

Abstract
Amyloids are highly ordered, cross-β-sheet-rich protein/peptide aggregates associated with both human diseases and native functions. Given the well established ability of amyloids in interacting with cell membranes, we hypothesize that amyloids can serve as universal cell-adhesive substrates. Here, we show that, similar to the extracellular matrix protein collagen, amyloids of various proteins/peptides support attachment and spreading of cells via robust stimulation of integrin expression and formation of integrin-based focal adhesions. Additionally, amyloid fibrils are also capable of immobilizing non-adherent red blood cells through charge-based interactions. Together, our results indicate that both active and passive mechanisms contribute to adhesion on amyloid fibrils. The present data may delineate the functional aspect of cell adhesion on amyloids by various organisms and its involvement in human diseases. Our results also raise the exciting possibility that cell adhesivity might be a generic property of amyloids.
Keywords: adhesion, amyloid, focal adhesions, integrin, protein self-assembly
Introduction
The extracellular matrix (ECM)5 composed of proteins, minerals, and carbohydrates (1) provides physical support to individual cells and facilitates interactions between cells and links them into functional tissue (2). Depending on the source of the tissue, the difference in the composition and organization of the proteins in the ECM dictate the physical properties of that particular tissue. For example, bones consist of highly mineralized ECM that supports and resist compression (3), whereas skin has dense interpenetrating matrix, which is elastic, strong, and regenerative (4). Cells within the tissue sense the physicochemical properties of the ECM and bind to each other through a gamut of different cell adhesion molecules, including integrins, cadherins, and trans-membrane proteoglycans, and regulate their own functions (5). These interactions between the cell and its surrounding ECM through integrins and other receptors directly control cell adhesion, cell proliferation, and tissue organization (6).
Amyloids are unique protein folds responsible for both disease and function in host organisms (7). Although the initial identification of amyloids emerged from their association with several human diseases, including Alzheimer disease and Parkinson disease, many amyloids with native functions in the host were also discovered from unicellular to multicellular organisms, including mammals (7, 8). For instance, in many organisms like Escherichia coli, yeast, and barnacle, amyloids help the host organism with their adhesion to surfaces (9–12). Recently in mammals, amyloids have been found to have a functional role in melanin synthesis and hormone storage (13, 14). Due to their self-replication capability (15, 16) and their function as biological and chemical catalysts (17), amyloid folds are now considered as one of the primitive protein folds (18, 19). Amyloid fibrils possess a highly repetitive structure along with unique surface properties consisting of a combination of charged and hydrophobic surfaces, depending on the sequence, making them sticky (20–22) and thereby enabling them to bind to both small molecules and large macromolecules/polymers (23–26). Various microorganisms, for their surface attachment and colonization, exploit this sticky property of amyloids. Amyloid fibrils have been reported to be a part of the biofilms in numerous microorganisms, including harpins of Xanthomonas campestris and Pseudomonas syringae (27), pili from Mycobacterium tuberculosis (28), chaplins from Streptomyces coelicolor (29), and hydrophobins from fungi (30). These amyloidogenic ECM components help these organisms to adhere onto the surface and form colonies. The above studies suggest that amyloids possess many ECM-like features and may be capable of supporting cell adhesion. In this context, amyloid fibrils functionalized with cell-adhesive RGD motifs were shown to support cell adhesion (31, 32). Recent studies also suggest that amyloid fibrils alone (without any functionalization) are also capable of supporting cell adhesion due to their unique nanotopographic features (33–37). However, it remains unclear whether this cell-adhesive property is dependent on the sequence composition or is a consequence of the amyloid nature. Here we demonstrate that irrespective of the sequence, amyloid fibrils are capable of supporting cell adhesion.
Experimental Procedures
Chemicals and Reagents
Unless specified, all chemicals and reagents were purchased from Sigma. Water was double-distilled and deionized using a Milli-Q system (Millipore Corp., Bedford, MA). All of the peptide hormones except human galanin and somatostatin were a kind gift from Prof. Roland Riek (ETH Zurich). Somatostatin was purchased from BACHEM, and human galanin peptides were custom synthesized by USV Ltd. (Mumbai, India) with >95% purity. Purity of all of these peptides was further confirmed by MALDI-TOF mass spectrometry.
Peptide/Protein Fibril Formation
To test the adhesion of cells on amyloid fibrils, the amyloid fibrils were prepared by dissolving the peptides of kassinin (2 mg/ml), GLP 1 (0.25 mg/ml), rat UCN (2 mg/ml), oCRF (2 mg/ml), glucagon (2 mg/ml), GIP (2 mg/ml), mouse UCN III (2 mg/ml), and Aβ(25–35) (1 mg/ml) in 5% d-mannitol with 0.01% sodium azide and incubated at 37 °C with slight rotation. The peptides of somatostatin (2 mg/ml), human GRF (2 mg/ml), bombesin (2 mg/ml), VIP (2 mg/ml), helodermin (2 mg/ml), GRP (2 mg/ml), galanin (1 mg/ml), β-endorphin (2 mg/ml), and Sub P (1 mg/ml) were also similarly dissolved in 5% d-mannitol with 0.01% sodium azide and incubated in the presence of 400 μm low molecular weight heparin at 37 °C in 1.5-ml Eppendorf tubes. α-Synuclein (α-Syn) protein was expressed and purified according to the protocol described by Volles and Lansbury (38) in E. coli BL21 (DE3) strain. For α-Syn, 30 mg/ml lyophilized protein was dissolved in 20 mm MES buffer, pH 6.0, and low molecular weight α-Syn was prepared by passing the dissolved protein through 100 kDa cut-off membrane as described before (39). The Eppendorf tubes containing peptide/protein solutions were placed into an EchoTherm model RT11 rotating mixture (Torrey Pines Scientific) at 50 rpm inside a 37 °C incubator. At suitable intervals, thioflavin T (ThT), circular dichroism (CD), and transmission electron microscopy (TEM) were performed to analyze the aggregation.
CD Spectroscopy
CD spectroscopy is a commonly used technique to monitor the secondary structural transitions during protein/peptide aggregation studies (40). To study the conformational changes during the aggregation of proteins/peptides, 15 μl of peptide solutions was diluted in 5% d-mannitol to 200 μl such that the final peptide concentration was of 20 μm. For α-Syn, 10 μl of protein solution was diluted in 20 mm MES buffer, pH 6.0, to 200 μl such that the final concentration was 15 μm. The protein/peptide solution was placed into a 0.1-cm path length quartz cell (Hellma, Forest Hills, NY), and the spectra were acquired using a JASCO 810 instrument. All measurements were done at 25 °C. Spectra were recorded over the wavelength range of 198–260 nm. Three independent experiments were performed with each sample. Raw data were processed by smoothing and subtraction of buffer spectra as per the manufacturer's instructions.
ThT Binding
ThT is an amyloid detection dye widely used to probe amyloid formation during protein aggregation (41). In order to track amyloid formation in the aggregating mixtures of proteins/peptides, a 10-μl aliquot of peptide/protein samples was diluted to 500 μl in 5% d-mannitol containing 0.01% (w/v) sodium azide such that the final concentration of the peptide/protein was of 8 μm. For α-Syn, 10 μl of protein solution was diluted to 500 μl such that the final concentration was of 6 μm. These solutions were then mixed with 2 μl of 1 mm ThT prepared in 10 mm Tris-HCl, pH 8.0. Fluorescence was measured immediately after the addition of ThT. The fluorescence experiment was carried out using a Fluoromax 4 spectrofluorometer (Horiba Jobin Yvon Inc.), with excitation at 450 nm and emission from 460 to 500 nm. The intensity values at 480 nm were plotted. Three independent experiments were performed for each sample.
TEM
To examine the morphology of the protein/peptide amyloid fibrils under EM, aliquots of peptide/protein samples were diluted in distilled water to obtain a final concentration of ∼50 μm. The diluted solutions were spotted on a glow-discharged, carbon-coated Formvar grid (Electron Microscopy Sciences, Fort Washington, PA) and incubated for 5 min. The grids were then washed with distilled water and stained with 1% (w/v) aqueous uranyl formate solution. Uranyl formate solution was freshly prepared and filtered through 0.22-μm sterile syringe filters (Millipore). EM analysis was performed using an FEI Tecnai G2 12-electron microscope at 120 kV with nominal magnifications in the range of 26,000–60,000. Images were recorded digitally by using the SIS Megaview III imaging system. At least two independent experiments were carried out for each sample. The images obtained were analyzed in ImageJ (National Institutes of Health) to calculate fibril diameter.
Aggregation and Amyloid Formation by BSA
It was suggested previously that BSA could form amyloid fibrils at low pH and in the presence of high salt conditions (42, 43). To further examine the conditions required for BSA amyloid formation, lyophilized BSA (Sigma) was dissolved in PBS, pH 7.4, with a concentration of 21 mg/ml, and the concentration was determined by UV absorption measurement at 280 nm, considering the molar absorptivity of BSA as 43,824 m−1 cm−1. The 300 μm stock solution of BSA was used for the further study. The 300 μm BSA and 5 m NaCl stock solutions were used to search the optimal conditions for BSA amyloid formation. The concentration of BSA in the reaction was varied from 5 to 100 μm using 20 mm glycine-NaOH buffer, pH 3.0, and the NaCl concentration was varied from 0 to 300 μm. All solutions were incubated at 65 °C, and the aggregation and amyloid formation were monitored using ThT binding studies (data not shown). The condition that showed the highest ThT binding was considered as the amyloid-forming condition and was used for further studies. The secondary structural changes were determined by CD spectroscopy, and morphological analysis was done using an electron microscope. Further amyloid formation was confirmed using Congo Red (CR) binding by UV spectral assay and birefringence study.
CR Binding Assay
CR is a dye routinely used for detecting amyloid formation in protein/peptide aggregation studies (44). In order to confirm the amyloid nature of BSA aggregates, CR binding studies were performed. For CR binding, a 10-μl aliquot of aggregated BSA (of 100 μm concentration) was mixed with 160 μl of PBS buffer containing 10% ethanol. Then 30 μl of 100 μm CR solution in PBS (containing 10% ethanol) was added. After a 5-min incubation in the dark at room temperature, CR absorbance was measured in the range of 300–700 nm using a JASCO V-650 spectrophotometer. Similarly, the CR alone spectrum was also recorded as a control. Three independent experiments were performed for each sample.
CR Birefringence
To further confirm the binding of CR to amyloid fibrils, CR birefringence was performed. 50 μl of incubated solutions of protein/peptide aggregates was subjected to ultracentrifugation (Optima Max-XP, Beckman Coulter) at 90,000 rpm for 1 h and was washed once by distilled water. The pellets obtained were then used for performing CR birefringence. To do that, pellets were stained with CR solution for 20 min during continuous vortexing. The mixtures were again centrifuged at 90,000 rpm for 1 h, and pellets were washed twice with 500 μl of 20% ethanol. The pellets were resuspended in Milli-Q water and then spread evenly onto glass slides and air-dried at room temperature. The slides were analyzed using a microscope (Olympus SZ61 stereo zoom) equipped with two polarizer equipped with a CCD camera.
Amyloid Formation by Polyamino Acids
The preparation of polyamino acid (PAA) amyloids was reported previously (45–47). Briefly, the 1 mg of lyophilized powder of positively charged poly-l-lysine (PLL) (Mr 70,000–150,000; Sigma) and negatively charged polyglutamic acid (PE) (Mr 2000–15,000; Sigma) were separately dissolved in 1 ml of sterile Milli-Q water. The pH of PLL solution was adjusted to 11.1 by adding a few drops of 10 mm NaOH, and the pH of PE was adjusted to pH 4.1 with dilute HCl. These freshly prepared PAAs were then incubated at 65 °C for 48 h. For preparing poly-l-alanine (PLA) (Mr 1000–5000; MP Biomedicals) amyloid, 2 mg of PLA was dissolved in 100 μl of trifluoroacetic acid (TFA) because it does not dissolve in most of the other solvents studied due to the highly hydrophobic nature of PLA. 10 μl of PLA solution was then gradually added into 190 μl of sterile Milli-Q water casted on a clean coverslip, and the entire setup was dried in steam. The resultant dried aggregates were collected and were used for the study. The secondary structure of PAA aggregates was analyzed using FTIR, and morphological analysis was done using an electron microscope. Further amyloid formation was confirmed using a CR binding by birefringence study and x-ray diffraction studies.
For secondary structural analysis of the aggregated PAAs, FTIR spectroscopy was performed. FTIR spectra of the PAAs were recorded using a Vertex-80 FTIR instrument (Bruker, Germany) equipped with a deuterated triglycine sulfate detector. 10 μl of each aggregated PAA was spotted and dried on translucent KBr pellet that was made prior to the experiment by compressing ground KBr powder. 10 μl of sterile Milli-Q water was spotted on another KBr pellet and used for the background spectrum. FTIR spectra (1500–1800 cm−1) were recorded as an average of 32 scans, and raw data corresponding to the amide-I region (1600–1700 cm−1) was deconvoluted by the Fourier self-deconvolution method. The deconvoluted spectra were then subjected to a Lorenzian curve-fitting procedure using Opus-65 software. Two independent experiments were performed for each sample.
For x-ray diffraction studies, sample solutions were dried either in glass capillary tubes or at the center of two glass rods arranged end to end. The resulting film was mounted parallel to the x-ray beam. The images were obtained using a Rigaku R Axis IV++ detector (Rigaku, Japan) mounted on a rotating anode. The sample to detector distance was 300 mm, and the image files were analyzed using Adxv software.
Amyloid Formation by Aβ Fibrils
Lyophilized powder of Aβ42 was a kind gift from Prof. Sudipta Maiti, Tata Institute of Fundamental Research (Navy Nagar, India). 1 mg of Aβ42 fibril was suspended in 50 mm sodium phosphate buffer, pH 7.4, and the peptide was dissolved by adding a few drops of 10 mm NaOH into it, after which the pH of the solution was brought back to pH 7.4. The final concentration of this solution was 300 μm.
Aβ(25–35) was purchased from BACHEM. For amyloid fibril preparation, 0.5 mg of this peptide was dissolved in 500 μl of sterile Milli-Q water to obtain 1 mm solution. Both of the Aβ solutions were incubated at 37 °C, and the aggregation and amyloid formation was monitored using ThT binding studies (data not shown). The secondary structural changes were determined by CD spectroscopy (data not shown), and morphological analysis was done using an electron microscope.
Biocompatibility Assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was carried out to evaluate the toxicity of all amyloid fibrils formed in presence and absence of heparin using neuronal cell line SH-SY5Y. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (HiMedia, Mumbai, India) medium supplemented with 10% FBS (Invitrogen), 100 units/ml penicillin, and 100 μg/ml streptomycin in a 5% CO2 humidified environment at 37 °C. Cells were seeded at a density of 1 × 104 cells/well on a 96-well plate. After 24 h of incubation, the old culture medium was replaced with fresh medium along with the addition of various amyloid fibrils at 10 μm final concentration, and cells were further incubated for 24 h at 37 °C. After incubation, 10 μl of a 5 mg/ml MTT stock in PBS was added to each well, and the incubation was continued for 4 h. Finally, 100 μl of a solution containing 50% dimethylformamide and 20% SDS (pH 4.7) was added to each well and incubated. After overnight incubation in a 5% CO2 humidified environment at 37 °C, absorption values at 560 nm were determined with an automatic microplate reader (Spectramax M2, Molecular Devices).
Surface Functionalization
For evaluation of cell adhesion on amyloid fibrils, the fibrils were coated on glass coverslips. To do so, round glass coverslips of 12-mm diameter were first washed with 0.1 n NaOH for 30 min and then rinsed with PBS and subsequently sterilized. All coverslips were kept in 24-well plates and coated subsequently with different substrates. Glass coverslips were coated with 10 μg/cm2 of collagen and each amyloid fibril. For coating, each fibril solution was diluted in PBS, pH 7.4, and applied to the surface of the coverslip and kept at 4 °C overnight. The next day, excess solution was removed, and the surfaces were washed with PBS and blocked with 2% F127 Pluronic (Sigma) for 15 min at room temperature before plating the cells.
Cell Culture
Mouse fibroblasts (L929 and NIH 3T3), neuroblastoma cell SH-SY5Y, and PC12 cells were obtained from the National Centre For Cell Science (Pune, India) and maintained as per established protocols. All cells were maintained at 37 °C with 5% CO2 and plated at a density of 4 × 104 cells/well.
For density-dependent experiments, glass coverslips were coated with kassinin at concentrations ranging from 0.5 to 7.5 μm at 37 °C for 2 h, blocked with 2% F127 Pluronic (Sigma) for 15 min at room temperature to prevent nonspecific cell attachment, and UV-sterilized. NIH 3T3 fibroblasts were cultured for 24 h and plated at a seeding density of 4500 cells/cm2 on the coated substrates. For drug studies, cells were cultured for 24 h and then exposed to 10 μm (for NIH 3T3) and 1 μm (for SH-SY5Y) Y27632 or ML7 (Calbiochem) for 1 h. For integrin-blocking experiments, ∼80 cells/μl of medium were treated with anti-integrin β1 antibody (1:100; clone P4C10, Millipore) for 15 min at 37 °C and subsequently plated on fibril-coated substrates. Cells were allowed to settle for 1 h at 37 °C and processed for imaging.
For RGD-mediated integrin blocking experiments, the 48-well plates were coated with fibronectin (5 μg/cm2), collagen, monomer, and fibril of kassinin at a density of 10 μg/cm2 and incubated overnight at 4 °C. On removing the solution, these wells were blocked with 2% Pluronic for 15 min and UV-sterilized. NIH 3T3 cells were harvested using 0.5% trypsin, seeded at a density of 4000 cells/cm2, and incubated for 2.5 h in DMEM containing 10% FBS at 37 °C with 5% CO2. After incubation, the cells were washed with 1× PBS and incubated with 0.2 mg/ml GRGDSP (Sigma) in DMEM for 2.5 h at 37 °C, after which the floating cells were removed using a PBS wash. All of the wells were imaged at ×10 magnification, cells from 10 random fields were counted, and the cell spread area for each condition was analyzed.
Quantification of Cell Shape and Cell Area
For evaluation of cell adhesion, the glass coverslips in 24-well plates were gently rinsed with PBS to remove unattached cells. Subsequently, the coverslips were imaged in phase contrast (Olympus XL51/IX71) at ×10 magnifications. The cell shape analysis was performed using ImageJ software. Cell morphology (spread area and circularity) was quantified by analyzing ∼100 cells/condition from two independent experiments.
For cell migration studies, cells were cultured on 35-mm Petri dishes with culture medium supplemented with 20 mm HEPES, pH 7.2. Petri dishes were placed on a custom-made temperature controller maintained at 37 °C. Phase-contrast images were captured at 10-min intervals under ×20 magnification for 3 h using a Nikon Ti Eclipse inverted microscope coupled with NIS Elements BR (version 4.20.00) image acquisition software. All of the frames were analyzed using the manual tracking plugin in ImageJ. For quantifying cell speeds, at least 30 cells were analyzed per condition across two independent experiments.
Immunocytochemistry
To study the focal adhesion complex formation on adhered cells, immunocytochemistry was performed. To do that, cells were cultured on different substrates, and after 24 h, cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min at 37 °C. After rinsing with PBS, cells were permeabilized by 0.2% Triton X-100, blocked with 5% BSA, and incubated with rabbit anti-focal adhesion kinase (anti-FAK) (Sigma; 1:350 dilution)/mouse anti-β1 integrin (Santa Cruz Biotechnology, Inc.; 1:250 dilution) antibody at 4 °C overnight for visualizing focal adhesions. After washing off the primary antibody, cells were incubated with either donkey anti-rabbit secondary antibody or donkey anti-mouse secondary antibody (Molecular Probes; 1:500 dilution) and Phallodin (Molecular Probes; 1:300 dilution) for 1 h at room temperature. Subsequently, samples were washed with PBS and stained with DAPI for nuclear staining (Molecular Probes; Invitrogen). Finally, the coverslips were mounted using Cytoseal (Sigma) and observed under fluorescence microscope (Zeiss Axiocam MRM). The distributions of focal adhesions/integrins were processed and calculated using ImageJ.
RT-PCR
For studying the integrin expression profile of cells grown on various substrates, RT-PCR experiments were performed. For this, 35-mm Petri dishes were coated with 10 μg/cm2 of each amyloid/ECM substrate and kept for overnight incubation at 4 °C. The next day, excess solution was removed, and the surfaces were washed with PBS before plating the cells. One million cells/dish were plated on each condition, and cells were harvested for RNA extraction after 24 h in culture. RNA was extracted by the TRIzol (Ambion) method according to the manufacturer's protocol. cDNA preparation was carried out using a first strand cDNA synthesis kit from Fermentas (Thermo Scientific), and PCR was performed using a kit from Novagen (Millipore) according to the manufacturer's instructions. GAPDH primers were used from the same kit. Integrin primers were custom-manufactured by Sigma with the following sequences: β1 integrin, 5′-ACG CCG CGC GGA AAAGAT GA-3′ (forward) and 5′-GCA CCA CCC ACA ATT TGG CCC T-3′ (reverse); β3 integrin, 5′-GGG GAC TGC CTG TGT GAC TC-3′ (forward) and 5′-CTT TTC GGT CGT GGATGG TG-3′ (reverse).
RBC Adhesion Study
For experiments with red blood cells (RBCs) (48), 1 μl of packed blood was collected from healthy individuals and added to 1.5 ml of 1× PBS (pH 7.4) and seeded on precoated wells at a seeding density of 30,000 cells/well in a 96-well plate. The wells that were coated with 0.01% poly-l-lysine and 2% Pluronic served as the positive and negative controls, respectively. After seeding the wells with the desired number of cells, the plates were incubated at room temperature for 30 min and taken for imaging. RBC adhesion was quantified by imaging 10–12 fields/well at ×40 magnifications. The morphology of the cells was analyzed, and representative images were used for calculating spreading area. The image analysis was done by ImageJ software.
De-adhesion Experiments
De-adhesion experiments were conducted as reported previously (49). Briefly, cells were mounted on the microscope stage (Olympus), washed with 1× PBS, and incubated with 0.25% trypsin-EDTA (HiMedia), and images were captured at 2-s intervals at ×10 magnification until the cells rounded up but remained attached to the substrate. Images were processed in ImageJ to obtain the experimental de-adhesion time. For quantifying the influence of fibril density on de-adhesion time, at least 30 cells across two independent experiments were analyzed.
Statistical Analysis
The statistical significance was determined by either Student's t test or one-way analysis of variance followed by the Newman-Keuls multiple comparison post hoc test. p values for each plot (* and **) are mentioned in the corresponding figure legends.
Results
Amyloids Support Mammalian Cell Adhesion
To address whether amyloid fibrils, irrespective of their sequence and composition, can support cell adhesion, more than 20 proteins/peptides possessing different sequences and length were chosen. These peptides were also previously shown to form non-toxic amyloids in vitro, many of which are suggested to be the storage state in secretory granules (14, 50, 51). Because most of these proteins/peptides were non-toxic, we chose these peptides for our cell adhesion study. All of the peptides were found to be unstructured in solution immediately after dissolution (day 0) as observed by CD. However, after 15 days of incubation, all of them underwent structural transition predominantly to β-sheet-rich structures except for Sub P and somatostatin (Fig. 1). Sub P and somatostatin were previously reported to form unusual structures after amyloid formation (14, 50, 52). Amyloid formation was probed using a ThT fluorescence study. ThT is an amyloid-detecting dye, which mostly binds to cross-β-sheet-rich amyloids but not their monomeric counterparts (41). An increase in ThT binding of 2-week incubated proteins/peptides suggests amyloid formation by these proteins/peptides (Fig. 2). Only kassinin did not show any significant ThT binding even after 15 days of incubation (ThT fluorescence at day 15 was almost identical to that of the day 0 sample) (Fig. 2). This observation is consistent with a previous study (52). The negligible ThT binding even after amyloid formation was also evident previously for many other proteins/peptides (53–55). The amyloid morphology of the aggregates was confirmed using electron microscopy, wherein all of the peptide/protein aggregates showed fibrillar morphology (Fig. 3). Thus, the fibril formation was verified by monitoring aggregation using CD spectroscopy, binding to amyloid-specific dye ThT (except for kassinin) and TEM imaging (Table 1). Finally, the toxicity of different fibrils was evaluated using an MTT assay. Most of the fibrils were found to be non-toxic to cells, consistent with the previous study (14) (Fig. 4). Only a few peptide fibrils showed higher than 30% cellular toxicity (Table 1).
FIGURE 1.

Secondary structural transition of peptides/proteins during amyloid formation. CD spectra of freshly solubilized peptide/protein showing secondary structure at day 0 (red) and conversion to mostly β-sheet-rich structure after 15 days of incubation (blue).
FIGURE 2.

Amyloid formation by peptide/proteins using ThT fluorescence. ThT binding of peptide/proteins showing increased ThT fluorescence after 15 days of incubation. Error bars, S.D. of two independent experiments.
FIGURE 3.

Morphology of peptide/protein aggregates. TEM images of protein/peptide aggregates formed after 15 days of incubation showing fibrillar morphology. Scale bars, 200 nm.
TABLE 1.
Sequences and physical properties of amyloid fibrils under study
| Peptide/Protein | Amino acid sequence | Secondary structure |
ThT Binding (D15) | Fibril Diameterc | Toxicity | Cell adhesion (cell spreading area)d |
||
|---|---|---|---|---|---|---|---|---|
| D0a | D15b | Monomers | Fibrils | |||||
| nm | μm2 | |||||||
| GLP 1 | HDEFERHAEGTFTSDVSSYL EGQAAKEFIAWLVKGRG | RCe | β-Sheet | + | 23 | Non-toxicf | 745 | 1308 |
| Kassinin | DVPKSDQFVGLM | RC | β-Sheet | + | 16 | Non-toxic | 1276 | 2205 |
| Sub P | RPKPQQFFGLM | RC | Helix | + | 14 | Non-toxic | 1699 | 2530 |
| Galanin | GWTLNSAGYLLGPHAVGNHRSFSDKNGLTS | RC | β-Sheet | + | 8 | Non-toxic | 1936 | 2484 |
| Vasoactive intestinal peptide | HSDAVFTDNYTRLRKQMAVKKYLNSILN | RC | β-Sheet | + | 7 | Non-toxic | 1401 | 2429 |
| Rat UCN | DDPPLSIDLTFHLLRTLLELARTQSQRERAEQNRIIFDSV | RC | β-Sheet | + | 25 | Non-toxic | 1409 | 2190 |
| Mouse UCNIII | FTLSLDVPTNIMNILFNIDKAKNLRAKAAA NAQLMAQI | RC | β-Sheet | + | 9 | Mildly toxicg | 1255 | 2516 |
| Ovine corticotropin-releasing factor | SQEPPISLDLTFHLLREVLEMTKADQLAQQAHSNRKLLDIA | RC | β-Sheet | + | 8 | Toxich | 1545 | 2272 |
| Somatostatin | AGCKNFFWKTFTSC | RC | β-Sheet | + | 11 | Non-toxic | 945 | 1088 |
| Helodermin | HSDAIFTQQYSKLLAKLALQKYLASILGSRTSPPP | RC | β-Sheet | + | 14 | Non-toxic | 1346 | 2158 |
| Glucagon | HSQGTFTSDYSKYLDSRRAQDFVQWLMNT | RC | β-Sheet | + | 11 | Toxic | 1289 | 2582 |
| Growth hormone releasing factor | YADAIFTNSYRKVLGQLSAR KLLQDIMSRQQGESNQERGA | RC | β-Sheet | + | 15 | Non-toxic | 1343 | 2321 |
| Glucose-dependent insulinotropic polypeptide | YAEGTFISDYSIAMDKIHQQDFVNWLLAQKGKKNDWKHNI TQ | RC | β-Sheet | + | 17 | Non-toxic | 1324 | 2295 |
| Bombesin | QRLGNQWAVG HLM | RC | β-Sheet | + | 12 | Non-toxic | 1288 | 2696 |
| Gastrin releasing peptide | VPLPAGGGTVLTKMYPRGNHWAVGHLM | RC | β-Sheet | + | 10 | Non-toxic | 1118 | 2104 |
| β-Endorphin | YGGFMTSEKSQTPLVTLFKNAIIKNAYKKG E | RC | β-Sheet | + | 13 | Non-toxic | 2064 | 2871 |
| α-Synuclein | See Ref. 104. | RC | β-Sheet | + | 18 | Mildly toxic | 1430 | 2610 |
| BSA | See NCBI Accession no. CAA76847. | Helix | β-Sheet | + | 11 | Non-toxic | 1354 | 1665 |
| Aβ42 | See NCBI Accession no. 2BEG_E) | β-Sheet | + | 9 | Toxic | NDi | 1924 | |
| Aβ(25–35) | GSNKGAIIGLM | β-Sheet | + | 12 | Toxic | ND | 1858 | |
| PLL | Poly-l-lysine | RC | β-Sheet | − | 10 | Non-toxic | 4269 | 5347 |
| PE | Polyglutamic acid | RC | β-Sheet | + | 3 | Non-toxic | 2347 | 3441 |
| PLA | Poly-l-alanine | RC | β-Sheet | − | 22 | Non-toxic | 1071 | 1291.4 |
a Day 0.
b Day 15.
c Averaged fibril diameter.
d Averaged spreading area.
e RC, random coil.
f Nontoxic, 85–100% viability.
g Mildly toxic, 70–85% viability.
h Toxic, <70% viability.
i ND, not done.
FIGURE 4.
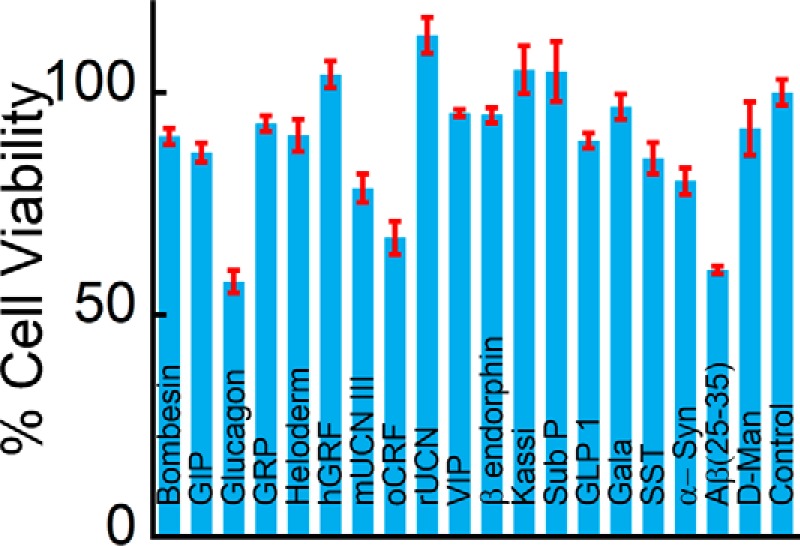
Cytotoxicity of the amyloid fibrils. MTT assay showing cytotoxicity of amyloid fibrils for SH-SY5Y cells. Most of the amyloid fibrils were non-toxic to the cells except for Aβ(25–35) and glucagon fibrils. Error bars, S.D.
The adhesiveness of the different fibrils was probed by plating an equal number of SH-SY5Y cells on coverslips coated with fibrils at identical density and quantifying cell attachment and spreading. Coverslips coated with the ECM protein collagen I at identical density served as control. After 24 h in culture, whereas cell attachment was observed across all the conditions (Fig. 5), higher attachment was observed on many of the amyloid fibrils compared with that on collagen (Fig. 6A). In particular, cell attachment was observed nearly 1.5-fold higher on kassinin fibrils compared with that on collagen. Further, the cell spreading area was calculated using ImageJ software to evaluate the cell adhesion on various surfaces. The study showed that on most of the amyloid fibrils, cell spreading was as efficient as that on collagen, with maximal spreading observed on β-endorphin (β-end) and bombesin fibril-coated surface (Fig. 6B). When we compared the cell spreading on monomer and fibril surfaces, we observed that cell spreading was significantly higher on fibrils than on their corresponding monomeric counterparts (Fig. 6B). Moreover, we also found that adhesion to amyloid fibrils was not specific to SH-SY5Y neuronal cells but was also observed with other cell types, including L929 fibrosarcoma cells, NIH 3T3 fibroblasts, and PC12 cells (Fig. 7, A and B). Although these cells were able to adhere and spread on kassinin amyloid fibrils similar to collagen, differences in spreading area were present among the different cell types (Fig. 7B). This variation could be due to differences in cell lineage and their different substrate requirement. However, the study indicates the favorable response of cells to the amyloid surface and suggests that adhesion to amyloid fibrils is not cell type-specific but rather is generic for most mammalian cell types.
FIGURE 5.

Cell adhesion on amyloid fibrils. Representative phase-contrast images of SH-SY5Y cells that were cultured on various protein/peptide amyloid fibrils showing robust cell adhesion on the substrate. Scale bar, 100 μm.
FIGURE 6.

Amyloid fibrils support cell adhesion and spreading. A, higher cell attachment of SH-SY5Y cells was observed on most of the amyloid fibril surfaces compared with that on collagen. Error bar, S.E. B, cell spreading analysis showing statistically higher spreading on different amyloid surfaces than on collagen and its corresponding monomers. Statistical significance was as follows: p < 0.005 (**) and p < 0.05 (*).
FIGURE 7.

Adhesion of various cell types on collagen and kassinin amyloid fibrils. A, phase-contrast images of PC12, NIH3T3, and L929 cells adhered on kassinin amyloid and collagen after 24 h of culture. Scale bars, 100 μm for both the top and bottom panels. B, spreading of PC12 cells, shown to be unchanged across the different conditions, whereas the other cell types spread more on kassinin substrates compared with collagen. *, statistical significance (p < 0.01).
Whereas the above studies were done at constant coating density, to understand the influence of fibril density on cell spreading and motility, NIH 3T3 fibroblast cells were seeded on varying coating density of fibril created by dilution of the kassinin fibril stock. The study suggests that, similar to the effect of ECM density on cell spreading, NIH 3T3 fibroblasts exhibited a biphasic spreading response on coverslips coated with kassinin at varying fibril density, with maximal spreading observed at a coating density of 1.5 μm (Fig. 8, A (top) and B).
FIGURE 8.

Adhesion and motility of NIH 3T3 fibroblasts on kassinin at different fibril densities. A, phase-contrast image of NIH3T3 cells adhered on kassinin amyloid at different fibril densities, in the presence and absence of serum after 24 h in culture. Scale bar, 100 μm. B, quantification of cell spreading on kassinin substrates in the presence (+S) and absence (−S) of serum. Whereas cells were found to spread extensively in the presence of serum across the different conditions, cells remained rounded when serum was absent, suggesting that although serum proteins are needed for cell spreading, amyloid topography by itself is sufficient for cell adhesion. C, random cell motility of fibroblasts on glass coverslips coated with kassinin at varying fibril densities. Cell speed was maximum on 1.5 μm kassinin-coated substrates.
Further, to probe the role of serum protein in cell adhesion on amyloids, glass coverslips were coated with varying density of kassinin fibrils, and 3T3 fibroblasts were cultured on them in serum-free medium. In serum-free medium, cells attached but remained rounded, suggesting that although the amyloid nanotopography is sufficient for initial cell adhesion (36), the serum proteins are required for cell spreading (Fig. 8, A (bottom) and B). Further, random cell motility on these substrates closely mirrored the cell spreading response, with maximum migration speeds observed on the same coating density corresponding to maximal spreading (Fig. 8C). Together, these results suggest that amyloid fibrils support attachment and spreading of a diverse range of cell types, with tuning of amyloid fibril density allowing for modulation of cell spreading and motility.
Lipid-Fibril Interactions and Integrin-based Adhesions Contribute to Cell Spreading on Amyloid Fibrils
To obtain further insights into the attachment and spreading of cells on amyloid surfaces, cytoskeletal organization and focal adhesion structures (56) were tracked in SH-SY5Y cells cultured on collagen and kassinin-coated substrates at identical coating density. Although cytoskeleton is important for cell shape and their spreading on ECM matrix (57), the extent and size of the focal adhesion complex will dictate the robustness in adhesion of cells on the surfaces (58). Our data showed that although no drastic differences were observed in the pattern of stress fibers visualized by F-actin staining, a difference in FAK staining was observed in cells attached on collagen and amyloid surfaces (Fig. 9A). FAK is known to be involved as a primary integrin effecter and is considered as a measure of cell spreading and migration (59–61). FAK and other focal adhesion proteins generally show a well distributed cytoplasmic staining pattern in adhered cells (62–64). A similar distribution of FAK was also observed in our study for the cells cultured on amyloid surfaces, indicating strong cell adhesion. In contrast, a strong perinuclear FAK staining was observed in cells cultured on monomers of these peptides (Fig. 9B). Further, quantitative analysis of FAK-positive focal adhesions of cells on collagen and amyloid surfaces using ImageJ software revealed the prevalence of larger adhesion complexes on selected amyloid fibrils of kassinin and Sub P compared with their corresponding monomers and on collagen (Fig. 9C). The presence of more and larger FAK-positive adhesions and the higher cell spreading area in cells cultured on amyloid fibrils suggest that the attachment of cells is integrin-mediated.
FIGURE 9.

Attachment and cytoarchitecture of SH-SY5Y cells on collagen and amyloid fibrils. A, cytoskeletal organization and focal adhesion formation in SH-SY5Y cells on collagen and amyloid fibrils. Phalloidin (green), FAK (red), and DAPI (blue) were used to visualize F-actin, focal adhesions, and nuclei, respectively. Scale bar, 20 μm. B, cytoskeletal organization and focal adhesion formation in SH-SY5Y cells on kassinin and Sub P monomers. Phalloidin (green), FAK (red), and DAPI (blue) staining showing organization of F-actin, focal adhesions, and nuclei in the immunofluorescence study, respectively. Scale bar, 20 μm. Perinuclear staining of FAK was observed in cells grown on both monomers of kassinin and Sub P. C, quantitative analysis of the size and number of FAK-positive focal adhesions. The higher number and larger size of focal adhesion complex are shown in the case of kassinin and Sub P fibrils relative to collagen. Statistical significance (*, p < 0.05) was determined by Student's t test. D, β1 and β3 integrin expression profile in SH-SY5Y cells cultured on uncoated, collagen-coated, and kassinin fibril-coated glass coverslips. The mRNA expression of β1 integrin was highest in cells cultured on kassinin fibrils. E, localization of β1 integrin of cells grown on uncoated glass, collagen, and kassinin amyloid surfaces. Scale bar, 20 μm. Increased β1 integrin staining and clustering (white arrows) were found in cells cultured on amyloid surfaces. F, clustering of β1 integrins in SH-SY5Y cells (quantified from fluorescence intensity) is shown to be higher on kassinin substrates compared with collagen. Error bars, S.D. **, p < 0.005.
To directly study the role of integrin in cell adhesions on amyloid fibrils, expression levels of β1 and β3 integrin were studied. The expression of β1 and β3 integrins and the localization of β1 integrin at focal adhesions in SH-SY5Y cells were found to be higher on kassinin amyloid surfaces (Fig. 9, D–F). To further probe whether integrin engagement is a consequence of attachment to amyloid fibrils or is required for cell spreading on amyloids, integrin-blocking experiments were performed in which fibroblasts were incubated for 30 min with integrin-blocking antibody P4C10 and then plated on collagen and kassinin amyloid substrates. After 1 h of incubation, cell spreading was reduced by ∼50% in P4C10-treated cells compared with untreated controls on both collagen and kassinin substrates (Fig. 10, A and B). The data indicate that β1 integrins at least partly modulate cell spreading on amyloid fibrils similar to collagen.
FIGURE 10.

Lipid-fibril interactions and integrin-based adhesions contribute to cell spreading on amyloid fibrils. A and B, spreading of NIH 3T3 fibroblasts on collagen and kassinin substrates after incubation with β1 integrin-blocking antibody (P4C10). Cell spreading was reduced in integrin-blocked samples on both collagen and kassinin. *, statistical significance (p < 0.05). Error bars, S.E. C, phase-contrast images of cells with and without RGD-treated cells. D and E, quantification of number of cells adhered (D) and spread area (E) of RGD-treated and untreated cells grown on various substrates. F, schematic of an experiment testing spreading of RBCs on kassinin-coated substrates. RBCs were allowed to attach and spread on kassinin-coated substrates for 1 h and then washed to remove the loosely attached cells. G, phase-contrast images of RBCs attached to kassinin-coated substrates. Scale bar, 50 μm. H, quantification of RBC attachment on coverslips coated with Pluronic (Plu), poly-l-lysine, and kassinin. RBCs spread on kassinin substrates in a density-dependent manner. Error bar, S.E.
To further confirm the role of integrins in cell adhesion, we studied the effect of RGD, an integrin ligand to block cell adhesion of adhered cells (65). RGD has a high affinity toward several integrin receptors and plays a major role in mediating cell adhesion (66–68). Treating adherent cells with RGD can temporarily disengage integrins and cause cells to de-adhere (69). In these experiments, NIH 3T3 fibroblasts were first allowed to adhere and spread on monomeric and amyloid forms of kassinin-coated surfaces for 2.5 h. Cells cultured on coated surface of collagen and fibronectin were used as positive controls. Subsequently, the adhered cells were treated with 200 μg/ml GRGDSP peptide for another 2.5 h. We found that more than half of cells de-adhered after this treatment when compared with the control where no RGD was added (Fig. 10, C–E). Additionally, for the cells that remained attached to the substrate, the cell spreading area was also reduced considerably when compared with untreated cells. The mean cell spreading area of RGD-treated cells adhered on kassinin amyloid fibrils was less than that of cells adhered on kassinin monomers (Fig. 10, C–E). Together, these results demonstrate the role of integrins in mediating cell adhesion and spreading on amyloid surfaces.
Further, to test whether amyloids can facilitate any mode of adhesion other than integrin-based cell adhesion, experiments were performed with human RBCs, which lack any integrin machinery (70, 71). Kassinin fibril-coated substrates were incubated with freshly isolated RBCs for 30 min, given a brief wash, and processed for imaging to check for the attachment (Fig. 10F). Pluronic (Plu)- and PLL-coated substrates were used as negative and positive controls, respectively. Whereas attachment on pluronic-coated substrates was negligible, cells attached robustly on PLL-coated substrates as expected, through charge-based interactions (71). Interestingly, RBCs attached to kassinin substrates in a fibril density-dependent manner (Fig. 10G), with attachment increasing with fibril density (Fig. 10H). Because RBCs are devoid of integrins, we propose that attachment of RBCs on amyloid could occur through cell membrane-amyloid interactions. Collectively, these data suggest that amyloids are capable of supporting the attachment of both adherent and non-adherent cells through a combination of membrane-fibril interactions and integrin-based focal adhesions.
Rho Kinase Signaling Modulates Cell Contractility on Amyloids
Given that the focal adhesions were observed in adherent cells both on collagen and on amyloid fibrils, we next probed whether the downstream signaling pathways that modulate focal adhesion dynamics were similar on collagen and amyloid fibrils. Specifically, we focused our attention on Rho-associated kinase (ROCK) and myosin light chain kinase (MLCK), both of which regulate cellular processes by modulating the activity of myosin II (72). To delineate the contributions of ROCK and MLCK pathways, experiments were performed with NIH 3T3 fibroblasts and SH-SY5Y cells cultured on collagen and kassinin amyloid fibrils in the presence of the ROCK inhibitor Y-27632 and the MLCK inhibitor ML7. For NIH 3T3 fibroblasts, ML7 and Y-27632 treatment led to significantly reduced cell spreading on kassinin fibrils but not on collagen (Fig. 11, A and B). However, drastic alterations in cell shape were observed in Y-27632-treated cells on both kassinin fibril and collagen substrates.
FIGURE 11.

Modulation of spreading and contractility by ROCK and MLCK signaling pathways in NIH 3T3 cells. A, phase-contrast images of NIH 3T3 fibroblast cells cultured on collagen and kassinin substrates for 24 h and subsequently treated with and without 10 μm ML7 (MLCK inhibitor) or 10 μm Y27632 (ROCK inhibitor) for 1 h. Scale bar, 40 μm. B, quantification of cell spreading area in drug-treated cells compared with controls. *, statistical significance (p < 0.05). Error bar, S.E. C, schematic of trypsin de-adhesion assay. Cells were washed with PBS, incubated with warm trypsin, and imaged by time lapse microscopy until cells became rounded (but remained attached to the substrate). De-adhesion time is an indirect measure of cell contractility. D, representative phase-contrast images of cells rounding up upon incubation with warm trypsin. Scale bar, 50 μm. E, de-adhesion times of control and drug-treated cells cultured on collagen and kassinin. Both ML7 and Y-27632 treatment are shown to delay de-adhesion on collagen substrates, whereas on kassinin substrates, cells were sensitive to Y-27632 treatment only. *, statistical significance (p < 0.05). Error bar, S.E.
Next, to test the relative contributions of the MLCK and ROCK pathways to cell contractility, trypsin-induced de-adhesion experiments were performed with SH-SY5Y and NIH 3T3 cells, where both cells were incubated with warm trypsin and imaged under a phase-contrast microscope until cells became rounded up but remained attached to the substrate (Fig. 11C), with the rounding off times directly set by actomyosin contractility (49). Interestingly, although both Y-27632 and ML7 treatment led to delayed de-adhesion on collagen substrates, only Y-27632 treatment led to delayed de-adhesion on kassinin substrates, with ML7 treatment causing no difference (Fig. 11, D and E). In SH-SY5Y neuronal cells, similar to NIH 3T3 fibroblasts, Y-27632 treatment also inhibited spreading of cells on collagen and kassinin substrates. However, ML7 treatment had no effect on cell spreading on both collagen and kassinin substrates (Fig. 12, A and B). Moreover, the cell contractility experiments on SH-SY5Y cells adhered on amyloid fibrils and collagen also showed results similar to those of NIH 3T3 cells (Fig. 12C), suggesting that cell adhesion on amyloid fibrils might be modulated by common signaling pathways in different cell lines. Together, these results indicate that although relative contributions of ROCK and MLCK pathways to cell contractility differ on collagen and amyloids, both are involved in cell spreading on collagen and amyloids.
FIGURE 12.

Modulation of spreading and contractility by ROCK and MLCK signaling pathways in SH-SY5Y cells. A, phase-contrast images of SH-SY5Y cells after 24 h of culture on collagen and kassinin substrates after treatment with 1 μm ML7 (MLCK inhibitor) or 1 μm Y27632 (ROCK inhibitor) for 1-h duration. Scale bar, 20 μm. B, quantification of cell spreading area in drug-treated cells compared with controls. Error bars, S.E. Statistical significance, **, p < 0.005. C, de-adhesion times of control and drug-treated cells cultured on collagen and kassinin. Error bars, S.E. (**, p < 0.005). Whereas both ML7 and Y-27632 treatment led to delayed de-adhesion on collagen substrates, cells were sensitive to ML7 treatment only on kassinin substrates.
Amyloid Version of a Non-adhesive Protein Is Also Capable of Supporting Cell Adhesion
If most of the peptides used in our studies do not possess direct integrin binding sites, then how do amyloid fibrils modulate these complex cellular processes in such a robust manner? We hypothesize that the amyloid form of any peptide/protein and higher order cross-β-sheet organization is sufficient to support cell adhesion. This may be achieved through both membrane-fibril interactions and integrin signaling. To probe this directly, cell studies were performed on monomer and amyloid forms of BSA, which, in its monomeric form, is routinely used to prevent nonspecific adhesion (73). To prepare BSA amyloids, 300 μm BSA was freshly prepared in PBS, pH 7.4, and the solution was further diluted to 100 μm in 20 mm glycine-HCl buffer, pH 3.0. The solution was treated with 300 mm NaCl and incubated at 65 °C for 1 h for BSA fibril formation. As before, formation of BSA fibrils was monitored using CD, ThT, and CR binding and electron microscopy (Fig. 13). The freshly dissolved BSA monomer showed CD spectrum characteristics of helical structure with two distinct minima, one at 220 nm and the other at 208 nm, whereas BSA incubated with 300 mm NaCl for 1 h at 65 °C showed a spectrum with a single minimum at ∼218 nm, indicating the presence of β-sheet (Fig. 13A). The amyloid formation by BSA was further analyzed by ThT and Congo Red binding studies. Both ThT and CR are known to bind mostly to the amyloid form of the protein, not to the corresponding monomeric counterpart (14). BSA aggregate formed in 20 mm glycine-HCl, pH 3.0, in the presence of NaCl showed higher ThT fluorescence at 480 nm compared with its monomer (Fig. 13B). Moreover, the BSA aggregates formed in the presence of 300 mm NaCl at pH 3.0 showed an increase in CR absorbance (Fig. 13C), and these aggregates showed green-gold birefringence when observed under cross-polarized light, which is typically observed in β-sheet-rich amyloid aggregates (Fig. 13D). Finally, the morphology of these BSA aggregates was analyzed using electron microscopic studies, which showed long fibrillar aggregates. However, BSA monomer showed only small amorphous aggregates (Fig. 14A, top). Thus, the CD spectra, ThT, and CR binding study and the fibrillar morphology of the incubated BSA aggregates under TEM are indicative of amyloid formation by BSA in the presence of salt at low pH.
FIGURE 13.
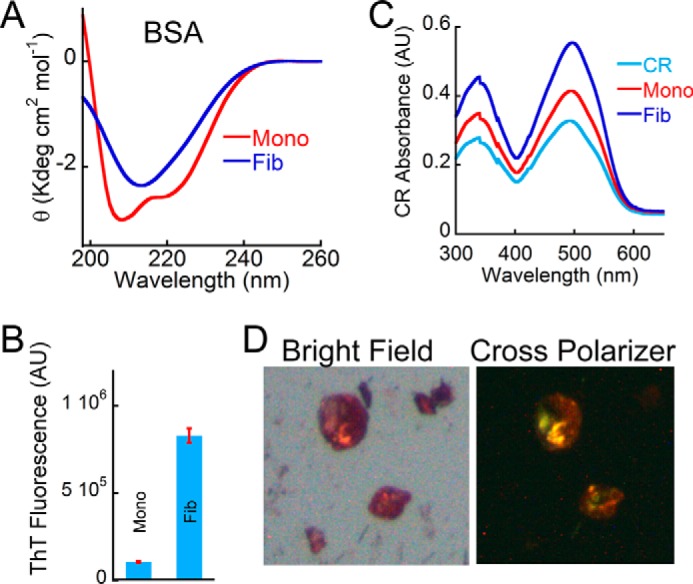
Biophysical characterization of BSA amyloids. A, far-UV CD spectrum of BSA monomer and BSA aggregates (formed in the presence of 300 mm NaCl at pH 3.0) showing helical and β-sheet-rich secondary structure, respectively. B, ThT fluorescence showing higher ThT binding for BSA incubated in the presence of salt and low pH. BSA monomer showed negligible ThT binding. Error bars, S.E. C, CR absorbance spectrum showing higher CR absorbance of BSA aggregate. CR alone was used as a control. D, BSA aggregate formed in the presence of salt and low pH showed greenish yellow CR birefringence under cross-polarized light, indicating the amyloidogenic nature of this aggregate. The corresponding bright field image is also shown on the left. AU, arbitrary units.
FIGURE 14.
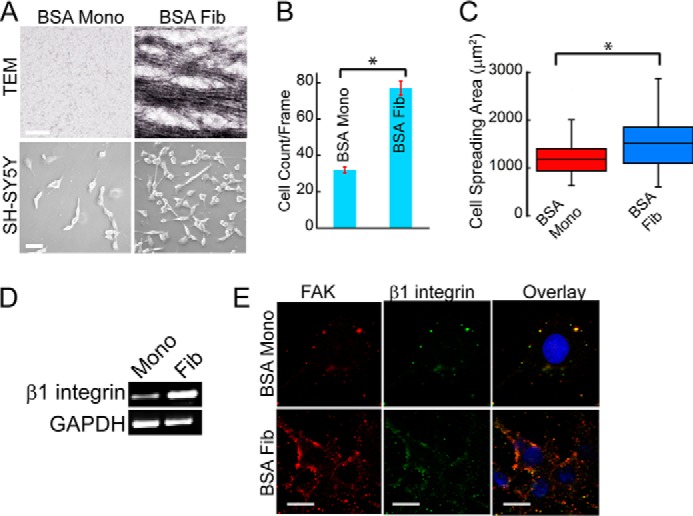
Amyloid versions of BSA support cell attachment. A, TEM images of soluble and fibrillar BSA (top). Scale bar, 200 nm. Phase-contrast images showing attachment of SH-SY5Y cells on BSA monomer and amyloid surfaces (bottom). Scale bar, 100 μm. B, quantification of SH-SY5Y attachment on BSA monomer/fibril surfaces, showing that cell attachment was higher on the fibril surfaces compared with the soluble protein. *, statistical significance (p < 0.05). Error bar, S.E. C, analysis of cell spreading area of SH-SY5Y cells on different substrates. *, statistical significance (p < 0.05). D, integrin expression profile of cells cultured on soluble and fibrillar BSA showing higher β1 integrin expression on BSA fibrils compared with BSA monomers. E, co-localization of β1 integrin and FAK in SH-SY5Y cells on different substrates. FAK (red), β1 integrin (green), and DAPI (blue) were used to visualize focal adhesion complex and nuclei, respectively. Scale bar, 20 μm.
Cell studies performed with SH-SY5Y cells showed 3-fold higher cell adhesion on BSA fibril surfaces compared with that on the monomeric surfaces (Fig. 14, A and B). On BSA-coated surfaces, cell spreading was also greater on fibril surfaces compared with that on their corresponding monomers (Fig. 14C). In line with increased attachment and spreading on BSA fibrils, mRNA expression analysis also revealed higher expression of β1 integrins on BSA fibrils (Fig. 14D). Further, the co-staining of SH-SY5Y cells with FAK and β1 integrin revealed a greater number of co-localized clusters on the fibril surfaces compared with the monomeric surfaces (Fig. 14E), suggesting the formation of active focal adhesion complexes. These results suggest that the amyloid form of a non-adhesive protein can also support cell adhesion.
Relative Contribution of Charge and Amyloid Form on Cell Adhesion
Further, to probe the role of cross-β structure in cell adhesion and also to decouple structure dependence and charge on cell adhesion, we expanded our study to three different charged PAAs and their amyloid counterparts. To do so, we used positively charged PLL, negatively charged PE, and neutral PLA. All of these PAAs have been previously reported to form amyloid in vitro (45–47).
The formation of PAA fibrils was characterized using FTIR (for secondary structure), electron microscopy (for morphology), CR birefringence, and x-ray diffraction. The freshly dissolved PLL and PE showed random coil structure in CD (data not shown). To examine the secondary structure of the incubated PAAs, FTIR spectroscopy of these aggregates was performed (74). For the analysis of peptide secondary structure, FTIR spectra in the range of 1600–1700 cm−1 (amide-I band) were deconvoluted and curve-fitted. The FTIR spectra of incubated PAAs showed peaks in the range of 1640–1650 cm−1 (amide-I band), corresponding to random coil conformation. In addition, the incubated PAAs also showed peaks at 1630 and 1617 cm−1 for PLL, 1633 and 1623 cm−1 for PE, and 1631 and 1615 cm−1 for PLA, indicating the β-sheet structure of incubated PAA (Fig. 15A). Further, the morphology of the aggregates was analyzed using TEM, which revealed fibrillar morphology for all of the aggregated PAAs. However, the fibril morphologies were different for each of the PAAs (Fig. 15B). To confirm the amyloid nature of these aggregates, CR birefringence was performed. Each of the PAA fibrils showed a greenish yellow birefringence under cross-polarized light, further confirming the amyloid nature of the PAA fibrils (Fig. 15C). Finally, using x-ray diffraction techniques, the fibrils were tested for the presence of the characteristic diffraction pattern found in amyloid fibrils. The cross-β-sheet of amyloid fibrils generally shows two reflections: meridional at 4.7 Å, which corresponds to the distance between two β-strands, and a variable equatorial reflection at ∼10 Å, which generally represents the distance between two β-sheets in a cross-β-sheet motif of amyloid fibrils (45, 75). All of the incubated PAAs also showed two reflections: one at ∼4.7 Å and another at 15 Å for PLL, 8.08 Å for PE, and 12.1 Å for PLA (Fig. 15D). This difference in cross-β-sheet packing could be due to the different amino acid composition and different solvation pattern of their corresponding amyloid fibrils by PAAs (45). It is important to note that the amyloid fibril formation by these PAAs was also consistent with a previous report (45). In each of these experiments, α-Syn was used as a positive control.
FIGURE 15.

Amyloid formation by PAAs. A, FTIR spectra of PLL, PE, and PLA showing peaks at 1630 and 1617 cm−1, 1633 and 1623 cm−1, and 1631 and 1615 cm−1, respectively, indicative of β-sheet-rich structure in incubated PAAs. B, TEM images of PAA aggregates showing fibrillar morphology. Scale bars, 200 nm. C, PAA fibrils showing greenish yellow CR birefringence under cross-polarized light, revealing their amyloidogenic nature. D, x-ray diffraction pictures of PAA fibrils showing meridional reflection at ∼4.7 Å and equatorial reflection at ∼8–15 Å. α-Syn fibrils were used as a positive control.
After characterizing the fibril formation by PAA, we performed adhesion studies using SH-SY5Y cells on the unstructured PAAs and their corresponding amyloid fibrils. Our results showed more cell adhesion and cell spreading on amyloid fibrils of PAA than in its respective unstructured state (Fig. 16, A–C). It is interesting to note that PLL is regularly used as a charge-based cell-adhesive substrate in cell culture studies (76). It is important to note that cell adhesion on unstructured PLA was much less (in terms of number of cells attached to substrate as well as spreading area of attached cells); however, upon conversion to amyloid fibril, the cell adhesion on PLA was enhanced significantly (Fig. 16, B and C). The results of this study clearly indicate that cells adhere more strongly to amyloid substrates than to its corresponding unstructured polyamino acid, although charges are known to be responsible for interacting with cells.
FIGURE 16.

Cell adhesion on PAAs and its corresponding amyloid fibrils. A, phase-contrast images of SH-SY5Y cells after 24 h of culture on freshly dissolved PAAs and their corresponding amyloid fibrils. Scale bars, 100 μm. B, quantification of cells adhered on PAA and their fibrils. Error bars, S.E. C, morphology of cells adhered on PAAs and their fibrils is quantified by calculating the spreading area of the cells. Increased cell number and spreading area on PAA fibrils suggest stronger cell adhesion on PAA amyloid fibril-coated surface. D, cytoskeletal organization and focal adhesion formation in SH-SY5Y cells on PLL and its corresponding amyloid fibril. Phalloidin (green), FAK (red), and DAPI (blue) were used to visualize F-actin, focal adhesions, and nuclei, respectively. Scale bar, 20 μm. E, β1 integrin expression profile in SH-SY5Y cells cultured on PLL and PLL fibril-coated glass coverslips. The higher integrin expression and large FAK clusters were observed in cells adhered on PLL fibrils, indicating stronger cell adhesion on amyloid fibrils than on their soluble counterparts.
To further characterize cell adhesion on PAA, we selected PLL fibrils and the corresponding soluble forms for cell adhesion. We stained the cells adhered on both monomeric and fibrillar PLL using anti-FAK antibody. The data showed that the greater number of FAK clusters were present on cells adhered to the fibril surfaces compared with the monomeric surfaces (Fig. 16D), suggesting the formation of active focal adhesion complexes. Further, we also quantified the integrin expression profile of cells grown on PLL monomer and amyloid, which revealed that integrins were expressed more in cells attached to PLL amyloid fibrils (Fig. 16E) compared with monomer. These findings clearly indicate that integrins play a significant role in amyloid-mediated cellular adhesion and suggest that although charge of the peptide/proteins helps in adhesion, the cross-β-structured state further promotes stronger adhesion.
Cell Adhesion on Amyloids Derived from the Disease-associated Protein/Peptide
Although the above experiments illustrate the cell-adhesive property of a range of different non-toxic amyloid fibrils, it remains unclear whether amyloids associated with human diseases also possess this cell-adhesive property. To study cell adhesion on disease amyloids, adhesivity of α-Syn, Aβ42, and Aβ(25–35) fibrils was assessed (Fig. 17A). Whereas α-Syn is associated with Parkinson disease (77), Aβ42 is associated with Alzheimer disease (78). Aβ(25–35) is a short peptide derived from Aβ42 that shows similar toxicity to the full-length Aβ42 and hence is used as a model peptide to study Aβ toxicity in cells (79). When SH-SY5Y cells were cultured on α-Syn, Aβ42, and Aβ(25–35) fibril-coated surface for 24 h, cell survival was found to be greater on α-Syn and Aβ42 fibrils compared with Aβ(25–35) fibrils (Fig. 17B). Moreover, SH-SY5Y cells (that survived) exhibited greater spreading on α-Syn fibrils than that on collagen, Aβ42, and Aβ(25–35) fibrils (Fig. 17C). Although β1 integrin expression and localization in cells cultured on α-Syn fibrils was similar to that on collagen (Fig. 17, D and E), in cells that survived on Aβ(25–35) fibrils, integrin staining was found to be very diffused.
FIGURE 17.

Cell adhesion on disease-associated amyloids. A, TEM images of α-Syn, Aβ42, and Aβ(25–35) amyloid fibrils used for cell adhesion (top). Scale bar, 200 nm. Bottom, cell adhesion of SH-SY5Y neuroblastoma cells on the amyloid surface. Scale bar, 100 μm. B, quantification of SH-SY5Y cell attachment on α-Syn, Aβ42, and Aβ(25–35) amyloid fibrils. Error bar, S.E. C, cell spreading area on different amyloid surfaces showing higher spreading on α-Syn amyloids compared with Aβ fibrils and collagen. *, statistical significance (p < 0.05). D, localization of β1 integrin of cells grown on α-Syn and Aβ(25–35) amyloid fibrils. Scale bar, 20 μm. E, β1 integrin expression profile in SH-SY5Y cells cultured on α-Syn amyloid fibrils and collagen. The mRNA expression of β1 integrin was highest in cells cultured on collagen. F, phase-contrast image showing cell death of attached cells after the addition of 4.5 μm Aβ42 fibrils in the cell culture medium.
Our cell adhesion data show that cells were able to adhere and spread on Aβ42 fibril-coated surfaces. This result is in contrast with cell adhesion on the Aβ(25–35) fibril-coated surface, where far fewer cells were found to adhere. We used 10 μg/cm2 Aβ42 fibrils for our study, which is ∼4.5 μm Aβ42 fibrils/well. Interestingly, when 4.5 μm Aβ42 fibrils was added directly to the medium of attached cells, cell death was observed (Fig. 17F). Such a difference in cell viability when fibrils are either precoated or added in solution phase in medium has also been reported for other amyloids (80). The reason for this phenomenon could be that the toxic epitopes, such as fibril ends, are exposed when Aβ42 fibrils are added to the medium and could result in membrane damage and cell death (80, 81). Alternatively, the precoating of Aβ42 fibrils on glass surfaces might also immobilize the fibrils and thereby restrict them from freely interacting with the cell membrane and inducing toxicity. The Aβ(25–35) fibrils are short peptides derived from full-length Aβ42 and form short amyloid fibrils. Due to this, the toxic epitope might be more available even after coating on a glass surface, leading to the resultant toxicity.
These results suggest that disease-associated amyloids also possess the cell-adhesive property. Moreover, the data further indicate that nanotopography of amyloid fibrils (both disease and functional) could support cell adhesion.
Discussion
Adhesion to the ECM is required by cells not only for their proliferation, differentiation, and survival but also for their organization into functional tissues (82). The ECM, an amalgamation of proteins secreted by cells, is composed primarily of a network of fibrils and components of the basement membrane (83). One of the common features shared by many ECM proteins with amyloids is their fibrillar nature. Moreover, amyloids possess a highly repetitive cross-β-sheet structure and a unique combination of charge and hydrophobic surfaces, which makes them sticky (22, 84, 85). It is perhaps these combinatorial features that enable amyloids to bind to membrane with high affinity (86). These properties of amyloids are utilized by lower organisms for their ECM formation (87). The fact that amyloid fibrils can behave analogously to ECM raises the exciting possibility that amyloids may have served as primitive cell-adhesive substrates. Therefore, we hypothesize that amyloid fibrils, irrespective of the primary structure, can support cell adhesion.
We show here that irrespective of sequence, composition, and absence of integrin binding sites, ∼20 different amyloid fibrils support mammalian cell adhesion (Fig. 5). Interestingly, BSA, a cell-repulsive protein (73), when converted into amyloid, is also able to support cell adhesion (Fig. 14). Moreover, the cell adhesion on PAA amyloids was higher than on their corresponding soluble counterparts. Interestingly, however, whereas the neutral PLA was mostly incapable of supporting cell adhesion, its amyloid counterpart showed robust adhesion of cells (Fig. 16, A–C). Further, the increased expression of FAK and β1 integrin in cells cultured on PLL amyloid fibrils compared with the PLL monomer further decouples the role of amyloid fibril structure versus charge in cell adhesion (Fig. 16, D and E). Taken together, these studies demonstrate the universal cell-adhesive capacity of amyloid fibrils. We therefore surmised that, similar to lower organisms, mammalian cells can also utilize the stickiness of amyloids for their adhesion. Both adherent and non-adherent cells can attach to amyloids through passive (i.e. lipid-fibril interactions) as well as active (i.e. integrin-based) modes (Fig. 18). The initial adhesion of cells to amyloids could be spontaneous, consistent with their membrane binding property (86) and as observed here with adhesion of RBCs to kassinin amyloid fibrils. Subsequent to the initial immobilization of cells on amyloid surfaces, cell spreading and motility require integrin engagement and formation/turnover of focal adhesions (88, 89). We have demonstrated this directly by quantifying integrin expression and localization at adhesion sites on amyloid fibrils similar to that on native ECM proteins (Fig. 9, D–F). Further, in line with this, an integrin-blocking experiment showed a marked reduction in cell spreading (Fig. 10, A–E). Interestingly, in SH-SY5Y cells, focal adhesion formation was more robust on amyloid fibrils compared with collagen (Fig. 9, A and C), suggesting that for some cell types, amyloid fibrils may be even more efficient than native ECM proteins in supporting cell adhesion.
FIGURE 18.
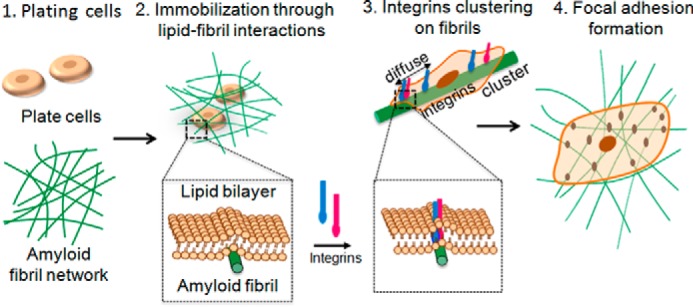
Schematic of cell spreading on amyloid fibrils. Proposed cell adhesion on amyloid fibrils mediated by lipid-fibril interactions. This is followed by integrin clustering and activation of downstream signaling cascades that lead to the formation of focal adhesions and cell spreading.
It has been suggested previously that amyloids may mediate cell adhesion by accelerating and stabilizing the deposition of serum proteins (33, 34). Although we observed a decrease in cell adhesion on amyloid surfaces in serum-free medium, complete abolition of the same was not observed (Fig. 8, A and B), suggesting that serum protein deposition is important but not necessary for cell adhesion on amyloid surfaces. Further, this implies that, similar to ECM, amyloid fibrils can also entrap proteins and small molecules that can facilitate spreading and growth of the cell. In addition, we and others have recently demonstrated that three-dimensional cell culture systems derived from amyloid networks can be used for long term cell culture and stem cell differentiation (35, 37).
Cell adhesion to the underlying matrix and to neighboring cells is mediated by transmembrane integrins (focal adhesions) (90), cadherins (cell-cell adhesions) (91, 92), and various other proteins. These proteins enable cells to sense and respond to external stimuli. Rho-based signaling is one of the prominent signaling cascades associated with reorganization of adhesion structures and the cytoskeleton following such stimuli (56). In neuronal cells, the ROCK pathway is involved in synaptic plasticity and activates MLCK for stimulating retraction of axonal and dendritic growth cones (93–95). Our results suggest that, similar to that on ECM proteins (72, 96), cell adhesion and spreading are mediated by both ROCK and MLCK pathways (Figs. 11 and 12).
Interestingly, we found that the disease-related amyloid fibrils Aβ42 (78, 97), Aβ(25–35) (79), and α-Syn (77) also support cell adhesion (Fig. 17). However, this was found to be less true for Aβ(25–35) than for α-Syn and Aβ42. Although both Aβ42 (78, 97) and Aβ(25–35) fibrils are known to be neurotoxic (79, 98–100), the differences in cell adhesion on both of these coated surfaces (Fig. 17) could be due to the availability of toxic epitopes (80). Moreover, both nanotopography and roughness of the coated fibrils can also play an important role in death/apoptosis of adherent cells on amyloid surfaces (80, 81). Specifically, increased roughness of amyloid plaques was shown to directly influence the cell survival (101).
The discovery of non-toxic functional amyloids and the ability of all proteins to form amyloids under specific conditions suggests that amyloids possess a much broader function than only being associated with diseases (7). Further, functional amyloid formation in organisms is tightly regulated, thus avoiding their possible toxicity (8, 102). Together, these results suggest that whereas cell adhesivity of amyloid fibrils is a generic property, the physio-chemical properties of the amyloid fibrils and their tight regulation may dictate the extent of toxicity that they may elicit, and adhesiveness and toxicity may represent two independent aspects of amyloid fibrils.
In conclusion, our study demonstrates several similarities between natural ECMs and amyloid fibrils, including their nanofibrillar morphology, integrin engagement and signaling, and density-dependent responses. These properties make amyloids a promising candidate for developing biomaterials (32, 35, 103). Cells can adhere to amyloids through a combination of interactions with the lipid membrane and activation of cell adhesion machinery. The present data suggest that cell adhesion could be a generic property of amyloids irrespective of amino acid sequences and thus may explain their association with diseases and utilization as an extracellular matrix by a wide variety of organisms.
Author Contributions
R. S. J., S. K. M., E. G., and S. Sen designed the experiments. R. S. J., E. G., P. K. S., A. A., and S. Salot performed experiments. All authors analyzed the data. S. K. M., R. S. J., E. G., and S. Sen wrote the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
We acknowledge the Industrial Research and Consultancy Centre (IRCC), Indian Institute of Technology (IIT) Bombay, Cell Biology Laboratory, Department of Biological Sciences and Engineering, Indian Institute of Technology Bombay for fluorescence imaging and the Centre for Research in Nanotechnology and Science and IRCC (IIT Bombay) for electron microscopy and protein crystallography, and we thank the IRCC (IIT Bombay) for use of the x-ray diffraction facility.
This work was supported by Department of Biotechnology, Ministry of Science and Technology, Government of India, Grant BT/PR9797/NNT/28/774/2014. The authors declare that they have no conflicts of interest with the contents of this article.
- ECM
- extracellular matrix
- TEM
- transmission electron microscopy
- CR
- Congo Red
- ThT
- thioflavin T
- FAK
- focal adhesion kinase
- Sub P
- substance P
- α-Syn
- α-synuclein
- PAA
- polyamino acid
- PLL
- poly-l-lysine
- PE
- polyglutamic acid
- PLA
- poly-l-alanine
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- RBC
- red blood cell
- ROCK
- Rho-associated kinase
- MLCK
- myosin light chain kinase
- VIP
- vasoactive intestinal peptide
- GRP
- gastrin releasing peptide
- GRF
- growth hormone releasing factor
- GIP
- glucose-dependent insulinotropic polypeptide
- oCRF
- ovine corticotropin-releasing factor.
References
- 1. Lord E. M., and Sanders L. C. (1992) Roles for the extracellular matrix in plant development and pollination: a special case of cell movement in plants. Dev. Biol. 153, 16–28 [DOI] [PubMed] [Google Scholar]
- 2. Daley W. P., and Yamada K. M. (2013) ECM-modulated cellular dynamics as a driving force for tissue morphogenesis. Curr. Opin. Genet. Dev. 23, 408–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sapir-Koren R., and Livshits G. (2011) Bone mineralization and regulation of phosphate homeostasis. IBMS BoneKEy 8, 286–300 [Google Scholar]
- 4. Watt F. M., and Fujiwara H. (2011) Cell-extracellular matrix interactions in normal and diseased skin. Cold Spring Harb. Perspect. Biol. 10.1101/cshperspect.a005124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi Y., Chung H., Jung H., Couchman J. R., and Oh E. S. (2011) Syndecans as cell surface receptors: unique structure equates with functional diversity. Matrix Biol. 30, 93–99 [DOI] [PubMed] [Google Scholar]
- 6. Geiger B., Bershadsky A., Pankov R., and Yamada K. M. (2001) Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2, 793–805 [DOI] [PubMed] [Google Scholar]
- 7. Chiti F., and Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 8. Fowler D. M., Koulov A. V., Balch W. E., and Kelly J. W. (2007) Functional amyloid: from bacteria to humans. Trends Biochem. Sci. 32, 217–224 [DOI] [PubMed] [Google Scholar]
- 9. Chapman M. R., Robinson L. S., Pinkner J. S., Roth R., Heuser J., Hammar M., Normark S., and Hultgren S. J. (2002) Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295, 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramsook C. B., Tan C., Garcia M. C., Fung R., Soybelman G., Henry R., Litewka A., O'Meally S., Otoo H. N., Khalaf R. A., Dranginis A. M., Gaur N. K., Klotz S. A., Rauceo J. M., Jue C. K., and Lipke P. N. (2010) Yeast cell adhesion molecules have functional amyloid-forming sequences. Eukaryot. Cell 9, 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sullan R. M., Gunari N., Tanur A. E., Chan Y., Dickinson G. H., Orihuela B., Rittschof D., and Walker G. C. (2009) Nanoscale structures and mechanics of barnacle cement. Biofouling 25, 263–275 [DOI] [PubMed] [Google Scholar]
- 12. Barlow D. E., Dickinson G. H., Orihuela B., Kulp J. L., Rittschof D., and Wahl K. J. (2010) Characterization of the adhesive plaque of the barnacle balanus amphitrite: amyloid-like nanofibrils are a major component. Langmuir 26, 6549–6556 [DOI] [PubMed] [Google Scholar]
- 13. Fowler D. M., Koulov A. V., Alory-Jost C., Marks M. S., Balch W. E., and Kelly J. W. (2006) Functional amyloid formation within mammalian tissue. PLoS Biol. 4, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maji S. K., Perrin M. H., Sawaya M. R., Jessberger S., Vadodaria K., Rissman R. A., Singru P. S., Nilsson K. P., Simon R., Schubert D., Eisenberg D., Rivier J., Sawchenko P., Vale W., and Riek R. (2009) Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 325, 328–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cohen F. E., and Prusiner S. B. (1998) Pathologic conformations of prion proteins. Annu. Rev. Biochem. 67, 793–819 [DOI] [PubMed] [Google Scholar]
- 16. Jarrett J. T., and Lansbury P. T. Jr. (1993) Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73, 1055–1058 [DOI] [PubMed] [Google Scholar]
- 17. Rufo C. M., Moroz Y. S., Moroz O. V., Stöhr J., Smith T. A., Hu X., DeGrado W. F., and Korendovych I. V. (2014) Short peptides self-assemble to produce catalytic amyloids. Nat. Chem. 6, 303–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greenwald J., and Riek R. (2012) On the possible amyloid origin of protein folds. J. Mol. Biol. 421, 417–426 [DOI] [PubMed] [Google Scholar]
- 19. Carny O., and Gazit E. (2005) A model for the role of short self-assembled peptides in the very early stages of the origin of life. FASEB J. 19, 1051–1055 [DOI] [PubMed] [Google Scholar]
- 20. Nelson R., and Eisenberg D. (2006) Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 16, 260–265 [DOI] [PubMed] [Google Scholar]
- 21. Wasmer C., Lange A., Van Melckebeke H., Siemer A. B., Riek R., and Meier B. H. (2008) Amyloid fibrils of the HET-s(218–289) prion form a β solenoid with a triangular hydrophobic core. Science 319, 1523–1526 [DOI] [PubMed] [Google Scholar]
- 22. Maji S. K., Wang L., Greenwald J., and Riek R. (2009) Structure-activity relationship of amyloid fibrils. FEBS Lett. 583, 2610–2617 [DOI] [PubMed] [Google Scholar]
- 23. Nilsson K. P. (2009) Small organic probes as amyloid specific ligands: past and recent molecular scaffolds. FEBS Lett. 583, 2593–2599 [DOI] [PubMed] [Google Scholar]
- 24. Calamai M., Kumita J. R., Mifsud J., Parrini C., Ramazzotti M., Ramponi G., Taddei N., Chiti F., and Dobson C. M. (2006) Nature and significance of the interactions between amyloid fibrils and biological polyelectrolytes. Biochemistry 45, 12806–12815 [DOI] [PubMed] [Google Scholar]
- 25. Ghosh D., Dutta P., Chakraborty C., Singh P. K., Anoop A., Jha N. N., Jacob R. S., Mondal M., Mankar S., Das S., Malik S., and Maji S. K. (2014) Complexation of amyloid fibrils with charged conjugated polymers. Langmuir 30, 3775–3786 [DOI] [PubMed] [Google Scholar]
- 26. Solomon J. P., Bourgault S., Powers E. T., and Kelly J. W. (2011) Heparin binds 8 kDa gelsolin cross-beta-sheet oligomers and accelerates amyloidogenesis by hastening fibril extension. Biochemistry 50, 2486–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oh J., Kim J. G., Jeon E., Yoo C. H., Moon J. S., Rhee S., and Hwang I. (2007) Amyloidogenesis of type III-dependent harpins from plant pathogenic bacteria. J. Biol. Chem. 282, 13601–13609 [DOI] [PubMed] [Google Scholar]
- 28. Alteri C. J., Xicohténcatl-Cortes J., Hess S., Caballero-Olín G., Girón J. A., and Friedman R. L. (2007) Mycobacterium tuberculosis produces pili during human infection. Proc. Natl. Acad. Sci. U.S.A. 104, 5145–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Claessen D., Rink R., de Jong W., Siebring J., de Vreugd P., Boersma F. G., Dijkhuizen L., and Wosten H. A. (2003) A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 17, 1714–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wösten H. A. (2001) Hydrophobins: multipurpose proteins. Annu. Rev. Microbiol. 55, 625–646 [DOI] [PubMed] [Google Scholar]
- 31. Gras S. L. (2009) Surface- and solution-based assembly of amyloid fibrils for biomedical and nanotechnology applications. Adv. Chem. Engineer 35, 161–209 [Google Scholar]
- 32. Gras S. L., Tickler A. K., Squires A. M., Devlin G. L., Horton M. A., Dobson C. M., and MacPhee C. E. (2008) Functionalised amyloid fibrils for roles in cell adhesion. Biomaterials 29, 1553–1562 [DOI] [PubMed] [Google Scholar]
- 33. Reynolds N. P., Charnley M., Mezzenga R., and Hartley P. G. (2014) Engineered lysozyme amyloid fibril networks support cellular growth and spreading. Biomacromolecules 15, 599–608 [DOI] [PubMed] [Google Scholar]
- 34. Reynolds N. P., Styan K. E., Easton C. D., Li Y., Waddington L., Lara C., Forsythe J. S., Mezzenga R., Hartley P. G., and Muir B. W. (2013) Nanotopographic surfaces with defined surface chemistries from amyloid fibril networks can control cell attachment. Biomacromolecules 14, 2305–2316 [DOI] [PubMed] [Google Scholar]
- 35. Li C., Born A. K., Schweizer T., Zenobi-Wong M., Cerruti M., and Mezzenga R. (2014) Amyloid-hydroxyapatite bone biomimetic composites. Adv. Mater. 26, 3207–3212 [DOI] [PubMed] [Google Scholar]
- 36. Yan H., Nykanen A., Ruokolainen J., Farrar D., Gough J. E., Saiani A., and Miller A. F. (2008) Thermo-reversible protein fibrillar hydrogels as cell scaffolds. Faraday Discuss. 139, 71–84; discussion 105–128, 419–120 [DOI] [PubMed] [Google Scholar]
- 37. Jacob R. S., Ghosh D., Singh P. K., Basu S. K., Jha N. N., Das S., Sukul P. K., Patil S., Sathaye S., Kumar A., Chowdhury A., Malik S., Sen S., and Maji S. K. (2015) Self healing hydrogels composed of amyloid nano fibrils for cell culture and stem cell differentiation. Biomaterials 54, 97–105 [DOI] [PubMed] [Google Scholar]
- 38. Volles M. J., and Lansbury P. T. Jr. (2007) Relationships between the sequence of α-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J. Mol. Biol. 366, 1510–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Singh P. K., Kotia V., Ghosh D., Mohite G. M., Kumar A., and Maji S. K. (2013) Curcumin modulates α-synuclein aggregation and toxicity. ACS Chem. Neurosci. 4, 393–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 41. LeVine H., 3rd (1993) Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 2, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bhattacharya M., Jain N., and Mukhopadhyay S. (2011) Insights into the mechanism of aggregation and fibril formation from bovine serum albumin. J. Phys. Chem. B 115, 4195–4205 [DOI] [PubMed] [Google Scholar]
- 43. Usov I., Adamcik J., and Mezzenga R. (2013) Polymorphism complexity and handedness inversion in serum albumin amyloid fibrils. ACS Nano 7, 10465–10474 [DOI] [PubMed] [Google Scholar]
- 44. Klunk W. E., Jacob R. F., and Mason R. P. (1999) Quantifying amyloid by Congo red spectral shift assay. Methods Enzymol. 309, 285–305 [DOI] [PubMed] [Google Scholar]
- 45. Fändrich M., and Dobson C. M. (2002) The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 21, 5682–5690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arnott S., Dover S. D., and Elliott A. (1967) Structure of β-poly-l-alanine: refined atomic co-ordinates for an anti-parallel β-pleated sheet. J. Mol. Biol. 30, 201–208 [DOI] [PubMed] [Google Scholar]
- 47. Fulara A., Lakhani A., Wójcik S., Nieznańska H., Keiderling T. A., and Dzwolak W. (2011) Spiral superstructures of amyloid-like fibrils of polyglutamic acid: an infrared absorption and vibrational circular dichroism study. J. Phys. Chem. B 115, 11010–11016 [DOI] [PubMed] [Google Scholar]
- 48. Subramanian S., Parthasarathy R., Sen S., Boder E. T., and Discher D. E. (2006) Species- and cell type-specific interactions between CD47 and human SIRPα. Blood 107, 2548–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sen S., and Kumar S. (2009) Cell-matrix de-adhesion dynamics reflect contractile mechanics. Cell Mol. Bioeng. 2, 218–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anoop A., Ranganathan S., Das Dhaked B., Jha N. N., Pratihar S., Ghosh S., Sahay S., Kumar S., Das S., Kombrabail M., Agarwal K., Jacob R. S., Singru P., Bhaumik P., Padinhateeri R., Kumar A., and Maji S. K. (2014) Elucidating the role of disulfide bond on amyloid formation and fibril reversibility of somatostatin-14: relevance to its storage and secretion. J. Biol. Chem. 289, 16884–16903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jha N. N., Anoop A., Ranganathan S., Mohite G. M., Padinhateeri R., and Maji S. K. (2013) Characterization of amyloid formation by glucagon-like peptides: role of basic residues in heparin-mediated aggregation. Biochemistry 52, 8800–8810 [DOI] [PubMed] [Google Scholar]
- 52. Singh P. K., and Maji S. K. (2012) Amyloid-like fibril formation by tachykinin neuropeptides and its relevance to amyloid β-protein aggregation and toxicity. Cell Biochem. Biophys. 64, 29–44 [DOI] [PubMed] [Google Scholar]
- 53. Sabaté R., Baxa U., Benkemoun L., Sanchez de Groot N., Coulary-Salin B., Maddelein M. L., Malato L., Ventura S., Steven A. C., and Saupe S. J. (2007) Prion and non-prion amyloids of the HET-s prion forming domain. J. Mol. Biol. 370, 768–783 [DOI] [PubMed] [Google Scholar]
- 54. Sabaté R., Lascu I., and Saupe S. J. (2008) On the binding of Thioflavin-T to HET-s amyloid fibrils assembled at pH 2. J. Struct. Biol. 162, 387–396 [DOI] [PubMed] [Google Scholar]
- 55. Pedersen J. S., Andersen C. B., and Otzen D. E. (2010) Amyloid structure: one but not the same: the many levels of fibrillar polymorphism. FEBS J. 277, 4591–4601 [DOI] [PubMed] [Google Scholar]
- 56. Geiger B., Spatz J. P., and Bershadsky A. D. (2009) Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 10, 21–33 [DOI] [PubMed] [Google Scholar]
- 57. Mooney D. J., Langer R., and Ingber D. E. (1995) Cytoskeletal filament assembly and the control of cell spreading and function by extracellular matrix. J. Cell Sci. 108, 2311–2320 [DOI] [PubMed] [Google Scholar]
- 58. Kim D. H., and Wirtz D. (2013) Predicting how cells spread and migrate: focal adhesion size does matter. Cell Adh. Migr. 7, 293–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mitra S. K., Hanson D. A., and Schlaepfer D. D. (2005) Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 [DOI] [PubMed] [Google Scholar]
- 60. Owen J. D., Ruest P. J., Fry D. W., and Hanks S. K. (1999) Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol. Cell Biol. 19, 4806–4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Michael K. E., Dumbauld D. W., Burns K. L., Hanks S. K., and García A. J. (2009) Focal adhesion kinase modulates cell adhesion strengthening via integrin activation. Mol. Biol. Cell 20, 2508–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kokkinos M. I., Brown H. J., and de Iongh R. U. (2007) Focal adhesion kinase (FAK) expression and activation during lens development. Mol. Vis. 13, 418–430 [PMC free article] [PubMed] [Google Scholar]
- 63. Sanborn S. L., Murugesan G., Marchant R. E., and Kottke-Marchant K. (2002) Endothelial cell formation of focal adhesions on hydrophilic plasma polymers. Biomaterials 23, 1–8 [DOI] [PubMed] [Google Scholar]
- 64. Torsoni A. S., Constancio S. S., Nadruz W. Jr., Hanks S. K., and Franchini K. G. (2003) Focal adhesion kinase is activated and mediates the early hypertrophic response to stretch in cardiac myocytes. Circ. Res. 93, 140–147 [DOI] [PubMed] [Google Scholar]
- 65. Ruoslahti E. (1996) RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 12, 697–715 [DOI] [PubMed] [Google Scholar]
- 66. D'Souza S. E., Ginsberg M. H., and Plow E. F. (1991) Arginyl-glycyl-aspartic acid (RGD): a cell adhesion motif. Trends Biochem. Sci. 16, 246–250 [DOI] [PubMed] [Google Scholar]
- 67. Ruoslahti E., and Pierschbacher M. D. (1986) Arg-Gly-Asp: a versatile cell recognition signal. Cell 44, 517–518 [DOI] [PubMed] [Google Scholar]
- 68. Ruoslahti E., and Pierschbacher M. D. (1987) New perspectives in cell adhesion: RGD and integrins. Science 238, 491–497 [DOI] [PubMed] [Google Scholar]
- 69. Hayman E. G., Pierschbacher M. D., and Ruoslahti E. (1985) Detachment of cells from culture substrate by soluble fibronectin peptides. J. Cell Biol. 100, 1948–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Trommler A., Gingell D., and Wolf H. (1985) Red blood cells experience electrostatic repulsion but make molecular adhesions with glass. Biophys. J. 48, 835–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hategan A., Sengupta K., Kahn S., Sackmann E., and Discher D. E. (2004) Topographical pattern dynamics in passive adhesion of cell membranes. Biophys. J. 87, 3547–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Totsukawa G., Wu Y., Sasaki Y., Hartshorne D. J., Yamakita Y., Yamashiro S., and Matsumura F. (2004) Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 164, 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kim S. H., Kim J. H., and Akaike T. (2003) Regulation of cell adhesion signaling by synthetic glycopolymer matrix in primary cultured hepatocyte. FEBS Lett. 553, 433–439 [DOI] [PubMed] [Google Scholar]
- 74. Barth A. (2007) Infrared spectroscopy of proteins. Biochim. Biophys. Acta 1767, 1073–1101 [DOI] [PubMed] [Google Scholar]
- 75. Serpell L. C., Fraser P. E., and Sunde M. (1999) X-ray fiber diffraction of amyloid fibrils. Methods Enzymol. 309, 526–536 [DOI] [PubMed] [Google Scholar]
- 76. Mazia D., Schatten G., and Sale W. (1975) Adhesion of cells to surfaces coated with polylysine: applications to electron microscopy. J. Cell Biol. 66, 198–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Goedert M. (2001) α-Synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2, 492–501 [DOI] [PubMed] [Google Scholar]
- 78. Hardy J., and Selkoe D. J. (2002) Medicine: the amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 79. Pike C. J., Walencewicz-Wasserman A. J., Kosmoski J., Cribbs D. H., Glabe C. G., and Cotman C. W. (1995) Structure-activity analyses of β-amyloid peptides: contributions of the β25–35 region to aggregation and neurotoxicity. J. Neurochem. 64, 253–265 [DOI] [PubMed] [Google Scholar]
- 80. Bongiovanni M. N., and Gras S. L. (2015) Bioactive TTR105–115-based amyloid fibrils reduce the viability of mammalian cells. Biomaterials 46, 105–116 [DOI] [PubMed] [Google Scholar]
- 81. Reynolds N. P., Charnley M., Bongiovanni M. N., Hartley P. G., and Gras S. L. (2015) Biomimetic topography and chemistry control cell attachment to amyloid fibrils. Biomacromolecules 16, 1556–1565 [DOI] [PubMed] [Google Scholar]
- 82. Ekblom P., Vestweber D., and Kemler R. (1986) Cell-matrix interactions and cell adhesion during development. Annu. Rev. Cell Biol. 2, 27–47 [DOI] [PubMed] [Google Scholar]
- 83. Hynes R. O. (2009) The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Greenwald J., and Riek R. (2010) Biology of amyloid: structure, function, and regulation. Structure 18, 1244–1260 [DOI] [PubMed] [Google Scholar]
- 85. Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gellermann G. P., Appel T. R., Tannert A., Radestock A., Hortschansky P., Schroeckh V., Leisner C., Lütkepohl T., Shtrasburg S., Röcken C., Pras M., Linke R. P., Diekmann S., and Fändrich M. (2005) Raft lipids as common components of human extracellular amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 6297–6302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Otzen D., and Nielsen P. H. (2008) We find them here, we find them there: functional bacterial amyloid. Cell Mol. Life Sci. 65, 910–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Carragher N. O., and Frame M. C. (2004) Focal adhesion and actin dynamics: a place where kinases and proteases meet to promote invasion. Trends Cell Biol. 14, 241–249 [DOI] [PubMed] [Google Scholar]
- 89. Mierke C. T. (2013) The role of focal adhesion kinase in the regulation of cellular mechanical properties. Phys. Biol. 10, 065005 [DOI] [PubMed] [Google Scholar]
- 90. Geiger B., and Yamada K. M. (2011) Molecular architecture and function of matrix adhesions. Cold Spring Harb. Perspect. Biol. 10.1101/cshperspect.a005033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Fukata M., and Kaibuchi K. (2001) Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat. Rev. Mol. Cell Biol. 2, 887–897 [DOI] [PubMed] [Google Scholar]
- 92. Aplin A. E., Howe A. K., and Juliano R. L. (1999) Cell adhesion molecules, signal transduction and cell growth. Curr. Opin. Cell Biol. 11, 737–744 [DOI] [PubMed] [Google Scholar]
- 93. Gallo G. (2004) Myosin II activity is required for severing-induced axon retraction in vitro. Exp. Neurol. 189, 112–121 [DOI] [PubMed] [Google Scholar]
- 94. Mokhtar S. H., Bakhuraysah M. M., Cram D. S., and Petratos S. (2013) The β-amyloid protein of Alzheimer's disease: communication breakdown by modifying the neuronal cytoskeleton. Int. J. Alzheimers Dis. 2013, 910502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Dityatev A., Schachner M., and Sonderegger P. (2010) The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 11, 735–746 [DOI] [PubMed] [Google Scholar]
- 96. Huttenlocher A., and Horwitz A. R. (2011) Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 3, a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Selkoe D. J. (2003) Folding proteins in fatal ways. Nature 426, 900–904 [DOI] [PubMed] [Google Scholar]
- 98. Pike C. J., Burdick D., Walencewicz A. J., Glabe C. G., and Cotman C. W. (1993) Neurodegeneration induced by β-amyloid peptides in vitro: The role of peptide assembly state. J. Neurosci. 13, 1676–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Stepanichev M., Lazareva N. A., Onufriev M. V., Mitrokhina O. S., Moiseeva Y. V., and Gulyaeva N. V. (1998) Effects of doses of fragment (25–35) of β-amyloid peptide on behavior in rats. Neurosci. Behav. Physiol. 28, 564–566 [DOI] [PubMed] [Google Scholar]
- 100. Stepanichev M. Y., Moiseeva Y. V., Lazareva N. A., and Gulyaeva N. V. (2005) Studies of the effects of fragment (25–35) of β-amyloid peptide on the behavior of rats in a radial maze. Neurosci. Behav. Physiol. 35, 511–518 [DOI] [PubMed] [Google Scholar]
- 101. Blumenthal N. R., Hermanson O., Heimrich B., and Shastri V. P. (2014) Stochastic nanoroughness modulates neuron: astrocyte interactions and function via mechanosensing cation channels. Proc. Natl. Acad Sci. 111, 16124–16129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Barnhart M. M., and Chapman M. R. (2006) Curli biogenesis and function. Annu. Rev. Microbiol. 60, 131–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mankar S., Anoop A., Sen S., and Maji S. K. (2011) Nanomaterials: Amyloids reflect their brighter side. Nano Rev. 10.3402/nano.v2i0.6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ghosh D., Singh P. K., Sahay S., Jha N. N., Jacob R. S., Sen S., Kumar A., Riek R., and Maji S. K. (2015) Structure based aggregation studies reveal the presence of helix-rich intermediate during α-synuclein aggregation. Sci. Rep. 5, 9228. [DOI] [PMC free article] [PubMed] [Google Scholar]