

Abstract
The N-terminal acetyltransferase NatA is a heterodimeric complex consisting of a catalytic subunit (Naa10/ARD1) and an auxiliary subunit (Naa15). NatA co-translationally acetylates the N termini of a wide variety of nascent polypeptides. In addition, Naa10 can act independently to posttranslationally acetylate a distinct set of substrates, notably actin. Recent structural studies of Naa10 have also revealed the molecular basis for N-terminal acetylation specificity. Surprisingly, recent reports claim that Naa10 may also acetylate lysine residues of diverse targets, including methionine sulfoxide reductase A, myosin light chain kinase, and Runt-related transcription factor 2. Here we used recombinant proteins to reconstitute and assess lysine acetylation events catalyzed by Naa10 in vitro. We show that there is no difference in lysine acetylation of substrate proteins with or without Naa10, suggesting that the substrates may be acetylated chemically rather than enzymatically. Together, our data argue against a role for Naa10 in lysine acetylation.
Keywords: acetyl-CoA, acetylation, acetyltransferase, posttranslational modification (PTM), transcription factor, ARD1, NAT, Naa10p, chemical acetylation
Introduction
Acetylation is a common and important protein modification in biology. It has roles in diverse biological processes including gene expression, metabolism, signal transduction, and protein degradation (1). In general, there are two forms of protein acetylation: lysine acetylation of the ϵ amino group and N-terminal acetylation, which occurs on the α amino group on the N termini of proteins. These two types of acetylation are catalyzed by two different groups of enzymes: the lysine acetyltransferases (KATs)2 and the N-terminal acetyltransferases (NATs) (2). Although lysine acetylation occurs on thousands of proteins to mediate diverse biological processes (3), N-terminal acetylation has also been shown more recently to be a ubiquitous and important modification with a variety of functional consequences for a diverse array of cellular processes, such as protein localization (4), mediating protein-protein interactions (5), and regulating protein degradation (6).
There are five conserved NATs found in eukaryotes, termed NatA-NatE. These enzymes differ in their N-terminal substrate profiles (2). NatA, which is responsible for the plurality of N-terminal acetylation (2), is a heterodimeric complex consisting of a catalytic subunit termed Naa10 (previously known as Ard1) and an auxiliary subunit termed Naa15 (previously known as NATH or Nat1) (7). NatA acetylates N termini containing an N-terminal residue that has a small radius of gyration: alanine, cysteine, glycine, serine, threonine, and valine (8). Structural and enzymatic studies reveal that the enzymatic subunit Naa10 has a different substrate profile when it is not in complex with the auxiliary subunit Naa15. As a monomer, Naa10 acetylates N termini in which the N-terminal residues are acidic, such as actin, whose first three N-terminal residues are glutamates (9, 10).
Recent reports claim to have identified lysine substrates for monomeric Naa10. These include the transcription factor Runt-related transcription factor 2 (Runx2) (11), the enzyme methionine sulfoxide reductase A (MSRA) (12), and myosin light chain kinase (MLCK) (13). These findings were surprising to us because the structures of all NATs determined to date (9, 14–17), including Naa10 (9), contain an extended loop that seems to occlude lysine side chains within a polypeptide from lying across the active site as they do in KATs (15–18). In addition, other reports demonstrating that Naa10 acetylates a lysine residue on Hif-1α have failed to be replicated (19–21), making the question of whether Naa10 is able to acetylate lysine residues controversial. We therefore performed a number of experiments to investigate this question. We found that the addition of acetyl-CoA did indeed promote the acetylation of MSRA and Runx2 but that this acetylation was Naa10-independent. On the basis of this observation, we conclude that lysine residues on these substrates are not acetylated by Naa10 but are, instead, acetylated by chemical means.
Materials and Methods
Cloning and Purification of Naa10
The gene encoding full-length human Naa10 (residues 1–235) was cloned into a pETDUET vector containing an N-terminal His6-SUMO tag. The resulting construct was transformed into Rosetta (DE3)pLysS-competent Escherichia coli. Cells were grown to A600 0.7–0.9 prior to inducing protein expression with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 16 °C for ∼16 h. All subsequent purification steps were carried out at 4 °C. Cells were isolated by centrifugation and lysed by sonication in lysis buffer containing 25 mm Tris (pH 8.0), 1 m NaCl, 10 mm β-mercaptoethanol (βME) and 10 μg/ml PMSF. The lysate was clarified by centrifugation and passed over 6 ml of Ni-NTA resin (Thermo Scientific) that was subsequently washed with 500 ml of lysis buffer supplemented with 25 mm imidazole. The protein was eluted in 50 ml of lysis buffer supplemented with 300 mm imidazole and dialyzed into ion exchange buffer containing 25 mm Tris (pH 8.5), 50 mm NaCl, and 10 mm βME and loaded onto a 5-ml Q ion exchange column (GE Healthcare). The protein was eluted in the same buffer with a salt gradient (50–750 mm NaCl) over the course of 20 column volumes. Peak fractions were pooled, concentrated to 500 μl, and loaded onto an s200 gel filtration column (GE Healthcare) in sizing buffer containing 25 mm HEPES (pH 7.0), 200 mm NaCl, and 1 mm DTT. Peak fractions were pooled and concentrated. Proteins were aliquoted, snap-frozen in liquid nitrogen, and stored at −80 °C for further use. A portion of the protein was then incubated with Ulp1 protease overnight to cleave off the His6-SUMO tag in sizing buffer. After cleavage, this solution was subjected to an additional Ni-NTA purification step to remove Ulp1, His6-SUMO, and any uncut Naa10 fusion protein. The resin was then washed with approximately 10 column volumes of dialysis buffer supplemented with 25 mm imidazole, which was pooled with the initial flow-through. The protein was concentrated to 500 μl and run on a Superdex 75 gel filtration column. Peak fractions were pooled and concentrated. Proteins were aliquoted, supplemented with 5% glycerol, snap-frozen in liquid nitrogen, and stored at −80 °C for further use.
Cloning and Purification of NatA and GST-Naa50
Human Naa10 (residues 1–160) and Naa15 (residues 1–866) constructs were engineered using a pFastBac dual vector. The Naa15 subunit contained an N-terminal His6 tag. A bacmid was generated by transposition into DH10 bac-competent E. coli cells using the Bac-to-bac system (Invitrogen). Spodoptera frugiperda (Sf9) cells cultured in serum-free media were transfected with the Naa10/Naa15 bacmid using Cellfectin reagent (Invitrogen). The resulting baculovirus was amplified until it reached a high titer. Sf9 cells were grown to a density of 1 × 106 cells/ml and infected using the amplified Naa10/Naa15 baculovirus to a multiplicity of infection of ∼1–2. The cells were grown at 27 °C and harvested 48 h post-infection. All subsequent purification steps were carried out at 4 °C. Cells were isolated by centrifugation and lysed by sonication in lysis buffer containing 25 mm Tris (pH 8.0), 1 m NaCl, 10 mm βME, 10 μg/ml PMSF, and DNase. The lysate was clarified by centrifugation and passed over 6 ml of Ni-NTA resin (Thermo Scientific) that was subsequently washed with 500 ml of lysis buffer supplemented with 25 mm imidazole. The protein was eluted in 50 ml of lysis buffer supplemented with 300 mm imidazole. The protein was concentrated to 500 μl and loaded onto a Superdex 200 gel filtration column (GE Healthcare) in sizing buffer containing 25 mm HEPES (pH 7.0), 200 mm NaCl, and 1 mm DTT. Peak fractions were pooled and concentrated. Proteins were aliquoted, snap-frozen in liquid nitrogen, and stored at −80 °C for further use. Human GST-Naa50 was purified as described previously (15) with the following changes. The protein was not cleaved with tobacco etch virus protease and was run on a Superdex200 gel filtration column.
Cloning and Purification of Runx2 and MSRA
The genes encoding the runt domain of mouse Runx2 (residues 186–315) and full-length human MSRA (residues 1–235) were ordered from Bio Basic. These were both cloned into a modified pETDUET vector containing a tobacco etch virus-cleavable, N-terminal GST tag. The resulting constructs were transformed into Rosetta (DE3)pLysS-competent E. coli. Cells were grown to A600 0.7–0.9 prior to inducing protein expression with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 16 °C for ∼16 h. All subsequent purification steps were carried out at 4 °C and were identical for both proteins except where noted. Cells were isolated by centrifugation and lysed by sonication in lysis buffer containing 25 mm Tris (pH 8.0), 1 m NaCl, 10 mm βME, and 10 μg/ml PMSF. The lysate was clarified by centrifugation and incubated with 6 ml glutathione resin (Thermo Scientific) for 1 h. The resin was subsequently washed with 500 ml of lysis buffer. The protein was eluted in 50 ml of lysis buffer supplemented with 20 mm reduced glutathione (Sigma) and dialyzed into ion exchange buffer containing 25 mm sodium citrate (pH 5.5), 25 mm NaCl, and 10 mm βME and loaded onto a 5-ml SP ion exchange column (GE Healthcare). The protein was eluted in the same buffer with a salt gradient (25–750 mm NaCl) over the course of 20 column volumes. Peak fractions were pooled, concentrated to 500 μl, and loaded onto an s200 gel filtration column (GE Healthcare) in sizing buffer containing 25 mm HEPES (pH 7.0), 200 mm NaCl, and 1 mm DTT. Peak fractions were pooled and concentrated. Half of the protein was then incubated on glutathione resin with tobacco etch virus protease overnight in sizing buffer to cleave off the GST tag. After cleavage, the resin was washed with approximately 10 column volumes of sizing buffer, and the untagged protein was collected. The protein was concentrated to 500 μl and run on a Superdex200 gel filtration column. Peak fractions were pooled and concentrated. Proteins were aliquoted, supplemented with 5% glycerol, snap-frozen in liquid nitrogen, and stored at −80 °C for further use.
Acetyltransferase Assays against Peptides
Acetyltransferase assays were carried out in sizing buffer. All peptides were ordered from GenScript. The peptides for the reported lysine substrates corresponded to ∼20 residues surrounding putative acetylated lysine. These peptides were ordered with acetylated N termini to ensure that the signal would not result from N-terminal acetyltransferase activity. Tryptophan was added to the C terminus to allow for accurate concentration determination. The peptides were as follows: MLCK, TVHEKKSSRKSEYLLPVAW; MSRA, RKEQTPVAAKHHVNGNRTVW; and Runx2, QVATYHRAIKVTVDGPRW. The actin substrate peptide used has been described previously (9). In the assays, 300 μm of radiolabeled [14C]acetyl-CoA (4 mCi/mmol, PerkinElmer Life Sciences), 1 μm Naa10 or NatA, and 1 mm peptide were reacted in 50 μl of total reaction volume for 4 h at 37 °C. To quench the reaction, 15 μl of the reaction mixture was added to negatively charged P81 paper (Millipore), and the paper disks were placed immediately into wash buffer (10 mm HEPES (pH 7.5)). The papers were washed three times, 5 min/wash, to remove unreacted acetyl-CoA. The papers were then dried with acetone and added to 4 ml of scintillation fluid, and the signal was measured with a Packard Tri-Carb 1500 liquid scintillation analyzer.
Acetyltransferase Assays against Protein Domains
Reactions were performed as above but with 50 μm substrate (Actin peptide, recombinant Runx2, or recombinant MSRA). 15 μl of 20% trichloroacetic acid (TCA) was added to 15 μl of reaction mixture to precipitate acetylated substrates. The substrates were allowed to precipitate on ice for 10 min. The precipitated substrates were pelleted by centrifuging for 5 min at 13,000 rpm. The supernatant was discarded, and 200 μl of cold acetone was added to wash the pellets. These were incubated on ice for 5 min before centrifuging at 13,000 rpm. This wash step was repeated twice more. The pellets were added to 4 ml of scintillation fluid, and the signal was measured with a Packard Tri-Carb 1500 liquid scintillation analyzer.
Acetyltransferase Assays with Increasing Concentrations of Acetyl-CoA
The assays were done as the TCA precipitation assays with the following changes. 1 mm of substrate (Actin peptide, recombinant Runx2, or recombinant MSRA) was used, and the amount of acetyl-CoA was varied (150–1000 μm). The reactions took place for 20 min at 37 °C.
Protein Pulldowns
All pulldowns were performed in sizing buffer. 1 μm of GST, GST-Runx2, GST-MSRA, or GST-Naa50 (positive control for NatA) was incubated with either 5 μm Naa10 or 5 μm NatA and 75 μl of glutathione resin in a total volume of 400 μl for 1 h at 4 °C. The resin was washed with 5 ml of sizing buffer and eluted with 100 μl of buffer supplemented with 20 mm glutathione.
Acetyltransferase Assay followed by Western Blotting
300 μm of acetyl-CoA (Sigma), 1 μm enzyme (Naa10 or hMOF), and 50 μm substrate (Runx2, MSRA, or the histone dimer H3/H4) were reacted in 20 μl of reaction volume for 4 h at 37 °C. The reaction was performed in a buffer containing 50 mm Tris (pH 8.0), 0.1 mm EDTA, 1 mm DTT, 10 mm sodium butyrate, 100 mm NaCl, and 10% glycerol. To quench the reaction, SDS loading dye was added. Reactions were diluted to 200 μl, and 20 μl of the dilution was run on the gel. Two different anti-acetyl-lysine antibodies were used (Cell Signaling Technology, catalog no. 9681; ImmuneChem, catalog no. ICP0380). hMOF and histones were prepared as described previously (22, 23).
Results
The Structure of Naa10 and Other NATs Is Incompatible with Lysine Acetylation
A superposition of Naa10 (9) and the Gcn5 KAT (24) bound to their cognate N-terminal and lysine peptide substrates, respectively, illustrates why these enzymes are tailored to acetylate their respective substrate (Fig. 1A). Specifically, the Gcn5 KAT bound to a histone tail peptide centered on the lysine 14 target reveals that the peptide sits across a wide surface groove of the enzyme, with the central lysine 14 residue inserting into a narrow active site cavity (24) (Fig. 1A). The Hat1 histone acetyltransferase KAT bound to a histone H4 peptide shows a similar substrate-binding configuration (25). The Naa10 NAT and Gcn5 KAT superimpose fairly well (root mean square deviation = 6.4) except for the substrate binding sites (root mean square deviation = 10.17). Most notably, Naa10 contains an extended loop between β strands β6 and β7 that is not present in Gcn5 and that blocks peptide binding across the surface groove that is present in Gcn5 (Fig. 1A). Instead, Naa10 contains an active site cavity that can only accommodate an N-terminal peptide (Fig. 1A). A superposition of all available NAT structures (9, 14–17) reveals that they contain substrate binding sites that are tailored for N-terminal substrate binding oriented similarly to Naa10 and incompatible with binding lysine-containing substrates oriented similarly to Gcn5 (Fig. 1B).
FIGURE 1.
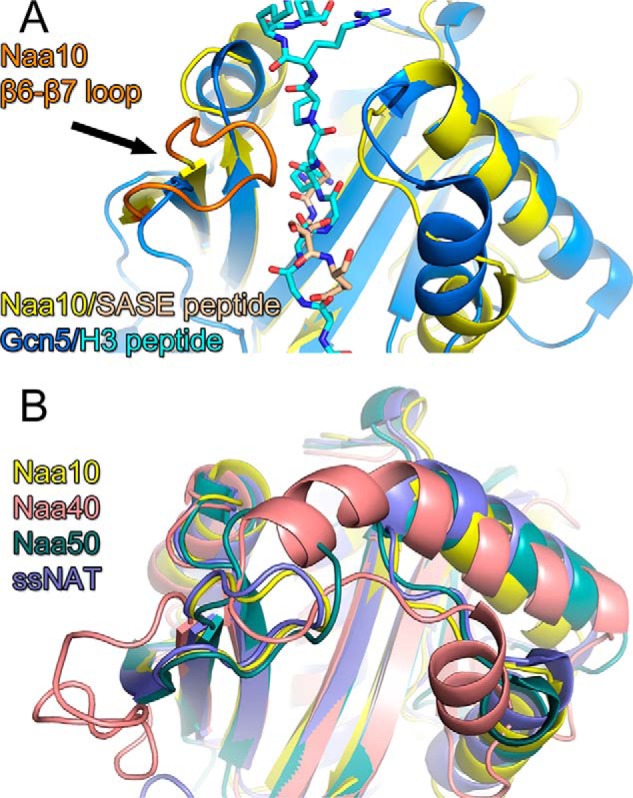
The structure of NATs is incompatible with lysine acetylation. A, superposition of the Gcn5 KAT-histone H3 peptide complex (dark and light blue, respectively) with the Naa10 N-terminal peptide complex (yellow and wheat, respectively). The proteins are shown as cartoon and the peptide ligands are shown as sticks. Naa10 is shown with its β6-β7 loop in orange. B, superposition of NAT/N-terminal peptide complexes with Naa10 (yellow), Naa40p (salmon) Naa50p (green), and the NAT from Sulfolobus solfataricus (ssNAT, purple).
Naa10 Does Not Acetylate Lysine Residues within Peptides from MLCK, MSRA, or RUNX2
Although the structural superpositions described above argue against Naa10 acetylation of lysine substrates, it remains possible that Naa10 may be able to adopt an alternative conformation to accommodate lysine substrate acetylation. To address this possibility, we first tested whether recombinant Naa10 was able to acetylate peptides corresponding to published lysine substrates (11–13). These peptides were ∼20 residues long and contained the reported lysine in the center of the peptide. We assayed three reported substrates: MLCK, MSRA, and Runx2, and a peptide corresponding to the N terminus of actin, a known Naa10 substrate, as a positive control (9, 10). We carried out acetyltransferase assays with a saturating concentration of peptide (1 mm) and radiolabeled acetyl-CoA and measured the signal by scintillation counting. Although Naa10 robustly acetylated its canonical actin substrate, there was no difference in the levels of acetylation in the three substrates in the absence or presence of Naa10 (Fig. 2A). These experiments reveal that Naa10 does not contribute to the acetylation of MLCK, MSRA, and Runx2 lysine peptide substrates.
FIGURE 2.
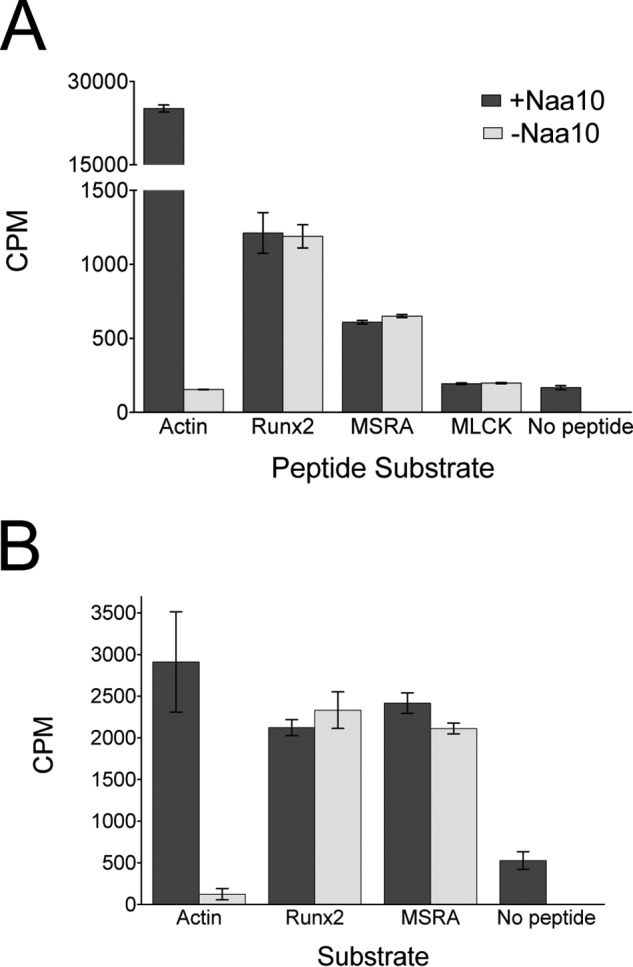
Naa10 does not acetylate peptides or proteins corresponding to reported lysine substrates. Recombinant human Naa10 was used for these experiments. A, radioactive filter binding assay of peptide acetylation in the presence or absence of Naa10. An N-terminal actin peptide was used as a positive control. B, radioactive TCA precipitation assay of protein acetylation with Runx2 and MSRA in the presence of absence of Naa10. The experiments were carried out in triplicate, and error bars are indicated. CPM, counts per minute.
Naa10 Does Not Acetylate Lysine Residues within MSRA and RUNX2 Proteins
To address the possibility that an expanded recognition motif on the substrates might be required for acetylation by Naa10, we subsequently performed the acetylation reactions using intact protein substrates or domains. To do this, we recombinantly expressed the Runt domain from Runx2 and full-length MSRA. We incubated the proteins or actin peptide (50 μm) with radiolabeled acetyl-CoA in the presence and absence of Naa10. After allowing the reaction to take place for 4 h, we added TCA to precipitate the protein and quantified acetylated protein in the pellet by scintillation counting. Again, we did not observe any difference in the level of acetylation of the substrates with or without Naa10 present, in contrast to the acetylation of actin, which showed enzyme dependence (Fig. 2B). We note that the difference in the counts for the actin control in figure 2 are due to the use of different substrate concentrations, 1 mm and 50 μm, used for the experiments represented in Fig. 2, A and B, respectively. The different concentrations reflect the nature of the experiments.
To evaluate the nature of acetylation of the substrates that was observed, we carried out reactions with increasing concentrations of acetyl-CoA in the presence and absence of Naa10. This analysis revealed that, although Naa10 displayed Michaelis-Menten kinetics against its actin substrate (Fig. 3A), it showed first-order kinetics against Runx2 and MSRA (Fig. 3, B and C). When Naa10 was omitted from the reaction, the acetylation against actin was negligible, whereas the acetylation of Runx2 and MSRA did not change (Fig. 3). These data strongly suggest that the acetylations of Runx2 and MSRA are independent of Naa10 and are, therefore, not enzymatic events but chemical events.
FIGURE 3.
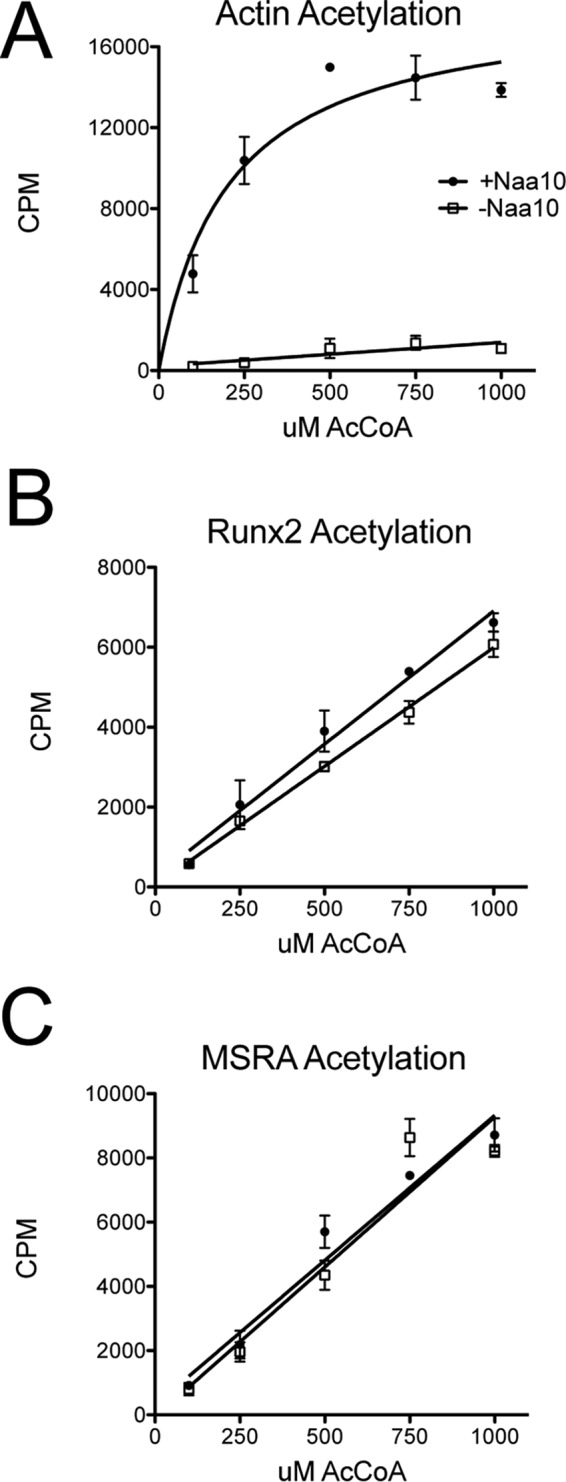
The kinetic profiles of Naa10 against MSRA and Runx2 are indicative of chemical acetylation. A–C, radioactive assay against (A) N-terminal actin peptide, (B) MSRA, and (C) Runx2 at increasing concentrations of acetyl-CoA in the presence (circles) and absence (squares) of Naa10. The experiments were carried out in triplicate, and error bars are indicated. CPM, counts per minute.
To perform an orthogonal assay for potential Naa10-mediated lysine acetylation, we also performed a Western blotting analysis with two different antibodies that recognize acetylated lysines (Cell Signaling Technology, catalog no. 9681; ImmuneChem, catalog no. ICP0380). In previous studies claiming Runx2 and MSRA acetylation by Naa10, the antibody from Cell Signaling Technology was employed (11, 12). We also used the same buffer conditions in the assay that were reported in the prior studies. Because there is no known lysine substrate of Naa10, we used acetylation of the histone tetramer H3/H4 by hMOF as a positive control because this is a well studied enzyme-dependent lysine acetylation event (23). In agreement with the radioactive assays, lysine acetylation was Naa10 independent for both Runx2 and MSRA (Fig. 4), confirming that the larger intact protein substrates or domains could not be acetylated by Naa10. In fact, the Cell Signaling Technology antibody detected a signal in the samples without any acetyl-CoA added, suggesting that the Runx2 and MSRA proteins may undergo chemical acetylation in the bacterial cells used for expression. This contrasts with H4, which was only acetylated in the presence of both hMOF and acetyl-CoA (Fig. 4).
FIGURE 4.

Western blotting detection of acetylated MSRA and Runx2 shows independence of Naa10. A and B, Western blotting analyses using two different antibodies from (A) Cell Signaling Technology (catalog no. 9681) and (B) ImmuneChem (catalog no. ICP0380) in the absence or presence of acetyl-CoA and Naa10 with MSRA and Runx2 as substrates. The hMOF KAT with histone H3/H4 substrate was used as a positive control. Coomassie staining of the gel is shown at the bottom.
Naa10 in the Context of the NatA Complex Cannot Acetylate Lysine Residues
To address the possibility that Naa10 requires assembly into the Naa10/Naa15 NatA complex for acetylating lysine substrates, we prepared a recombinant human NatA complex in insect cells and assayed its ability to acetylate lysine residues in the context of peptide and protein substrates. As shown in Fig. 5, these results are identical to those with Naa10, indicating that Naa10 in the NatA complex is unable to acetylate Runx2 and MSRA.
FIGURE 5.
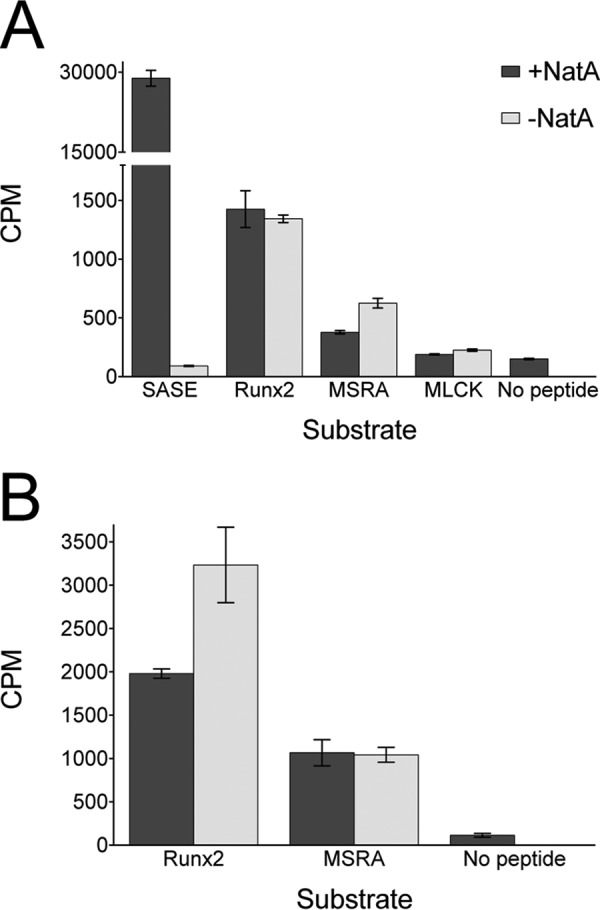
NatA does not acetylate peptides or proteins corresponding to reported lysine substrates. Recombinant human Naa10/Naa15 (NatA) was used for these experiments. A, radioactive filter binding assay of peptide acetylation in the presence or absence of Naa10. An N-terminal SASE peptide was used as a positive control as reported previously (9). B, radioactive TCA precipitation assay of protein acetylation with Runx2 and MSRA in the presence of absence of Naa10. The experiments were carried out in triplicate, and error bars are indicated.
Naa10 and NatA Do Not Interact with MRSA and Runx2
Previous reports have claimed that Naa10 interacts directly with MRSA and Runx2 (11, 12). To address the possibility that Naa10 or NatA may at least interact with the putative lysine-containing substrates in MRSA and Runx2, we carried out pulldown studies. We performed the pulldown by using GST-tagged MSRA and Runx2. In contrast to previous reports, we were unable to detect an interaction between Naa10 and either substrate (Fig. 6A). Due to the presence of a faint band which may have corresponded to Naa10 in the GST-Runx2 elution lane, we performed gel filtration on a mixture of the two proteins. They exhibited the same elution profiles as the pure protein and did not co-elute as one would expect from a stable complex (data not shown). Similar studies with the NatA complex also failed to show an interaction. Specifically, although Naa50 successfully pulled down NatA, as reported previously (26), MSRA and Runx2 did not (Fig. 6, B and C). Taken together, these studies failed to detect a direct interaction of Naa10 and NatA with MRSA or Runx2.
FIGURE 6.

Naa10 and NatA do not interact with MSRA or Runx2. A, GST pulldown assays of Naa10 using GST tagged Runx2 and MSRA. Coomassie staining of SDS-PAGE is shown for both the input (left) and elution (right). Corresponding migrating proteins in the gels are labeled. B, GST pulldown assays of Naa10/Naa15 (NatA) using GST-tagged Runx2 and MSRA. The experiment was carried out as described in A. C, as a positive control, pulldowns of NatA were carried out using GST-tagged Naa50. Asterisks indicate free hydrolyzed GST.
Discussion
Acetylation has long been recognized as an important protein modification (27). Although much of the early research examined its role as a histone modification (1), studies have found that thousands of non-histone proteins are acetylated in cells, showing that the modification is much more widespread than initially thought (3). Despite the expansion in the number of lysines targeted for acetylation, the enzymes responsible for, and the functional consequences of the majority of these marks remain unknown.
The data presented here suggest that the acetylation of Runx2 and MSRA is not an enzymatic event but, rather, a chemical one. As a moderately reactive high-energy acetyl donor, acetyl-CoA can acetylate lysines in the absence of an enzyme under the right conditions. This is particularly true when the lysine is located in close proximity to other basic residues, which lowers the pKa of the ϵ amino group to allow for a nucleophilic attack on the acetyl group (28, 29). Indeed, the lysines reported for both Runx2 and MSRA cluster within a basic patch on the proteins (30, 31) (Fig. 7).
FIGURE 7.
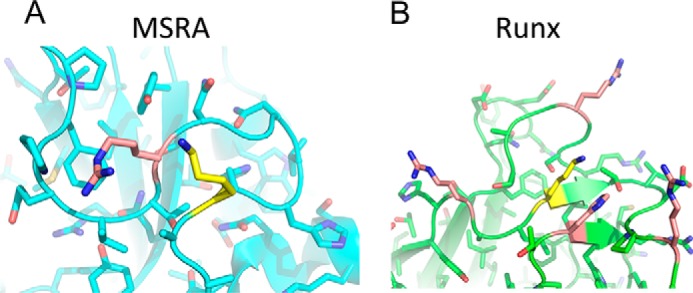
The lysines in MSRA and Runx2 are found in proximity to basic residues that could facilitate chemical acetylation. A and B, crystal structures of (A) bovine MSRA and (B) murine Runx1 (90% identity to Runx2), highlighting the lysines within these proteins reported to be acetylated by Naa10 (yellow) as well as proximal basic residues (salmon).
The results presented here are in contrast to previous reports claiming that Naa10 is able to directly interact with and acetylate lysine residues within several protein targets, including Runx2, MSRA, and MLCK (11–13). This discrepancy can be explained by a number of factors. The authors of the previous studies generated knockouts of Naa10 in mice and tissue culture for many of the assays performed. However, because the NatA complex targets hundreds of proteins for acetylation, pleiotropic effects can occur upon depletion or overexpression of the protein, the causes of which can be difficult to decipher. This is reflected in the diverse phenotypes found in NatA-null yeast (7). In addition, immunoprecipitation from cells cannot distinguish between direct and indirect protein-protein interactions, so, given that Naa10 has multiple N-terminal substrates in cells, it is likely that the observed Naa10 interactions in cells were not direct. Therefore, the in vivo and cell-based results must be interpreted with caution. In the in vitro system described here, neither Naa10 alone nor Naa10 in the context of the NatA complex is observed to acetylate or interact with these putative lysine substrates. Instead, we found that these substrates are acetylated chemically. Chemical acetylation of lysines has been shown to be an important modification in its own right, and it may be significant for regulating these proteins as well.
Author Contributions
R. S. M. and Z. M. M. performed the experiments. R. S. M. and Z. M. M. prepared the figures and text. R. M. designed and supervised the experiments by R. S. M. and Z. M. M., revised the text, and advised regarding figure presentation. All authors read and approved the manuscript.
Acknowledgments
We acknowledge the support of the DNA Sequencing Facility at the Perelman School of Medicine, University of Pennsylvania (National Institutes of Health Grant P30 CA016520).
This work was supported by National Institutes of Health Grants R01 GM060293 (to R. M.) and T32 GM071339 (to R. S. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- KAT
- lysine acetyltransferase
- NAT
- N-terminal acetyltransferase
- MSRA
- methionine sulfoxide reductase A
- MLCK
- myosin light chain kinase
- SUMO
- small ubiquitin-like modifier
- βME
- β-mercaptoethanol
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Verdin E., and Ott M. (2015) 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 16, 258–264 [DOI] [PubMed] [Google Scholar]
- 2. Starheim K. K., Gevaert K., and Arnesen T. (2012) Protein N-terminal acetyltransferases: when the start matters. Trends Biochem. Sci. 37, 152–161 [DOI] [PubMed] [Google Scholar]
- 3. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., and Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 4. Forte G. M., Pool M. R., and Stirling C. J. (2011) N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS Biol. 9, e1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scott D. C., Monda J. K., Bennett E. J., Harper J. W., and Schulman B. A. (2011) N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science 334, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hwang C. S., Shemorry A., and Varshavsky A. (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mullen J. R., Kayne P. S., Moerschell R. P., Tsunasawa S., Gribskov M., Colavito-Shepanski M., Grunstein M., Sherman F., and Sternglanz R. (1989) Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 8, 2067–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Polevoda B., and Sherman F. (2003) N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 325, 595–622 [DOI] [PubMed] [Google Scholar]
- 9. Liszczak G., Goldberg J. M., Foyn H., Petersson E. J., Arnesen T., and Marmorstein R. (2013) Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat. Struct. Mol. Biol. 20, 1098–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Damme P., Evjenth R., Foyn H., Demeyer K., De Bock P. J., Lillehaug J. R., Vandekerckhove J., Arnesen T., and Gevaert K. (2011) Proteome-derived peptide libraries allow detailed analysis of the substrate specificities of N(α)-acetyltransferases and point to hNaa10p as the post-translational actin N(α)-acetyltransferase. Mol. Cell. Proteomics 10.1074/mcp.M110.004580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoon H., Kim H. L., Chun Y. S., Shin D. H., Lee K. H., Shin C. S., Lee D. Y., Kim H. H., Lee Z. H., Ryoo H. M., Lee M. N., Oh G. T., and Park J. W. (2014) NAA10 controls osteoblast differentiation and bone formation as a feedback regulator of Runx2. Nat. Commun. 5, 5176. [DOI] [PubMed] [Google Scholar]
- 12. Shin S. H., Yoon H., Chun Y. S., Shin H. W., Lee M. N., Oh G. T., and Park J. W. (2014) Arrest defective 1 regulates the oxidative stress response in human cells and mice by acetylating methionine sulfoxide reductase A. Cell Death Dis. 5, e1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shin D. H., Chun Y. S., Lee K. H., Shin H. W., and Park J. W. (2009) Arrest defective-1 controls tumor cell behavior by acetylating myosin light chain kinase. PloS ONE 4, e7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vetting M. W., Bareich D. C., Yu M., and Blanchard J. S. (2008) Crystal structure of RimI from Salmonella typhimurium LT2, the GNAT responsible for N(α)-acetylation of ribosomal protein S18. Protein Sci. 17, 1781–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liszczak G., Arnesen T., and Marmorstein R. (2011) Structure of a ternary Naa50p (NAT5/SAN) N-terminal acetyltransferase complex reveals the molecular basis for substrate-specific acetylation. J. Biol. Chem. 286, 37002–37010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liszczak G., and Marmorstein R. (2013) Implications for the evolution of eukaryotic amino-terminal acetyltransferase (NAT) enzymes from the structure of an archaeal ortholog. Proc. Natl. Acad. Sci. U.S.A. 110, 14652–14657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Magin R. S., Liszczak G. P., and Marmorstein R. (2015) The molecular basis for histone H4- and H2A-specific amino-terminal acetylation by NatD. Structure 23, 332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dörfel M. J., and Lyon G. J. (2015) The biological functions of Naa10: from amino-terminal acetylation to human disease. Gene 567, 103–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jeong J. W., Bae M. K., Ahn M. Y., Kim S. H., Sohn T. K., Bae M. H., Yoo M. A., Song E. J., Lee K. J., and Kim K. W. (2002) Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell 111, 709–720 [DOI] [PubMed] [Google Scholar]
- 20. Murray-Rust T. A., Oldham N. J., Hewitson K. S., and Schofield C. J. (2006) Purified recombinant hARD1 does not catalyse acetylation of Lys532 of HIF-1α fragments in vitro. FEBS Lett. 580, 1911–1918 [DOI] [PubMed] [Google Scholar]
- 21. Arnesen T., Kong X., Evjenth R., Gromyko D., Varhaug J. E., Lin Z., Sang N., Caro J., and Lillehaug J. R. (2005) Interaction between HIF-1 α (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 α. FEBS Lett. 579, 6428–6432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Daniel Ricketts M., Frederick B., Hoff H., Tang Y., Schultz D. C., Singh Rai T., Grazia Vizioli M., Adams P. D., and Marmorstein R. (2015) Ubinuclein-1 confers histone H3.3-specific-binding by the HIRA histone chaperone complex. Nat. Commun. 6, 7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yuan H., Rossetto D., Mellert H., Dang W., Srinivasan M., Johnson J., Hodawadekar S., Ding E. C., Speicher K., Abshiru N., Perry R., Wu J., Yang C., Zheng Y. G., Speicher D. W., Thibault P., Verreault A., Johnson F. B., Berger S. L., Sternglanz R., McMahon S. B., Côté J., and Marmorstein R. (2012) MYST protein acetyltransferase activity requires active site lysine autoacetylation. EMBO J. 31, 58–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rojas J. R., Trievel R. C., Zhou J., Mo Y., Li X., Berger S. L., Allis C. D., and Marmorstein R. (1999) Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nature 401, 93–98 [DOI] [PubMed] [Google Scholar]
- 25. Li Y., Zhang L., Liu T., Chai C., Fang Q., Wu H., Agudelo Garcia P. A., Han Z., Zong S., Yu Y., Zhang X., Parthun M. R., Chai J., Xu R. M., and Yang M. (2014) Hat2p recognizes the histone H3 tail to specify the acetylation of the newly synthesized H3/H4 heterodimer by the Hat1p/Hat2p complex. Genes Dev. 28, 1217–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gautschi M., Just S., Mun A., Ross S., Rücknagel P., Dubaquié Y., Ehrenhofer-Murray A., and Rospert S. (2003) The yeast N(α)-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol. Cell Biol. 23, 7403–7414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Allfrey V. G., Faulkner R., and Mirsky A. E. (1964) Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 51, 786–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baeza J., Smallegan M. J., and Denu J. M. (2015) Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem. Biol. 10, 122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Olia A. S., Barker K., McCullough C. E., Tang H. Y., Speicher D. W., Qiu J., LaBaer J., and Marmorstein R. (2015) Nonenzymatic protein acetylation detected by NAPPA protein arrays. ACS Chem. Biol. 10, 2034–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tahirov T. H., Inoue-Bungo T., Morii H., Fujikawa A., Sasaki M., Kimura K., Shiina M., Sato K., Kumasaka T., Yamamoto M., Ishii S., and Ogata K. (2001) Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFβ. Cell 104, 755–767 [DOI] [PubMed] [Google Scholar]
- 31. Lowther W. T., Brot N., Weissbach H., and Matthews B. W. (2000) Structure and mechanism of peptide methionine sulfoxide reductase, an “anti-oxidation” enzyme. Biochemistry 39, 13307–13312 [DOI] [PubMed] [Google Scholar]