

Abstract
Vascular cell responses to exogenous heparin have been documented to include decreased vascular smooth muscle cell proliferation following decreased ERK pathway signaling. However, the molecular mechanism(s) by which heparin interacts with cells to induce those responses has remained unclear. Previously characterized monoclonal antibodies that block heparin binding to vascular cells have been found to mimic heparin effects. In this study, those antibodies were employed to isolate a heparin binding protein. MALDI mass spectrometry data provide evidence that the protein isolated is transmembrane protein 184A (TMEM184A). Commercial antibodies against three separate regions of the TMEM184A human protein were used to identify the TMEM184A protein in vascular smooth muscle cells and endothelial cells. A GFP-TMEM184A construct was employed to determine colocalization with heparin after endocytosis. Knockdown of TMEM184A eliminated the physiological responses to heparin, including effects on ERK pathway activity and BrdU incorporation. Isolated GFP-TMEM184A binds heparin, and overexpression results in additional heparin uptake. Together, these data support the identification of TMEM184A as a heparin receptor in vascular cells.
Keywords: ERK, heparin-binding protein, receptor, signal transduction, vascular smooth muscle cells
Introduction
For more than 30 years, heparin has been known to specifically bind to cells in the vasculature and alter their physiology in addition to its well recognized function as an anticoagulant. Heparin binds to many proteins, including numerous growth factors, cytokines, coagulation factors, cell adhesion molecules, growth factor receptors, matrix glycoproteins, and others (for a review, see Ref. 1). In fact, heparin and the closely related glycosaminoglycan heparan sulfate (HS),4 interact with more than 400 proteins (2). Heparin decreases endothelial cell (EC) inflammatory gene expression and slows vascular smooth muscle cell (VSMC) proliferation (reviewed in Ref. 3). Specifically, ECs bind and endocytose heparin (4, 5), which is followed by decreased inflammatory signaling through NF-κB (6) and stress kinase activity (7, 8). Heparin binding in VSMCs (9) results in decreases in growth factor-induced ERK signaling (10, 11), inhibition of downstream transcription factor activity (12–14), changes in cell cycle inhibitory factors (15), and decreased proliferation (10, 16).
Reports of fluorescent heparin uptake into cells, where it modulated transcription factor function (17), and the requirements of HSPGs for basic growth factor delivery to the nucleus (18) indicate that receptor-mediated uptake of heparin or HS may also be critical for some heparin effects. Similarly, shed HSPG syndecan-1 can be taken up by cells and transported to the nucleus, where it alters histone acetylation (19). HS chains are required for uptake, and this uptake can be inhibited by exogenously added heparin. It is likely that the uptake of highly charged heparin and HS chains involves a receptor to manage transport across the membrane. Although many heparin-interacting proteins have been linked to specific functions, a receptor responsible for heparin uptake has not been identified.
In addition to heparin uptake into vascular cells, there is clear evidence for heparin/HS binding to growth factors, growth factor receptors, and matrix proteins. A systems analysis of heparin binding proteins suggests that many of the heparin-interacting proteins fall into clusters of proteins that are involved with interactions between cells and the extracellular space. Within that broad area, researchers identified clusters involved in control of the immune system, cell-cell communication, and control of proliferation (2).
Heparin and HS are structurally similar, having the same core repeating disaccharide and modifications. Heparin is a glycosaminoglycan, whereas HS typically remains bound to protein cores embedded in the membrane or the matrix (1). In addition, heparin chains have higher levels of sulfation than HS, although both HS and heparin have regions of differing sulfation levels (1). The interactions of heparin or HS with specific extracellular proteins are important in many functions. Therefore, it has often been difficult to assign effects of heparin/HS interactions to any one of these proteins without specific modifications to the heparin-interacting protein in question. In vivo, functions characterized as heparin-induced are actually likely to be carried out by one or more HSPGs. Because the many different HSPGs are ubiquitous and the presence of HS is evolutionarily conserved, it has been suggested that heparin binding proteins have actually evolved to interact with HSPGs and have, therefore, been termed HS binding proteins (1).
In the vasculature, interactions of HSPGs with growth factors and their receptors are crucial in angiogenesis as well as in vascular disease (20). HS/heparin interacts with both growth factors and their receptors and scaffolds proteins, facilitating growth factor dimerization and interaction between growth factors and their receptors (1). The hypothesis that heparin could block HSPG interactions with growth factors and thereby decrease responses to growth factors provides one logical explanation for some of the heparin responses in vascular cells. This hypothesis is supported by studies that indicate transactivation of heparin-binding EGF receptors in response to G protein-coupled receptor activation (21). Heparin has even been shown to inhibit angiotensin II-induced VSMC responses (22). Continuing reports of HSPG requirements for cell proliferation in response to activation of additional G protein-coupled receptor systems further support the hypothesis that heparin effects are simply due to displacement of HSPG co-receptors (23).
However, heparin interference with HSPG-growth factor interactions cannot explain all of the heparin effects on VSMCs. Specifically, it does not account for the fact that heparin treatment results in rapidly turning down cell responses rather than blocking them from starting (e.g. Refs. 10, 11). Nor does it address the fact that heparin decreases signaling even when initiated by phorbol esters (12). Heparin induction of p27kip synthesis (15) and DUSP1 (MKP1) expression (24) also occurs directly in response to heparin treatment, and high-affinity heparin binding sites and heparin uptake likely involve interactions with a receptor other than growth factors.
To identify and characterize a heparin binding protein(s) that could facilitate heparin uptake and other responses, we created mAbs that specifically block heparin binding to ECs (5). These mAbs (HRmAbs) mimic many heparin effects, including blocking VSMC ERK activation and proliferation and inducing DUSP1 synthesis (10, 24). These antibodies are able to immunoprecipitate a membrane protein from both ECs and VSMCs that is ∼45–50 kDa (5, 10).
We have determined recently that both HRmAbs and heparin induce signaling through a cGMP-dependent protein kinase pathway to alter VSMC responses to growth factors (14). The antibodies and heparin also alter EC physiology by decreasing JNK and p38 activity and downstream signaling because of JNK and p38 activity (see the accompanying report (8)). These studies all suggest that the antibodies recognize and stimulate a receptor for heparin that exists on both VSMCs and ECs. To determine the identity of the protein to which the HRmAbs bind, we hypothesized that HRmAb immunoprecipitates of membrane proteins from vascular cells would contain the protein responsible for heparin effects. We employed both heparin affinity and HRmAb affinity chromatography of membrane proteins and then identified the immunoprecipitated protein. Here we report evidence that this procedure isolates the transmembrane protein identified as TMEM184A. Prior studies on TMEM184A are limited, but evidence indicates involvement of the protein in vesicle transport in exocrine cells and Sertoli cells of mice (25, 26). Our data presented here and in the accompanying report (8) indicate that heparin effects on vascular cell physiology are blocked when TMEM184A on the surface is decreased significantly, supporting the hypothesis that heparin responses are mediated, at least partially, through TMEM184A, which acts as a receptor for heparin.
Experimental Procedures
Materials
Cell culture chemicals, DMEM and minimum Eagle's medium, 2.5% trypsin/EDTA, porcine gelatin, heparin, penicillin/streptomycin, PDGF, and glutamine were obtained from Sigma. Pretested FBS was obtained from Invitrogen, Atlanta Biologicals (Atlanta, GA), or Biowest (St. Louis MO) and heat-inactivated for 1 h at 55 °C or purchased as heat-inactivated. Anti-active ERK (catalog no. 4370), anti-BrdU (catalog no. 5292), and anti-phospho ELK-1 (pELK, catalog no. 9181) antibodies were from Cell Signaling Technology (Beverly, MA). Anti-DUSP1 (MKP-1, catalog no. sc1199), anti-caveolin-1 (catalog no. sc53564), and anti-TMEM184A (catalog no. sc292006, N-terminal domain, NTD, rabbit; catalog no. sc163460, internal domain, INT, goat) were from Santa Cruz Biotechnology (La Jolla, CA). Anti-TMEM184A (C-terminal domain, CTD, rabbit) was obtained from ProSci Inc. (Poway, CA). Biotinylated anti-GFP (MA5–15256-BTIN) was obtained from Thermo Fisher Scientific (Waltham, MA). Secondary antibodies with fluorescent tags or Biotin-labeled (donkey or bovine for goat primary antibodies, minimal cross-reactivity) were obtained from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Extra-avidin-alkaline phosphataseTM, 5-bromo-4-chloro-3-indolyl phosphate, and nitro blue tetrazolium were obtained from Sigma.
Cell Culture
A7r5 rat smooth muscle cells were obtained from the ATCC (Manassas, VA). Bovine aortic endothelial cells (BAOECs), bovine aortic smooth muscle cells (BAOSMCs), and rat aortic smooth muscle cells were obtained from Cell Applications, Inc. (San Diego, CA). Commercially available cells were grown as recommended by the supplier and exchanged into minimum Eagle's medium over time before experiments. Human brain microvascular endothelial cells were obtained from Cell Systems (Kirkland, WA) and cultured using Cell Systems complete medium before exchange into minimum Eagle's medium supplemented identically as the BAOEC culture. All vascular cells were cultured on plates or coverslips coated with 0.2% porcine gelatin (Sigma). Hybridomas were cultured as described previously, and the monoclonal antibodies were purified using affinity chromatography (5, 10).
Isolation of the Heparin Receptor
In a typical experiment, several (12–16) 150-mm dishes of BAOECs were washed with PBS and harvested by scraping with a rubber policeman in a 10% PBS solution supplemented with two protease inhibitor mixtures (Sigma, catalog nos. P8340 and P2714, used at the concentrations recommended by the manufacturer) and homogenized in a Dounce homogenizer, and then the supernatant was collected after centrifugation at 2000 × g for 10 min to remove nuclei and debris. The supernatant was centrifuged further at 28,000 × g for 1 h at 4 °C. All remaining purification steps also contained the protease inhibitor mixtures. The resulting membrane pellet was resuspended in PBS + 0.2% CHAPS. Three to four pellets were combined, and the 0.2% CHAPS/PBS-solubilized pellet was subjected to heparin affinity chromatography on heparin-agarose. The heparin column was washed with at least five column volumes of 0.2% CHAPS/PBS until the A280 reached a baseline absorbance. Heparin-binding proteins were eluted with 1 m NaCl in 0.2% CHAPS/PBS. The eluate was concentrated using an Amicon Ultra-15 centrifugal filter unit (Millipore, Billerica, MA) with a molecular weight cutoff of 10,000 and exchanged into 0.2% CHAPS/PBS, and then the protein concentration was determined using a Lowry assay.
To generate the affinity resin for precipitation, HRmAbs (5, 10) were incubated at 25 °C for 30 min with a 50-fold molar excess of EZ-link Sulfo-NHS-LC-Biotin (Pierce). Excess biotin was removed by washing several times with PBS. The biotinylated antibodies were allowed to bind 1.0 ml of streptavidin-modified magnetic beads (Pierce) with gentle shaking at 25 °C for 1 h. To remove any unbound material, the antibody-magnetic bead solution was washed several times with 0.2% CHAPS/PBS. 1 ml of the eluted material from the heparin affinity column was added to the antibody-modified magnetic beads and incubated with gentle shaking at 25 °C for 1 h. The beads were localized to the side of the incubation tube using a magnet, and the supernatant was removed. After several washings with 0.2% CHAPS/PBS (removal of all unbound protein was verified by measuring baseline absorbance at A280 in the washings), the magnetic beads were eluted with a 1 m NaCl, 0.2% CHAPS/PBS solution. The eluted material was concentrated using a Centricon-20 plus or an Amicon Ultra-15 centrifugal filter unit (Millipore) with a molecular weight cutoff of 10,000 and exchanged into 0.2% CHAPS/PBS. To exchange receptor protein out of CHAPS, trichloroacetic acid was added to 10% (v/v), and the precipitated protein was pelleted by centrifugation. TCA precipitates were processed as noted below.
PAGE
Approximately 10 μg of TCA-precipitated protein was solubilized in 2× sample buffer and brought to a pH of 8.5 by addition of 1 m Tris buffer (pH 8.8). SDS-PAGE was carried out using the Laemmli system. Direct staining of PAGE gels was accomplished with Coomassie Brilliant Blue R250 or with SYPRO® Ruby (Sigma). For immunoblotting, gels were transferred, and total protein was stained with colloidal gold (Bio-Rad). For preparative gels, entire TCA-precipitated samples were solubilized and separated as above, and gels were stained with SYPRO Ruby, which does not interfere with additional digestion or analysis.
MALDI Mass Analysis
For analysis of the intact protein from an acrylamide gel slice, the gel slice was destained at room temperature (22–23 °C) in aqueous 50% methanol/5% acetic acid overnight. The gel slice was then dehydrated with acetonitrile until the slice curled. The slice was then crushed in 100 μl of 100 mm NH4HCO3 and incubated at room temperature for 1 h. The supernatant was removed to a new tube, the extraction of the crushed slice was repeated, and the combined extracts were lyophilized to dryness. Matrix A (a saturated solution of sinapinic acid (Sigma) in 50% acetonitrile, 0.1% trifluoroacetic acid) was added to the dried sample, and the resulting solution was spotted (2 × 0.75 μl) on a ground stainless steel target and analyzed using a Bruker Microflex MALDI-TOF mass spectrometer (positive/linear ion mode, at least 100 laser shots, minimum laser power of 60%, 500-ns delayed ion extraction).
MALDI was also performed on a solution sample of the detergent-solubilized, antibody-purified heparin receptor. A heparan receptor sample (500 μl) was subjected to precipitation with cold acetone (−20 °C, overnight). The pellet was redissolved in 10 μl of matrix A. The supernatant from the acetone precipitation was lyophilized to dryness and resuspended in 10 μl of matrix A. Each matrix-resuspended sample (1 μl) was spotted on a ground stainless steel target and subjected to on-plate washing with cold water and acetone. In separate experiments, a solution sample of antibody-purified heparin receptor was precipitated with TCA, resuspended in 100 μl of PBS, and neutralized by adding Tris buffer (pH 8.8). These samples were then extracted four times with a 10-fold excess of water-saturated ethyl acetate to remove any excess CHAPS detergent. The extracted samples were lyophilized, and the pellets were resuspended in 20 μl of matrix A and analyzed as above. For publication, spectra were visualized using MoyerZ (Proteometrics), and the peak masses were determined from the smoothed spectra (smoothing factor, 10). Data were exported and plotted in Microsoft Excel.
For digestion of isolated receptor to peptides, TCA-precipitated samples were suspended in 100 μl of water and brought to pH 8.5 by addition of Tris buffer (pH 8.8). Samples were reduced with 10 mm DTT for 30 min at room temperature and acetylated with iodoacetamide to prevent disulfide bonds from reforming. Chymotrypsin (Pierce, MS grade) digestion was carried out in suspension at 37 °C overnight in 100 mm Tris-HCl (pH 8) with 2 mm calcium chloride (as recommended by the supplier). A portion of the sample was mixed with equal parts of matrix B (saturated α-cyano-4-hydroxy-cinnamic acid (Sigma) in 50% aqueous acetonitrile, 0.1% trifluoroacetic acid) and analyzed by MALDI. The spectra were calibrated using protein standards that included insulin (5734.52 m/z), cytochrome c [M+H] (12360.97 m/z) and [M+2H] (6181.05 m/z), myoglobin [M+H] (16952.31 m/z) and [M+2H] (8476.66 m/z), and ubiquitin I (8565.76 m/z) and analyzed using MASCOTTM (peptide mass fingerprint) to identify generated peptides. For the in-gel chymotrypsin digestion, treatment included reduction with100 mm tris[2-carboxyethyl]phosphine (1 h, at room temperature) and alkylation with 100 mm iodoacetamide (1 h at room temperature in the dark).
For trypsin digestion, gel slices were washed in 100% acetonitrile for 10 min. Upon removal of the acetonitrile, slices were rehydrated and reduced in 10 mm DTT at room temperature for 30 min. The DTT solution was removed, and proteins were alkylated with 50 mm iodoacetamide in 25 mm NH4OAc at room temperature for 30 min. The alkylation solution was discarded, trypsin (20 ng/μl in 50 mm NH4OAc) was added to the gel slice, and the slice was crushed using a pipette tip. The suspension was incubated for 30 min at 37 °C. The solution containing the released peptides was removed to a clean tube, and the gel fragments were again digested with fresh protease at 37 °C for 3 h. Solutions from the digests were combined, lyophilized to dryness, and suspended in 12.5 μl of 20% aqueous acetonitrile. Peptides were prepared for MALDI using Zebra spin columns (Pierce). MS Digest and Peptide Cutter were used to prepare theoretical digests of TMEM184A for comparison with experimental masses.
Immunoprecipitation
Confluent 150-mm dishes of BAOECs, BAOSMCs, or A7r5 cells were harvested in radioimmune precipitation assay buffer (150 mm NaCl, 10 μm Tris (pH 7.2), 0.1% SDS, 0.1% Triton-X-100, and 0.5% deoxycholate; Sigma) supplemented with two protease inhibitor cocktails (Sigma, catalog nos. P8340 and P2714) used at the concentrations recommended by the manufacturer. Briefly, 150-mm dishes were rinsed with cold PBS twice and incubated with 1 ml of radioimmune precipitation assay buffer for 30 min at 4 °C with rocking. Following this incubation, cells were scraped off the plates, placed in a microcentrifuge tube, and centrifuged for 10 min at 10,000 × g. The supernatant was mixed with 2 μg/ml of HRmAbs or commercial TMEM184A antibodies, as noted in the text, and incubated overnight at 4 °C on a rocker. After antibody incubation, 75 μl of equilibrated EZview red protein G affinity gel beads (Sigma) were added and incubated overnight at 4 °C on a rocker. After incubation, the beads were rinsed with radioimmune precipitation assay buffer three times, and bound protein was released by boiling the beads in SDS sample buffer for 5 min. Western blotting was performed as described below.
Western Blotting
Western blotting for specific proteins was carried out after solubilizing cell samples into 2× sample buffer, boiling, and separating by SDS-PAGE as above. Primary antibodies as noted in the text, were diluted 1:1000, except for α-tubulin (1:8000). The blots in shown Figs. 3A and 7A employed biotin-conjugated secondary antibodies (1:2000) and were developed using the ABC peroxidase staining kit and Pierce ECL Western blotting substrate (Thermo Scientific) development system according to the recommended protocol. The blots shown in Fig. 9, A and D, employed secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647 (diluted 1:5000), and visualization of bands was accomplished by fluorescence detection using the Bio-Rad ChemiDoc MP system (catalog no. 170-8280). The blots shown in Fig. 7, B and C, employed biotin-conjugated secondary antibodies (1:2000) and were developed with an alkaline phosphatase, nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate system as described previously (10, 14).
FIGURE 3.
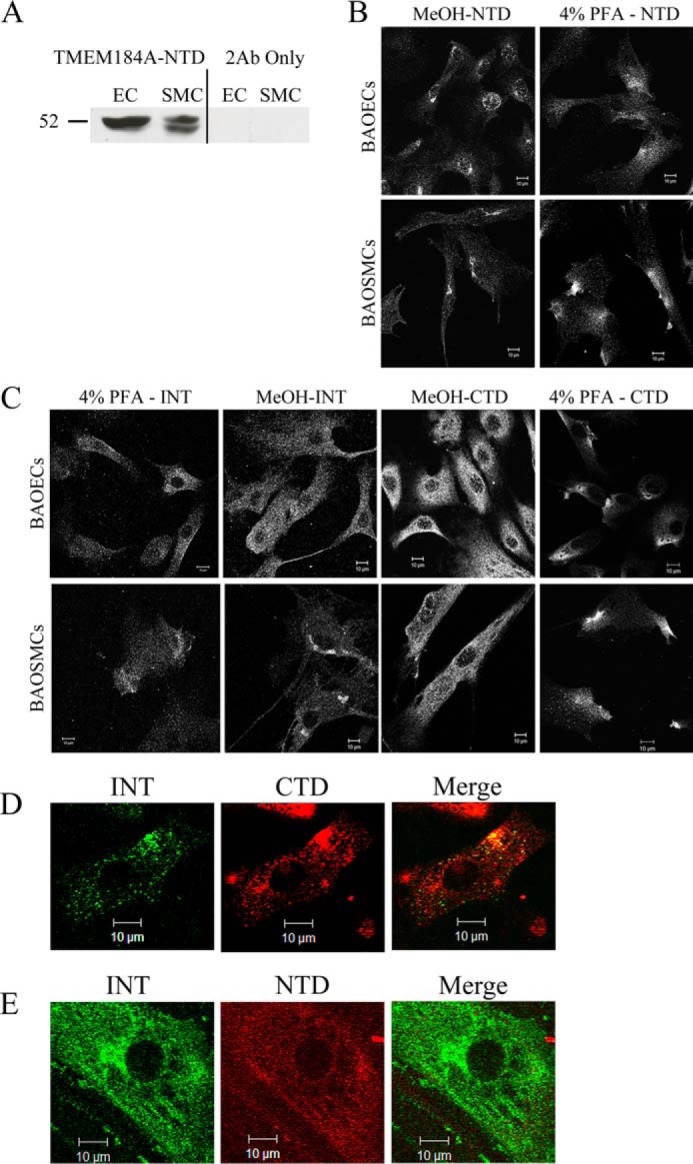
TMEM184A is expressed in vascular cells. A, whole cell lysates were immunoblotted for TMEM184A-NTD or compared with corresponding secondary antibody-only (2Ab Only) controls. B, cells were fixed with ice-cold methanol or 4% PFA without permeabilization, as noted, and stained for TMEM184A-NTD. C, cells were stained with the TMEM184A-INT and CTD antibodies. D, BAOECs were fixed with 4% PFA without permeabilization and stained concurrently with TMEM184A-INT and TMEM184A-CTD primary antibodies. The contrast of the INT antibody image was increased in FireworksTM, and the brightness of the CTD image was decreased to better visualize the areas of colocalization. E, BAOECs were fixed as in D and stained with TMEM184A-INT and TMEM184A-NTD. However, the NTD primary antibodies were added 8 h after the INT antibodies. The INT image brightness was decreased and the NTD image contrast was increased in FireworksTM to visualize areas of colocalization. All images were obtained using confocal microscopy. Western blotting and images are all representative of at least three repeats. Scale bars = 10 μm.
FIGURE 7.

Immunoprecipitation of the heparin receptor detectsTMEM184A. A, BAOECs were harvested for immunoprecipitation (IP) as described under “Experimental Procedures.” Cell lysates were incubated with the HRmAbs 12B1 (B1) or 18E9 (E9) and precipitated using EZview affinity beads. One-fourth of the final sample was developed with NTD, CTD, or secondary-only antibodies. IB, immunoblot. B and C, similar BAOEC lysates (B) and A7r5 lysates (C) were divided into identical fractions and incubated with 12B1, commercial NTD TMEM antibody, or beads alone (B). A whole cell (WC) sample was also used in B. Blots were developed using INT TMEM184A antibody and are representative of three repeats. Bands near the bottom of the blots are also seen with secondary antibodies only. All blots were developed and converted to grayscale for post-hoc analysis as described under “Experimental Procedures.”
FIGURE 9.

TMEM184A knockdown A7r5 cells do not respond to heparin. A7r5 cells were electroporated with control shRNA or TMEM184A shRNA as under “Experimental Procedures.” A, Western blotting indicating TMEM184A levels. NT, untreated cells; KD, knockdown. B and C, cells were treated with PDGF with or without heparin (Hep) pretreatment and fixed with 4% PFA, permeabilized with 0.3% Triton X-100, and stained for pERK (B) or pElk-1 (C). D, stable knockdown A7r5 cells were starved for 48 h, treated with heparin, fixed with 4% PFA, permeabilized with 0.3% Triton X-100, and stained for DUSP1 (D1). Some cells were also stained for TMEM184A and compared with A7r5 parent cells (D1). Heparin treatments were run in duplicate, and at least 500 cells were examined at each point (D2). Western blotting (D3) indicates DUSP1 levels for stable knockdown cells and parent A7r5 cells after 20 min heparin treatment. E, TMEM184A shRNA was used to knock down TMEM184A before addition of BrdU. Treatment with PDGF with or without heparin pretreatment was performed for 24 h, and BrdU incorporation was measured. All graphs represent at least 200 cells from at least three separate experiments (mean ± S.E.). *, p < 0.05.
Indirect Immunofluorescence
For characterization of TMEM184A, cells were fixed with 4% paraformaldehyde (PFA) for 15 min, washed three times in PBS, and permeabilized with 0.3% Triton-X-100 in PBS for 5 min. As noted, some experiments involved ice-cold methanol (MeOH) fixation instead of PFA. To stain for TMEM184A mostly on the cell surface, cells were fixed with methanol-free 4.0% PFA and not permeabilized. The cover-slips were incubated with primary antibodies against TMEM184A-NTD (dilution, 1:200) specific for amino acid residues 11–63 near the N terminus of human TMEM184A, TMEM184A-INT (dilution, 1:100) specific for an internal region of TMEM184A developed for 20 amino acids between residues 250 and 300, or TMEM184A-CTD (dilution, 1:200) specific for the C-terminal region developed for a 16-amino acid sequence near the C terminus of TMEM184A overnight at 4 °C. Co-staining with TMEM184A-CTD or TMEM184A-NTD with TMEM184A-INT antibodies was done by incubating primary antibodies together or, in some cases, incubating with one of the primary antibodies for up to 12 h before adding the second primary antibody. Because of species identity, the TMEM184A-CTD and TMEM184A-NTD primary antibodies could not be used together. Coverslips were washed with PBS and incubated with minimal cross-reactivity secondary antibodies (1:200, except 1:100 for anti-goat antibodies) conjugated to Alexa Fluor 488, tetramethylrhodamine isothiocyanate (TRITC), or Alexa Fluor 647 (Jackson ImmunoResearch Laboratories) for 2 h at room temperature or 45 min at 37 °C. Secondary antibody-only controls were performed and showed virtually no nonspecific interactions (data not shown). Cells were imaged using a Zeiss LSM 510 Meta microscope with a ×63 oil immersion lens at room temperature. Some A7r5 images were collected on a Nikon C2+ confocal microscope with a ×60 oil immersion lens at room temperature. All images in a set were taken at approximately the same Z plane of the cell where intensity was the greatest. Gain intensity was set just below saturating levels for each protein of interest, and those settings were used to image the remaining slides within a replicate.
GFP-TMEM184A Expression
A7r5 cells and BAOSMCs were transfected with 20 μg/ml of GFP-tagged TMEM184A plasmid (Origene, Rockville, MD) in the Bio-Rad Gene Pulser X-Cell system. Briefly, 100-mm confluent plates of cells were trypsinized, rinsed with PBS, suspended in HEPES-buffered saline, electroporated with a preset HeLa protocol (160 V modified to 170 V, 500 microfarad capacitance, ∞Ω resistance) and seeded onto glass coverslips in 6-well culture dishes, and then fresh medium was added after ∼24 h. BAOECs were transfected with GFP-TMEM184A using the same protocol with a preset CHO protocol (160 V, 1 pulse of 15.0-ms pulse length). Exogenous GFP-TMEM184A expression was monitored by confocal microscopy after 24 h (BAOECs and BAOSMCs) and 48 h (A7r5 cells).
Rhodamine-Heparin Binding and Uptake
Incubation of GFP-TMEM184A-transfected cells with 100 μg/ml rhodamine-conjugated heparin (Creative PEG Works, Winston-Salem, NC) for up to 10 min was carried out in culture medium, followed by immediate fixation in 4% PFA without permeabilization as above. Staining for TMEM184A, GFP-TMEM184A, and/or rhodamine-heparin was imaged by confocal microscopy as above. To quantify rhodamine-conjugated heparin uptake, cells were imaged as for the functional studies below. Fluorescence in individual cells was determined using ImageJ, and the average uptake in A7r5 cells (more than 50) in a given experiment was set as 1. Knockdown and transfected cell uptake was reported compared with A7r5 cells. For live imaging of heparin uptake, cells were grown in MatTek (Ashland, MA) 35-mm dishes as above for imaging after fixation. Individual dishes were moved to the confocal microscope after at least 24 h of culture, and heparin was added after cells were in focus. If uptake was not visible immediately, new regions were examined, and the time post-heparin addition was noted.
TMEM184A Knockdown
For siRNA-based confirmation of TMEM184A expression, A7r5 cells were transfected using 20 μg/ml rat TMEM184A siRNA or control siRNA (Santa Cruz Biotechnology) using the Bio-Rad Gene Pulser X-Cell system. Briefly, 100-mm confluent plates of cells were trypsinized, rinsed with PBS, suspended in HEPES-buffered saline, electroporated with the preset HeLa protocol (with preset voltage at 170), and seeded onto 100-mm dishes or glass coverslips in 6-well plates as described above.
For functional studies, A7r5 cells were transfected using 20 μg/ml of shRNA designed for rat TMEM184A in the pGFP-V-RS plasmid or control nonsense shRNA in the same plasmid (Origene) using the Bio-Rad Gene Pulser X-Cell system as for the siRNA transfection. Four shRNA constructs were tested for knockdown ability. Constructs B and C were identified as the most effective in the A7r5 cells. Thereafter, all knockdown experiments employed one of these constructs. Fresh medium was added after ∼24 h. Identification of TMEM184A knockdown involved staining for TMEM184A in cells that were fixed with 4% PFA, but without detergent or methanol, to observe surface protein specifically. For some experiments, knockdown was also analyzed with Western blotting. Approximately 72 h after electroporation, cells were treated with heparin at 200 μg/ml heparin for 20 min. Cells were then activated with PDGF with or without heparin as above. Untreated cells served as controls. Each condition was tested with cells containing control shRNA and with cells containing the shRNA against TMEM184A. Cells treated with PFA and permeabilized with Triton-X-100 were stained for pERK or pElk as described previously (14) to determine heparin effects.
Stable transfection of A7r5 cells was accomplished by transfecting the TMEM184A shRNA construct (which contains a puromycin selection gene) as above and plating cells onto 100-mm dishes for normal growth. 48 h post-electroporation, fresh minimum Eagle's medium containing 3 μg/ml puromycin was added. Medium containing 3 μg/ml puromycin was changed every 2 days for a total of 8 days. Viability was low, but several small colonies remained and were transferred to a 35-mm dish. Cells were maintained in puromycin selection medium for 7 more days. Cells were then removed from puromycin and analyzed for GFP and TMEM184A expression. All cells expressed varying levels of GFP and limited TMEM184A compared with control A7r5 cells. Stably transfected cells were employed in rhodamine-heparin uptake and DUSP1 induction as described.
DUSP1 induction was determined with cells stably transfected for TMEM184A knockdown. After seeding, stable knockdowns were transferred to FBS-free medium to facilitate optimal DUSP1 induction. Heparin was added to cells after 48 h in FBS-free medium at 200 μg/ml for 10, 20, or 30 min, and nuclear DUSP1 levels were determined as described previously (14). TMEM184A levels were confirmed by immunofluorescence and Western blotting.
BrdU Incorporation Assay
To evaluate cell proliferation in knockdown cells, A7r5 cells were electroporated with TMEM184A or control shRNA as above and seeded onto glass coverslips in 6-well plates. Fresh medium was not added for these experiments. 72 h after electroporation, 0.03 mg/ml BrdU was added to the medium for each well. 6 h after BrdU addition, cells were treated with PDGF with or without a 20-min heparin (200 μg/ml) pretreatment. Control cells were left untreated. After 24 h, coverslips were washed with cold 70% ethanol for 5 min at room temperature. Coverslips were washed with PBS three times and incubated with 1.5 m HCl for 30 min at room temperature. Coverslips were washed with PBS and incubated in a blocking buffer (1× PBS/5% bovine serum albumin/0.3% Triton X-100) for at least 1 h at room temperature. Coverslips were incubated with anti-BrdU primary antibody (1:50) overnight at 4 °C. Coverslips were washed with PBS and incubated with anti-mouse secondary antibody (1:200) for 45 min at 37 °C, washed again, mounted with Mowiol, and observed using a Nikon eclipse TE 2000-U fluorescence microscope.
Quantitative RT-PCR
A7r5 cells were transfected with TMEM184A or control siRNA as described and seeded onto 100-mm dishes. RNA was isolated using the Qiagen RNeasy total RNA isolation kit with optional on-column DNase digestion. Reverse transcription of 5 μg of total RNA was performed with SuperScript III First-Strand Synthesis Super Mix reagents (Invitrogen). RT-PCR reactions were set up using the Qiagen QuantiTect SYBR Green PCR kit. GAPDH was used as an internal control for normalization (GAPDH and TMEM184A primers were from Qiagen). RT-PCR cycling was performed with Qiagen RotorGene. RT-PCR data were analyzed using the 2−ΔΔCt method to determine relative TMEM184A expression.
Heparin Binding Assay
BAOECs were electroporated with 20 μg/ml GFP-TMEM184A vector and seeded into a 150-mm plate. 72 h after electroporation, cells were harvested in 0.2% CHAPS/PBS (as above) on ice with protease inhibitor mixture (catalog no. P2714, Sigma). To purify GFP-TMEM184A, 2 μg/ml biotinylated anti-GFP antibody was added to the solution and incubated overnight at 4 °C with rocking. Streptavidin-agarose beads (Sigma, catalog no. S1638) were washed three times with 10 column volumes of 0.2% CHAPS/PBS before being added to the solution and incubated overnight at 4 °C with rocking. Following this incubation, the solution was washed three times with 0.2% CHAPS/PBS before incubation with 0.2 m glycine/0.2% CHAPS/PBS on ice for 3 min with occasional gentle mixing. Beads were removed by centrifugation. The supernatant was neutralized with 1 m sodium bicarbonate. Sample was concentrated and exchanged into 0.2% CHAPS/PBS as above for large-scale preparations of wild-type TMEM184A. For comparison, GFP was purified from BAOECs in the same way as GFP-TMEM184A. Preparations were made under bright light to bleach GFP fluorescence.
Black NeutrAvidin 96-well dishes (Thermo Fisher Scientific) and black non-coated plates were washed with CHAPS/PBS. Biotinylated anti-GFP (60 pmol/well) was added to appropriate wells in the NeutrAvidin plate and incubated for 2 h at room temperature with shaking. Wells were washed three times for 5 min with 0.2% CHAPS/PBS with shaking. GFP-TMEM184A (∼5 nmol/well) was added to appropriate wells and incubated for 1 h at room temperature with shaking. Wells were washed three times, as above, with 0.2% CHAPS/PBS. Identical wells were prepared with GFP. Various concentrations of fluorescein-conjugated heparin (Creative PEG Works) were prepared in 0.2% CHAPS/PBS, added to the appropriate wells, and incubated for 10 min at room temperature with shaking. All samples were prepared in triplicate. Fluorescein emission (λexcitation, 494 nm; λemission, 519 nm) from the plate with bound GFP-TMEM184A was read initially after addition of heparin (complete wells) to obtain total fluorescence (TECAN Infinite® m200 Pro plate reader). The instrument gain was adjusted to ensure detection of heparin between 10 and 200 μg/ml heparin added. Unbound fluorescein-conjugated heparin was removed from wells coated with immobilized GFP-TMEM184A and placed into wells in the non-coated black plates. 0.2% CHAPS/PBS was added back into the wells from which heparin was removed. Fluorescein emission from the plate with bound GFP-TMEM184A was read, the 0.2% CHAPS/PBS containing residual unbound fluorescein-heparin was removed, and fresh 0.2% CHAPS/PBS was added to the GFP-TMEM184A-bound wells. Unbound fluorescein-heparin was placed in empty wells, and those wells were read to facilitate calculations of bound and unbound heparin. Wells without antibody and GFP-TMEM184A were prepared with the fluorescein-conjugated heparin dilutions to serve as standards. Fluorescein emission from the plate with bound GFP-TMEM184A was read after the final wash to obtain values for bound heparin. Emission values for triplicate samples were plotted after correction for protein without heparin background readings in control wells in each plate. Emission values from the complete wells were used to determine total heparin added, and the bound value was determined from the emission value after the wash. Free heparin was calculated from the difference between the original and final fluorescence in the plate. These calculations were necessary because there was more quenching of fluorescence in the presence of antibody or antibody and antigen than in the wells of either dish without added proteins.
Statistical Analysis
Mean nuclear fluorescence intensities are shown as mean ± S.E. Comparisons between groups were analyzed using analysis of variance followed by a Tukey post hoc test. Comparisons between two groups were analyzed using Student's t test. Differences with p < 0.05 were considered significant. Refer to the figure legends for statistical significance.
Results
TMEM184A Is Identified by HRmAb-based Purification
Heparin affinity chromatography and HRmAb-based affinity chromatography were employed to isolate the heparin receptor from BAOECs, where the HRmAb ability to block heparin binding was first identified (5). Detailed procedures for isolation of the putative heparin receptor are described under “Experimental Procedures.” In brief, BAOECs were removed from culture dishes by scraping in ice-cold PBS supplemented with protease inhibitors. The cells were homogenized, debris was cleared, and membrane fractions were obtained after high-speed centrifugation. 0.2% CHAPS/PBS was then employed to solubilize membrane proteins, and a heparin binding fraction was obtained. The proteins that bound to a heparin column in 0.2% CHAPS/PBS buffer were then subjected to a second affinity step involving HRmAbs that remained associated with streptavidin magnetic beads. The isolated putative receptor was analyzed by SDS-PAGE and stained for total protein (Fig. 1A). The approximate molecular weights of various receptor preparations were determined by SDS-PAGE to be 52–58 kDa. Isolated protein was subjected to SDS-PAGE under reducing conditions, and the band of interest was excised from the gel after Coomassie staining of an equivalent separate lane or, in some instances, after staining with SYPRO Ruby. The MALDI spectrum of protein from a gel slice shows a molecular weight in the 47.4-kDa range (Fig. 1B), whereas a solution sample of affinity-purified protein, not run on a gel but extracted with cold acetone (to remove CHAPS and any tightly bound lipid) showed a molecular weight of ∼45.5 kDa. MALDI spectra of solution (detergent-solubilized) heparin receptor could not be obtained without solvent extraction. When extracted with acetone, the protein shown in Fig. 1C stayed soluble in the acetone. Additional experiments with ethyl acetate-extracted solution samples gave m/z values of 45,877 and 48,149.3 (data not shown). Although the weights obtained by MALDI were much lower than the molecular mass of ∼53 kDa determined by SDS-PAGE, the MALDI weights are closer to those obtained by SDS-PAGE of prior analytical preparations (5, 10).
FIGURE 1.

Analysis of protein isolated with HRmAbs. A, analytical amounts of protein isolated as described under “Experimental Procedures” were separated by SDS-PAGE and stained with Coomassie Blue (lane 1) or transferred to nitrocellulose and stained for protein with colloidal gold (lane 2). A complete preparation was loaded onto a gel and stained with SYPRO Ruby (lane 3). B, MALDI analysis of affinity-purified protein recovered from an acrylamide gel slice. C, MALDI analysis of detergent-solubilized, affinity-purified protein prepared for analysis by acetone extraction (the spectrum is of acetone-soluble material).
The intact receptor was separated by gel electrophoresis and stained with SYPRO Ruby, and stained receptor bands were cut out of the gel. These gel slices were crushed and digested with chymotrypsin as under “Experimental Procedures” and analyzed by MALDI followed by MASCOTTM analysis for possible protein matches. Two separate analytical runs returned peptide matches for TMEM184A (Table 1). Similar gel samples and soluble receptor samples were digested with trypsin. MS Digest and ExPASY Peptide Cutter (see “Experimental Procedures”) were used to prepare theoretical digests of TMEM184A for comparison with experimental masses (Table 2). Together, several different analyses resulted in peptides that covered 75% of the TMEM184A sequence. Fig. 2 summarizes all of the chymotrypsin and trypsin digest data.
TABLE 1.
Comparison of Theoretical Peptides from TMEM184A to Chymotryptic Peptides from Isolated Receptor
Peptides were generated from bovine endothelial protein isolated using monoclonal antibodies against the heparin receptor. These peptides were identified by MASCOT as matching in TMEM184A (bovine) that had been alkylated prior to digestion.
| Chymotrypsin-generated Peptides | Locationa | Observed m/z | Theoretical m/z |
|---|---|---|---|
| TVPHEQRYIIRL | 72–83 | 1523.84 | 1522.8327 |
| QFCIVKPIMALVTIVL | 178–193 | 1847.10 | 1846.0927 |
| CIVKPIMAL | 180–188 | 1046.70 | 1045.6927 |
| VYNASVSLALYALFLF | 216–231 | 1790.10 | 1789.0927 |
| FLFYSATRELLQPF | 229–242 | 1733.10 | 1632.0927 |
| SEKTESSPAPSAPMQSISSGL | 320–340 | 2091.39 | 2090.3827 |
a Refers to location in TMEM184A.
TABLE 2.
Comparison of Theoretical Peptides from TMEM184A to Tryptic Peptides from Isolated Receptor Protein
Peptides were generated from bovine endothelial protein isolated using monoclonal antibodies against the heparin receptor. This table shows only those peptides with 0 or 1 missed cleavage. Samples were carboxymethylated and analysis includes the possibility of modification. *, peptides from in-gel digest; masses identified by MS Digest.; **, peptides from in-gel digest; masses identified by PeptideCutter; ***, peptides from expressed solution digest; masses identified by PeptideCutter; ****, peptides from expressed solution digest; masses identified by MS Digest.
| Trypsin-generated Peptides | Locationa | Observed m/z | Theoretical m/z | Notes |
|---|---|---|---|---|
| TSCFHGTCCLR | 150–160 | 1285.35b | 1285.5021 | * |
| GMTYSIGFLR | 161–170 | 1144.11b | 1145.3741 | *, ** |
| TSCFHGTCCLRGMTYSIGFLR | 150–170 | 2469.72b | 2470.8903 | **** |
| QATLQFCIVKPIMALVTIVLQAFGK | 174–198 | 2733.10b | 2734.4471 | ***, **** |
| ETMSPQDIVQDAIHNFSPAYQK | 342–363 | 2518.66b | 2520.7974 | ***, **** |
| NVEKRMLIPAEEL | 402–414 | 1542.01b | 1542.8335 | ** |
| AGNGSQGPGPLFLTSPLARGVSGVFVWAALVLTGHQIYLHLR | 28–69 | 4356.05c | 4360.0665 | **, *** |
| SYTVPHEQR | 70–78 | 1114.84c | 1117.2091 | ** |
| DCYEAFVIYSFLSLCFQYLGGESAI MAEIRGKPVR | 115–149 | 4093.54c | 4094.7040 | ***, **** |
| IHDGDFNVR | 199–207 | 1121.73c | 1123.1694 | *, **, ***, **** |
| SGYLYITLVYNASVSLALYALFLFYSATR | 208–236 | 3277.81c | 3281.8183 | ***, **** |
| ELLOPFEPVLK | 237–247 | 1310.42c | 1313.5793 | ** |
| FLTIKAVIFLSFWQGLLLAILER | 248–270 | 2690c | 2693.3335 | **, ***, **** |
| YAFTCQVYSEKTESSPAPSAPMQSISSGLK | 312–341 | 3196.94c | 3196.5707 | ** |
| TESSPAPSAPMQSISSGLKETMSPQDIVQDAIHNFSPAYQK | 323–363 | 4373.82c | 4377.8412 | **, ***, **** |
| YTQQATQEAPRPGQGSVPSPR | 364–384 | 2256.63c | 2256.4394 | ***, **** |
| TPTHSPDGGPGGGR | 385–398 | 1291.26c | 1293.1769 | *, **, **** |
| RMLIPAEEL | 406–414*** | 1069.28c | 1072.3075 | ***, **** |
a Location refers to their location in TMEM184A.
b Identified by Protein Prospector as high confidence matches in one or more samples; many of these peptides were also seen in additional samples but with lower confidence.
c Identified by Protein Prospector as lower confidence matches in one or more samples.
FIGURE 2.

Receptor peptide analysis in the TMEM184A gene sequence. Chymotrypsin- and trypsin-generated peptides were mapped onto the bovine TMEM184A gene sequence to examine the extent of gene coverage. Red and green underlines indicate chymotrypsin-generated peptides from Table 1. Blue and purple underlines represent matches from the trypsin digests described in Table 2.
Primary and Cloned Vascular Cells Express TMEM184A
Protein analyses suggested TMEM184A as a heparin receptor candidate. Therefore, the expression of TMEM184A and its location(s) in cells were investigated in BAOECs and BAOSMCs. Whole cell Western blotting was carried out for BAOECs and BAOSMCs using the NTD commercial antibody. As shown in Fig. 3A, distinct bands are present in the predicted molecular weight range of TMEM184A that are absent from secondary antibody-only controls. The doublet bands in BAOSMC samples could be due to posttranslational modifications (e.g. glycosylation) that have been identified in mouse TMEM184A proteins (25). However, modifications in mouse samples resulted in high molecular weight protein. High molecular weight TMEM184A was not typically identified in our studies. NTD antibody staining of primary BAOECs and BAOSMCs showed cell surface staining with regions of varying intensity in cells fixed with PFA and not permeabilized (Fig. 3B). In permeabilized cells, the antibody stained mostly a perinuclear region.
Two additional commercially available polyclonal antibodies raised to different regions of the human TMEM184A protein were employed to confirm TMEM184A presence and localization in vascular cells. As shown in Fig. 3C, cells that were fixed with PFA (but not permeabilized) show specific staining patterns with the CTD antibody as well, consistent with a presence at the plasma membrane. In permeabilized cells (Fig. 3C), TMEM184A was detectable by confocal microscopy in the perinuclear region, consistent with reports of TMEM184A in Sertoli cells and exocrine cells (25, 26). These studies also indicated colocalization with vesicle-associated membrane protein. Our vascular cells express a large amount of vesicle-associated membrane protein, which is diffuse in vesicles throughout the cytoplasm with intense clusters in defined perinuclear regions, where it colocalized with TMEM184A (data not shown). The CTD staining was also localized to the membrane and to cytoplasmic vesicles, with broader distribution than the NTD staining. The INT antibody stained in a pattern similar to both NTD and CTD staining. This antibody stained the membrane, the perinuclear region, and cytoplasmic vesicles (Fig. 3C).
To further examine the differences in staining between the three commercial antibodies, cells were stained with pairs of these antibodies. One example of each antibody pair used to stain BAOECs is illustrated in Fig. 3, D and E. Obtaining clear images using co-staining with either NTD and INT antibodies or CTD and INT antibodies was difficult because the staining by each antibody appeared to be affected negatively by the other antibody used. When added together, either the NTD or the CTD antibody resulted in limited staining by the INT antibody. When the INT antibody was added first, the staining of the CTD or NTD antibody declined somewhat. Because the NTD and CTD antibodies were both produced in rabbit, this pair was not tested. These results suggest that the antibody pairs evaluated interfere with each other, presumably by steric hindrance between the primary (and/or secondary) antibodies. Differences in staining could be due to differential antibody accessibility, localization of epitopes on different faces of the membrane, or some combination of these factors. Nevertheless, significant colocalization of antibody pairs is seen in dually stained cells. The colocalization observed represents evidence for TMEM184A localization at the cell surface and in the perinuclear region along with some staining of other vesicles. Therefore, we identified the candidate heparin receptor in bovine vascular cells.
To confirm the presence of TMEM184A in vascular cells of other species, primary and cloned rat VSMCs were examined. Similar staining occurred in both rat aortic smooth muscle cells and cloned A7r5 cells, indicating that the staining was not specific to bovine cells or primary cells (Fig. 4A). In addition, human brain microvascular endothelial cells express TMEM184A (Fig. 4B). In all of these cells, localization appears to be similar to that observed in BAOECs and BAOSMCs. The presence of TMEM184A in vascular cells is consistent with its identification as a heparin receptor.
FIGURE 4.

Rat and human vascular cells express TMEM184A. A, primary rat aortic smooth muscle cells and cloned A7r5 cells were stained with the NTD and CTD antibodies after either methanol or PFA fixation as described in Fig. 3. B, human brain microvascular endothelial cells were stained with the NTD antibodies. Images are representative of at least three repeats. Scale bars = 10 μm.
To confirm specificity for the antibody detection of TMEM184A, A7r5 cells were treated with either species-specific siRNA for TMEM184A or control siRNA and stained for TMEM184A (Fig. 5A). The TMEM184A staining in PFA-fixed cells decreased in knockdown cells compared with control siRNA-treated cells. To ensure that TMEM184A mRNA was decreased by this technology, mRNA was prepared, and quantitative RT-PCR was performed as described under “Experimental Procedures.” As expected, Fig. 5B illustrates that there was a significant decrease in the level of TMEM184A mRNA after knockdown, indicating that mRNA for TMEM184A was also present. The knockdown of message as well as surface staining supports the specific antibody detection of TMEM184A at the cell surface.
FIGURE 5.

Validation of TMEM184A using TMEM184A siRNA and GFP-TMEM184A expression. A, A7r5 cells were electroporated with control siRNA or TMEM184A siRNA, and cells were fixed with 4% PFA, stained for TMEM184A-INT on the surface of cells, and imaged with confocal microscopy. B, A7r5 cells were treated with scrambled or siRNA for TMEM184A. After 24 h, RNA was prepared, and RT-PCR was carried out as described. The graph indicates differences in RNA. C, A7r5s, BAOECs, and BAOSMCs were electroporated with 20 μg/ml of GFP-TMEM184A and allowed to proliferate for 24 h (BAOECs and BAOSMCs) or 48 h (A7r5 cells), and then they were fixed with 4% PFA. Images in the right column are magnified ×2 from the boxed areas on the left. All images are representative of at least three repeats. Scale bars = 10 μm.
The subcellular distribution of TMEM184A in vascular cells determined by antibody staining was confirmed by transfection of A7r5 cells, BAOECs, and BAOSMCs with GFP-TMEM184A. Significant expression of GFP-TMEM184A in the bovine cells occurred at about 24 h after transfection. The subcellular distribution of GFP-TMEM184A is similar to the membrane and perinuclear location seen with antibody staining (Fig. 5C), providing further evidence for the subcellular distribution of TMEM184A. Perinuclear staining in bovine cells was more limited than in A7r5 cells, where increased expression was observed at 48 h. The images of cells in Fig. 5 indicate the range of GFP-TMEM184A localization observed in transfected cells. These results confirm that TMEM184A localizes to the plasma membrane and perinuclear regions in cultured vascular cells and indicate a range of intracellular distribution as seen with TMEM184A antibodies (Fig. 3) rather than appearing identical to staining with any one of the antibodies. These data are consistent with TMEM184A identification as a heparin receptor.
TMEM184A Colocalizes with Caveolin in Vascular Cells
The patchy surface staining suggested that plasma membrane TMEM184A might be clustered in membrane regions also containing caveolin, a protein with well documented roles in membrane trafficking (reviewed in Ref. 27). We investigated the possibility that patches of TMEM184A were in regions containing caveolin by co-staining with commercial antibodies against both proteins. As predicted, we found co-localizing patches of both proteins in both ECs and VSMCs (Fig. 6).
FIGURE 6.
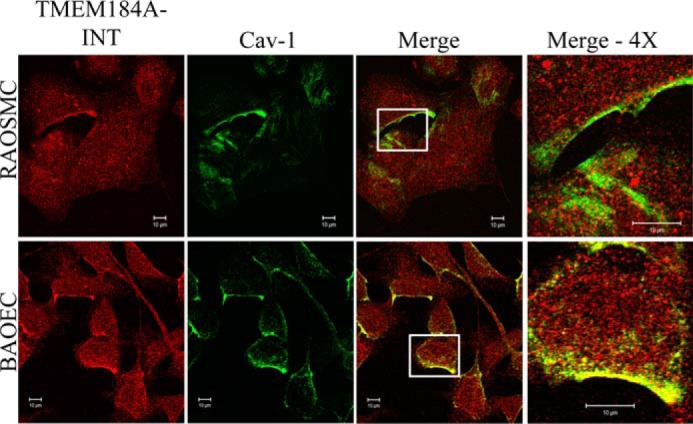
TMEM184A colocalizes with caveolin-1. Rat aortic smooth muscle cells and BAOECs were fixed and permeabilized with 4% PFA and 0.3% Triton X-100. Cells were treated with antibodies specific for TMEM184A-NTD (red) and cav-1 (green). The images in the fourth column are magnified ×4 from the original boxed areas in the third column. Images are representative of at least three repeats. Scale bars = 10 μm.
HRmAbs Immunoprecipitate TMEM184A
TMEM184A is clearly expressed in cultured vascular cells and localized appropriately for a receptor that participates in endocytosis. To examine further whether the protein isolated with the HRmAbs was TMEM184A, vascular cell protein immunoprecipitated using the HRmAbs as described under “Experimental Procedures” was analyzed by Western blotting using commercial antibodies. HRmAb-precipitated samples from BAOECs (Fig. 7A) were recognized using commercial TMEM184A antibody preparations. Affinity beads alone and beads with secondary antibody alone did not yield TMEM184A staining. Parallel immunoprecipitation from BAOECs (Fig. 7B) and A7r5 cells (Fig. 7C) with the HRmAbs and NTD antibodies resulted in identical immunostaining with INT antibodies. Therefore, the HRmAbs do bind to the TMEM184A protein, supporting its identification as the heparin receptor.
Internalized Heparin Localizes with TMEM184A
To support the hypothesis that TMEM184A is a receptor involved in heparin uptake, cells expressing GFP-TMEM184A were treated with rhodamine-heparin. Extensive colocalization was evident between GFP-TMEM184A and rhodamine-heparin at puncta throughout the cytoplasm (Fig. 8). Cells that internalized rhodamine-heparin appeared to exhibit less characteristic surface GFP, suggesting that TMEM184A on the surface was internalized with the labeled heparin (Fig. 8A). A7r5 cells expressing GFP-TMEM184A but not treated with rhodamine-heparin were used as a control and showed insignificant levels of red autofluorescence (data not shown). To show that the rhodamine-heparin uptake depended on TMEM184A, A7r5 cells were stably transfected with the rat-specific TMEM184A shRNA construct as described under “Experimental Procedures.” Rhodamine-heparin uptake was decreased significantly in A7r5 cells stably transfected compared with normal A7r5 cells (Fig. 8B). In addition, live imaging of A7r5 cells transfected with GFP-TMEM184A indicated that vesicles enclosed in GFP-TMEM184A stain contain a rhodamine label, and these moved together (Fig. 8C). Taken together, these results support the subcellular localization of TMEM184A in VSMCs and also provide evidence further supporting the identification of TMEM184A as a receptor for heparin. To evaluate the ability of TMEM184A to enhance heparin uptake, wild-type A7r5 cells, stable knockdown cells, and both of these cell types transfected with the GFP-TMEM184A construct were treated with rhodamine-heparin and analyzed for heparin uptake. Fig. 8D illustrates an average rhodamine label/cell from three separate experiments. As in Fig. 8B, stable knockdown cells took up significantly less labeled heparin. Transfection of the GFP-TMEM184A construct resulted in significant increases in rhodamine-heparin uptake for both the stable knockdown and A7r5 cells. Heparin uptake into the two transfected cell types was not significantly different.
FIGURE 8.

GFP-tagged TMEM184A colocalizes with rhodamine-heparin. A, A7r5 cells electroporated with GFP-TMEM184A as in Fig. 5 were treated with 100 μg/ml rhodamine-heparin for the indicated times, and then they were fixed with 4% PFA. Images are representative of two separate experiments. Scale bars = 10 μm. B, A7r5 cells and stable knockdowns for TMEM184A were treated with 100 μg/ml rhodamine-heparin for 10 min and fixed with 4% PFA. Images are representative of duplicates from two separate experiments. Scale bars = 10 μm. C, GFP-TMEM184A-transfected A7r5 cells were incubated with 200 μg/ml rhodamine-heparin, and cells were imaged immediately without fixing. Time-lapse confocal microscopy, initiated at about 4 min after heparin addition, identified colocalization of rhodamine-heparin (white vertical line) with a cluster of the GFP-TMEM 184A construct (scan zoom, 6.9; objective, ×63/1.4 oil differential interference contrast). The reference white bar points to the initial location and is a reference for concurrent movement of both labels. D, A7r5 cells and stable knockdowns for TMEM184A (KD) were either transfected with the GFP-TMEM184A construct or not transfected and treated as in B. At least 50 cells/condition (in each of three experiments) were analyzed for heparin uptake. **, p < 0.0001.
Knockdown of TMEM184A Eliminates Heparin Effects in VSMCs
To further examine the hypothesis that TMEM184A acts as a receptor for heparin, shRNA specific for rat TMEM184A was used to decrease TMEM184A expression in A7r5 cells. Transfection efficiencies of ∼75% were obtained consistently in the A7r5 cell line (on the basis of GFP expression also induced by the shRNA vectors). As shown in Fig. 9A, TMEM184A levels are reduced significantly in 72-h TMEM184A shRNA-transfected cells relative to control shRNA-transfected cells.
Control shRNA-transfected cells exhibited a PDGF-induced increase in nuclear phosphorylated ERK (pERK) as we have seen previously in VSMC (14). Similarly, TMEM184A shRNA cells responded equally well to PDGF (Fig. 9B). However, only control shRNA cells responded to heparin pretreatment with a significant decrease in nuclear pERK at both 10 and 20 min of PDGF (Fig. 9B). Knockdown cells did not respond to heparin and had levels of nuclear pERK similar to those without heparin treatment (Fig. 9B). PDGF treatment also induced a significant increase in pElk by 20 min, which was attenuated robustly by a 20 min heparin pretreatment prior to PDGF treatment (Fig. 9C). PDGF induced a smaller but significant increase in pElk-1 staining intensity in TMEM184A shRNA-transfected cells. After knockdown of TMEM184A, the heparin-induced attenuation of PDGF-activated pElk-1 staining was lost (Fig. 9C). Therefore, when TMEM184A levels were decreased, the cells no longer appeared to be sensitive to heparin. These results support the conclusion that TMEM184A is involved in mediating the heparin responses in VSMCs by serving as a receptor for heparin.
These data suggest that knockdown of TMEM184A would also decrease heparin induction of DUSP1 (24), which is required for heparin effects on ERK activity. Optimal heparin-induced expression of DUSP1 occurs in starved cells (24). However, no conditions to obtain significant knockdown of TMEM184A and the low levels of DUSP1 optimal for evaluating DUSP1 expression were found. Therefore, cells stably transfected with the TMEM184A shRNA construct were evaluated. TMEM184A staining is shown in Fig. 9D1. No evidence for heparin increases in DUSP1 was found in stable TMEM184A knockdown cells by evaluating nuclear DUSP1 levels (Fig. 9D2). Western blotting comparing stable knockdown cells to A7r5 cells treated identically also identified no heparin-induced increase in DUSP1 in cells lacking TMEM184A (Fig. 9D3), whereas there was an increase in A7r5 cells consistent with what we have observed previously in A7r5 cells (24). Again, these results indicate that TMEM184A functions as a receptor for heparin.
Because heparin decreases VSMC proliferation (e.g. 10), the effect of TMEM184A knockdown on heparin attenuation of PDGF-induced cell proliferation was examined using a BrdU incorporation assay. As shown in Fig. 9E, a 20-min heparin treatment prior to PDGF stimulation greatly reduces BrdU incorporation in control cells. The antiproliferative effect of heparin is eliminated in TMEM184A knockdown cells, where BrdU incorporation is actually increased above levels induced by PDGF alone. As with the previous results, these data indicate that the protein product of TMEM184A is needed for cells to respond typically to heparin. Taken together, the data indicate that decreasing TMEM184A expression results in decreased heparin sensitivity.
Evidence That TMEM184A Binds to Heparin
Together, the preceding evidence indicates a role for TMEM184A in heparin uptake and heparin responses. Several pieces of evidence also suggest that TMEM184A binds to heparin. First, heparin affinity chromatography in PBS was used in the purification of the putative receptor protein(s) for MALDI mass analysis. Further, HRmAb affinity-purified protein bound heparin in PBS and the binding decreased when added sodium chloride reached ∼0.5 m (5).
We examined the protein sequence of TMEM184A for sequences consistent with heparin binding. Although there is no specific single heparin binding sequence, heparin and HS binding proteins typically have small clusters of basic amino acids and often have additional basic amino acids further away in the linear sequence that are involved in heparin and/or HS binding (1). In fact, different sulfation patterns appear to be able to bind different sequences in HS binding proteins (e.g. 28). A portion of the bovine sequence for TMEM184A (amino acids 392 to the carboxyl terminus) is shown in Fig. 10A. A comparison to the human and rat sequences shows the presence of conserved basic amino acid clusters that are conserved in many other species as well. There are additional basic amino acids between the last predicted transmembrane region and the C-terminal region shown and in other regions of the protein that are not predicted to be membrane-spanning. Therefore, the sequence data are also consistent with identification of TMEM184A as a heparin/HS receptor.
FIGURE 10.
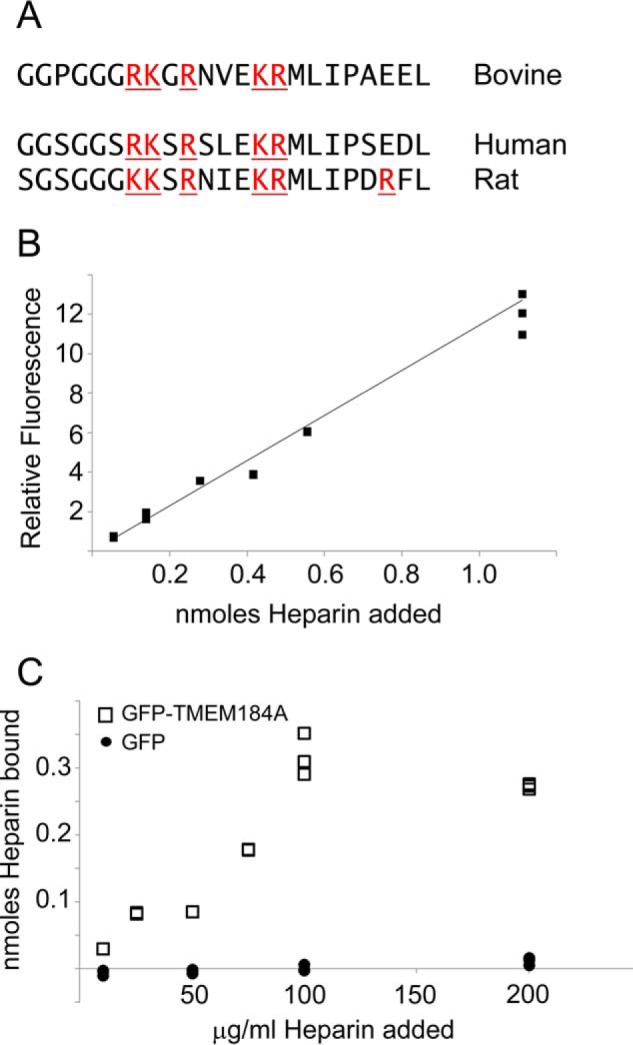
Heparin binds to TMEM184A. A, the sequence of bovine TMEM184A was investigated for basic amino acid clusters. The most extensive cluster was found near the C terminus (amino acids 392–414). The corresponding human and rat sequences are shown below. GFP-TMEM184A was isolated from transfected BAOECs in CHAPS/PBS and bound to NeutrAvidin-coated wells using biotinylated anti-GFP as described under “Experimental Procedures.” GFP was prepared identically and used as a control. Various concentrations of fluorescein-conjugated heparin, from 10–200 μg/ml (10 μg/ml = 0.056 nmol, 25 μg/ml = 0.1389 nmol, 50 μg/ml = 0.2778 nmol, 75 μg/ml = 0.4167 nmol, 100 μg/ml = 0.5556 nmol, 200 μg/ml = 1.1111 nmol), were added (100-μl total volume), unbound heparin was removed, and wells were washed as described. Total heparin, bound heparin, and unbound heparin were determined by measuring emission at 519 nm. B and C, triplicate samples for total heparin fluorescence readings (B) and for heparin binding to GFP-TMEM184A (C, squares) and to GFP control (circles).
Published evidence suggests some heparin effects in VSMC at 10 μg/ml, but most studies find that even 100 μg/ml does not completely saturate the heparin effects (9, 29). We have previously reported significant heparin effects on VSMC DUSP1 synthesis as low as 50 μg/ml (24), but we typically employed 200 μg/ml to match the responses to HRmAbs (10, 24). To further examine the ability of TMEM184A to bind heparin, we used anti-GFP antibodies to pull down GFP-TMEM184A from transfected ECs. GFP for controls was obtained from transfected ECs using identical protocols. To perform a heparin binding assay, we treated the wells of NeutrAvidin-coated plates with biotin-tagged anti-GFP antibodies. The wells were incubated with GFP-TMEM184A or GFP control and washed several times. Fluorescein-heparin was added to the wells, and the total fluorescent signal was determined for each well. Fig. 10B illustrates the signal over the heparin range from 10–20 μg/ml heparin from the wells containing bound GFP-TMEM184A. After washing to remove unbound heparin, the fluorescence was again determined. Fig. 10C illustrates the heparin that bound to the immobilized GFP-TMEM184A or GFP control. In GFP-TMEM184A wells, the amount of bound heparin plateaued at ∼100 pmol/well and between 25 and 50 μg/ml added heparin but showed no binding to GFP-only (control) wells (Fig. 10C). Interestingly, above 50 μg/ml heparin, an increase in fluorescent heparin binding in GFP-TMEM184A-containing wells was detected. Heparin binding leveled off at ∼100 μg/ml added heparin (Fig. 10C). Heparin concentrations of ∼70 μg/ml were required to achieve half-maximal binding.
An explanation for higher than expected heparin binding includes the possibility that GFP-TMEM184A associates into dimers or higher-order oligomers. Future studies aimed at determining the specific amino acid residues in TMEM184A required for heparin binding will be useful for a better understanding of heparin interactions with cells.
Discussion
The very large number of proteins with which heparin interacts has made deciphering the specific protein(s) responsible for a given heparin response difficult. Our production of HRmAbs that were identified by their ability to block heparin binding to ECs facilitated studies characterizing cellular responses to heparin (5, 10, 14, 24). The HRmAbs mimicked heparin-induced decreases in ERK activation and VSMC proliferation (10) as well as induction of DUSP1 and activation of cGMP-dependent protein kinase in the VSMC responses to heparin (14, 24). Most recently, these antibodies have confirmed that at least some aspects of the anti-inflammatory effects of heparin on ECs are due to the same heparin binding site (8).
We report here the use of heparin affinity chromatography followed by affinity purification of the HRmAb-interacting protein, our heparin receptor candidate. Mass spectral analysis of proteolytic peptides led us to identify the HRmAb-interacting protein as TMEM184A. Subsequently, we showed that TMEM184A is present in both VSMCs and ECs, colocalizes with endocytosed heparin, binds heparin when isolated, and is required for VSMC responses to heparin. On the basis of our previous signaling studies (10, 14, 24) and the heparin binding and knockdown data reported here, we propose a signaling pathway within VSMCs beginning with TMEM184A heparin binding (Fig. 11). Several steps, including whether the heparin receptor must be endocytosed for signaling, the protein(s) with which TMEM184A interacts, and the players involved in transmitting the information from activated cGMP-dependent protein kinase to the nucleus, will require additional investigations. Several previous reports have suggested that trans-activation of the heparin binding-epidermal growth factor receptor and heparin blocking of that receptor function could account for heparin effects on VSMC (e.g. 21). However, some effects do not appear to occur through modification of growth factor signaling (15, 24), and HRmAbs that mimic heparin effects also modulate intracellular signaling (e.g. 14). The data reported here support the identification of TMEM184A as the vascular cell receptor for heparin. To our knowledge, this is the first identification of a receptor for heparin that is required for receptor-mediated heparin uptake and physiological effects in vascular cells.
FIGURE 11.
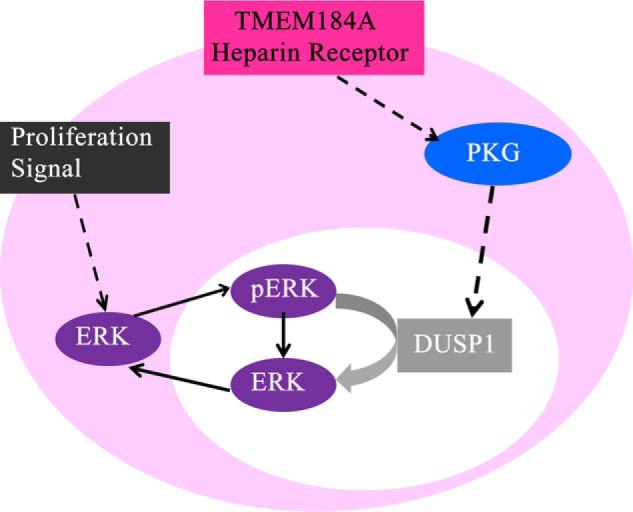
Predicted signal pathway from the heparin receptor. Shown is signaling from heparin through TMEM184A to the induction of DUSP1 and decrease in ERK activation.
There are very little published data on TMEM184A protein function. It has been identified in mice as a tissue specific, male germ line cell membrane protein (26) that appears to be important in exocrine function (25). Its role in vesicle transport is consistent with the transport activities in ECs. Interestingly, the mouse protein is of high molecular weight because of significant glycosylation (25, 26). However, sequence data indicate an N-terminal extension in the mouse gene that may contain a glycosylation site that is not available or not used extensively in other species. Our data, with protein from several species, indicate that molecular weights of affinity-purified heparin receptor are close to the predicted molecular weight of TMEM184A without glycosylation.
Heparin binding is critical to the isolation of the heparin receptor. An analysis of several predicted TMEM184A protein sequences (Fig. 10) indicates that the C-terminal region is enriched in basic amino acids that could participate in heparin binding. There are other regions of the protein that, like the C terminus, are also not predicted to exist in transmembrane segments that contain multiple basic residues and may also participate in heparin binding. However, it will be necessary to determine binding of these regions to heparin before specific regions of the protein can be identified as critical for heparin interactions. In vivo, it seems likely that the heparin receptor binds to HS chains because it is true that heparin binding proteins often function in vivo by interacting with HS chains from HSPGs (1). Documentation of such interactions will also require additional investigations.
HSPGs in vascular cells have numerous functions that could involve the heparin receptor identified here. For example, abluminal HSPGs are critical for processing of lipoproteins by facilitating lipoprotein lipase transport (30), and HS is critical for clearance of triglyceride-rich lipoproteins in mice (31). HSPGs in ECs move in response to acute flow and accumulate near cell boundaries. In doing so, they facilitate signal transduction. These changes are followed by longer-term adjustment of other membrane molecules and cell alignment (32–34). Among the most studied are HS interactions with growth factors, cytokines, and their receptors (1), which are likely critical for the cell proliferation changes illustrated in Fig. 9. In vivo, HSPGs might also normally interact with a heparin receptor, resulting in modulation of growth factor responses in addition to functioning as a co-receptor for growth factors. Additional studies will be required to demonstrate that HS interactions with TMEM184A modulate vascular cell proliferation in vivo. However, decreased signaling through TMEM184A might well allow heparin to enhance cell proliferation through growth factor interactions, as suggested by our results in the BrdU incorporation assay (Fig. 9E).
Caveolae are non-clathrin-coated invaginations of the plasma membrane containing caveolin-1, are present in most mammalian cell types, and are particularly prominent in endothelial cells (35). Caveolins act as scaffolds in caveolae, which concentrate numerous receptors and signaling molecules involved in transport and signal transduction (27, 35, 36). The presence of TMEM184A in caveolae, as suggested by our data, would be consistent with our data indicating signaling from heparin in VSMCs (this work and Ref. 14) and ECs (8). Endothelial nitrous oxide synthase is also present in caveolae and plays a role in control of vascular reactivity and inflammation (36, 37). Interestingly, cellular HSPGs are critical for endothelial nitrous oxide synthase activation in response to flow (34). It is also interesting that the HSPG, syndecan-1, requires lipid rafts (of which caveolae are a subset) for internalization (38). Involvement of the heparin receptor identified here, which results in cGMP-dependent protein kinase activation (14), could be part of the mechanism by which the system works.
Recently, an analysis of the heparin/HS glycomic interactome for angiogenesis has been published (20). This publication evaluates the large number of vascular proteins with roles in angiogenesis that can interact with heparin and HSPGs. The newly identified heparin receptor TMEM184A represents another relevant protein in this system. It will be important to determine whether HS chain association with the heparin receptor is required in any of these functions. The functions and dynamics of HSPGs are complicated. Their protein cores can be type I transmembrane proteins linked to the actin cytoskeleton with matrix metalloproteinase-sensitive domains (which allow shedding of soluble ectodomains carrying HS chains), glycosylphosphatidylinositol-linked proteins requiring association with other membrane components to signal to the inside of the cell, or extracellular matrix proteins, which must link to the membrane through the HS chains and/or the protein core (reviewed in Ref. 39). The possibility that each of these HSPGs could also function through interactions between the HS chains and the heparin receptor, in cell types where it is expressed, significantly increases the possible mechanisms by which HSPGs can function. Ultimately, it will be important to determine which HSPG functions in vascular cells involve the heparin receptor identified here.
Studies of VSMC responses to heparin have long focused on the effects of heparin and EC HS on VSMC proliferation and migration, key events during the development of atherosclerosis (40, 41). HS chains are decreased by heparanase up-regulation in atherosclerosis (reviewed in Ref. 42). In addition, glycocalyx HSPGs are expressed at higher levels in vascular areas subjected to laminar shear stress, which is known to impart atherosclerosis resistance, than in regions of disturbed flow (43). Recent knockdown studies have indicated that both syndecan-1 and -4 HSPGs are critical in the flow-induced protection of ECs (44, 45). In this light, signaling through the TMEM184A heparin receptor provides one mechanism by which HS levels would elicit cellular change during the development of atherosclerosis. This first identification of a heparin receptor on VSMCs and its role in ECs (8) will now inform basic research to further understand the mechanisms by which TMEM184A contributes to vascular function and, importantly, should facilitate a more directed approach to designing therapeutics that take advantage of vascular cell physiological changes in response to heparin.
Author Contributions
L. J. L. K. conceived and coordinated the study and led the writing of the paper. R. J. P. and W. A. P. conceived and carried out the experiments shown in Figs. 1 and 2 and Tables 1 and 2. J. B. S. conceived and carried out the experiments shown in Figs. 3–8. S. L. N. F. conceived and carried out the experiments shown in Figs. 3–5, 9, and 10 and helped to write the paper. Y. L. carried out experiments shown in Figs. 7 and 8. T. B. carried out the experiments shown in Fig. 9. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Charles R. Schmidt and Haley Washburn (Lebanon Valley College) for technical help with MALDI-MS work and Michael Pavio (Lehigh University) for help with the initial TMEM184A immunoprecipitations.
This work was supported by Public Health Service Contract Grant HL54269 (to L. J. L. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- HS
- heparan sulfate
- EC
- endothelial cell
- VSMC
- vascular smooth muscle cell
- HSPG
- heparan sulfate proteoglycan
- HRmAb
- heparin receptor monoclonal antibody
- NTD
- commercial antibody to the amino-terminal domain of TMEM184A
- CTD
- commercial antibody to a carboxy-terminal domain of TMEM184A
- INT
- commercial antibody to the internal sequence of TMEM184A
- BAOEC
- bovine aortic endothelial cell
- BAOSMC
- bovine aortic smooth muscle cell
- TCA
- trichloroacetic acid
- PFA
- paraformaldehyde
- TRITC
- tetramethylrhodamine isothiocyanate.
References
- 1. Xu D., and Esko J. D. (2014) Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 83, 129–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ori A., Wilkinson M. C., and Fernig D. G. (2011) A systems biology approach for the investigation of the heparin/heparan sulfate interactome. J. Biol. Chem. 286, 19892–19904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Slee J. B., Pugh R., and Lowe-Krentz L. (2012) Beyond anticoagulation: roles for heparin in the vasculature. in Heparin: Properties, Uses and Side Effects (Liang R. H., and Piyathilake D. E., eds.), pp. 59–81, Nova Sciences Publishers, Inc., New York [Google Scholar]
- 4. Bârzu T., Molho P., Tobelem G., Petitou M., and Caen J. (1985) Binding and endocytosis of heparin by human endothelial cells in culture. Biochim. Biophys. Acta 845, 196–203 [DOI] [PubMed] [Google Scholar]
- 5. Patton W. A. 2nd, Granzow C. A., Getts L. A., Thomas S. C., Zotter L. M., Gunzel K. A., and Lowe-Krentz L. J. (1995) Identification of a heparin-binding protein using monoclonal antibodies that block heparin binding to porcine aortic endothelial cells. Biochem. J. 311, 461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thourani V. H., Brar S. S., Kennedy T. P., Thornton L. R., Watts J. A., Ronson R. S., Zhao Z. Q., Sturrock A. L., Hoidal J. R., and Vinten-Johansen J. (2000) Nonanticoagulant heparin inhibits NF-κB activation and attenuates myocardial reperfusion injury. Am. J. Physiol. Heart Circ. Physiol 278, H2084–H2093 [DOI] [PubMed] [Google Scholar]
- 7. Li X., Zheng Z., Mao Y., and Ma X. (2012) Unfractionated heparin promotes LPS-induced endothelial barrier dysfunction: a preliminary study on the roles of angiopoietin/Tie2 axis. Thromb. Res. 129, e223–e228 [DOI] [PubMed] [Google Scholar]
- 8. Farwell S., Kanyi D., Hamel M., Slee J. B., Miller E., Cipolle M., and Lowe-Krentz L. (2016) Heparin decreases in TNFα-induced endothelial stress responses require TMEM184A and induction of DUSP1. J. Biol. Chem. 291, 5342–5354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Castellot J. J. Jr., Wong K., Herman B., Hoover R. L., Albertini D. F., Wright T. C., Caleb B. L., and Karnovsky M. J. (1985) Binding and internalization of heparin by vascular smooth muscle cells. J. Cell. Physiol. 124, 13–20 [DOI] [PubMed] [Google Scholar]
- 10. Savage J. M., Gilotti A. C., Granzow C. A., Molina F., and Lowe-Krentz L. J. (2001) Antibodies against a heparin receptor slow cell proliferation and decrease MAPK activation in vascular smooth muscle cells. J. Cell. Physiol. 187, 283–293 [DOI] [PubMed] [Google Scholar]
- 11. Pukac L. A., Carter J. E., Ottlinger M. E., and Karnovsky M. J. (1997) Mechanisms of inhibition by heparin of PDGF stimulated MAP kinase activation in vascular smooth muscle cells. J. Cell. Physiol. 172, 69–78 [DOI] [PubMed] [Google Scholar]
- 12. Ottlinger M. E., Pukac L. A., and Karnovsky M. J. (1993) Heparin inhibits mitogen-activated protein kinase activation in intact rat vascular smooth muscle cells. J. Biol. Chem. 268, 19173–19176 [PubMed] [Google Scholar]
- 13. Pukac L. A., Castellot J. J. Jr., Wright T. C. Jr., Caleb B. L., and Karnovsky M. J. (1990) Heparin inhibits c-fos and c-myc mRNA expression in vascular smooth muscle cells. Cell Regul. 1, 435–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gilotti A. C., Nimlamool W., Pugh R., Slee J. B., Barthol T. C., Miller E. A., and Lowe-Krentz L. J. (2014) Heparin responses in vascular smooth muscle cells involve cGMP dependent protein kinase. J. Cell. Physiol. 229, 2142–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fasciano S., Patel R. C., Handy I., and Patel C. V. (2005) Regulation of vascular smooth muscle proliferation by heparin: inhibition of cyclin-dependent kinase 2 activity by p27kip1. J. Biol. Chem. 280, 15682–15689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reilly C. F., Kindy M. S., Brown K. E., Rosenberg R. D., and Sonenshein G. E. (1989) Heparin prevents vascular smooth muscle cell progression through the G1 phase of the cell cycle. J. Biol. Chem. 264, 6990–6995 [PubMed] [Google Scholar]
- 17. Busch S. J., Martin G. A., Barnhart R. L., Mano M., Cardin A. D., and Jackson R. L. (1992) Trans-repressor activity of nuclear glycosaminoglycans on Fos and Jun/AP-1 oncoprotein-mediated transcription. J. Cell Biol. 116, 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hsia E., Richardson T. P., and Nugent M. A. (2003) Nuclear localization of basic fibroblast growth factor is mediated by heparan sulfate proteoglycans through protein kinase C signaling. J. Cell. Biochem. 88, 1214–1225 [DOI] [PubMed] [Google Scholar]
- 19. Stewart M. D., Ramani V. C., and Sanderson R. D. (2015) Shed syndecan-1 translocates to the nucleus of cells delivering growth factors and inhibiting histone acetylation: a novel mechanism of tumor-host cross-talk. J. Biol. Chem. 290, 941–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chiodelli P., Bugatti A., Urbinati C., and Rusnati M. (2015) Heparin/heparan sulfate proteoglycans glycomic interactome in angiogenesis: biological implications and therapeutical use. Molecules 20, 6342–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kalmes A., Daum G., and Clowes A. W. (2001) EGFR transactivation in the regulation of SMC function. Ann. N.Y. Acad. Sci. 947, 42–54; discussion 54–45 [DOI] [PubMed] [Google Scholar]
- 22. Xie-Zukauskas H., Das J., Short B. L., Gutkind J. S., and Ray P. E. (2013) Heparin inhibits angiotensin II-induced vasoconstriction on isolated mouse mesenteric resistance arteries through Rho-A- and PKA-dependent pathways. Vascul. Pharmacol. 58, 313–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rauch B. H., Millette E., Kenagy R. D., Daum G., Fischer J. W., and Clowes A. W. (2005) Syndecan-4 is required for thrombin-induced migration and proliferation in human vascular smooth muscle cells. J. Biol. Chem. 280, 17507–17511 [DOI] [PubMed] [Google Scholar]
- 24. Blaukovitch C. I., Pugh R., Gilotti A. C., Kanyi D., and Lowe-Krentz L. J. (2010) Heparin treatment of vascular smooth muscle cells results in the synthesis of the dual-specificity phosphatase MKP-1. J. Cell. Biochem. 110, 382–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Best D., and Adams I. R. (2009) Sdmg1 is a component of secretory granules in mouse secretory exocrine tissues. Dev. Dyn. 238, 223–231 [DOI] [PubMed] [Google Scholar]
- 26. Best D., Sahlender D. A., Walther N., Peden A. A., and Adams I. R. (2008) Sdmg1 is a conserved transmembrane protein associated with germ cell sex determination and germline-soma interactions in mice. Development 135, 1415–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parton R. G., and Simons K. (2007) The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 8, 185–194 [DOI] [PubMed] [Google Scholar]
- 28. Singh A., Kett W. C., Severin I. C., Agyekum I., Duan J., Amster I. J., Proudfoot A. E., Coombe D. R., and Woods R. J. (2015) The interaction of heparin tetrasaccharides with chemokine CCL5 is modulated by sulfation pattern and pH. J. Biol. Chem. 290, 15421–15436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Castellot J. J. Jr., Addonizio M. L., Rosenberg R., and Karnovsky M. J. (1981) Cultured endothelial cells produce a heparinlike inhibitor of smooth muscle cell growth. J. Cell Biol. 90, 372–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Obunike J. C., Lutz E. P., Li Z., Paka L., Katopodis T., Strickland D. K., Kozarsky K. F., Pillarisetti S., and Goldberg I. J. (2001) Transcytosis of lipoprotein lipase across cultured endothelial cells requires both heparan sulfate proteoglycans and the very low density lipoprotein receptor. J. Biol. Chem. 276, 8934–8941 [DOI] [PubMed] [Google Scholar]
- 31. Stanford K. I., Bishop J. R., Foley E. M., Gonzales J. C., Niesman I. R., Witztum J. L., and Esko J. D. (2009) Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J. Clin. Invest. 119, 3236–3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zeng Y., Waters M., Andrews A., Honarmandi P., Ebong E. E., Rizzo V., and Tarbell J. M. (2013) Fluid shear stress induces the clustering of heparan sulfate via mobility of glypican-1 in lipid rafts. Am. J. Physiol. Heart Circ. Physiol. 305, H811–H820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zeng Y., and Tarbell J. M. (2014) The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS ONE 9, e86249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ebong E. E., Lopez-Quintero S. V., Rizzo V., Spray D. C., and Tarbell J. M. (2014) Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr. Biol. (Camb.) 6, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sowa G. (2012) Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front. Physiol. 2, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rath G., Dessy C., and Feron O. (2009) Caveolae, caveolin and control of vascular tone: nitric oxide (NO) and endothelium derived hyperpolarizing factor (EDHF) regulation. J. Physiol. Pharmacol. 60, 105–109 [PubMed] [Google Scholar]
- 37. Chidlow J. H. Jr., and Sessa W. C. (2010) Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc. Res. 86, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen K., and Williams K. J. (2013) Molecular mediators for raft-dependent endocytosis of syndecan-1, a highly conserved, multifunctional receptor. J. Biol. Chem. 288, 13988–13999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Couchman J. R., Gopal S., Lim H. C., Nørgaard S., and Multhaupt H. A. (2015) Syndecans: from peripheral coreceptors to mainstream regulators of cell behaviour. Int. J. Exp. Pathol. 96, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Clowes A. W., and Karnowsky M. J. (1977) Suppression by heparin of smooth muscle cell proliferation in injured arteries. Nature 265, 625–626 [DOI] [PubMed] [Google Scholar]
- 41. Marcum J. A., Atha D. H., Fritze L. M., Nawroth P., Stern D., and Rosenberg R. D. (1986) Cloned bovine aortic endothelial cells synthesize anticoagulantly active heparan sulfate proteoglycans. J. Biol. Chem. 261, 7507–7517 [PubMed] [Google Scholar]
- 42. Vlodavsky I., Blich M., Li J.-P., Sanderson R. D., and Ilan N. (2013) Involvement of heparanase in atherosclerosis and other vessel wall pathologies. Matrix Biol. 32, 241–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koo A., Dewey C. F. Jr., and García-Cardeña G. (2013) Hemodynamic shear stress characteristic of atherosclerosis-resistant regions promotes glycocalyx formation in cultured endothelial cells. Am. J. Physiol. Cell Physiol 304, C137–C146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baeyens N., Mulligan-Kehoe M. J., Corti F., Simon D. D., Ross T. D., Rhodes J. M., Wang T. Z., Mejean C. O., Simons M., Humphrey J., and Schwartz M. A. (2014) Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc. Natl. Acad. Sci. 111, 17308–17313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Voyvodic P. L., Min D., Liu R., Williams E., Chitalia V., Dunn A. K., and Baker A. B. (2014) Loss of syndecan-1 induces a pro-inflammatory phenotype in endothelial cells with a dysregulated response to atheroprotective flow. J. Biol. Chem. 289, 9547–9559 [DOI] [PMC free article] [PubMed] [Google Scholar]