

Abstract
Dysregulation of endoplasmic reticulum (ER) Ca2+ homeostasis triggers ER stress leading to the development of insulin resistance in obesity and diabetes. Impaired function of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) has emerged as a major contributor to ER stress. We pharmacologically activated SERCA2b in a genetic model of insulin resistance and type 2 diabetes (ob/ob mice) with a novel allosteric activator, CDN1163, which markedly lowered fasting blood glucose, improved glucose tolerance, and ameliorated hepatosteatosis but did not alter glucose levels or body weight in lean controls. Importantly, CDN1163-treated ob/ob mice maintained euglycemia comparable with that of lean mice for >6 weeks after cessation of CDN1163 administration. CDN1163-treated ob/ob mice showed a significant reduction in adipose tissue weight with no change in lean mass, assessed by magnetic resonance imaging. They also showed an increase in energy expenditure using indirect calorimetry, which was accompanied by increased expression of uncoupling protein 1 (UCP1) and UCP3 in brown adipose tissue. CDN1163 treatment significantly reduced the hepatic expression of genes involved in gluconeogenesis and lipogenesis, attenuated ER stress response and ER stress-induced apoptosis, and improved mitochondrial biogenesis, possibly through SERCA2-mediated activation of AMP-activated protein kinase pathway. The findings suggest that SERCA2b activation may hold promise as an effective therapy for type-2 diabetes and metabolic dysfunction.
Keywords: AMP-activated kinase (AMPK), diabetes, endoplasmic reticulum stress (ER stress), glucose metabolism, lipid metabolism, Ca2+ homeostasis, SERCA2b, hepatosteatosis, insulin sensitivity, mitochondria efficiency
Introduction
Obesity and insulin resistance are major causes of type 2 diabetes (T2D),2 which represents an enormous health burden to societies worldwide. T2D is now one of the most prevalent diseases globally and is the fourth leading cause of death in many developed countries (1). Endoplasmic reticulum stress (ER stress) has emerged as an important cause of the metabolic syndrome and T2D. ER stress and the unfolded protein response have now been described in organs playing key roles in metabolic homeostasis such as liver, pancreatic β-cells, adipose tissue, and hypothalamus in both obese and/or diabetic humans and rodents (2–5) and have recently emerged as key pathophysiological pathways triggering insulin resistance and T2D (4). Amelioration of ER stress through chemical chaperones has been demonstrated to be a promising pharmacological strategy for treatment of T2D (6–10).
The ER is the main storage site of intracellular Ca2+, and alterations in Ca2+ homeostasis have been demonstrated to trigger ER stress and activation of the unfolded protein response (11, 12). The sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps Ca2+ from the cytoplasm into the ER. Recent studies demonstrate that SERCA dysfunction leads to elevation of cytoplasmic calcium and triggers ER stress. SERCA2 activity and expression is diminished in islets (13), liver (2, 14), and heart (15) in animal models of obesity/diabetes, highlighting a potential pathological role for SERCA2 dysfunction and disturbed ER Ca2+ homeostasis in the development of metabolic abnormalities in insulin resistance and diabetes. Enhancing ER Ca2+-loading capacity by increasing SERCA2 function may ameliorate over-nutrition-induced ER stress and in turn improve metabolic control. Indeed, restoration of SERCA2b expression via short term gene transfer in the liver of obese mice reduces ER stress and improves glucose homeostasis (2, 14), whereas SERCA2b silencing has the opposite effects (2). Furthermore, inhibition of SERCA2 results in the activation of the ER stress response with concurrent activation of apoptotic pathways within the ER and the mitochondria.
Developing therapies that directly target defective endogenous SERCA enzyme and correct the concomitant Ca2+ imbalance in the ER may constitute a novel approach to treat diabetes and metabolic disorders. Here we assessed the metabolic effects of a small molecule SERCA2 activator, CDN1163, that acts directly on SERCA enzyme probably via allosteric mechanism to activate SERCA2's Ca2+-ATPase activity and improve Ca2+ homeostasis (16). We demonstrate that SERCA2b activation via CDN1163 attenuates ER stress, ameliorates mitochondrial efficiency, and improves glucose and lipid metabolism. The data obtained provide proof-of-concept that SERCA2 agonists may represent promising pharmacological agents to treat T2D and the metabolic syndrome.
Experimental Procedures
Animals
Male 8–10-week old ob/ob mice (n = 20) and lean ob/+ mice (n = 10) were obtained from The Jackson Laboratory (Bar Harbor, ME). Obese and lean mice were divided into four groups and treated with either vehicle (10% DMSO, 10% Tween 80 in 0.9% NaCl) or CDN1163 (50 mg/kg) intraperitoneally for 5 consecutive days (day 0 to day 4). Animals were obtained and handled as approved by the Mount Sinai Institutional Animal Care and Use Committee in accordance with the Principles of Laboratory Animal Care by the National Society for Medical research and the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication no. 86-23, revised 1996).
ER Stress Cell Viability Assay
Cells were grown in 96-well plates (n = 6) and exposed to CDN1163 (10 μm) or vehicle (DMSO) for 2 h followed by the addition of 200 μm H2O2 compared with untreated controls. H2O2 activates the unfolded protein response and is an inducer of ER stress-promoted apoptosis. After 16 h of incubation, cell viability was measured using Promega CellTiter-Glo® Luminescent Cell Viability Assay (catalogue no. G7570).
Glucose and Insulin Tolerance Tests
Ten-hour fasting blood glucose levels were measured in whole blood drawn from the tail vein using the OneTouch Ultra 2 Meter (LifeScan, Inc. New Brunswick, NJ). Both the glucose and insulin tolerance tests were performed after a 10-h fast and an additional 2-h CDN1163 injection, with baseline blood glucose measurement taken before the start of the test. For the glucose tolerance testing, d-glucose (Sigma) dissolved in 0.9% NaCl was delivered intraperitoneally at a dose of 1 g/kg. For the insulin tolerance testing, insulin (Humulin R; Lilly) was administered intraperitoneally at a dose of 1 IU/kg. Blood glucose was measured at the indicated time points.
Metabolic Chamber and Calorimetry Analysis
Animals (male mice 8–10 weeks old, n = 8) were placed in gas-tight metabolic cages individually for indirect calorimetry and measurement of food intake and locomotor activity (TSE Systems, Inc., Chesterfield, MO). Data collection started after the first 3 days of the acclimatization period and lasted for 4 days. Air flow rate in each chamber was 0.45 liters/min, which is in great excess of minimal requirements. Ad libitum intake of food was automatically monitored. The O2 and CO2 gas exchange was measured with a sampling rate of 1 min, and the chamber temperature was adjusted to the ambient temperature. Physical locomotor activity was assessed concurrently using a one-dimensional infrared light beam system installed on the cage bottom.
Magnetic Resonance Imaging
Live animals were analyzed for total body fat, lean tissue, and body water content using an EchoMRI quantitative magnetic resonance system (Echo Medical Systems, Houston, TX).
Metabolic Parameters
Plasma levels of insulin, triglyceride (TG, catalogue no.10010303), free fatty acids (FFA, catalogue no.700310), cholesterol (catalogue no. 10007640), and malondialdehyde (MDA, catalogue no.10009055) were assayed with commercially available kits (Cayman Chemicals, Ann Arbor, MI). Hepatic TG and MDA were extracted using the chloroform-methanol method and lysed in radioimmune precipitation assay buffer and quantified using the corresponding assay kits.
Clinical Chemistry
A separate cohort of C57BL/6 mice (10 weeks old, n = 10–20) were either untreated or treated with vehicle or 50 mg/kg CDN1163 as indicated above for a total of 6 weeks. Animals were euthanized, and terminal blood samples were collected (∼0.5 ml); sera were analyzed for a panel of clinical chemistry parameters.
Histology of Liver Sections
Frozen specimens were sectioned (5 μm), fixed with 4% paraformaldehyde, and stained with hematoxylin/eosin (Sigma) for histological examination. For Oil Red O staining, frozen liver sections were fixed with 4% paraformaldehyde, rinsed with 100% propylene glycol (PEG) and stained with fresh Oil Red O (Sigma) dissolved in PEG for 30 min, rinsed with 80% PEG, and then counterstained with hematoxylin. To detect apoptotic cells, TUNEL assays were performed on liver sections (8 μm) fixed in 10% neutral-buffered formalin and stained with the ApopTag staining kit (catalogue no. S7165, EMD Millipore, Billerica MA) according to the manufacturer's instructions. Tissue sections were then counterstained with DAPI stain (catalogue no. H1200, Vector Laboratories, Burlingame, CA) and viewed with a micro-optics microscope (Carl Zeiss, Oberkochen, Germany) equipped with filter sets for rhodamine and DAPI staining. To quantify apoptosis, six to seven randomly selected microscopic fields per section zone of each liver sample were examined. The percentage of apoptotic cells was determined by counting the total number of nuclei (DAPI) and TUNEL-positive nuclei (apoptotic cells).
Real-time Quantitative PCR
RNA was isolated from liver tissues using TRIzol (Invitrogen, ThermoFisher), and cDNA was synthesized using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, ThermoFisher). Real-time PCR was performed with the iTaq Fast SYBR Green Supermix with ROX (catalogue no. 172-5100, Bio-Rad) in a 7500 Real-time PCR (Applied Biosystems, ThermoFisher). Gene expression was normalized to 18S. Data obtained by qPCR were analyzed by the ΔΔCT method. Primers used in this study are available upon request.
Western Blotting
Isolated liver tissues were homogenized in radioimmune precipitation assay buffer containing protease and phosphatase inhibitors (Roche Applied Science). 30–50 μg of protein were applied to SDS-PAGE and transferred onto the nitrocellulose membrane. Antibodies used were phospho- or total against protein kinase RNA-like ER kinase (PERK), eukaryotic initiation factor 2α (eIF2α), inositol-requiring kinase 1α (IRE1α), JNK, Bip, activating transcription factor 4 (ATF4), C/EBP homologous protein (CHOP), Bax, caspase 3, caspase 12, peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α), and AMPK (catalogue nos. 5683, 3398/9722, 3294, 4668P/9252, 3177, 11815S, 2895S, 9665S, 2202, 2178, and 2535S/2603S, respectively; all from Cell Signaling Technology, Danvers, MA), ATF6α (catalogue no. IMG-273, Novus Biologicals, Littleton, CO), SERCA (catalogue no. sc-8094, Santa Cruz biotechnology), ATP5A, UQCRC2, SDHB, MTCO1, and NADH dehydrogenase (ubiquinone) 1 β subcomplex subunit 8 (NDUFB8) (catalogue no. MS604, MitoSciences, Eugene, OR). GAPDH (catalogue no. G8795, Sigma) expression verified protein loading.
XBP1 Splicing
To assess the splicing of XBP1, PCR primers were designed to flank the 26-bp splicing sequence of mouse XBP1 (forward 5′-AGTTAAGAACACGCTTGGGAAT-3′ and reverse-5′-AAGATGTTCTGGGGAGGTGAC-3′). L32 was used as a loading control (forward (5′-GAGCAACAAGAAAACCAAGCA-3′) and reverse (5′-TGCACACAAGCCATCTACTCA-3′)). The PCR reaction was performed as follows: 94 °C for 3 min; 32 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s; 72 °C for 5 min. PCR products were separated by electrophoresis on a 2% agarose gel.
Mitochondrial DNA Copy Number
The mitochondrial (mtDNA) and nuclear DNA contents were determined by real-time quantitative PCR using specific primers for the mtDNA genes (mtCytB, mtCOX1, mtND1) and the nuclear H19 gene. The mtDNA copy number is calculated from the difference in threshold cycle numbers of mtDNA and nuclear DNA.
Ca2+ Uptake
Ca2+ transport activity by liver microsomal preparations was measured using the 45Ca2+ uptake assay. Briefly, fresh liver tissues were rinsed in PBS and homogenized in a lysis buffer containing 0.25 m sucrose, 2 mm Tris, pH 7.4, and 1 mm DTT and EDTA-free protease inhibitor. The microsomes were obtained after a series of centrifugation (800 × g for 15 min, 6000 × g for 15 min, 100,000 × g for 1 h) and then resuspended in 250 mm sucrose. The reaction medium contained 150 μg of protein and 100 mm KCl, 30 mm imidazole hydrochloride, 5 mm NaN3, 5 mm MgCl2, 5 mm K2C2O4, 50 μm of CaCl2, 1 μm Ruthenium Red, and 1 μCi/μl 45Ca2+ (PerkinElmer Life Sciences) and was preincubated for 5 min at 37 °C. The uptake process was started by the addition of 5 mm ATP to the reaction mixture for 10 min at 37 °C and stopped by the addition of 0.15 m KCl, 1 mm LaCl3. The radioactive mixture was then filtered through a 0.22-μm Millipore glass fiber filters and washed with distilled water, and radioactivity on the filters was measured by liquid scintillation counting. Background counts, obtained in aliquots before the addition of ATP, were subtracted from subsequent counts. The amount of SERCA independent calcium transport was quantified in the presence of 10 μm thapsigargin and subtracted from the calculation and normalized to total protein content measured by Micro BCA protein assay kit (Pierce).
Ca2+-ATPase Activity and ATP Content
Ca2+-dependent ATPase activity of SERCA2 was assessed by a colorimetric ATPase assay kit (catalogue no. 601-0120, Novus Biologicals, Littleton, CO). Liver homogenates (50 μg) were preincubated with the ionophore A23187 and EGTA, final concentration 1 μg/ml each, for 5 min to prevent a buildup of Ca2+ inside the vesicles that might inhibit the Ca2+-ATPase activity. The activity rates were read at 650 nm and normalized to total protein content measured by MicroBCA protein assay (Pierce). Total cellular ATP levels were assayed using an ATP determination kit (catalogue no. A22066, Invitrogen, ThermoFisher). The luminescence was measured at 560 nm.
Statistics
Data are expressed as the means ± S.E. The significance of the differences in mean values was evaluated by using Student's t test or one way analysis of variance where appropriate from at least three independent experiments. Values of p < 0.05 were considered to be statistically significant.
Results
CDN1163 Increases SERCA2 Ca2+-ATPase Activity, Decreases ER Stress-induced Cell Death in Vitro and Improves Liver Ca2+ Transport Activity in ob/ob Mice in Vivo
CDN1163 is a small molecular allosteric SERCA2 activator that directly binds to and perturbs SERCA2 structure leading to its activation (16). The chemical structure of the CDN1163 compound is shown in Fig. 1A. Here we have tested if CDN1163 increases SERCA2 function. Indeed, CDN1163 dose-dependently increased the Vmax of SERCA2 Ca2+-ATPase activity in ER microsomes (Fig. 1B). We then tested the effects of CDN1163 on the accumulation of ER Ca2+ in vitro as SERCA2 regulates Ca2+ transport and uptake into the ER. CDN1163 significantly enhanced Ca2+ uptake into the ER (Fig. 1C). As a control, we also overexpressed SERCA2b via adenoviral gene transfer and observed a similar increase in ER Ca2+ (Fig. 1C). Next we assessed the ability of CDN1163 to rescue cells from ER stress-induced cell death. To this end, we treated HEK cells with hydrogen peroxide (H2O2), a known inducer of ER stress, in the presence or absence of CDN1163 and assessed cell viability. CDN1163 largely attenuated H2O2-stimulated cell death (Fig. 1D), indicating that at least in this commonly employed model of ER stress CDN1163 is able to rescue cells from cell death.
FIGURE 1.

CDN1163 is an allosteric activator of SERCA2b and enhances Ca2+ transport activity in the liver of obese mouse. A, chemical structure of CDN1163 SERCA activator (patents on file). B, the Ca2+-ATPase Vmax (limiting activity at 10 μm Ca2+) was measured in ER vesicles from liver tissue after 20 min of incubation in the presence of various concentrations of CDN1163 as indicated (n = 6). C, effect of CDN1163 on ER Ca2+ accumulation in HEK cells that were either transfected with an adenovirus encoding Serca2b (Ad.Serca2b, multiplicity of infection 50) versus control or treated with CDN1163 (10 μm) versus vehicle from a minimum of three determinations. A representative of Western blot of Serca2b expression is shown. *, p < 0.05; and **, p < 0.01 versus control. D, CDN1163 rescues cells from ER stress-induced cell death. ER stress-induced cell death was assessed in HEK cells either untreated or treated with vehicle (DMSO) or with hydrogen peroxide (H2O2) in the presence or absence of 10 μm CDN1163 using CellTiter-Glo® luminescent cell viability assay. The data are expressed as the mean ± S.E. from at least three determinations. *, p < 0.05 versus H2O2; **, p < 0.01versus vehicle. E, SERCA2b Ca2+ transport activity in vivo. Ca2+-ATPase activity (left) and the rate of Ca2+ uptake (right) were determined in liver ER microsomes purified from vehicle-treated obese (Ob) or CDN1163-treated obese mice (Ob+CDN) after treatment with 50 mg/kg CDN1163 for 5 days (n = 5 animals/group). Data are expressed as the means ± S.E. #, p < 0.05, obese versus CDN1163-treated obese mice.
We next determined if CDN1163 increases Ca2+-ATPase activity in vivo and concurrently stimulates ER Ca2+ transport in ob/ob mouse livers. Indeed, CDN1163 increased Ca2+-ATPase activity and rates of Ca2+ uptake (Fig. 1E) in liver ER microsomes purified from ob/ob mice treated with CDN1163 compared with vehicle-treated animals. These results show that CDN1163 can target and activate existing SERCA2 pumps in the ER and correct the ER Ca2+ imbalance associated with SERCA2b dysfunction, a fundamental abnormality seen in obesity and insulin resistance conditions.
CDN1163 Reduces Blood Glucose Levels and Improves Metabolic Parameters in ob/ob Mice
Short term gene transfer of SERCA2b in the liver of obese mice has recently been shown to regulate glucose homeostasis (14). Our observation that CDN1163 increases SERCA2 activity and regulates ER Ca2+ content similar to SERCA2b overexpression in vitro prompted us to examine whether pharmacological activation of SERCA2b with CDN1163 would improve glucose homeostasis in ob/ob mice. To this end, 10-week-old ob/ob male mice were intraperitoneally injected with 300 μl of 50 mg/kg CDN1163 or vehicle once a day for a total of 5 days. Fasting glucose was measured at baseline and 10 h after CDN1163 administration. One day after commencement of treatment, fasting glucose levels of CDN1163-treated ob/ob mice were reduced to 302.0 ± 39.7 mg/dl compared with vehicle-treated obese mice (438.4 ± 30.4 mg/dl) (Fig. 2A). Fasting glucose levels continued to fall after CDN1163 administration, and at day 50, 6 weeks after the last administered dose, CDN1163-treated mice exhibited sustained lower glucose levels (129.6 ± 6.38 mg/dl) compared with controls (365.4 ± 25.16 mg/dl) (Fig. 2B). Importantly, the reduction in glucose levels were not due to CDN1163-induced suppression in food consumption as there were no differences in caloric intake between treated and untreated mice (Fig. 2D), and there was no change in body weight in CDN1163-treated mice (Fig. 2C). Furthermore, treatment with CDN1163 was associated with a 16% decrease in adipose tissue weight with no change in lean mass (Table 1), also confirmed by magnetic resonance imaging analysis (Fig. 2E).
FIGURE 2.

CDN1163 improves glucose homeostasis and metabolic parameters in ob/ob mice. CDN1163 was administrated for 5 consecutive days (D0 to D4). A, fasting blood glucose levels after first injection of CDN1163 at day 1 (D1). Fasting blood glucose levels (B) and body weight (C) were determined weekly until day 50. D, average food intake in lean, vehicle-treated obese (Ob) and CDN1163-treated obese (Ob+CDN) mice (n = 10/group). E, body composition analyzed for 7 days in a separate cohort (n = 8) before and after 5 days of CDN1163 treatment in vehicle-treated obese (Ob) and CDN1163-treated obese (Ob+CDN) mice. Indirect calorimetry conducted for the last 4 days was assessed oxygen consumption (F), respiratory exchange ratio (RER) (G), and physical activity and caloric intake (H). J, mRNA expression of UCP1, UCP2, and UCP3 in brown tissue in lean, vehicle-treated obese (Ob) and CDN1163-treated obese (Ob+CDN) mice (n = 5). I, glucose levels and body weight after CDN1163 treatment of lean mice. CDN1163 was administrated for 5 consecutive days (D0 to D4). Fasting blood glucose levels and body weight were determined weekly until day 30 in lean plus vehicle (LN+Veh) and lean plus treatment (LN+CDN) mice (n = 6/group). Body weight values for day 1 (D1) and day 31 (D31) are shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001, lean versus obese; #, p < 0.05; ##, p < 0.01; ###, p < 0.001, obese versus CDN1163-treated obese mice.
TABLE 1.
Mouse characterization of metabolic parameters at end of study
Data are the means ± S.E. n = 6 per group. Data were collected at end of study (i.e. day 50).
| Parameters | Lean | Obese + vehicle | Obese + CDN1163 |
|---|---|---|---|
| Glucose (mg/dl) | 85.2 ± 2.60 | 365.4 ± 25.16a | 129.6 ± 6.38b |
| Insulin (ng/ml) | 1.08 ± 0.03 | 7.70 ± 0.89c | 6.30 ± 0.28d |
| Food intake (g/day) | 4.78 ± 0.41 | 5.45 ± 0.26e | 5.18 ± 0.18f |
| Body weight (g) | 27.2 ± 0.73 | 57.8 ± 1.32a | 55.3 ± 0.60 |
| Liver weight (g) | 1.06 ± 0.18 | 3.13 ± 0.19a | 2.69 ± 0.26f |
| Liver/body (%) | 2.96 ± 0.12 | 5.61 ± 0.33e | 4.57 ± 0.26 |
| Epididymal fat (g) | 0.52 ± 0.07 | 3.89 ± 0.10a | 3.26 ± 0.07f |
| Plasma TG (mg/dl) | 96.7 ± 7.95 | 371.7 ± 37.68e | 188.3 ± 20.28f |
| Liver TG (mg/g) | 31.5 ± 2.75 | 208.9 ± 16.12e | 147.3 ± 26.47f |
| Plasma free fatty acids (mmol/liter) | 1.65 ± 0.09 | 1.90 ± 0.17 | 1.76 ± 0.08 |
| Plasma cholesterol (mg/dl) | 71.65 ± 8.67 | 180.73 ± 11.64e | 118.20 ± 0.08f |
| Plasma MDA (μm) | 1.80 ± 0.37 | 6.83 ± 2.20 | 3.50 ± 3.97f |
| Liver MDA (μmol/g) | 6.88 ± 2.82 | 13.92 ± 1.11e | 8.89 ± 1.39 |
a p < 0.001 lean vs. obese.
b p < 0.001 obese vs. obese + CDN1163.
c p < 0.01 lean vs. obese.
d p < 0.01, obese vs. obese + CDN1163.
e p < 0.05, lean vs. obese.
f p < 0.05, obese vs. obese + CDN1163.
We next measured the metabolic rate of CDN1163-treated ob/ob mice using indirect calorimetry. Oxygen consumption (VO2) in CDN1163-treated mice was significantly increased in comparison to vehicle-treated mice (Fig. 2F) although the respiratory exchange ratio (RER) was similar (Fig. 2G), suggesting that the ratio of carbohydrate and fatty acids used for β-oxidation was not altered. Of note, metabolic chamber assessment showed that both groups of ob/ob mice had comparable physical activity levels and similar caloric intakes (Fig. 2H). The increase in energy expenditure was accompanied by increased expression of uncoupling protein 1 (UCP1) and UCP3 in brown adipose tissue (Fig. 2J), indicative of increased thermogenesis in brown adipose tissue, which may account for the reduction in fat mass and increase in energy expenditure. Furthermore, CDN1163 treatment of normal lean mice did not alter either fasting glucose level or body weight (Fig. 2I), indicating that pharmacological activation of SERCA2b is unlikely to induce hypoglycemia or impair energy homeostasis in metabolically healthy animals. We have also tested the effects of CDN1163 in a rat model of obesity and diabetes, ZDF. Similar to ob/ob mice, CDN1163-treated ZDF rats displayed significant reduction in blood glucose compared with vehicle treatment with no apparent changes in body weight in both groups (not shown). CDN1163 also had no effect on blood glucose or body weight in lean control rats (not shown).
To further analyze the effect of CDN1163 on carbohydrate metabolism, we performed glucose tolerance testing on day 8. CDN1163-treated mice exhibited improvement in glucose clearance as demonstrated by a 21.8% reduction in the area under the curve (AUC) compared with vehicle-treated mice (Fig. 3A). Administration of CDN1163 resulted in a moderate but significant reduction in the circulating levels of insulin, which could be a consequence of the lower fasting glucose levels (Fig. 3C). Insulin tolerance testing on day 11 indicated that insulin sensitivity did not significantly change (Fig. 3B) despite the finding that administration of CDN1163 resulted in a moderate reduction in the circulating levels of insulin, which again is likely a consequence of the lower fasting glucose levels. We next explored whether CDN1163 treatment changed the phosphorylation state of important insulin signaling molecules in liver tissues after an acute insulin bolus. Consistent with the lack of improvement in insulin sensitivity, insulin-stimulated Akt phosphorylation was not different between CDN1163-treated and non-treated samples (Fig. 3D). Thus, although key aspects of the metabolic syndrome were improved through SERCA2b activation, these metabolic improvements appear to be mostly insulin-independent.
FIGURE 3.

CDN1163 increases glucose tolerance in ob/ob mice. A, intraperitoneal glucose tolerance test (GTT) assessed on day 8 in lean, vehicle-treated obese (Ob) and CDN1163-treated obese (Ob+CDN) mice (n = 10/group); glucose was measured at times shown after 1 g/kg of glucose injection and calculated as the area under the curve (AUC). B, the intraperitoneal insulin tolerance test was assessed on day 11 after 1 IU/kg of insulin injection 2 h after CDN injection, and glucose clearance is expressed as % reduction from basal levels, and (B) plasma insulin levels at end of study (day 50) are shown for lean and vehicle- and CDN1163-treated obese mice (n = 10) (C). D, representative from at least three experiments of Western blot analysis of insulin-stimulated Akt phosphorylation at serine 473 (pS-Akt) and threonine 308 (pT-Akt) in the liver after an acute insulin bolus (36.3 μg/ml in 0.9% saline (1 units/kg, assuming potency of 27.5 units/mg)). Protein loading was verified with total Akt and GAPDH. Also shown is Western blot liver SERCA2b expression in the three different groups of mice with GAPDH as a loading control. E, quantitative real-time PCR analysis of genes involved in gluconeogenesis in the liver in vehicle-treated obese (Ob) and CDN1163-treated (Ob+CDN) mice. Data are expressed as the means ± S.E. from at least 3–5 determinations. *, p < 0.05; **, p < 0.01; ***, p < 0.001, lean versus obese; #, p < 0.05 and ###, p < 0.001, obese versus CDN1163-treated obese mice. Data in C–E were performed at the end of the study (i.e. day 50). G6Pase, glucose-6-phosphatase.
CDN1163 Reduces Gluconeogenic Gene Expression in ob/ob Mice
Hepatic gluconeogenesis is commonly increased in T2D and contributes to the unrestrained hepatic glucose production and glucose intolerance (17). Thus, the lowered fasting blood glucose levels could be reflective of lower hepatic gluconeogenesis. We, therefore, examined the mRNA levels of key enzymes involved in hepatic gluconeogenesis such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase. Indeed glucose-6-phosphatase (G6Pase) and PEPCK expression was significantly reduced in the livers of CDN1163-treated ob/ob mice compared with vehicle-treated mice (Fig. 3E). Furthermore, the level of the transcription factor, hepatocyte nuclear factor 4α (HNF4α), which regulates G6Pase and PEPCK expression, was also down-regulated in CDN1163-treated mice (Fig. 3E).
CDN1163 Reverses Hepatic Steatosis in ob/ob Mice
Lipid accumulation in the liver is a consequence of an imbalance between hepatic triglyceride production, triglyceride utilization (mitochondrial β-oxidation), and triglyceride mobilization (VLDL secretion). Excessive accumulation of hepatic lipids results in nonalcoholic fatty liver diseases and is closely associated with insulin resistance in humans (18) and represents a major adverse health consequence of the metabolic syndrome (19, 20). We examined hepatic lipid accumulation by histological examination of liver sections. H&E and Oil Red O staining showed marked decreases both in the number and size of lipid droplets in CDN1163-treated liver sections (Fig. 4A). This was confirmed by biochemical analyses that demonstrated significantly reduced hepatic and plasma triglycerides as well as cholesterol levels in CDN1163-treated ob/ob mice (Table 1). Furthermore, there is no suggestion of toxicity with CDN1163. A panel of serum markers for hepatotoxicity (including markers of synthetic and clearance activities as well as hepatocellular lysis), renal toxicity, metabolic changes, and electrolyte abnormalities revealed no CDN1163-related pathology (Table 2).
FIGURE 4.

CDN1163 reduces lipid accumulation and decreases lipogenesis in obese mice livers. A, lipid accumulation measured with H&E (top) and Oil red O staining (bottom) in the liver of lean, vehicle-treated obese (Obese) and CDN1163-treated obese (Obese+CDN) mice (n = 5). qRT-PCR analysis of genes involved in liver de novo lipogenesis (B) and lipid oxidation (C) in obese (Ob) versus obese plus CDN1163 (Ob+CDN) mice. Bar, 50 μm. Data are expressed as the means ± S.E. (n = 5). #, p < 0.05; ##, p < 0.01; ###, p < 0.001, obese versus CDN1163-treated obese mice. All determinations were performed on harvested liver tissues at the end of the study (i.e. day 50).
TABLE 2.
Clinical chemistry and toxicity parameters
SGPT, serum glutamic-pyruvic transaminase; ALT, alanine transaminase; SGOT, serum glutamic-oxaloacetic transaminase; AST, aspartate transaminase; BUN, blood urea nitrogen; ND, not detected.
| Group 1: untreated | Group 2: vehicle | Group 3: CDN1163 | |
|---|---|---|---|
| Hepatic: synthetic | |||
| Albumin (g/dl) | 3.04 ± 0.05 | 3.20 ± 0.10 | 2.85 ± 0.25 |
| Globulin (g/dl) | 2.34 ± 0.07 | 2.64 ± 0.05 | 2.65 ± 0.05 |
| A/G ratio | 1.30 ± 0.03 | 1.24 ± 0.05 | 1.10 ± 0.10 |
| Total protein (g/dl) | 5.38 ± 0.11 | 5.84 ± 0.10 | 5.28 ± 0.21 |
| Hepatic: lysis | |||
| Alkaline phosphatase (units/liter) | 88.4 ± 3.6 | 92.0 ± 8.8 | 78.4 ± 10.2 |
| SGPT (ALT) (units/liter) | 30.4 ± 4.5 | 25.2 ± 2.2 | 27.4 ± 4.6 |
| SGOT (AST) (units/liter) | 61.6 ± 10.6 | 77.4 ± 12.3 | 70.8 ± 6.8 |
| Creatine phosphokinase (units/liter) | 119.4 ± 46.4 | 223.6 ± 54.0 | 186.6 ± 33.3 |
| Hepatic: clearance | |||
| Total bilirubin (mg/dl) | 0.18 ± 0.02 | 0.15 ± 0.05 | 0.50 ± 0.06 |
| Direct bilirubin (mg/dl) | 0.02 ± 0.02 | 0.00 ± 0.00 | ND |
| Indirect bilirubin (mg/dl) | 0.16 ± 0.02 | 0.15 ± 0.05 | ND |
| Renal: clearance | |||
| BUN (mg/dl) | 16.6 ± 0.6 | 20.2 ± 1.2 | 19.0 ± 0.9 |
| Creatinine (mg/dl) | 0.20 ± 0.00 | 0.20 ± 0.00 | 0.20 ± 0.01 |
| B/C ratio | 83.0 ± 3.0 | 101.0 ± 0.0 | 95.0 ± 1.5 |
| Metabolic | |||
| Cholesterol (mg/dl) | 102.6 ± 8.1 | 112.8 ± 6.3 | 105.0 ± 7.0 |
| Glucose (mg/dl) | 164.69 ± 21.75 | 173.32 ± 8.25 | 163.83 ± 21.33 |
| Bicarbonate (mg/dl) | 23.2 ± 0.8 | 21.4 ± 0.9 | 22.0 ± 1.2 |
| Electrolyte | |||
| Calcium (mmol/liter) | 9.84 ± 0.12 | 9.58 ± 0.15 | 9.10 ± 0.38 |
| Phosphorus (mmol/liter) | 8.54 ± 0.52 | 7.52 ± 0.44 | 7.52 ± 0.29 |
| Chloride (mmol/liter) | 110.8 ± 0.2 | 107.0 ± 1.0 | 110.3 ± 071 |
| Potassium (mmol/liter) | 7.64 ± 0.22 | 6.80 ± 0.00 | 12.31 ± 0.81 |
| Sodium (mmol/liter) | 151.6 ± 0.7 | 151.0 ± 0.0 | 147 ± 1.08 |
To dissect the underlying molecular mechanisms by which CDN1163 treatment reduced hepatic steatosis, we evaluated the hepatic expression of key genes involved in de novo lipogenesis. Stearoyl-CoA desaturase-1 (SCD1), diacylglycerol acyltransferase-2 (DGAT2), fatty acid synthase (FASn), and acetyl-CoA carboxylase-1 and -2 (ACC1 and ACC2) as well as a key transcription factor regulating these genes, sterol regulatory element binding protein-1c (SREBP1c), were appreciably down-regulated in CDN1163-treated ob/ob mice (Fig. 4B). Furthermore, we also evaluated the expression of a number of genes involved in lipid oxidation such as fatty acid translocase (FAT), carnitine palmitoyltransferase-1β and -2 (CPT1β and CPT2β), peroxisome proliferator-activated receptor-α (PPARα), and malonyl coenzyme A decarboxylase (MCD). Although the expression of these genes was not statistically significant, they tend to be up-regulated in the livers of CDN1163-treated obese mice (Fig. 4C). In aggregate, these data suggest that CDN1163 decreases hepatic lipid accumulation by primarily decreasing de novo lipogenesis.
CDN1163 Inhibits ER Stress and ER Stress-induced Apoptosis in ob/ob Mice
Inadequate or depleted ER Ca2+ content and other perturbations such as decreased expression of chaperone proteins, oxidative stress, and redox imbalance lead to unfolded protein response and ER stress (12). ER stress induces the dissociation of glucose regulated protein 78 (GRP78/BiP) from three ER stress signaling mediators: PERK, IRE1α, and ATF6. PERK phosphorylates and activates eIF2α and increases the translation of transcription factor ATF4 and its downstream target CHOP. A decreased phosphorylation of PERK or elF2α is indicative of alleviation of ER stress. IRE1α having kinase and endonuclease activities phosphorylates JNK and splices XBP1 mRNA, respectively (21).
We assessed the effect of CDN1163-mediated SERCA2b activation on ER stress by evaluating the expression and activity of the three ER stress master regulators PERK, IREα, and ATF6 and their downstream targets by Western blot using phospho-specific antibodies. Obesity-induced phosphorylation of PERK (p-PERK) and its downstream target elF2α (p-elF2α) and the expression of the pro-apoptotic transcription factor CHOP was significantly decreased in the livers of CDN1163-treated obese mice (Fig. 5A). Likewise, CDN1163 treatment markedly reduced the obesity-induced phosphorylation of IRE1α and its downstream target JNK, a known mediator of apoptosis (Fig. 5A). This was further validated by the fact that XBP1 splicing was reduced in the livers of CDN1163-treated mice (Fig. 5B), largely suggesting that CDN1163 abrogates ER stress and ER stress-induced apoptosis. Examination of expression of ER protein chaperones after CDN1163 treatment revealed no changes in the mRNA expression levels of multiple ER chaperones shown in Fig. 5C, indicating that amelioration of ER stress is due to the inactivation of kinases mediating ER stress signaling rather than the up-regulation of chaperones, which aid protein folding. These findings support the hypothesis that SERCA2b activation improved ER homeostasis, leading to a reduction in the level of the ER stress.
FIGURE 5.

CDN1163 relieves ER stress and attenuates apoptosis in the liver of ob/ob mice. Shown are Western blot analyses and densitometry quantification of ER stress markers (A) and qRT-PCR analyses of ER chaperones in vehicle-treated obese (Ob) versus CDN1163-treated obese (Ob+CDN) mice (C). B, determination and quantification of spliced (s) and unspliced (u) XBP1 in lean (L), vehicle-treated obese (O), and CDN1163-treated obese (OC) mice. D, TUNEL staining of apoptotic cells in liver tissues from vehicle-treated obese (ob/ob-vehicle) versus CDN1163-treated obese (ob/ob-CDN1163) mice (bar = 50 μm) and quantification of apoptosis shown as a percentage of apoptotic nuclei (red by TUNEL) versus total nuclei (blue by DAPI) (n = 5). E, representative Western blot analyses and quantification of apoptosis markers in liver tissues from vehicle-treated obese (Ob) versus CDN1163-treated obese (Ob+CDN) mice. Data are expressed as the means ± S.E. from at least 3–5 determinations. **, p < 0.01, lean versus obese; #, p < 0.05, ##, p < 0.01, and *, p < 0.05, obese versus CDN1163-treated obese mice. All determinations were performed on harvested liver tissues at the end of the study (i.e. day 50).
To further evaluate ER Stress-induced apoptosis in the liver directly, we used the TUNEL assay on liver tissue sections which showed a greater reduction in TUNEL-positive cells with CDN1163 treatment (3.55% CDN versus 11.88% vehicle, p < 0.05) (Fig. 5D). To elucidate the mechanism underlying the anti-apoptotic effect of CDN1163, we performed Western blot analysis of the apoptosis marker proteins Bax, caspase 12 and caspase 3. CDN1163-treated obese livers showed a decrease in Bax expression and in the protein levels of cleaved caspase 3 (Casp3) and caspase 12 (Casp12), active mitochondrial elements involved in the apoptotic pathway, compared with vehicle-treated samples (Fig. 5E). Combined with the down-regulation of CHOP and dephosphorylation of JNK—positive indicators of apoptosis, these results demonstrate that CDN1163 relieved ER stress via ameliorating the pro-apoptotic pattern through the PERK/eIF2/CHOP and IRE1α/JNK/XBP1 pathways.
CDN1163 Improves Mitochondrial Efficiency in ob/ob Mice
ER stress and mitochondrial dysfunction are closely associated and constitute two major defects of T2D (22). Because the ER and mitochondria both store and functionally depend on Ca2+, recent evidence indicates that disruption of Ca2+ homeostasis, as it occurs in diabetic conditions, induces ER stress and mitochondrial dysfunction (23–26). Notably, mitochondrial DNA copy number, mitochondrial mass, and mitochondrial activity are all decreased in ob/ob mouse model (27). Our observation that CDN1163 treatment relieved ER stress and attenuated ER stress-induced cell death prompted us to explore whether CDN1163 also confers beneficial effects on obese mice-compromised mitochondrial function. CDN1163-treated liver of obese mice showed increased expression of molecules known to control mitochondrial biogenesis such as peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) and estrogen-related receptor α (ERRα), molecules involved in transcription such as nuclear respiratory factor 1 (NRF1), and molecules involved in mitochondrial DNA replication/translation such as mitochondrial transcription factor A (TFAm) (Fig. 6A) along with increased mitochondrial DNA content (mtDNA) as evidenced by significant increases in cytochrome B (mtCytB), cytochrome c oxidase-1 (mtCOX1), and NADH-ubiquinone oxidoreductase-1 (mtND1) expression levels (Fig. 6B), consistent with CDN1163-induced up-regulation of hepatic mitochondrial biogenesis and replication control markers (Fig. 6A) and supporting a role for CDN1163 in reversing obesity/diabetes-induced hepatic mitochondrial defect.
FIGURE 6.

CDN1163 improves mitochondrial efficiency in the liver of ob/ob mice. All determinations were performed on harvested liver tissues at the end of the study (i.e. day 50). qRT-PCR analyses of genes involved in mitochondrial biosynthesis (A) and mitochondrial DNA contents (B). C, Western blot analyses and quantification of proteins involved in OXPHOS and qRT-PCR analyses of OXPHOS genes (D), ATP content (E), and antioxidant enzymes (G) in vehicle (Ob)- versus CDN1163-treated obese (Ob+CDN) mice. F, AMPK phosphorylation (pAMPK) and densitometry (normalized to total AMPK (tAMPK)) in liver samples from lean, vehicle-treated (Ob) versus CDN1163-treated obese (Ob-CDN) mice. GAPDH is a loading control. Data are expressed as the means ± S.E. from at least three experiments; #, p < 0.05 and ##, p < 0.01 obese versus CDN-treated obese mice; **, p < 0.01 versus lean and ***, p < 0.001 CDN1163 versus vehicle.
To further characterize the impact of CDN1163 treatment on mitochondrial efficiency in hepatic cells, we analyzed the relative levels of mitochondrial-encoded proteins involved in the OXPHOS pathway by Western blotting and quantitative real-time PCR. Protein levels of ATP synthase, H+ transporting, mitochondrial F1 complex, α subunit (Atp5A) of complex V, ubiquinol cytochrome c reductase core protein 2 (UQCRC2) of complex III, succinate dehydrogenase (ubiquinone) iron sulfur subunit (SDHB) of complex II and PGC1α were increased in mitochondria from CDN1163-treated ob/ob mice compared with ob/ob vehicle-treated (Fig. 6C). Likewise, CDN1163 treatment up-regulated the mRNA levels of uncoupling protein 2 (UCP2), UCP3, and COX4 encoding subunit 4 of cytochrome c oxidase (Fig. 6D). However, the protein levels of mitochondrially encoded cytochrome c oxidase I (MTCO1) of complex IV and NADH dehydrogenase (ubiquinone) 1 β subcomplex subunit 8 (NDUFB8), a representative of complex I (Fig. 6C), or the mRNA expression levels of NADH dehydrogenase (ubiquinone) flavoprotein 1 (NDUFV1) and NADH dehydrogenase (ubiquinone) 1α subcomplex subunit 9 (NDUFA9) were unchanged (Fig. 6D). We also determined whether ATP levels were altered by CDN1163 treatment as it stimulated mitochondrial capacity. As a result, one would have expected ATP to be increased, but total ATP contents were not different between the groups (Fig. 6E); however, elevated energy supply due to increased mitochondrial activity might have been offset by energy expenditure due to increased SERCA2b activity induced by CDN1163.
We next attempted to determine the potential molecular mechanism(s) underlying CDN1163-SERCA2b stimulation of mitochondrial biogenesis. We provide evidence that this process appears to be regulated in part by AMP-activated protein kinase (AMPK)-dependent pathway possibly through activation of PGC1α given the implication of this latter molecule in the direct regulation of mitochondrial biogenesis (28). CDN1163 reversed and increased obesity-compromised phosphorylation of AMPK (Fig. 6F) in parallel with the up-regulation of PGC1α reported above (Fig. 6, A and C), suggesting that SERCA2 activation may regulate PGC1α and by extension promote mitochondrial biogenesis through modulation of AMPK signaling.
The apparent improvement in mitochondrial efficiency led us to explore the effects of CDN1163 on mitochondrial oxidant defense. Although OXPHOS is an essential process to generate ATP, higher rates can be damaging because of excess reactive oxygen species. Excessive reactive oxygen species are promptly eliminated from the cell by a variety of antioxidant enzymes. Interestingly, CDN1163-treated livers showed enhanced levels of the antioxidant enzymes glutathione peroxidase (GPX), superoxide dismutase (SOD2), and peroxiredoxin (PRDX3) (Fig. 6G). These findings dovetail with the observed increase in UCP2 and UCP3, as uncoupling proteins have been implicated in the reduction of reactive oxygen species (29). Furthermore, and in agreement with the antioxidant enzymes activation, lipid peroxidation assessed by measuring the concentration of plasma and liver MDA was correspondingly decreased in CDN1163-treated ob/ob mice compared with vehicle-treated littermates (Table 1).
Discussion
Here we demonstrate that direct pharmacological activation of SERCA2b through administration of a novel allosteric SERCA2 modulator, CDN1163, decreased blood glucose and fasting insulin levels, resulting in improved glucose tolerance in ob/ob mice even in the absence of a reduction in food intake. We also observed that CDN1163 administration to ob/ob mice for 5 days had a sustained blood glucose lowering effect for a period well beyond cessation of the compound administration. Importantly, CDN1163 treatment of normal lean mice did not alter either circulating glucose levels or body weight, indicating that pharmacological activation of SERCA2b is unlikely to induce hypoglycemia or impair energy homeostasis in metabolically healthy animals. These findings are important as they imply that targeting SERCA2b is effective only under disease state (i.e. diabetes in our case) as normal ER Ca2+ levels shut down SERCA2b function overriding any allosteric modulation by CDN1163. SERCA2b enzyme is calcium-regulated, and when Ca2+ storage in the ER is normal the compound has little to no effect outside of the physiological range for SERCA2b variability. This is indeed consistent with a previous study that reported overexpression of SERCA2b by adenoviral gene transfer in the liver of euglycemic lean mice does not lead to hypoglycemia; however, overexpression of SERCA2b significantly reduces the blood glucose levels in genetically matched ob/ob mice (14).
To gain molecular mechanistic insights into CDN1163 glucose lowering action, we examined gene expression of proteins mediating gluconeogenesis, as increased gluconeogenesis can be a cause for elevated hepatic glucose production in diabetic patients and animal models of diabetes (17). Two key gluconeogenic enzymes, PEPCK and G6Pase, and one of the transcription factors that control their expression, HNF4α, were down-regulated by CDN1163, suggesting that SERCA2b activation may reduce diabetes-induced gluconeogenesis with a concomitant reduction in hyperglycemia. This apparent CDN1163 effect on glucose metabolism was associated with a reduction in insulin levels but not with a change in insulin sensitivity, indicating that CDN1163 induces improvement in glucose homeostasis independent of insulin activation and insulin signaling. This is somehow similar to the action of FGF19, which has also been reported to promote insulin-independent glucose disposal (30).
CDN1163 administration also improved hepatic lipid metabolism in ob/ob mice and was associated with a reduction in whole body fat mass. Several lines of evidence in our study demonstrate CDN1163-dependent lipid lowering effects: reduction in cholesterol and free fatty acids levels, marked reduction in plasma and liver triglycerides contents, striking improvement of obesity-induced hepatic steatosis accompanied by significant decreases in lipogenic gene expression, and a trend toward increased lipid oxidation markers. These results indicate that CDN1163 possibly reduces dyslipidemia by inhibiting SREBP1, as SREBP1 is a transcription factor that controls the expression of lipogenic genes (31) leading to reduced hepatosteatosis, lipogenic gene expression, and conceivably the rate of de novo fatty acid and triglyceride synthesis. These findings are in agreement with recent demonstration that short term SERCA2b gene transfer (14) or alleviation of ER stress by chemical or molecular chaperones in ob/ob mice (6, 8) decreased SREBP1c activation and lipogenesis and markedly improved liver fat accumulation and insulin sensitivity in these animals. The decrease in adiposity and improvement of hepatosteatosis after CDN1163 treatment is consistent with our observation that CDN1163 stimulates energy expenditure and increased metabolic rate with no change in lean mass or physical activity. Consistent with increased energy expenditure, obese mice treated with CDN1163 had increased mRNA expression levels of UCP1 and UCP3 in brown adipose tissue, suggesting that increased thermogenesis in brown adipose tissue contributes to the enhanced energy expenditure after CDN1163 administration.
The metabolic benefits of CDN1163 SERCA2b activation seem in part to be mediated through offsetting obesity-induced ER stress and mitochondrial compromise. Here we show that pharmacologically targeting SERCA2b dysfunction re-establishes ER Ca2+ levels, alleviates ER stress, and reduces ER stress-induced cell death in ob/ob mice, possibly through inhibition of PERK/eIF2α/CHOP and IRE1α/JNK/XBP1 signaling pathways. Interestingly, the protection against ER stress through CDN1163 was associated with marked improvement of bioenergetics mRNA expression suggestive of improved mitochondrial efficiency. Mitochondria require Ca2+ to maintain the tricarboxylic acid cycle and ATP production. ER and mitochondria are physically and functionally interconnected, and recent studies demonstrated that disruption of ER homeostasis triggers ER stress and induces mitochondrial dysfunction, which feeds back and amplifies ER stress (22, 24, 32). Impaired mitochondrial function and capacity could potentially affect multiple cellular functions including mitochondrial biogenesis, mitochondrial DNA copy number, levels of mitochondrial DNA-encoded proteins, and mitochondrial OXPHOS capacity. Our findings demonstrate that normalizing ER Ca2+ mobilization could interrupt this vicious cycle of mitochondria/ER negative cross-talk. It is, therefore, our hypothesis that CDN1163-dependent enhancement of SERCA2b function and restoration of ER Ca2+ homeostasis and signaling in ob/ob mice is associated with attenuation of ER stress and a concomitant enhancement of ER function. The re-establishment of proper ER function reciprocally improves mitochondrial function and contributes to the control of systemic glucose and lipid metabolism. CDN1163-induced beneficial effects on mitochondrial efficiency in this study were demonstrated by improvement in mitochondrial biogenesis and replication, as evidenced by the marked elevation in the mitochondrial biogenesis gene expression program, i.e. NRF1, ERRα, TFAm, and PGC1α, and stimulation of mitochondrial OXPHOS capacity along with increased levels of mitochondrial DNA contents (mtDNA). PGC1α is a key regulator of mitochondrial content and function. In the liver, mitochondrial biogenesis, fatty acid oxidation, and mtDNA copy number are controlled by PGC1α through its physical interactions with the transcription factors ERRα and NRF1 (33, 34). In this case, increased expression of PGC1α provoked by CDN1163 induces the transcription of ERRα and NRF1, leading to the increased expression of TFAm as well as other mitochondrial subunits of the electron transport chain complexes. TFAm translocates to the mitochondria where it is necessary for mtDNA stability and also initiates mtDNA transcription that is essential for mtDNA replication and mitochondrial-encoded gene transcription (35). Besides regulation of mitochondrial biogenesis and respiration, PGC1α is also known to stimulate hepatic gluconeogenesis (36). In this study we observed that CDN1163 inhibited the gluconeogenesis genetic program despite the up-regulation of PGC1α. However, because gluconeogenic gene expression driven by PGC1α requires the liver-enriched transcription factor, HNF4α (37, 38), inhibition of HNF4α after treatment with CDN1163 may explain PGC1α differential stimulation of fatty acid oxidation and electron transport in the mitochondria without concertedly inducing gluconeogenesis. This uncoupling scenario of hepatic gluconeogenesis from fatty acid oxidation has recently been proposed in the liver where it was shown that the p70 ribosomal protein S6 kinase 1, for instance, can phosphorylate PGC1α directly on two sites within its arginine/serine-rich domain and attenuates the coactivation of the gluconeogenic target genes by interfering with the ability of PGC1α to bind to HNF4α while leaving undisturbed its interactions with factors regulating mitochondrial biogenesis and fatty acid oxidation, such as ERRα and peroxisome proliferator-activated receptor-α (39).
To further explore the molecular mechanisms that elicit the metabolic benefits of CDN1163, we examined the activation of AMPK, as it has been widely reported to target PGC1α and HNF4α signaling. AMPK activation has also been shown to down-regulate the expression of the transcription factor SREBP1c (40), which we also show was decreased by CDN1163 in this study. The beneficial effects of AMPK in the setting of insulin resistance and T2D were attributed in part to its ability to reduce dyslipidemia and stimulate fatty acid oxidation in many tissues by inhibiting SREBP1c and up-regulating PGC1α and mitochondrial biogenesis (41), findings that were also evoked by CDN1163 activation of SERCA2b. Also, AMPK activation has been demonstrated to repress the expression of gluconeogenesis enzymes, PEPCK, and glucose-6-phosphatase (42) possibly through degradation of the transcription factor HNF4α (43) and to ameliorate palmitate-induced ER stress and cell death (44). The interplay between SERCA2 and AMPK has been further highlighted by the findings that pharmacological or genetic inhibition of AMPK reduces SERCA2b activity, inhibits SERCA2b-dependent Ca2+ clearance and storage, and triggers ER stress response, whereas overexpression and/or activation of AMPK produced the opposite phenotype (45). These observations strongly align with our current findings and led us to propose that CDN1163 may confer its protective metabolic benefits through SERCA2b activation of AMPK, which then triggers amelioration of glucose homeostasis, ER stress, and mitochondrial dysfunction and subsequent improvement of diabetes and metabolic disorders (Fig. 7). How and whether the SERCA2b-AMPK axis promotes some sort of a “metabolic reset” that confers long term protection is tentative and warrants further investigation.
FIGURE 7.
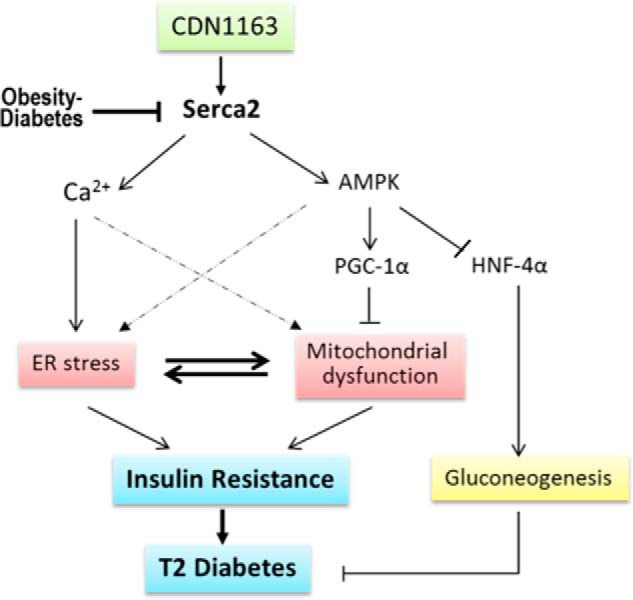
Schematic diagram depicting proposed molecular mechanism underlying CDN1163/SERCA2b metabolic benefits. A fundamental abnormality of obesity and diabetes is down-regulation and dysfunction of Serca2b causing intracellular Ca2+ imbalance with a concomitant induction of ER stress and mitochondrial dysfunction, as these two events are reciprocally related. ER stress and mitochondrial dysfunction then trigger insulin resistance and development of diabetes. Pharmacological restoration of Serca2b activity by CDN1163 (a) normalizes intracellular Ca2+ dyshomeostasis (which reestablishes ER homeostasis) and (b) activates AMPK, which in turn up-regulates PGC1α (which improves mitochondrial biogenesis) and down-regulates HNF4α leading to suppression of gluconeogenesis. Evidence also shows that AMPK can improve ER stress and Ca2+ and has long played a major role in mitochondria (dashed lines). Attenuation of gluconeogenesis and ER stress and improvement of mitochondrial efficiency altogether then ameliorate insulin resistance and diabetes.
It is noteworthy to point out that we have primarily focused in this study on the liver as an insulin-responsive tissue; however, other tissues such as the pancreas/β-cells, adipose tissue, skeletal muscle, or the central nervous system could also be targeted by the SERCA2 small molecules, and the effects on these peripheral tissues would likely contribute to the overall beneficial metabolic effects evoked by SERCA2b activation. For instance, parallel to the conferred beneficial effects on the liver, we also documented similar bioactivity of CDN1163 in the hearts of ob/ob mice where we observed significant improvement of cardiac performance and mitochondrial function.3 This is of interest as cardiovascular co-morbidities are common in diabetes, and therefore, it is possible that dual pharmacological activities of SERCA2 agonism may also have beneficial effects on the cardiovascular system by increasing cardiac muscle contractility and improving vascular tone (46–48).
In summary, the current study has validated for the first time that pharmacological activation of SERCA2b in an animal model of insulin resistance and prediabetes provided efficient and durable glucose control and improved dyslipidemia. In addition, CDN1163 normalized ER Ca2+ dyshomeostasis in vivo, improved hepatic steatosis, and corrected multiple metabolic abnormalities associated with ER stress and mitochondrial efficiency, possibly through a SERCA2b-mediated activation of AMPK pathway. This study provides a proof-of-concept that SERCA2 agonists have the potential to become an effective therapy for the treatment of diabetes and metabolic dysfunction.
Author Contributions
S. K and D. L. conceived and designed this study, researched the data, and wrote the manuscript. R. D. researched the data. A. S., W. H., and C. B. advised on the experimental design of and performed analysis of energy expenditure and body composition, wrote and edited the manuscript, and contributed to the scientific discussion. K. M. Z. and R. H. contributed reagents and contributed to the scientific discussion and reviewed and edited manuscript.
Acknowledgments
We thank Scott Friedman, Derek LeRoith, and Andrew Stewart for helpful discussions.
This work was supported by National Institutes of Health Grants HL097375 and DK020541 (to D. L.), DK083658 (to C. B.), and K01 DK099463 (to A. S.). The authors declare that they have no conflicts of interests with the content of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
S. Kang and D. Lebeche, manuscript in preparation.
- T2D
- type 2 diabetes
- ER
- endoplasmic reticulum
- SERCA
- sarco/endoplasmic reticulum Ca2+-ATPase
- TG
- triglyceride
- MDA
- malondialdehyde
- eIF2α
- eukaryotic initiation factor 2α
- PERK
- protein kinase RNA-like ER kinase
- CHOP
- C/EBP homologous protein
- AMPK
- AMP-activated kinase
- mt-
- mitochondrial
- PEPCK
- phosphoenolpyruvate carboxykinase
- HNF4α
- hepatocyte nuclear factor 4α
- SREBP1c
- sterol regulatory element binding protein-1c
- IRE1
- inositol-requiring kinase 1
- PGC-1α
- peroxisome proliferator-activated receptor-γ coactivator 1α
- UCP
- uncoupling protein
- NDUFB8
- NADH dehydrogenase (ubiquinone) 1 β subcomplex subunit 8
- ERRα
- estrogen-related receptor α
- TFAm
- transcription factor A
- ATF4
- activating transcription factor 4.
References
- 1. Disdier-Flores O. M., Rodríguez-Lugo L. A., Pérez-Perdomo R., and Pérez-Cardona C. M. (2001) The public health burden of diabetes: a comprehensive review. P. R. Health Sci. J. 20, 123–130 [PubMed] [Google Scholar]
- 2. Fu S., Yang L., Li P., Hofmann O., Dicker L., Hide W., Lin X., Watkins S. M., Ivanov A. R., and Hotamisligil G. S. (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Williams L. M. (2012) Hypothalamic dysfunction in obesity. Proc. Nutr. Soc. 71, 521–533 [DOI] [PubMed] [Google Scholar]
- 4. Back S. H., and Kaufman R. J. (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 81, 767–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fonseca S. G., Gromada J., and Urano F. (2011) Endoplasmic reticulum stress and pancreatic beta-cell death. Trends Endocrinol. Metab. 22, 266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kammoun H. L., Chabanon H., Hainault I., Luquet S., Magnan C., Koike T., Ferré P., and Foufelle F. (2009) GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest. 119, 1201–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kars M., Yang L., Gregor M. F., Mohammed B. S., Pietka T. A., Finck B. N., Patterson B. W., Horton J. D., Mittendorfer B., Hotamisligil G. S., and Klein S. (2010) Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 59, 1899–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., and Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 9. Xiao C., Giacca A., and Lewis G. F. (2011) Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and β-cell dysfunction in humans. Diabetes 60, 918–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Engin F., and Hotamisligil G. S. (2010) Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes. Metab. 12, 108–115 [DOI] [PubMed] [Google Scholar]
- 11. Ozcan L., and Tabas I. (2012) Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 63, 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ron D., and Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 13. Kulkarni R. N., Roper M. G., Dahlgren G., Shih D. Q., Kauri L. M., Peters J. L., Stoffel M., and Kennedy R. T. (2004) Islet secretory defect in insulin receptor substrate 1 null mice is linked with reduced calcium signaling and expression of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)-2b and -3. Diabetes 53, 1517–1525 [DOI] [PubMed] [Google Scholar]
- 14. Park S. W., Zhou Y., Lee J., Lee J., and Ozcan U. (2010) Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc. Natl. Acad. Sci. U.S.A. 107, 19320–19325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wold L. E., Dutta K., Mason M. M., Ren J., Cala S. E., Schwanke M. L., and Davidoff A. J. (2005) Impaired SERCA function contributes to cardiomyocyte dysfunction in insulin resistant rats. J. Mol. Cell Cardiol. 39, 297–307 [DOI] [PubMed] [Google Scholar]
- 16. Cornea R. L., Gruber S. J., Lockamy E. L., Muretta J. M., Jin D., Chen J., Dahl R., Bartfai T., Zsebo K. M., Gillispie G. D., and Thomas D. D. (2013) High-throughput FRET assay yields allosteric SERCA activators. J. Biomol. Screen 18, 97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Consoli A., Nurjhan N., Capani F., and Gerich J. (1989) Predominant role of gluconeogenesis in increased hepatic glucose production in NIDDM. Diabetes 38, 550–557 [DOI] [PubMed] [Google Scholar]
- 18. Bechmann L. P., Hannivoort R. A., Gerken G., Hotamisligil G. S., Trauner M., and Canbay A. (2012) The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 56, 952–964 [DOI] [PubMed] [Google Scholar]
- 19. Kotronen A., Seppälä-Lindroos A., Bergholm R., and Yki-Järvinen H. (2008) Tissue specificity of insulin resistance in humans: fat in the liver rather than muscle is associated with features of the metabolic syndrome. Diabetologia 51, 130–138 [DOI] [PubMed] [Google Scholar]
- 20. Yki-Järvinen H. (2005) Fat in the liver and insulin resistance. Ann. Med. 37, 347–356 [DOI] [PubMed] [Google Scholar]
- 21. Tabas I., and Ron D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rieusset J. (2011) Mitochondria and endoplasmic reticulum: mitochondria-endoplasmic reticulum interplay in type 2 diabetes pathophysiology. Int. J. Biochem. Cell Biol. 43, 1257–1262 [DOI] [PubMed] [Google Scholar]
- 23. Ashby M. C., and Tepikin A. V. (2001) ER calcium and the functions of intracellular organelles. Semin. Cell Dev. Biol. 12, 11–17 [DOI] [PubMed] [Google Scholar]
- 24. Lim J. H., Lee H. J., Ho Jung M., and Song J. (2009) Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: a molecular mechanism leading to hepatic insulin resistance. Cell. Signal. 21, 169–177 [DOI] [PubMed] [Google Scholar]
- 25. Schröder M., and Kaufman R. J. (2005) The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789 [DOI] [PubMed] [Google Scholar]
- 26. Vangheluwe P., Raeymaekers L., Dode L., and Wuytack F. (2005) Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: cell biological implications. Cell Calcium 38, 291–302 [DOI] [PubMed] [Google Scholar]
- 27. García-Ruiz I., Rodríguez-Juan C., Díaz-Sanjuan T., del Hoyo P., Colina F., Muñoz-Yagüe T., and Solís-Herruzo J. A. (2006) Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology 44, 581–591 [DOI] [PubMed] [Google Scholar]
- 28. Spiegelman B. M. (2007) Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found Symp. 287, 60–63; discussion 63–69 [PubMed] [Google Scholar]
- 29. Jaburek M., Miyamoto S., Di Mascio P., Garlid K. D., and Jezek P. (2004) Hydroperoxy fatty acid cycling mediated by mitochondrial uncoupling protein UCP2. J. Biol. Chem. 279, 53097–53102 [DOI] [PubMed] [Google Scholar]
- 30. Morton G. J., Matsen M. E., Bracy D. P., Meek T. H., Nguyen H. T., Stefanovski D., Bergman R. N., Wasserman D. H., and Schwartz M. W. (2013) FGF19 action in the brain induces insulin-independent glucose lowering. J. Clin. Invest. 123, 4799–4808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Browning J. D., and Horton J. D. (2004) Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leem J., and Koh E. H. (2012) Interaction between mitochondria and the endoplasmic reticulum: implications for the pathogenesis of type 2 diabetes mellitus. Exp. Diabetes Res. 2012, 242984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mootha V. K., Handschin C., Arlow D., Xie X., St Pierre J., Sihag S., Yang W., Altshuler D., Puigserver P., Patterson N., Willy P. J., Schulman I. G., Heyman R. A., Lander E. S., and Spiegelman B. M. (2004) Errα and Gabpa/b specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. U.S.A. 101, 6570–6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schreiber S. N., Emter R., Hock M. B., Knutti D., Cardenas J., Podvinec M., Oakeley E. J., and Kralli A. (2004) The estrogen-related receptor α (ERRα) functions in PPARγ coactivator 1α (PGC-1α)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. U.S.A. 101, 6472–6477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gaspari M., Larsson N. G., and Gustafsson C. M. (2004) The transcription machinery in mammalian mitochondria. Biochim. Biophys. Acta 1659, 148–152 [DOI] [PubMed] [Google Scholar]
- 36. Yoon J. C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C. R., Granner D. K., Newgard C. B., and Spiegelman B. M. (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138 [DOI] [PubMed] [Google Scholar]
- 37. Puigserver P., Rhee J., Donovan J., Walkey C. J., Yoon J. C., Oriente F., Kitamura Y., Altomonte J., Dong H., Accili D., and Spiegelman B. M. (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423, 550–555 [DOI] [PubMed] [Google Scholar]
- 38. Rhee J., Inoue Y., Yoon J. C., Puigserver P., Fan M., Gonzalez F. J., and Spiegelman B. M. (2003) Regulation of hepatic fasting response by PPARγ coactivator-1α (PGC-1): requirement for hepatocyte nuclear factor 4α in gluconeogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 4012–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lustig Y., Ruas J. L., Estall J. L., Lo J. C., Devarakonda S., Laznik D., Choi J. H., Ono H., Olsen J. V., and Spiegelman B. M. (2011) Separation of the gluconeogenic and mitochondrial functions of PGC-1α through S6 kinase. Genes Dev. 25, 1232–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., and Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zong H., Ren J. M., Young L. H., Pypaert M., Mu J., Birnbaum M. J., and Shulman G. I. (2002) AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. U.S.A. 99, 15983–15987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lochhead P. A., Salt I. P., Walker K. S., Hardie D. G., and Sutherland C. (2000) 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 49, 896–903 [DOI] [PubMed] [Google Scholar]
- 43. Hong Y. H., Varanasi U. S., Yang W., and Leff T. (2003) AMP-activated protein kinase regulates HNF4α transcriptional activity by inhibiting dimer formation and decreasing protein stability. J. Biol. Chem. 278, 27495–27501 [DOI] [PubMed] [Google Scholar]
- 44. Borradaile N. M., Han X., Harp J. D., Gale S. E., Ory D. S., and Schaffer J. E. (2006) Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 47, 2726–2737 [DOI] [PubMed] [Google Scholar]
- 45. Dong Y., Zhang M., Liang B., Xie Z., Zhao Z., Asfa S., Choi H. C., and Zou M. H. (2010) Reduction of AMP-activated protein kinase α2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 121, 792–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sakata S., Lebeche D., Sakata Y., Sakata N., Chemaly E. R., Liang L., Nakajima-Takenaka C., Tsuji T., Konishi N., del Monte F., Hajjar R. J., and Takaki M. (2007) Transcoronary gene transfer of SERCA2a increases coronary blood flow and decreases cardiomyocyte size in a type 2 diabetic rat model. Am. J. Physiol. Heart Circ. Physiol. 292, H1204–H1207 [DOI] [PubMed] [Google Scholar]
- 47. Sakata S., Lebeche D., Sakata Y., Sakata N., Chemaly E. R., Liang L. F., Padmanabhan P., Konishi N., Takaki M., del Monte F., and Hajjar R. J. (2006) Mechanical and metabolic rescue in a type II diabetes model of cardiomyopathy by targeted gene transfer. Mol. Ther. 13, 987–996 [DOI] [PubMed] [Google Scholar]
- 48. Hadri L., Bobe R., Kawase Y., Ladage D., Ishikawa K., Atassi F., Lebeche D., Kranias E. G., Leopold J. A., Lompré A. M., Lipskaia L., and Hajjar R. J. (2010) SERCA2a gene transfer enhances eNOS expression and activity in endothelial cells. Mol. Ther. 18, 1284–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]