay was used to demonstrate exosome-mediated uptake of extracellular Aβ by microglia (46). The transwell assay was used to examine the relationship between exosome release and uptake of Aβ by microglia using N2a cells as exosome-producing donor cells and murine microglial BV-2 cells as recipient cells. Exosome release from N2a cells was also modulated by targeting the nSMase pathway. The cells were either treated with siRNA against nSMase2 to decrease exosome release or with siRNA against sphingomyelin synthase 2 to increase exosome release. Through the transwell assay, a linear relationship was also revealed between exosome release and uptake of Aβ by microglia (46).
The ability of exosomes to successfully mediate the transfer of prions between two cell lines of distinct origins, one mouse and one rabbit, reveals a potential role for exosomes in interspecies dissemination of prions. Although both GT1-7 and MoRK13 cells express mouse PrP, the results obtained from the transwell assays indicate that exosomes of murine origin can interact with or be taken up by a cell of rabbit origin and facilitate prion transmission. Exosomes, therefore, not only represent vesicles that can facilitate the spread of prions between different cell types but can also assist spreading between cells from different species. Obtaining a better understanding of exosome biogenesis, release, and mode of interaction with recipient cells could therefore provide much needed insights into prion disease pathogenesis.
In addition to increasing prion transfer by stimulating exosome release, monensin was also found to enhance transmission by hindering trafficking of PrPC through the Golgi, resulting in aberrant glycosylation of PrPC, which consequently led to an increase of PrPSc over time. Glycosylation plays an important role in promoting the correct folding of PrPC, and glycosylation patterns have been found to contribute toward prion strain specificity (41, 42). It has also been shown that aberrantly glycosylated PrPC displays an intrinsic tendency to adopt PrPSc-like properties such as decreased solubility in detergents, increased resistance to release from the cell surface by phosphatidylinositol-specific phospholipase C treatment, and increased hydrophobicity (47). Similar properties were also observed for wild-type PrPC in cells that had been treated with tunicamycin to inhibit glycosylation (47). It was therefore concluded that glycosylation plays an important role in suppressing the intrinsic tendency of PrPC to adopt PrPSc-like properties. Indeed, a familial form of prion disease has been attributed to a mutation at codon 183 that abolishes glycosylation at codon 181 (48). Taken together, these findings suggest that aberrant glycosylation of PrPC can have profound effects on the stability and susceptibility of PrPC to conversion.
The increase of PrPSc in prion-infected cells following treatment with monensin suggests that the ionophore could increase the level of infectivity associated with monensin-treated cells. However, contrary to this, monensin-treated cell lysates were not found to display higher levels of infectivity than control cell lysates. Monensin therefore stimulated the production of PrPres, which was not infectious. Indeed, there is a growing body of evidence in the literature suggesting that the level of PrPSc present does not always correlate with infectivity or disease (49–51). Because of conformational heterogeneity arising from posttranslational modifications, multiple isoforms of PrPC and, therefore, PrPSc could exist, and the different isoforms could display different biochemical properties and affect prion infectivity and disease differently. One example of such a case is the HYPER (HY) and DROWSY (DY) strains of transmissible mink encephalopathy in Syrian golden hamsters (52). Although both the HY and DY strains originated from the same transmissible mink encephalopathy strain, serial passage in the hamsters led to isolation of two biologically distinct strains. The HY and DY strains exhibited differences in sedimentation property in Sarkosyl, sensitivity to PK digestion, and electrophoretic mobility on polyacrylamide gels. Although the HY strain displayed properties distinct from that of PrPC, the DY strain has been found to share similarities in properties with both PrPC and the HY strain. Most prominently, the DY strain displayed higher sensitivity to PK digestion than the HY strain, indicating that PrPSc levels did not correlate with infectivity. This has also been shown to be the case in another study in which brain tissue from mice with prion disease has been found to display high levels of infectivity despite showing only low levels of PrPSc (49). Similar findings of low PrPSc levels in infected animals have also been reported by other studies (50, 51). Together, these findings highlight the limitation associated with the use of PrPSc as a sole indicator of infectivity and emphasize the need to include additional analyses, such as PICA, to specifically and definitively assay for prion infectivity.
Given the effect of monensin on exosome release and prion conversion presented in this study, comprehensive in vivo analyses testing the effect of monensin on prion disease pathogenesis are required, especially in light of the commercial application of monensin as a dietary supplement for ruminant livestock. Although the PrPres produced was not tested to be infectious using the PICA, it cannot be excluded that this result could be different in vivo as many additional variables will be introduced, such as the route of administration of monensin, length of exposure, and mechanism of challenge with prions. Furthermore, cell culture models lack the ability to provide a measure of the neurotoxicity of the PrPres produced, and it could be possible for PrPres to be non-infectious but highly toxic. In contrast, if monensin treatment indeed promotes the formation of non-infectious and non-toxic PrPres, then treatment with the ionophore could provide a method for potential disruption of disease transmission and pathogenesis because PrPres would act as a “sink” to trap PrPC and prevent the formation of PrPSc. Further in vivo characterization is therefore imperative for a definitive understanding of the effect of monensin on PrP conversion and disease pathogenesis.
Together, the data presented in this study provide support for and highlight exosomes as extracellular vesicles that facilitate intercellular prion transmission. Much additional work is required to fully characterize how this pathway contributes to prion dissemination, how the exosomes interact with recipient cells, and how this pathway could be blocked to impair disease progression. Evaluating the effects of modulating exosome release and uptake on prion transmission would not only help to increase our understanding of prion disease pathogenesis but could also have implications on other neurodegenerative diseases. Furthermore, understanding and identifying mechanisms for manipulating exosome release/uptake will also prove to be invaluable for the development and application of exosomes as vesicles for targeted delivery of therapeutic compounds.
Author Contributions
B. B. G., S. A. B., and A. F. H. conceived the study. B. B. G. designed and performed experiments, and analyzed the data with guidance from S. A. B. and A. F. H. B. B. G., S. A. B., and A. F. H. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Victoria Lawson (University of Melbourne) for mouse brain homogenates and the α-PrP 03R19 antibody. We also thank Dr. Bradley Coleman (University of Melbourne) for assistance with the PMCA assay and Dr. Peter Kaub and Dr. Simon Hawke (University of Sydney) for the monoclonal L3 antibody.
This study was funded by Australian National Health and Medical Research Council Grants 628946 and 400202. The authors declare that they have no conflicts of interest with the contents of this article.
- PrP
- prion protein
- AChE
- acetylcholinesterase
- PK
- proteinase K
- PMCA
- protein misfolding cyclic amplification
- PICA
- prion-infected cell assay
- MON
- monensin
- BMDC
- bone marrow-derived dendritic cell.
References
- 1. Zitvogel L., Regnault A., Lozier A., Wolfers J., Flament C., Tenza D., Ricciardi-Castagnoli P., Raposo G., and Amigorena S. (1998) Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat. Med. 4, 594–600 [DOI] [PubMed] [Google Scholar]
- 2. Izquierdo-Useros N., Naranjo-Gómez M., Archer J., Hatch S. C., Erkizia I., Blanco J., Borràs F. E., Puertas M. C., Connor J. H., Fernández-Figueras M. T., Moore L., Clotet B., Gummuluru S., and Martinez-Picado J. (2009) Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 113, 2732–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bellingham S. A., Guo B. B., Coleman B. M., and Hill A. F. (2012) Exosomes: vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front. Physiol. 3, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caby M. P., Lankar D., Vincendeau-Scherrer C., Raposo G., and Bonnerot C. (2005) Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 17, 879–887 [DOI] [PubMed] [Google Scholar]
- 5. Pisitkun T., Shen R. F., and Knepper M. A. (2004) Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. U.S.A. 101, 13368–13373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raposo G., Nijman H. W., Stoorvogel W., Liejendekker R., Harding C. V., Melief C. J., and Geuze H. J. (1996) B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 183, 1161–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O'Neill H. C., and Quah B. J. (2008) Exosomes secreted by bacterially infected macrophages are proinflammatory. Sci. Signal. 1, e8. [DOI] [PubMed] [Google Scholar]
- 8. Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J. J., and Lötvall J. O. (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659 [DOI] [PubMed] [Google Scholar]
- 9. Bellingham S. A., Coleman B. M., and Hill A. F. (2012) Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res. 40, 10937–10949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fevrier B., Vilette D., Archer F., Loew D., Faigle W., Vidal M., Laude H., and Raposo G. (2004) Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 101, 9683–9688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vella L. J., Sharples R. A., Lawson V. A., Masters C. L., Cappai R., and Hill A. F. (2007) Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 211, 582–590 [DOI] [PubMed] [Google Scholar]
- 12. Rajendran L., Honsho M., Zahn T. R., Keller P., Geiger K. D., Verkade P., and Simons K. (2006) Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharples R. A., Vella L. J., Nisbet R. M., Naylor R., Perez K., Barnham K. J., Masters C. L., and Hill A. F. (2008) Inhibition of γ-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 22, 1469–1478 [DOI] [PubMed] [Google Scholar]
- 14. Alvarez-Erviti L., Seow Y., Schapira A. H., Gardiner C., Sargent I. L., Wood M. J., and Cooper J. M. (2011) Lysosomal dysfunction increases exosome-mediated α-synuclein release and transmission. Neurobiol. Dis. 42, 360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gomes C., Keller S., Altevogt P., and Costa J. (2007) Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 428, 43–46 [DOI] [PubMed] [Google Scholar]
- 16. Vilette D., Laulagnier K., Huor A., Alais S., Simoes S., Maryse R., Provansal M., Lehmann S., Andreoletti O., Schaeffer L., Raposo G., and Leblanc P. (2015) Efficient inhibition of infectious prions multiplication and release by targeting the exosomal pathway. Cell. Mol. Life Sci. 72, 4409–4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanu N., Imokawa Y., Drechsel D. N., Williamson R. A., Birkett C. R., Bostock C. J., and Brockes J. P. (2002) Transfer of scrapie prion infectivity by cell contact in culture. Curr. Biol. 12, 523–530 [DOI] [PubMed] [Google Scholar]
- 19. Gousset K., Schiff E., Langevin C., Marijanovic Z., Caputo A., Browman D. T., Chenouard N., de Chaumont F., Martino A., Enninga J., Olivo-Marin J. C., Männel D., and Zurzolo C. (2009) Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 11, 328–336 [DOI] [PubMed] [Google Scholar]
- 20. Deleault N. R., Lucassen R. W., and Supattapone S. (2003) RNA molecules stimulate prion protein conversion. Nature 425, 717–720 [DOI] [PubMed] [Google Scholar]
- 21. Deleault N. R., Piro J. R., Walsh D. J., Wang F., Ma J., Geoghegan J. C., and Supattapone S. (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 109, 8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deleault N. R., Walsh D. J., Piro J. R., Wang F., Wang X., Ma J., Rees J. R., and Supattapone S. (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. U.S.A. 109, E1938–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang F., Wang X., Yuan C. G., and Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lawson V. A., Vella L. J., Stewart J. D., Sharples R. A., Klemm H., Machalek D. M., Masters C. L., Cappai R., Collins S. J., and Hill A. F. (2008) Mouse-adapted sporadic human Creutzfeldt-Jakob disease prions propagate in cell culture. Int. J. Biochem. Cell Biol. 40, 2793–2801 [DOI] [PubMed] [Google Scholar]
- 25. Guo B. B., Bellingham S. A., and Hill A. F. (2015) The neutral sphingomyelinase pathway regulates packaging of the prion protein into exosomes. J. Biol. Chem. 290, 3455–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coleman B. M., Hanssen E., Lawson V. A., and Hill A. F. (2012) Prion-infected cells regulate the release of exosomes with distinct ultrastructural features. FASEB J. 26, 4160–4173 [DOI] [PubMed] [Google Scholar]
- 27. Ellman G. L., Courtney K. D., Andres V. Jr., and Feather-Stone R. M. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95 [DOI] [PubMed] [Google Scholar]
- 28. Cantin R., Diou J., Bélanger D., Tremblay A. M., and Gilbert C. (2008) Discrimination between exosomes and HIV-1: purification of both vesicles from cell-free supernatants. J. Immunol. Methods 338, 21–30 [DOI] [PubMed] [Google Scholar]
- 29. Rabesandratana H., Toutant J. P., Reggio H., and Vidal M. (1998) Decay-accelerating factor (CD55) and membrane inhibitor of reactive lysis (CD59) are released within exosomes during in vitro maturation of reticulocytes. Blood 91, 2573–2580 [PubMed] [Google Scholar]
- 30. Harrison C. F., Lawson V. A., Coleman B. M., Kim Y.-S., Masters C. L., Cappai R., Barnham K. J., and Hill A. F. (2010) Conservation of a glycine-rich region in the prion protein is required for uptake of prion infectivity. J. Biol. Chem. 285, 20213–20223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rasband W. S. (1997) ImageJ, National Institutes of Health, Bethesda, MD [Google Scholar]
- 32. Schätzl H. M., Laszlo L., Holtzman D. M., Tatzelt J., DeArmond S. J., Weiner R. I., Mobley W. C., and Prusiner S. B. (1997) A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J. Virol. 71, 8821–8831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mellon P. L., Windle J. J., Goldsmith P. C., Padula C. A., Roberts J. L., and Weiner R. I. (1990) Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5, 1–10 [DOI] [PubMed] [Google Scholar]
- 34. Savina A., Furlán M., Vidal M., and Colombo M. I. (2003) Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J. Biol. Chem. 278, 20083–20090 [DOI] [PubMed] [Google Scholar]
- 35. Trajkovic K., Hsu C., Chiantia S., Rajendran L., Wenzel D., Wieland F., Schwille P., Brügger B., and Simons M. (2008) Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247 [DOI] [PubMed] [Google Scholar]
- 36. Saborio G. P., Permanne B., and Soto C. (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411, 810–813 [DOI] [PubMed] [Google Scholar]
- 37. Mollenhauer H. H., Morre D. J., and Rowe L. D. (1990) Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity. Biochim. Biophys. Acta 7, 225–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. LeBlanc A. C., and Goodyer C. G. (1999) Role of endoplasmic reticulum, endosomal-lysosomal compartments, and microtubules in amyloid precursor protein metabolism of human neurons. J. Neurochem. 72, 1832–1842 [DOI] [PubMed] [Google Scholar]
- 39. Bäck N., and Soinila S. (1996) Effect of monensin on secretory granules and basal β-endorphin secretion in the melanotroph of the rat pituitary. Histochem. J. 28, 591–597 [DOI] [PubMed] [Google Scholar]
- 40. Kuismanen E., Saraste J., and Pettersson R. F. (1985) Effect of monensin on the assembly of Uukuniemi virus in the Golgi complex. J. Virol. 55, 813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cancellotti E., Mahal S. P., Somerville R., Diack A., Brown D., Piccardo P., Weissmann C., and Manson J. C. (2013) Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J. 32, 756–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Collinge J., Sidle K. C., Meads J., Ironside J., and Hill A. F. (1996) Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383, 685–690 [DOI] [PubMed] [Google Scholar]
- 43. DeArmond S. J., Sánchez H., Yehiely F., Qiu Y., Ninchak-Casey A., Daggett V., Camerino A. P., Cayetano J., Rogers M., Groth D., Torchia M., Tremblay P., Scott M. R., Cohen F. E., and Prusiner S. B. (1997) Selective neuronal targeting in prion disease. Neuron 19, 1337–1348 [DOI] [PubMed] [Google Scholar]
- 44. Cancellotti E., Bradford B. M., Tuzi N. L., Hickey R. D., Brown D., Brown K. L., Barron R. M., Kisielewski D., Piccardo P., and Manson J. C. (2010) Glycosylation of PrPC determines timing of neuroinvasion and targeting in the brain following transmissible spongiform encephalopathy infection by a peripheral route. J. Virol. 84, 3464–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taraboulos A., Raeber A. J., Borchelt D. R., Serban D., and Prusiner S. B. (1992) Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell 3, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yuyama K., Sun H., Mitsutake S., and Igarashi Y. (2012) Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. J. Biol. Chem. 287, 10977–10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lehmann S., and Harris D. A. (1997) Blockade of glycosylation promotes acquisition of scrapie-like properties by the prion protein in cultured cells. J. Biol. Chem. 272, 21479–21487 [DOI] [PubMed] [Google Scholar]
- 48. Nitrini R., Rosemberg S., Passos-Bueno M. R., da Silva L. S., Iughetti P., Papadopoulos M., Carrilho P. M., Caramelli P., Albrecht S., Zatz M., and LeBlanc A. (1997) Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann. Neurol. 42, 138–146 [DOI] [PubMed] [Google Scholar]
- 49. Barron R. M., Campbell S. L., King D., Bellon A., Chapman K. E., Williamson R. A., and Manson J. C. (2007) High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. J. Biol. Chem. 282, 35878–35886 [DOI] [PubMed] [Google Scholar]
- 50. Lasmézas C. I., Deslys J. P., Robain O., Jaegly A., Beringue V., Peyrin J. M., Fournier J. G., Hauw J. J., Rossier J., and Dormont D. (1997) Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 275, 402–405 [DOI] [PubMed] [Google Scholar]
- 51. Manson J. C., Jamieson E., Baybutt H., Tuzi N. L., Barron R., McConnell I., Somerville R., Ironside J., Will R., Sy M. S., Melton D. W., Hope J., and Bostock C. (1999) A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J. 18, 6855–6864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bessen R. A., and Marsh R. F. (1992) Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 66, 2096–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]