

Abstract
Hydrogen sulfide (H2S) is an endogenously produced gaseous molecule with important roles in cellular signaling. In mammals, exogenous H2S improves survival of ischemia/reperfusion. We have previously shown that exposure to H2S increases the lifespan and thermotolerance in Caenorhabditis elegans, and improves protein homeostasis in low oxygen. The mitochondrial SQRD-1 (sulfide quinone oxidoreductase) protein is a highly conserved enzyme involved in H2S metabolism. SQRD-1 is generally considered important to detoxify H2S. Here, we show that SQRD-1 is also required to maintain protein translation in H2S. In sqrd-1 mutant animals, exposure to H2S leads to phosphorylation of eIF2α and inhibition of protein synthesis. In contrast, global protein translation is not altered in wild-type animals exposed to lethally high H2S or in hif-1(ia04) mutants that die when exposed to low H2S. We demonstrate that both gcn-2 and pek-1 kinases are involved in the H2S-induced phosphorylation of eIF2α. Both ER and mitochondrial stress responses are activated in sqrd-1 mutant animals exposed to H2S, but not in wild-type animals. We speculate that SQRD-1 activity in H2S may coordinate proteostasis responses in multiple cellular compartments.
Keywords: Caenorhabditis elegans (C. elegans), eukaryotic initiation factor 2 (eIF2), hydrogen sulfide, proteostasis, translation initiation
Introduction
Hydrogen sulfide (H2S)3 is an endogenously produced gas molecule with roles in signaling, neuromodulation, and vasodilation (reviewed in Ref. 1–4). Treatment with exogenous H2S improves outcome in multiple mammalian models of ischemia/reperfusion injury (5). However, H2S is also toxic at high concentrations, provoking immediate apnea and loss of consciousness that can result in death (6). Industrial exposure to H2S is the second-leading cause of death by inhalation, behind only carbon monoxide. The mechanistic differences between beneficial and toxic effects of H2S are poorly understood.
Sulfide-quinone oxidoreductase (SQRD) is a highly conserved mitochondrial protein that oxidizes cellular H2S by transferring electrons to the mitochondrial electron transport chain and adding sulfane sulfur atoms to free sulhydryl moieties (Fig. 1A) (7–9). Isolated mitochondria from chicken liver and human cells can generate ATP when exposed to H2S as a result of SQRD activity, which is considered an important aspect of cellular sulfide detoxification (10–12). However, it is now clear that protein activity can be regulated by post-translational modification by sulfide, and this may be an important aspect of the cellular signaling roles of H2S (2, 4). SQRD is therefore positioned to modulate both signaling and toxicity of H2S in animals.
FIGURE 1.
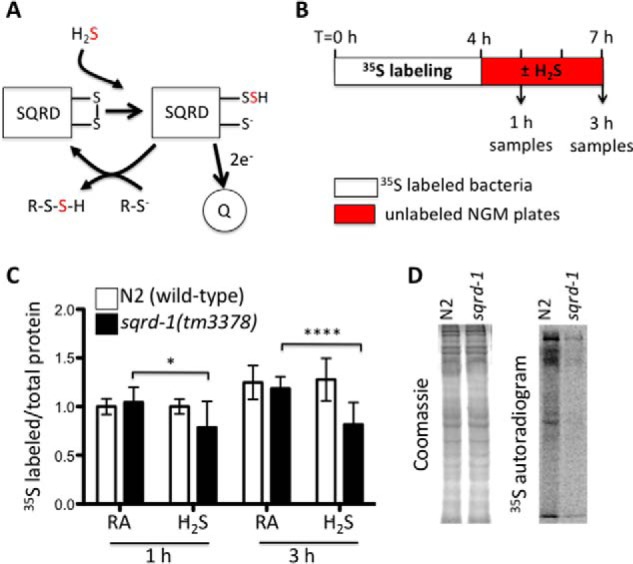
SQRD-1 is required for optimal protein translation in H2S. A, SQRD catalyzes the oxidation of H2S at the mitochondria. H2S is oxidized, resulting in the sulfur atom from H2S (red) forming a persulfite intermediate on SQRD. Electrons from H2S are fed into the quinone pool of the electron transport chain. The SQRD persulfite intermediate is resolved by oxidation with another cellular sulfur moiety to form the final -R-S-S-H species. R can include a variety of species, including sulfhydryl residues of cellular proteins (8, 9). B, experimental strategy. Worms were fed [35S]methionine labeled OP50 in liquid culture for 4 h to label cellular amino acid precursor pools and then transferred to solid NGM plates seeded with unlabeled OP50 for exposure to either H2S or room air. C, mutants lacking SQRD-1 do not efficiently incorporate [35S]methionine into protein when exposed to H2S. Incorporation of [35S]methionine was measured by autoradiograms from three independent experiments. All samples were normalized to room air exposed wild-type animals (N2). Plot shows average ± standard deviation. D, representative autoradiogram of proteins from animals exposed to H2S. Proteins were extracted from wild-type (N2) and sqrd-1(tm3378) mutant animals 3 h after transfer to NGM plates, and separated by SDS-PAGE. Coomassie-stained gels (left) show total protein and autoradiogram (right) shows proteins with incorporated [35S]methionine.
The nematode Caenorhabditis elegans has a single orthologue of SQRD, sqrd-1. SQRD-1 localizes to mitochondria and is essential for animals to survive exposure to even low concentrations of H2S (13). Here, we show SQRD-1 activity is required to prevent activation of the integrated stress response upon exposure to H2S. We found that the translation initiation factor eIF2α is phosphorylated by both PEK-1 and GCN-2 kinases in sqrd-1 mutant animals exposed to H2S. These kinases are activated in response to stress in the ER or mitochondria, respectively. Our results suggest that SQRD-1 coordinates cellular stress responses in at least two different cellular compartments in H2S.
Experimental Procedures
Strains
C. elegans strains were cultured at 20 °C on NGM plates with OP50 Escherichia coli (14). Alleles used were: sqrd-1(tm3378) V, pek-1(ok275) X, gcn-2(ok886) II, and hif-1(ia04) V. Strains were obtained from the Caenorhabditis Genetics Center at the University of Minnesota or the National BioResource Project (Tokyo, Japan). Double and triple mutants were generated using standard genetic techniques, and genotypes were verified by PCR genotyping. Primer sequences are available upon request.
H2S Exposure
C. elegans were exposed to H2S in atmospheric chambers perfused with H2S continuously diluted into room air, as described (15). Concentrated tanks of compressed H2S gas (5,000 ppm balanced with N2) were purchased from Airgas. Mixing was achieved using SmartTrak mass flow controllers (Sierra Instruments). Experiments were conducted at room temperature. Matched control environments were perfused with room air and maintained at the same temperature.
[35S]Methionine Labeling
OP50 bacteria were grown overnight at 37 °C in defined medium with [35S]methionine (20 mm NH4Cl, 0.2% glucose, 2 mm MgSO4, 4 μg/ml uracil, 2.72 μm mixed amino acids without methionine, and 3.75 μCi/ml [35S]methionine in M9 buffer). For each sample, 1500 L4/young adult C. elegans were collected and washed with M9, then added to 200 μl of radioactive OP50 bacterial culture. Samples were incubated for 4 h at 20 °C while rotating. Animals were allowed to settle by gravity, moved to non-radioactive NGM plates seeded with OP50 food, and then exposed to H2S as indicated. At each time point, worms were rinsed from plates, washed two times with M9 buffer, and the settled worm pellet was flash frozen in an equal volume SDS-PAGE loading buffer with 4% SDS and 0.01% β-mercaptoethanol. Samples were boiled for 15 min, centrifuged to pellet cellular debris, and then proteins were separated on a 10% polyacrylamide gel. The gel was stained with Coomassie Blue, dried between cellophane sheets using a Promega gel drying kit, placed on a storage phosphor screen for 5 days, and imaged on a STORM 860 phosphorimager. Coomassie-stained gels were imaged with a Bio-Rad Gel Doc XR imager. Coomassie and 35S autoradiograms were quantitated using Image J (NIH), using the upper portion of the gel.
Polysome Profiling
Polysomes were run from a protocol optimized from Martin, 1973 (16). Briefly, C. elegans were grown on high-growth plates with NA22 bacteria food. For each sample, 80,000 animals were grown to L4/young adult and exposed to 50 ppm H2S or room air for 1 h. Animals were rinsed from the plates in M9, pelleted by centrifugation, and flash frozen in liquid N2. Samples were lysed with 60 strokes with each pestle in a Dounce homogenizer in 2× lysis buffer (50 mm Tris-HCl pH 8.0, 300 mm NaCl, 10 mm MgCl2, 1 mm NaEGTA, 0.2 mg/ml heparin, 2.5 mm PMSF, 0.2 mg/ml cycloheximide, 800 units/ml, 1% Triton X-100, 0.1% Na DOC, RNase free H2O to 5 ml final volume), and the lysate was centrifuged at 13,200 rpm at 4 °C for 18 min to pellet insoluble fraction. 20 OD (A260) of the supernatant was brought to 1 ml total volume with 1× lysis buffer, then floated on top of a 7.5%-47.5% sucrose gradient. Sucrose gradients were centrifuged at 39,000 rpm in a Beckman Coulter SW41 rotor at 4 °C for 2 h under vacuum. The samples were analyzed with a Brandel fractionator, and absorbance at A260 recorded as a function of retention time.
Quantitative RT-PCR
Total RNA was isolated from ∼9000 young adult C. elegans after exposure to 50 ppm H2S for 3 h. Animals were harvested in M9 buffer, added to 1 ml of TRIsol RNA isolation reagent (Life Technologies), and flash frozen in liquid nitrogen. mRNA was isolated following the manufacturer's protocol, and then cDNA was synthesized from 5 μg RNA using polyT primers included with Superscript III Reverse Transcriptase (Invitrogen) according to the manufacturer's instructions. Each 10 μl qPCR reaction contained 1 μl of cDNA and 5 μl of 2× Sybr Green Master Mix (Kappa Biosystems). Primers were added using a 0.2 μl pin tool. Absorbance was measured over 40 cycles using a Mastercycler RealPlex 2 (Eppendorf). The threshold cycle (Ct) for each sample was measured using the provided software, and normalized to hil-1 and irs-2 controls to generate ΔCt values as described (17). ΔΔCt was calculated as the change in ΔCt between animals exposed to H2S and room air controls. Average ΔΔCt ± S.D. are presented.
Western Blot
For SDS-PAGE, 3000 young adult C. elegans were harvested after a 2h exposure to 50 ppm H2S or room air. Animals were rinsed off plates with M9, pelleted by centrifugation and 50 μl of worm pellet was transferred into an equal volume of SDS-PAGE loading buffer. Samples were flash frozen in liquid N2. Before gel electrophoresis, samples were boiled for 15 min, and centrifuged to pellet debris. Proteins were separated on a 10% polyacrylamide gel, then transferred to a nitrocellulose membrane. Membranes were blocked in 5% Carnation nonfat dry milk in TBS for at least 1 h, and then incubated with primary antibody for 16 h at 4 °C. Membranes were washed for 5 min three times with TBST and then incubated with secondary antibody for at least 1 h at 4 °C and washed again as above. All antibodies were diluted in 5% BSA in TBS. Primary antibodies used were: α-phospho-eIF2α (S51) from Cell Signaling Technology (9721) at 1:2500; α-eIF2α from Cell Signaling Technology (9722) at 1:2500. Secondary donkey α-rabbit was conjugated to AlexFluor 680 or 790 (Invitrogen Life Technologies) used at 1:20,000 dilution.
Results and Discussion
C. elegans exposed to low concentration H2S are long-lived and better able to maintain proteostasis in hypoxia (18, 19). One key aspect of the proteostasis network is control of protein translation. Genetic perturbations that decrease global protein translation increase lifespan and prevent the age-associated decline of proteostasis (20–22). H2S has been shown to decrease translation in glucose-stressed rat kidney cells (23), raising the possibility that decreased global translation underlies the beneficial effects of H2S in C. elegans. Arguing against this possibility, however, C. elegans grown in H2S develop and produce embryos at the same rate as untreated controls, unlike animals in which global protein translation has been reduced (18).
To resolve whether H2S has effects on protein translation in C. elegans, we used a metabolic labeling approach. In these experiments, animals were labeled with [35S]methionine, then exposed to 50 ppm H2S (Fig. 1B). We reasoned that this approach would enrich the amino acid precursor pool with [35S]Met and enable us to measure translation during acute exposures to H2S on solid plates. As expected, the abundance of 35S-labeled protein increased over the three-hour exposure to H2S. There was no difference in 35S incorporation in wild-type (N2) animals exposed to H2S relative to untreated controls, indicating that H2S does not decrease protein synthesis (Fig. 1B). These data suggest that the beneficial effects of H2S on lifespan and proteostasis effects do not derive from global effects on translation.
SQRD-1 is the C. elegans orthologue of the conserved sulfide-quinone oxidoreductase (7). SQRD-1 is essential to survive in H2S, and its expression is rapidly up-regulated upon exposure to H2S (13). We observed less [35S]methionine incorporation in sqrd-1(tm3378) mutant animals exposed to low concentration of H2S, suggesting that translation had arrested in these animals (Fig. 1, C and D). The tm3378 allele of sqrd-1 is a 445 bp deletion that removes exon two and is a predicted molecular null (13). We confirmed the previous observation that sqrd-1(tm3378) mutant animals die when exposed to H2S (13), though we found that it takes at least 10 h before sqrd-1(tm3378) mutant animals succumb in 50 ppm H2S. For this reason, we only measured translation for up to three hours of H2S exposure, at which time sqrd-1(tm3378) animals were mobile and visibly indistinguishable from untreated animals and wild-type controls.
Our metabolic labeling experiments suggest that SQRD-1 is necessary to maintain global translation in H2S. To corroborate this observation, we performed polysome profiling experiments. These experiments measure the distribution of ribosomes engaged with mRNA and can help distinguish different mechanisms of altering translation, such as effects on translational initiation or termination (24, 25). Polysome profiles of wild-type worms exposed to H2S were indistinguishable from untreated controls, consistent with our assertion that H2S does not change translation in wild-type animals (Fig. 2A). In contrast, polysome profiles of sqrd-1(tm3378) mutant animals exposed to H2S show an increase in free 40S and 60S ribosomal subunits and a reduction in the translating fractions (Fig. 2B). This result supports our conclusion that translation is reduced in sqrd-1 mutants exposed to H2S. Moreover, the alterations in the sqrd-1 polysome profiles we observe are consistent with a reduction in the early steps of translational initiation.
FIGURE 2.

Decrease in translation in H2S is associated with sqrd-1 deficiency. A, polysome profile of wild-type (N2) animals exposed to H2S (solid red line) compared with controls that remained in room air (black dotted line). Arrows point to peaks containing free 40S and 60S ribosome subunits. The 80S monosome peak is marked, and polysome fractions are bracketed. B, polysome profile of sqrd-1(tm3378) mutant animals exposed to H2S (solid red line) compared with controls that remained in room air (black dotted line). Annotations as in A. C, quantification of change in percent of ribosomes actively translating after exposure to H2S. In addition to exposure to 50 ppm H2S (first three bars), the change in translation was also measured for wild-type (N2) animals exposed to 150 ppm H2S or hypoxia (far right). ΔTranslation = (% active H2S) − (%active room air). Number of independent replicates: N2, n = 5; sqrd-1, n = 3; hif-1, n = 7; N2 in 150 ppm H2S, n = 3. N2 in hypoxia n = 3
One possibility is that translation arrest in H2S is simply a result of cellular damage due to H2S toxicity. At high concentration, H2S binds to cytochrome oxidase and inhibits mitochondrial respiration (26). Our earlier experiments show that 50 ppm H2S does not diminish metabolic output in wild-type animals, even in combination with hypoxic conditions that inhibit respiration (18). Moreover, there is a 4000-fold excess of O2 (210,000 ppm) over H2S (50 ppm) in our experiments. Finally, C. elegans survive in anoxia, where the lack of O2 which severely limits mitochondrial respiration, for several days (29), whereas sqrd-1 mutant animals die within hours when exposed to H2S (13). For these reasons, we do not favor a model in which protein translation arrests due to H2S inhibition of respiration in sqrd-1 mutant animals, though we cannot exclude the possibility that inhibition of mitochondrial function does not contribute to the sqrd-1 mutant phenotype.
H2S toxicity is multifactorial and the organismal effects of excess H2S are not only due to the inhibition of respiration (27). We reasoned that if the effect of H2S on protein translation in sqrd-1 mutant animals resulted from nonspecific cytotoxicity then we would also observe an arrest of translation in other situations where exposure to H2S is lethal. To test this idea, we measured the effects of H2S on protein translation in wild-type animals exposed to lethally high concentrations of H2S (150 ppm; Fig. 2C). We observed no decrease in global translation in these experiments. We similarly found little change in global translation when hif-1(ia04) mutant animals, which are also sensitive to H2S, were exposed to 50 ppm H2S (Fig. 2C). These results indicate that the H2S-induced decrease in protein translation is associated with loss of SQRD-1 activity, rather than being a nonspecific effect that occurs when animals die from exposure to H2S. HIF-1 is required to survive exposure to low H2S and for increased expression of sqrd-1 in H2S (13, 17). This suggests that even basal expression of SQRD-1 is sufficient for sustained protein translation in H2S, even in conditions where H2S exposure is lethal.
One common mechanism of regulating translation is through phosphorylation of eIF2α. When phosphorylated, eIF2α sequesters translation initiation factors, which leads to a rapid arrest of global protein translation (30). We investigated whether the translational arrest in H2S was associated with increased phosphorylation of eIF2α. Consistent with this hypothesis, we observed a significant increase in phosphorylation of eIF2α when sqrd-1(tm3378) mutant animals were exposed to H2S (Fig. 3, A and B). In contrast, H2S exposure did not increase phosphorylation of eIF2α in wild-type controls. Thus, in H2S, phosphorylation of eIF2α is correlated with reduced global protein synthesis. We conclude that SQRD-1 activity is required to maintain translation in H2S by inhibiting phosphorylation of eIF2α.
FIGURE 3.
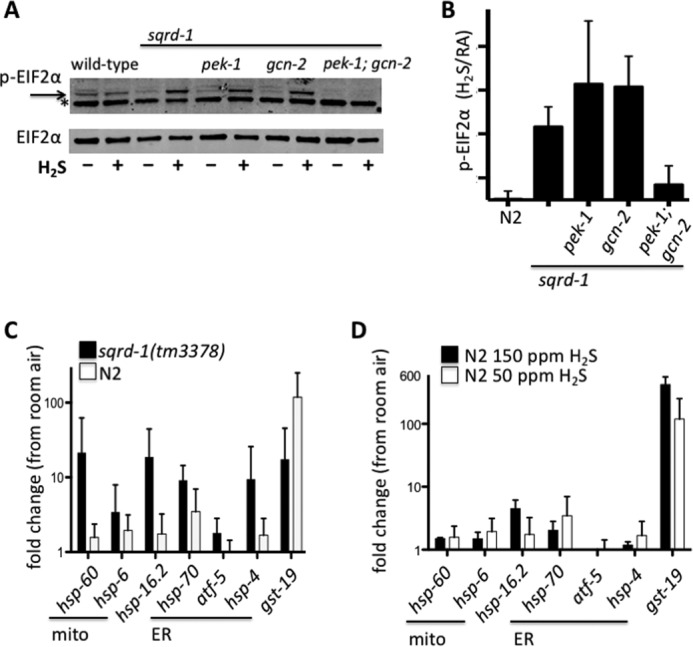
SQRD-1 prevents ER and mitochondrial stress in H2S. A, phosphorylation of eIF2α is stimulated in sqrd-1(tm3378) mutant animals exposed to H2S. Western blots to detect phosphorylated eIF2α. All strains except wild-type (N2) have the sqrd-1(tm3378) allele. In top blot, phosph-eIF2α is indicated by arrow, the * is a nonspecific band present in all samples. Bottom blot shows total eIF2α staining as a loading control. B, relative quantification of phospho-eIF2α staining from replicate Western blot experiments. Data shown are average of five independent biological replicates (error bars show S.D.) for each genotype. C, change in transcript abundance of gene products measured by qRT-PCR after exposure to H2S. Avg fold change calculated from ΔΔCt (ΔCtH2S - ΔCtRA), error bars show S.D. N2, n = 4; sqrd-1 n = 5 independent experiments. D, fold-change of stress response genes, measured by qRT-PCR of wild-type (N2) animals exposed to 150 ppm H2S for 3 h (n = 3 independent biological replicates). For comparison, data for N2 in 50 ppm is same as in panel C.
We hypothesized that H2S would inhibit translation in sqrd-1 mutant animals by activating one of the known eIF2α kinases. Phosphorylation of eIF2α is mediated by at least four kinases in mammals, PEK/PERK, GCN2, HRI, and PKR (31). C. elegans has orthologues of two of these kinases, GCN2 (gcn-2) and PERK (pek-1) (32). PEK-1 is an ER resident kinase that is activated by the accumulation of misfolded or unfolded proteins in the ER (33, 34). GCN-2 kinase binds to and is activated by uncharged tRNAs that accumulate during amino acid deprivation, and in response to mitochondrial stress (31, 32). In C. elegans, pek-1 is not required for the appropriate response to mitochondrial stress and gcn-2 is not activated in conditions that cause ER stress, suggesting that these two kinases act in distinct stress-response pathways (32).
To evaluate whether either GCN-2 or PEK-1 kinases are required for H2S-dependent phosphorylation of eIF2α, we introduced gcn-2(ok886) or pek-1(ok275) deletion alleles into sqrd-1(tm3378) mutant animals. When exposed to H2S, we observed robust phosphorylation of eIF2α in both pek-1; sqrd-1 and gcn-2; sqrd-1 double mutant animals (Fig. 3, A and B). This result suggests that either these kinases act redundantly to phosphorylate eIF2α in H2S, or that neither of these eIF2α kinases are involved in this response to H2S. To distinguish these possibilities, we generated pek-1; gcn-2; sqrd-1 triple mutant animals. H2S-dependent phosphorylation of eIF2α was abrogated in these animals (Fig. 3, A and B). We conclude that both PEK-1 and GCN-2 phosphorylate eIF2α when sqrd-1 animals are exposed to H2S.
The fact that both GCN-2 and PEK-1 phosphorylate eIF2α in sqrd-1 mutant animals exposed to H2S suggests that these animals are experiencing both mitochondrial and ER stress. We have previously shown that H2S does not induce either ER or mitochondrial stress responses in wild-type animals (17, 18). This suggests the possibility that these H2S induced cellular stresses only occur in the absence of SQRD-1 activity. To evaluate this possibility, we measured expression of genes that are up-regulated in response to mitochondrial or ER stress. We observed a significant increased in the abundance of transcripts encoding ER stress response genes as well as markers of mitochondrial stress when sqrd-1(tm3378) mutant animals were exposed to 50 ppm H2S (35–37) (Fig. 3C). As we previously reported, none of these transcripts were more abundant after H2S exposure of wild-type animals. Other stress-induced gene products, such as sod-3, a marker of oxidative stress, were not induced in either wild-type or sqrd-1(tm3378) mutant animals exposed to H2S (data not shown). Moreover, we did not observe increased expression of ER or mitochondrial stress response gene products in wild-type animals exposed to lethally high levels of H2S (Fig. 3D). These data show that H2S triggers a general unfolded protein response in the absence of SQRD-1 activity. We conclude that SQRD-1 activity normally protects the animals from unfolded protein stress in the ER and mitochondria when exposed to H2S.
Together, our data suggest that one activity of SQRD-1 in H2S is to prevent activation of the unfolded protein response in multiple cellular compartments. Our observation that phosphorylation of GCN-2 and PEK-1 occur only in the absence of SQRD-1 activity supports the idea that this protein is involved in normal cellular signaling in response to H2S. Consistent with our assertion that the inhibition of translation in H2S is not simply a consequence of nonspecific cytotoxicity of H2S, we found that unfolded protein response genes were not up-regulated in wild-type animals even when exposed to lethally high concentrations of H2S. (Fig. 3D). However, we cannot rule out the possibility that there may be fundamental differences between the nature of H2S toxicity at low and high H2S concentrations or in different mutant backgrounds.
One intriguing possibility is that SQRD-1 mediates hydrogen sulfide signaling to promote proteostasis, in addition to its function to oxidize and thereby detoxify H2S. We speculate that SQRD-1 could use H2S to generate a polysulfide, or sulfane sulfur, species (Fig. 1A) that could act as a cellular signal. This putative signal could be the sulfhydration of specific protein(s) (for example, as in 2, 4), though other reactive sulfur species can also be generated by SQRD-1 (42). Further studies are required to conclusively determine whether SQRD-1 promotes signaling in H2S in addition to detoxification.
The coordination of proteostasis across cellular compartments could be a conserved mechanism that underlies beneficial effects of H2S. We have found that treatment with H2S enhances proteostasis in C. elegans (19). Similarly, H2S alleviates protein aggregation in the forebrain of Zucker Diabetic Fatty Rats (38). Recently, H2S signaling has also been shown to mediate at least some aspects of dietary restriction, which reduces the age-associated decline in proteostasis (39–41). Understanding the role of SQRD-1 in these situations could provide new insight into fundamental cellular mechanisms of maintaining homeostasis in changing conditions.
Author Contributions
J. W. H. performed experiments. Both authors designed experiments, analyzed, and interpreted data, wrote the manuscript, and approve the final version.
Acknowledgments
We thank Dr. Vivien McKay for expert advice that was essential for us to complete polysome profiling experiments, and Dr. Kate Stoll, who performed initial polysome profile experiments. Dr. Malene Hansen (Sanford Burnham Institute) provided valuable advice for metabolic labeling experiments. We would like to thank the Miller lab for insightful comments on the manuscript. Some strains were provided by the Caenorhabditis Genetic Stock Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
This work was supported by National Institutes of Health Grants R00 AG033050, R01 ES024958, and R01 AG0044378 (to D. L. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- H2S
- hydrogen sulfide
- SQRD
- sulfide quinone oxidoreductase.
References
- 1. Olson K. R., DeLeon E. R., and Liu F. (2014) Controversies and conundrums in hydrogen sulfide biology. Nitric Oxide 41, 11–26 [DOI] [PubMed] [Google Scholar]
- 2. Kimura H. (2014) Hydrogen sulfide and polysulfides as biological mediators. Molecules 19, 16146–16157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bos E. M., van Goor H., Joles J. A., Whiteman M., and Leuvenink H. G. D. (2015) Hydrogen sulfide - physiological properties and therapeutic potential in ischaemia. Br. J. Pharmacol. 172, 1479–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paul B. D., and Snyder S. H. (2012) H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 13, 499–507 [DOI] [PubMed] [Google Scholar]
- 5. Nicholson C. K., and Calvert J. W. (2010) Hydrogen sulfide and ischemia-reperfusion injury. Pharmacol. Res. 62, 289–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Milby T. H., and Baselt R. C. (1999) Hydrogen sulfide poisoning: clarification of some controversial issues. Am. J. Ind. Med. 35, 192–195 [DOI] [PubMed] [Google Scholar]
- 7. Theissen U., Hoffmeister M., Grieshaber M., and Martin W. (2003) Single eubacterial origin of eukaryotic sulfide:quinone oxidoreductase, a mitochondrial enzyme conserved from the early evolution of eukaryotes during anoxic and sulfidic times. Mol. Biol. Evol. 20, 1564–1574 [DOI] [PubMed] [Google Scholar]
- 8. Libiad M., Yadav P. K., Vitvitsky V., Martinov M., and Banerjee R. (2014) Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 289, 30901–30910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jackson M. R., Melideo S. L., and Jorns M.S. (2012) Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51, 6804–6815 [DOI] [PubMed] [Google Scholar]
- 10. Tu B. P., and Weissman J. S. (2002) The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 10, 983–994 [DOI] [PubMed] [Google Scholar]
- 11. Goubern M., Andriamihaja M., Nübel T., Blachier F., and Bouillaud F. (2007) Sulfide, the first inorganic substrate for human cells. The FASEB J. 21, 1699–1706 [DOI] [PubMed] [Google Scholar]
- 12. Lagoutte E., Mimoun S., Andriamihaja M., Chaumontet C., Blachier F., and Bouillaud F. (2010) Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta 1797, 1500–1511 [DOI] [PubMed] [Google Scholar]
- 13. Budde M. W., and Roth M. B. (2011) The response of Caenorhabditis elegans to hydrogen sulfide and hydrogen cyanide. Genetics 189, 521–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Riddle D. L., Blumenthal T., Meyer B. J., Preiss J. R., and Pettitt J. (1998) C. elegans II. Trends Cell Biol. 8, 92 [Google Scholar]
- 15. Fawcett E. M., Horsman J. W., and Miller D. L.. 2012. Creating defined gaseous environments to study the effects of hypoxia on C. elegans. J. Vis. Exp. e4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martin T. E. (1973) A simple general method to determine the proportion of active ribosomes in eukaryotic cells. Exp. Cell Res. 80, 496–498 [DOI] [PubMed] [Google Scholar]
- 17. Miller D. L., Budde M. W., and Roth M. B. (2011) HIF-1 and SKN-1 coordinate the transcriptional response to hydrogen sulfide in Caenorhabditis elegans. PLoS ONE 6, e25476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miller D. L., and Roth M. B. (2007) Hydrogen sulfide increases thermotolerance and lifespan in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 104, 20618–20622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fawcett E. M., Hoyt J. M., Johnson J. K., and Miller D. L. (2015) Hypoxia disrupts proteostasis in Caenorhabditis elegans. Aging Cell 14, 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kennedy B. K., and Kaeberlein M. (2009) Hot topics in aging research: protein translation, 2009. Aging Cell 8, 617–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim H., and Strange K. (2013) Changes in translation rate modulate stress-induced damage of diverse proteins. Am. J. Physiol. Cell Physiol. 305, C1257–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sherman M. Y., and Qian S-B. (2013) Less is more: improving proteostasis by translation slow down. Trends Biochem. Sci. 38, 585–591 [DOI] [PubMed] [Google Scholar]
- 23. Lee H. J., Mariappan M. M., Feliers D., Cavaglieri R. C., Sataranatarajan K., Abboud H. E., Choudhury G. G., and Kasinath B. S. (2012) Hydrogen sulfide inhibits high glucose-induced matrix protein synthesis by activating AMP-activated protein kinase in renal epithelial cells. J. Biol. Chem. 287, 4451–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gebauer F., and Hentze M. W. (2004) Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 5, 827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sonenberg N., and Hinnebusch A. G. (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nicholls P., and Kim J. K. (1982) Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can. J. Biochem. 60, 613–623 [DOI] [PubMed] [Google Scholar]
- 27. Truong D. H., Eghbal M. A., Hindmarsh W., Roth S. H., and O'Brien P. J. (2006) Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab. Rev. 38, 733–744 [DOI] [PubMed] [Google Scholar]
- 28. Deleted in proof
- 29. Padilla P. A., Nystul T. G., Zager R. A., Johnson A. C. M., and Roth M. B. (2002) Dephosphorylation of cell cycle-regulated proteins correlates with anoxia-induced suspended animation in Caenorhabditis elegans. Mol. Biol. Cell 13, 1473–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leroux A., and London I. M. (1982) Regulation of protein synthesis by phosphorylation of eukaryotic initiation factor 2α in intact reticulocytes and reticulocyte lysates. Proc. Natl. Acad. Sci. U.S.A. 79, 2147–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Donnelly N., Gorman A. M., Gupta S., and Samali A. (2013) The eIF2α kinases: their structures and functions. Cell Mol. Life Sci. 70, 3493–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baker B. M., Nargund A. M., Sun T., and Haynes C. M.. 2012. Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genetics 8:e1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harding H. P., Zhang Y., and Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 [DOI] [PubMed] [Google Scholar]
- 34. Shen X., Ellis R. E., Lee K., Liu C. Y., Yang K., Solomon A., Yoshida H., Morimoto R., Kurnit D. M., Mori K., and Kaufman R. J. (2001) Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107, 893–903 [DOI] [PubMed] [Google Scholar]
- 35. Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., and Ron D. (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 [DOI] [PubMed] [Google Scholar]
- 36. Patil C. K., Li H., and Walter P. (2004) Gcn4p and novel upstream activating sequences regulate targets of the unfolded protein response. PLos Biol. 2, E246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoneda T., Benedetti C., Urano F., Clark S. G., Harding H. P., and Ron D. (2004) Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 117, 4055–4066 [DOI] [PubMed] [Google Scholar]
- 38. Talaei F., Van Praag V. M., Shishavan M. H., Landheer S. W., Buikema H., and Henning R. H. (2014) Increased protein aggregation in Zucker diabetic fatty rat brain: identification of key mechanistic targets and the therapeutic application of hydrogen sulfide. BMC Cell Biol. 15, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uthus E. O., and Brown-Borg H. M. (2006) Methionine flux to transsulfuration is enhanced in the long living Ames dwarf mouse. Mech. Ageing Dev. 127, 444–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kabil H., Kabil O., Banerjee R., Harshman L. G., and Pletcher S. D. (2011) Increased transsulfuration mediates longevity and dietary restriction in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 108, 16831–16836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hine C., Harputlugil E., Zhang Y., Ruckenstuhl C., Lee B. C., Brace L., Longchamp A., Treviño-Villarreal J. H., Mejia P., Ozaki C. K., Wang R., Gladyshev V. N., Madeo F., Mair W. B., and Mitchell J. R. (2015) Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160, 132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mishanina T. V., Libiad M., and Banerjee R. (2015) Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 11, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]