

Abstract
Despite the large number of heparin and heparan sulfate binding proteins, the molecular mechanism(s) by which heparin alters vascular cell physiology is not well understood. Studies with vascular smooth muscle cells (VSMCs) indicate a role for induction of dual specificity phosphatase 1 (DUSP1) that decreases ERK activity and results in decreased cell proliferation, which depends on specific heparin binding. The hypothesis that unfractionated heparin functions to decrease inflammatory signal transduction in endothelial cells (ECs) through heparin-induced expression of DUSP1 was tested. In addition, the expectation that the heparin response includes a decrease in cytokine-induced cytoskeletal changes was examined. Heparin pretreatment of ECs resulted in decreased TNFα-induced JNK and p38 activity and downstream target phosphorylation, as identified through Western blotting and immunofluorescence microscopy. Through knockdown strategies, the importance of heparin-induced DUSP1 expression in these effects was confirmed. Quantitative fluorescence microscopy indicated that heparin treatment of ECs reduced TNFα-induced increases in stress fibers. Monoclonal antibodies that mimic heparin-induced changes in VSMCs were employed to support the hypothesis that heparin was functioning through interactions with a receptor. Knockdown of transmembrane protein 184A (TMEM184A) confirmed its involvement in heparin-induced signaling as seen in VSMCs. Therefore, TMEM184A functions as a heparin receptor and mediates anti-inflammatory responses of ECs involving decreased JNK and p38 activity.
Keywords: actin, JNK, endothelial cell, heparin-binding protein, inflammation, p38 MAPK
Introduction
For almost 100 years heparin has been used as an anticoagulant. Specific heparin interactions with proteins important in the anti-clotting system are now well understood. Many heparin binding proteins, including quite a few involved in modulating vascular function, inflammation, and angiogenesis, have been identified (reviewed in Ref. 1). The large number of reports indicating evidence of decreased endogenous heparin and heparan sulfates (HS)3 in atherosclerosis (in model animals and human disease) led to a proposal that decreases in endogenous heparins might be important in the development of atherosclerosis (2). More recent evidence in support of that hypothesis includes increased heparanase expression in atherosclerosis (reviewed in Ref. 3) and increased levels of glycocalyx heparan sulfate in regions of the vasculature where laminar flow decreases the likelihood of atherosclerosis development (4).
In addition to heparin, heparin binding proteins typically also bind HS chains on HS proteoglycans (HSPGs). Although the carbohydrate backbones of heparin and HS are identical, modifications and sulfation patterns vary. HS chains have fewer sulfate residues per disaccharide and a lower overall charge, but their widespread expression suggests that HS may provide many in vivo functions identified originally as heparin functions (reviewed in Ref. 5). Heparin binding to growth factors modulates their activity and appears to protect them from degradation, whereas HSPGs in the extracellular matrix serve as a reservoir of protected growth factors believed to facilitate wound repair after cellular damage (1, 5). Interactions of matrix glycoproteins with cell HSPGs (syndecans and glypicans) in cell membranes contribute to appropriate cellular interactions with the matrix (6, 7). Heparin interactions with any of these molecules might contribute to the overall responses to heparin treatment.
Heparin also binds specifically to both VSMCs and ECs, suggesting the presence of a receptor for heparin (8–10). A number of studies have identified physiological changes in heparin-treated ECs, including the production and secretion of proteins involved in coagulation (11, 12) and changes in inflammatory responses (13–17). In fact, a study by Li et al. (17) provides evidence that heparin effects involve modulation of p38 activity. Numerous reports have identified VSMC responses to heparin. These responses include decreases in cell proliferation (18, 19), ERK pathway activity (19–21), and activation of specific transcription factors (21–23). Heparin binding results in increased levels of DUSP1 protein that are required for decreases in ERK activity (24). The heparin-induced increases in VSMC DUSP1 suggest that heparin-induced decreases in EC inflammatory responses might also involve DUSP1 expression. In support of this idea, DUSP1 induction by anti-inflammatory glucocorticoid hormones does increase DUSP1 expression (25, 26), and low molecular weight heparin has been reported to decrease peroxide-induced JNK and p38 activity (27). Heparin uptake and many heparin functions likely depend on a heparin/HS receptor.
Monoclonal antibodies that block heparin binding to endothelial cells (HRmAbs) are able to mimic heparin responses in VSMCs (10, 19, 22, 24), providing evidence that the protein to which they bind functions as a heparin receptor. The accompanying report identifies TMEM184A as the heparin-interacting protein to which the HRmAbs bind (28). Knockdown of TMEM184A in VSMCs eliminates heparin responses (28). Here we report evidence that unfractionated heparin treatment of ECs results in decreased JNK and p38 activity and that HRmAbsmimic heparin effects on JNK and p38 activity. The heparin effects on JNK and p38 depend on increased DUSP1 expression. Heparin effects on TNFα-induced stress fiber formation also depend on the induction of DUSP1. Furthermore, knockdown of TMEM184A blocks EC heparin responses and indicates that TMEM184A also serves as a receptor for heparin in ECs.
Experimental Procedures
Materials
TNFα was obtained from GenScript (Piscataway, NJ). Primary antibodies against JNK1/3 (catalog no. sc-474), phosphorylated JNK (pJNK; catalog no. sc-6254-mouse, used in microscopy and Western blotting; catalog no. sc-12882-goat, used in Western blotting), p38 (catalog no. sc-535), DUSP1 (MKP-1, catalog nos. sc-370 and sc-1199, used interchangeably), α-tubulin (catalog no. sc-398103), phosphorylated HSP27 (pHSP27, catalog no. sc-12923), phosphorylated c-jun (pcJun, catalog no. sc-31675), phosphorylated MAPK-activated protein kinase 2 (pMK2, sc-31675), and TMEM184A against an amino-terminal domain (NTD, catalog no. sc-292006) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to phosphorylated p38 (pp38, catalog nos. 9211 and 9216) were obtained from Cell Signaling Technology (Beverly, MA). HRmAbs were isolated and purified as reported previously (10). Secondary antibodies conjugated to tetramethylrhodamine isothiocyanate (TRITC), Alexa Fluor 488, Alexa Fluor 594, Cy3, and Alexa Fluor 647 (in donkey or bovine with minimal cross-reactivity) were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Unfractionated heparin, non-IgG endotoxin-tested BSA, and TRITC-phalloidin were obtained from Sigma. Alexa Fluor 488 phalloidin was obtained from Invitrogen.
Cell Culture
Bovine aortic endothelial cells (BAOECs) and rat aortic endothelial cells (RAOECs), obtained from Cell Applications (San Diego, CA), were cultured using Cell Applications media according to their recommendations on 0.2% porcine gelatin and exchanged into minimum Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen or Atlanta Biologicals, Atlanta, GA), 5% l-glutamine, 1% sodium pyruvate, 1% minimum non-essential amino acids, and 1% penicillin/streptomycin antibiotics (Sigma). Human brain microvascular endothelial cells (HBMECs) were obtained from Cell Systems (Kirkland, WA) and cultured using Cell Systems complete medium according to their recommendations or minimum Eagle's medium after exchange into minimum Eagle's medium supplemented identically to the BAOEC culture. Typically, primary cells between passages 4 and 25 were used for these experiments. For microscopy, cells were cultured to between 70% and 90% confluence on glass coverslips coated with 0.2% porcine gelatin (Sigma) in 6-well culture dishes.
SDS-PAGE and Western Blotting
Cells were grown to the desired density on 100-mm tissue culture dishes and treated with stressing agents, heparin, and/or HRmAbs, as noted in the text. Cell samples were harvested directly into 2× sample buffer (11, 19) for SDS-PAGE. Primary antibody dilutions were 1:5000 except for α-tubulin (1:8000) and TMEM184A antibodies (1:1000). Secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594, Alexa Fluor 647, or TRITC (diluted 1:5000) were incubated with the blots, and visualization of the bands was accomplished by fluorescence detection using the Bio-Rad ChemiDoc MP system (catalog no. 170-8280). Bands of interest were identified by comparison with lanes developed using only secondary antibodies and by molecular weight on the basis of migration of prestained RainbowTM molecular weight markers (GE Healthcare LifeSciences). The blots are representative of at least three experiments.
Immunofluorescence Staining
BAOECs, RAOECs, and HBMECs were fixed with 4.0% paraformaldehyde (Pierce) for 20 min at room temperature with gentle shaking and permeabilized with 0.2% Triton-X-100 (Sigma) for 5 min at room temperature with gentle shaking. Coverslips were incubated with 1% BSA for 30 min at room temperature with gentle shaking. Coverslips were incubated with appropriate primary antibodies overnight at 4 °C, washed with 1× PBS, and incubated with appropriate secondary antibodies for 1 h at 37 °C. Primary and secondary antibodies were used at the recommended optimized dilutions (1:200 or 1:100 for bovine anti-goat conjugated with Alexa Fluor 488, TRITC, and Alexa Fluor 594 or 1:50 for bovine anti-goat conjugated to Alexa Fluor 647). Phalloidin staining used a dilution 1:200. Coverslips were washed with 1× PBS and mounted on Mowiol (Calbiochem, Billerica, MA) to minimize photobleaching. Secondary antibody-only controls were performed to rule out nonspecific staining (data not shown).
Confocal Microscopy
Fluorescent labels were visualized using a Zeiss laser-scanning microscope (510 Meta confocal microscope with a ×63 oil immersion lens) at room temperature. Stress fiber images in TMEM184A knockdown cells were taken with a Nikon C2+ confocal microscope (×60 oil immersion lens). The gain intensity and amplifier settings for each channel were set at a level below saturation and appropriate to provide a dynamic range of intensity for pp38, pJNK, pcJun, pMK2, pHSP27, and DUSP1 using control slides from initial experiments. The gain intensity and amplifier settings for Alexa Fluor 488-phalloidin were set by scanning to approximately the same Z plane in control cells where both the intensity and resolution of stress fibers were optimal. All settings were saved and used to image each sample slide within an experiment and for slides in all subsequent repeats of experiments. Z stacks were taken for all slides. All images of DUSP1 and actin stress fibers represent approximately the same Z plane of each cell.
DUSP1 Knockdown
BAOECs were cultured to confluence in 100-mm plates. Cells were trypsinized, washed with 1× PBS, washed with HEPES-buffered saline, and resuspended in HEPES-buffered saline. Cells were electroporated with 20 μg/ml of scrambled control or bovine-specific DUSP1 siRNA (Santa Cruz Biotechnology) in the Bio-Rad Gene Pulser X-Cell system using a modified, preset HeLa protocol (170 V for 7.0 ms). Cells were seeded onto 0.2% porcine gelatin-coated glass coverslips in a 6-well culture dish with Cell Systems BAOEC medium for immunofluorescence staining and quantitative microscopy of pJNK, pp38, DUSP1, and actin stress fibers at 12–24 h post-electroporation.
TMEM184A Knockdown
RAOECs were prepared for knockdown as above for DUSP1 siRNA treatment. Cells were transfected using 20 μg/ml of shRNA designed for rat TMEM184A in the pGFP-V-RS plasmid or control shRNA in the same plasmid (Origene, Rockville, MD) using the Bio-Rad Gene Pulser X-Cell system. Cells were seeded and processed as above for the DUSP1 knockdown system. Experimental treatments were accomplished 72 h after electroporation. Imaging of nuclear staining in TMEM184A knockdown cells was accomplished using a Nikon Eclipse TE 2000-U fluorescence microscope. Stress fibers in TMEM184A knockdown cells were visualized as above.
Quantitative Immunofluorescence Image Analysis
To quantify pJNK, pp38, pcJun, and DUSP1, National Institutes of Health ImageJ software was used to determine the mean nuclear fluorescence intensity of at least 75 cells/treatment from each of at least three independent experiments. The nucleus of each cell was outlined accurately with the Oval tool by using the differential interference contrast images taken for each slide on the laser-scanning confocal or Nikon Eclipse microscope, and the nuclear fluorescence intensity for each cell was recorded. The nuclear fluorescence intensity measurements were corrected against minimal background fluorescence for each slide, and untreated controls were set to 100 to standardize between experiments. The averaged nuclear fluorescence intensity for each treatment was compared with untreated samples. To quantitate pMK2 and pHSP27, ImageJ software was used to determine the mean whole-cell fluorescence intensity of at least 75 cells/treatment/experiment and at least three independent experiments. The outline of each cell was determined and outlined with the Freehand Selections tool by referencing the differential interference contrast images, and the whole-cell fluorescence intensity for each cell was recorded. All measurements were corrected against minimal background fluorescence for each slide. The whole-cell fluorescence intensity was averaged for each treatment, and untreated control values were set to 100.
ImageJ software was used to quantitate stress fibers in at least 100 cells/treatment/experiment from at least three independent experiments. The Threshold tool was used to eliminate background fluorescence. Perpendicular line profiling through the cytoplasm of individual cells was used to generate average fluorescence intensity profiles at regular intervals along the freehand line selection, where the y axis represented the level of fluorescence and the x axis represented the distance across each cell. Sharp, distinct peaks above the threshold were counted for each profile and averaged. Cortical actin peaks were excluded from the counts because their dynamics responded differently than the fibers that spanned the cells.
Statistical Analysis
All experiments are representative of at least three independent trials. Graphical data are shown as mean ± S.E. Comparisons between groups were analyzed using analysis of variance followed by a Tukey post hoc test. Comparisons between two groups were analyzed using a Student's t test. Differences with p < 0.05 were considered significant (†, p < 0.05; *, p < 0.01; ** p < 0.001).
Results
Heparin Decreases TNFα-induced p38 Activity
To test the hypothesis that unfractionated heparin alters p38 activation in ECs, whole-cell p38 activity was first evaluated in TNFα- treated BAOECs with or without heparin pretreatment. TNFα treatment (25 ng/ml) resulted in increased p38 activation, measured as pp38 by Western blotting. In cells pretreated with heparin, the level of pp38 was consistently lower than in TNFα-treated cells without heparin (Fig. 1A). There was little decrease in pp38 at low heparin concentrations. In fact, as shown in Fig. 1A, it sometimes appeared that, at low concentrations of heparin, there was a slight increase in whole-cell pp38. However, by 50 μg/ml heparin, it always appeared that there was less pp38 than in cells treated with TNFα alone. Neither heparin nor TNFα affected the levels of total p38. Activated p38 typically moves to the nucleus, where it phosphorylates numerous substrates (reviewed in Ref. 29). In the nucleus, pp38 can be inactivated by DUSP1 (reviewed in Ref. 30). Therefore, it was important to understand whether there were heparin-induced changes in nuclear pp38. Quantitative analysis of nuclear pp38 using immunofluorescence was determined for a total of more than 200 cells at each time point from at least three experiments per condition. TNFα alone resulted in increased active p38 in the nucleus. Heparin treatment for 20 min without TNFα caused no change in pp38 levels (Fig. 1B). Heparin pretreatment decreased the TNFα-induced nuclear pp38 levels over at least 2 h (Fig. 1B). Longer incubations with heparin also resulted in no change in active p38. There were no changes in nuclear total p38 (data not shown). To be certain that the levels of active p38 in human ECs were also sensitive to heparin treatment, we evaluated nuclear pp38 in HBMECs (Fig. 1C). As with the BAOECs, TNFα treatment resulted in significant activation of p38, and heparin pretreatment caused a significant decrease in pp38 relative to TNFα treatment alone. To investigate the idea that heparin effects on ECs were mediated through a heparin receptor similar to heparin effects in VSMCs, HRmAbs effects on TNFα-induced responses in ECs were investigated. HRmAbs were able to mimic heparin in BAOECs, resulting in decreased pp38 compared with TNFα treatment without heparin or HRmAbs (Fig. 1D). Together, these data indicated that heparin treatment caused a decrease of pp38 in the nucleus and that this response depended on the heparin receptor to which the HRmAbs bind.
FIGURE 1.

Active p38 is decreased in heparin-treated endothelial cells. BAOECs were treated with 25 ng/ml TNFα for varying times. Identical samples were pretreated with 1–200 μg/ml heparin 20 min before TNFα addition. A, cells treated with varying heparin concentrations were harvested into sample buffer and analyzed by Western blotting. Images are representative of more than three experiments. B, to evaluate pp38 in the nuclei, BAOECs were cultured on coverslips, treated as cells in A, and processed for immunofluorescence as described under “Experimental Procedures.” Representative images for one time point are shown. Mean nuclear staining of more than 200 cells/point (from three separate experiments) is shown, with error bars as mean ± S.E. C, HBMECs were treated and analyzed, as were BAOECs. Differences were significant at all times after TNFα addition. **, p < 0.001. D, BAOECs were treated as for A, but some samples contained HRmAb 12B1 at 1.0 μg/ml or 200 μg/ml heparin. The graph is an analysis of four identical experiments. **, p < 0.001.
Heparin Decreases TNFα-induced JNK Activation
To evaluate whether the decreases in activated p38 would also be observed with JNK, BAOECs were treated as for the p38 studies above and analyzed for JNK activity. Western blotting analysis indicated that BAOECs pretreated with a range of heparin concentrations up to 200 μg/ml heparin before TNFα showed a decrease in pJNK at higher heparin concentrations compared with cells treated with TNFα alone (Fig. 2A). As with heparin effects on pp38, the pJNK response was concentration-dependent. Also, as observed in Western blotting analyses for total pp38, total cell pJNK appeared to be slightly higher at low heparin concentrations, as shown in Fig. 2A. Neither heparin nor TNFα treatment caused a change in total JNK levels. As expected from the literature (e.g. Ref. 31), TNFα treatment of BAOECs significantly increased nuclear pJNK (Fig. 2B). Compared with TNFα only, nuclear pJNK was decreased significantly in BAOECs treated with heparin prior to TNFα. As with pp38, heparin alone caused no change in pJNK (Fig. 2B) at 20 min (or longer, data not shown). We again used HBMECs to confirm that heparin sensitivity was applicable to both human ECs as well as microvessel ECs (Fig. 2C) and found that TNFα-induced pJNK levels were decreased significantly by pretreatment with heparin. Pretreatment with HRmAbs also decreased pJNK, comparable with the effect of heparin (Fig. 2D). The results indicate that heparin pretreatment resulted in decreased active JNK in the nuclei and that these effects likely occurred through heparin interactions with its receptor.
FIGURE 2.

Active JNK is decreased in heparin-treated endothelial cells. BAOECs were treated with 25 ng/ml TNFα for various times. Identical samples were pretreated with 1–200 μg/ml heparin 20 min before TNFα addition. A, cells treated with varying heparin concentrations were harvested into sample buffer and analyzed by Western blotting. Data are representative of more than three repeats. B, to evaluate pJNK in the nuclei, BAOECs were cultured on coverslips and treated as cells in A, and processed for immunofluorescence as described under “Experimental Procedures.” Representative images for one time point are shown. Mean nuclear staining of more than 200 cells/point (from three separate experiments) is shown, with error bars as mean ± S.E. **, p < 0.001. C, HBMECs were treated and analyzed, as were BAOECs. Differences at all times after TNFα addition were significant. **, p < 0.001. D, BAOECs were treated as in A, but some cells were treated with HRmAb 12B1 at 1.0 μg/ml or 200 μg/ml heparin. The space between time points in the image is due to removal of a marker lane and different loading patterns. The graph represents an analysis from four different experiments for the p46 JNK band only. †, p < 0.05.
Heparin Decreases Activation of Downstream p38 and JNK Substrates
To investigate whether heparin-induced decreases in JNK and p38 activation also decreased downstream signaling, the activation status of several JNK and p38 substrates was examined using Western blotting and immunofluorescence microscopy (Fig. 3). Heparin effects on the JNK transcription factor target, cJun, were analyzed by Western blotting (data not shown) and immunofluorescence. Fig. 3A illustrates typical pcJun from at least three independent experiments. As expected, TNFα increased nuclear pcJun after 30 min (Fig. 3A). Heparin pretreatment significantly decreased nuclear pcJun, consistent with the heparin-induced decrease in pJNK. Similar results were observed in HBMECs (data not shown). Substrates for p38 include transcription factors and other kinase enzymes, including MK2. Activated MK2 has been shown to be involved in signaling responses to growth factors and inflammation (32). MK2 activity plays an important role in EC actin remodeling (e.g. Ref. 33), making it an important p38 target to evaluate. Therefore, MK2 was tested using quantitative immunofluorescence (Fig. 3B). pMK2 increased rapidly in response to TNFα treatment. In cells pretreated with heparin, there were significant decreases in MK2 phosphorylation compared with cells treated with TNFα only (Fig. 3B). HSP27, a downstream target of MK2 involved in modulating actin remodeling in ECs (34) and other cell types, was similarly responsive to heparin. Fig. 3C illustrates significant heparin-induced decreases in whole-cell pHSP27 relative to cells treated with TNFα alone. These results indicate that both transcription factors and other downstream targets are phosphorylated at lower levels in heparin-treated cells.
FIGURE 3.

Downstream targets of JNK and p38 are also decreased in response to heparin treatment. BAOECs were treated with 25 ng/ml TNFα (for 5–30 min) 20 min after heparin addition and examined by quantitative immunofluorescence. A, nuclear pcJun was quantified in more than 200 cells from three separate experiments. **, p < 0.001. B and C, whole-cell staining was performed for pMK2 (B) and pHSP27 (C) in more than 200 cells from three separate experiments. *, p < 0.01; †, p < 0.05.
DUSP1 Is Critical for Heparin-induced Decreases in JNK and p38 Activity
In VSMCs, heparin has been shown to induce concentration- and time-dependent expression of DUSP1, a dual-specificity phosphatase that localizes to the nucleus, where it regulates MAPK signaling (24). To test the hypothesis that increased DUSP1 is required for heparin-induced decreases in JNK and p38 activation, we first evaluated whether increased DUSP1 is observed in heparin-treated ECs (Fig. 4). BAOECs were treated with heparin, and cells were harvested for Western blotting analysis to determine the relative levels of DUSP1 (Fig. 4A). DUSP1 levels increased by 10 min, and maximal DUSP1 expression was achieved by 30 min in cells treated with 200 μg/ml heparin. Quantitative immunofluorescence showed that HBMECs (Fig. 4B) also exhibited a rapid increase in DUSP1.
FIGURE 4.

Heparin-induced DUSP1 is critical for effects on JNK and p38 activity. A, BAOECs were treated with 200 μg/ml heparin for various times and harvested for Western blotting. The blot is representative of at least five similar experiments. B, HBMECs were grown on coverslips, treated with 200 μg/ml heparin for the times noted, and stained for DUSP1, and then nuclear DUSP1 levels were determined for at least 200 cells/point. DUSP1 levels are plotted relative to control samples set at 100. †, p < 0.05; **, p < 0.001. C–F, BAOECs were transfected with DUSP1 siRNA (light columns) or scrambled siRNA (dark columns) and seeded on coverslips as noted under “Experimental Procedures.” Some coverslips were stained for DUSP1 to confirm knockdown (e.g. C and D are representative of more than 200 cells/treatment, scale bars = 20 μm). After 24 h, cells were treated with TNFα (25 ng/ml) with or without a 20 min pretreatment of 200 μg/ml heparin, fixed, and stained for pJNK (E) or pp38 (F). Nuclear levels of the activated enzymes were determined. *, p < 0.01; **, p < 0.001; −, not significant.
We then questioned whether the observed decreases in JNK and p38 activation required this heparin-induced DUSP1. BAOECs were electroporated with DUSP1 siRNA as described under “Experimental Procedures,” treated, and evaluated using quantitative immunofluorescence assays. Treatment with species-specific DUSP1 siRNA consistently resulted in a significant (p < 0.05) decrease in nuclear DUSP1 (e.g. Fig. 4C). Whole-cell knockdown averaged 54% of control siRNA-treated cells, whereas nuclear DUSP1 levels decreased to 65% of control siRNA-treated cells. Calculated levels of cytoplasmic knockdown indicate that DUSP1 siRNA-treated cells had 45% as much DUSP1 in their cytoplasm as controls, consistent with the fact that this nuclear enzyme is likely in the cytoplasm primarily shortly after synthesis. Knockdown cells treated with heparin did not significantly increase DUSP1 expression (Fig. 4D). TNFα treatments activated JNK and p38 in both control and DUSP1 siRNA-treated BAOECs (Fig. 4, E and F). Interestingly, knockdown of DUSP1 resulted in higher levels of nuclear pJNK without any other treatment (Fig. 4E). As expected, heparin pretreatment in control siRNA-treated cells resulted in significantly decreased TNFα-induced JNK and p38 activity. However, in DUSP1 knockdown cells pretreated with heparin, this heparin-induced reduction in JNK and p38 activation was not observed (Fig. 4, E and F). Therefore, heparin effects on JNK and p38 activation require the induction of DUSP1 in a manner similar to VSMCs (24).
Heparin Decreases TNFα-induced Stress Fiber Formation and Organization
Activated JNK and p38 have been shown to play roles in actin remodeling and subsequent EC morphology changes (reviewed in Ref. 35). JNK activation is linked with newly organized cortical actin and EC adaptation, whereas p38 appears to be associated with stress fibers and actin events at focal adhesions (35). After observing significant heparin-induced decreases in JNK and p38 activation, it became important to investigate whether these heparin-induced JNK and p38 activity decreases could reduce TNFα-induced stress fiber formation. BAOECs treated with TNFα for at least 30 min exhibited increased stress fibers (e.g. compare Fig. 5, B, c–f, with A). In cells pretreated with heparin (Fig. 5B, g–j), there was an overall decrease in the number of stress fibers per cell. There was individual variability in stress fiber formation between cells, as suggested by the two control actin images (Fig. 5A, a and b). Therefore, stress fibers were counted using ImageJ software as described under “Experimental Procedures” (illustrated in Fig. 5C). Peripheral cortical actin was not included in the line profiling because changes in cortical actin do not correspond to changes in stress fibers traversing the whole cell (36, 37). An example of the line profile is shown in Fig. 5C. As illustrated in Fig. 5A and quantified in Fig. 5D (time 0), few stress fibers were found in untreated BAOECs stained with Alexa Fluor 488-phalloidin.
FIGURE 5.

Heparin decreases TNFα-induced stress fiber levels. ECs were seeded on coverslips and treated for various times with 25 ng/ml TNFα with or without 200 μg/ml heparin pretreatment. A and B, BAOECs were fixed and stained with Alexa Fluor 488-phalloidin. Images are representative of more than 100 cells examined from at least three different experiments per condition. The untreated cells in A (a and b) are examples from two experiments. The cells in B were treated with TNFα only (top row, c–f) or TNFα after heparin (bottom row, g–j). Top/bottom row pairs are from the same experiment. C, example quantitative analysis of fluorescent phalloidin staining in BAOECs. The line across the cell image results in the graph shown. Arrows depict the fluorescence peaks identified. Note that neither cortical actin at two edges nor peaks below a threshold are counted (refer to “Experimental Procedures”). D and E, more than 100 BAOECs (D) or HBMECs (E) for each condition were analyzed for the number of peaks above basal intensity. Error bars represent the mean number of peaks with S.E. *, p < 0.01. F, BAOECs in k and l were treated as in A and B. Cells in m and n were treated with 1 μg/ml HRmAbs H6 or B1 instead of heparin. Cells in k–n were stained with rhodamine-phalloidin. The images are representative of three independent experiments and were analyzed as in D and E. †, p < 0.05. G, BAOECs were treated with DUSP1 siRNA (light columns) or control siRNA (dark columns) before 30 min of TNFα and heparin, and stress fibers were analyzed. **, p < 0.001. Scale bars = 20 μm.
The average number of fluorescence peaks was increased significantly with 30–120 min of TNFα treatment in both BAOECs (Fig. 5, B and D) and HBMECs (Fig. 5E). However, in cells treated with heparin before TNFα, the average number of fluorescence peaks was significantly lower than in cells treated with TNFα alone. In BAOECs, this heparin effect was observed as early as 30 min of TNFα treatment and persisted through 120 min (Fig. 5D). In HBMECs, heparin effects on stress fiber levels were also evident by 30 min of TNFα treatment and were maintained through 120 min of TNFα treatment (Fig. 5E). In cells pretreated with HRmAbs in place of heparin (Fig. 5F), there was also a decrease in the number of stress fibers compared with cells treated with TNFα alone. In these experiments, the measured increase in stress fibers induced by TNFα was not as great as in the other experiments, possibly because of differences in the batch of TNFα or the visualization system (rhodamine-phalloidin).
To test whether the effects of heparin on stress fiber formation were also dependent on DUSP1, stress fibers were quantified in BAOECs transfected with either control or DUSP1 siRNA (Fig. 5G). A 30-min TNFα treatment increased stress fibers in both control and DUSP1 siRNA-treated cells. In control siRNA cells, heparin pretreatment resulted in a significant decrease in stress fibers. In cells with DUSP1 knocked down, heparin pretreatment did not decrease stress fibers; rather, an increase was observed. These data indicate that heparin treatment decreases stress fiber responses to TNFα, which depends on heparin-induced DUSP1 increases that lead to decreased JNK and p38 activity.
Knockdown of TMEM184A Interferes with Heparin Responses
We have recently identified TMEM184A to be a heparin-responsive transmembrane protein required for heparin effects in VSMCs (28). The identification of TMEM184A also involved the use of HRmAbs that mimic effects of heparin in ECs (Figs. 1, 2, and 5). Knockdown of TMEM184A in rat VSMCs eliminated heparin responses (28). To determine whether this heparin-responsive protein identified with our HRmAbs and confirmed in VSMC is also responsible for the heparin effects in ECs reported here, we first confirmed that TMEM184A is present in RAOECs across the range of cell density we studied (Fig. 6A). We then studied RAOEC responses in knockdown compared with control shRNA-treated cells. Knockdown of TMEM184A was typically around 50% on the basis of staining of cells from each knockdown experiment and quantification of the TMEM184A staining. Representative images and data from more than 200 cells/condition are shown in Fig. 6B. Both cell surface TMEM184A and total cell TMEM184A are decreased to a similar extent. We employed an immunofluorescence assay for nuclear JNK and p38 (Figs. 1 and 2) to characterize TMEM184A knockdown effects on heparin-induced JNK and p38 activity decreases. Heparin treatment of TMEM184A knockdown cells did not decrease nuclear pp38 levels (Fig. 6C), although control shRNA-transfected cells responded to heparin normally, with a significant decrease at 15 min (Fig. 6C). At 30 min, the level of TNFα-induced nuclear pp38 was already much lower than at 15 min, and the heparin-induced decrease in control cells was not significant. Again, there was no heparin-induced change in knockdown cells. Heparin-induced decreases in nuclear pJNK levels were significant at both 15 and 30 min in TNFα-activated RAOECs. Knockdown cell nuclear pJNK remained high at both time points despite heparin treatment (Fig. 6D). Knockdown of TMEM184A also blocked the ability of heparin treatment to induce DUSP1 expression (Fig. 6E), consistent with the requirement of heparin-induced DUSP1 expression for resultant decreases in JNK and p38 phosphorylation. Stress fiber formation was significant after 60 min of TNFα treatment in control shRNA-treated cells and TMEM184A knockdown cells. In cells pretreated with heparin, control cells exhibited significantly fewer stress fibers. However, there was no stress fiber decrease in heparin-treated knockdown cells (Fig. 6F). The data from the TMEM184A knockdown experiments support the hypothesis that TMEM184A is required for heparin responses in ECs and VSMCs and indicate its involvement in anti-inflammatory EC responses to heparin.
FIGURE 6.

TMEM184A knockdown blocks heparin responses. A, RAOECs (not transfected) were evaluated for TMEM184A staining (NTD antibody) over a range (a–d) of cell densities. Scale bars = 10 μm. RAOECs were transfected with control or TMEM184A shRNA constructs and seeded on coverslips as described under “Experimental Procedures.” TMEM knockdown was confirmed by staining some cells from each experiment for TMEM184A using the NTD antibody (e.g. B, images from permeabilized cells). Results from three experiments with more than 200 cells for each condition are shown in the graph. After 72 h, cells were treated with TNFα (25 ng/ml) with or without a 20 min pretreatment of 200 μg/ml heparin, fixed, and stained for pp38 (C) or pJNK (D). Nuclear levels of the activated enzymes were determined. E, TMEM184A or control shRNA-transfected cells were treated with 200 μg/ml heparin for up to 30 min, fixed, and stained for DUSP1, and then nuclear levels of DUSP1 were determined. F, TMEM184A or control shRNA-transfected cells were treated for 60 min with TNFα (25 ng/ml) with or without a 20 min pretreatment of 200 μg/ml heparin and stained with Alexa Fluor 488-phalloidin. Fluorescence peaks were determined as in Fig. 5. All graphical results are from at least three repeat experiments per condition with at least 50 cells analyzed for each experiment. †, p < 0.05.
Discussion
Inflammatory mediators such as TNFα and other cytokines have been implicated in the chronic pathogenesis of vascular disease. They induce EC p38 and JNK activation, increase immune cell adhesion molecule expression, promote VSMC migration, and cause EC barrier dysfunction, all of which further accelerate local inflammation (38, 39). A number of studies have determined that heparin exhibits anti-inflammatory properties and modulates angiogenesis (1). Low molecular weight heparin has been shown to reduce JNK and p38 activation, downstream transcription factor activation, and induction of cell adhesion molecule expression (27). Here we present evidence that unfractionated heparin decreases nuclear JNK and p38 activity and also alters TNFα effects on actin remodeling that depend on JNK and p38 activity. Interestingly, at low heparin levels, there appears to be a slight increase in JNK and p38 activation. Because heparin-induced inactivation of these enzymes depends on DUSP1 synthesis and movement of the pJNK and pp38 to the nucleus for dephosphorylation, differences in movement to the nucleus or other cytoplasmic signaling initiated in response to heparin might explain these results and are interesting possibilities to investigate further. The heparin effects in ECs are mimicked by HRmAbs that bind to a heparin receptor. The observations that pretreatment with antibodies to a putative heparin receptor decreases TNFα-induced JNK and p38 activation and decreases stress fiber formation are consistent with previous reports showing that these antibodies mimic the effects of heparin in VSMCs (19, 22, 24). Here we provide functional evidence that the anti-inflammatory effects of heparin in ECs are mediated through the heparin receptor TMEM184A, as seen in VSMCs (28). TMEM184A knockdown interferes with heparin responses, confirming our HRmAb evidence. Our data strongly suggest that the TNFα and heparin pathways, when activated simultaneously in ECs, engage in cross-talk dependent on the heparin receptor TMEM184A. A schematic for this cross-talk between the TNFα and heparin signals is shown in Fig. 7.
FIGURE 7.
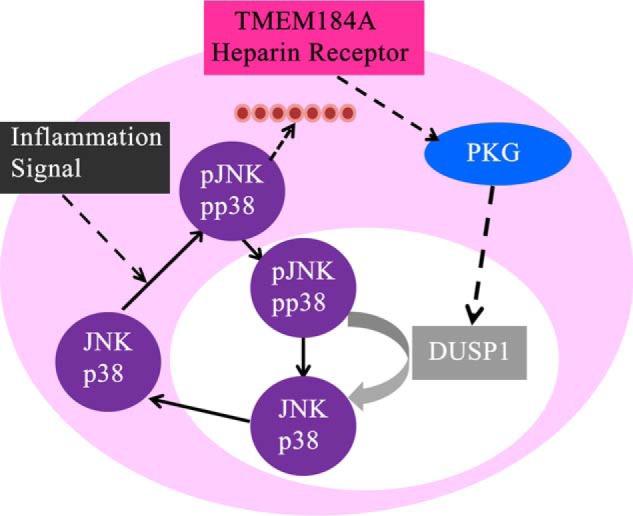
Predicted signal pathway from the heparin receptor. Heparin interacts with TMEM184A and results in increased DUSP1 and decreases in active JNK and p38, which impact stress fiber formation. The red circles indicate actin fibers.
EC dysfunction in response to inflammation results in the formation of pericellular gaps, altered interactions between ECs and matrix proteins, decreased calcium-dependent signaling at EC barriers, and changes in mechanosensitivity (39–41). Reorganization of F-actin into stress fibers through the MAPK signaling cascades in response to TNFα and low or disturbed fluid shear stress has also been shown to increase EC permeability (39, 41, 42). JNK associates with stress fibers in ECs and plays a role in actin remodeling (43, 44). Regions of the vasculature with disturbed flow have higher levels of active JNK (45, 46). Similarly, p38 plays a role in actin remodeling (41, 47, 48) and facilitates actin remodeling in response to changes in shear stress (49). This likely requires the p38 substrate MK2 and phosphorylation of its substrate HSP27 (34, 47). Our results indicating heparin-induced decreases in pMK2 and pHSP27 are consistent with the evidence that heparin-treated ECs show lower levels of stress fibers when stressed with TNFα.
Natural inhibitors of inflammation and stress signaling include glucocorticoid hormones, which can decrease signaling through stress kinase pathways by inducing the synthesis of DUSP1 (25, 26). JNK and p38, as targets of DUSP1, have been well characterized in vascular cell types, and our data agree with previous reports showing that, after p38 and JNK activation, DUSP1 is effective in decreasing activation and protecting cells from actin stress fiber formation, which often leads to EC dysfunction (39, 48, 50). In this regard, our finding that heparin treatment of ECs after DUSP1 knockdown resulted in increased TNFα-induced pJNK and stress fibers indicates that DUSP1 may be critical for routine modulation of stress fiber levels and their remodeling in ECs. ECs express higher levels of DUSP1 in atheroprotective regions of the vasculature, resulting in the lower levels of active JNK and p38 found in those regions (50). The lower levels of DUSP1 in the areas of disturbed flow therefore contribute to increased inflammation in these regions. Although heparin is convenient for cell studies, HSPGs likely interact with most heparin binding proteins in vivo (5). It is interesting that EC HSPGs are involved in EC remodeling in response to shear stress (51). In fact, knocking down syndecan-1 or -4 decreases the laminar shear stress protection of endothelial layers from inflammatory damage (52, 53).
We have recently identified TMEM184A colocalization with caveolin-1 in vascular cells (28). Caveolae organize numerous players in signaling pathways (54). The role of caveolae in endothelial nitrous oxide synthase activation, which results in increased NO production and resultant increases in cGMP (55), and the colocalization of the heparin receptor with caveolin-1 (28) suggest that these players could be linked in modulating healthy endothelial function. The HSPG syndecan-1 requires caveolae for uptake (56), indicating that it might interact with TMEM184A in caveolae. TNFα has been shown to inhibit endothelial nitrous oxide synthase activity and, as a result, decrease NO production, which would lead to lower levels of active cGMP-dependent protein kinase. This causes ECs to become vasoconstrictive and, eventually, dysfunctional (38, 57). Recent studies have shown that the effects of heparin in VSMCs involve cGMP-dependent protein kinase (22). We hypothesize that the effects of heparin in ECs might also be dependent on cGMP-dependent protein kinase and that the heparin-induced increased cGMP prior to exposure to TNFα could prevent the effects resulting from changes in NO production. Elucidating the signaling dynamics between these pathways will be important to understand the protective effects of heparin on the endothelium.
In addition, the significant effects of HSPGs and heparin/HS binding proteins in angiogenesis (1) suggest that the heparin receptor might be a player in that complex process. Endogenous heparin and HS are crucial for vascular health. Decreased glycocalyx HSPGs and increased heparanase, which degrades endogenous HS chains in atherosclerosis (3, 4), indicate critical roles for heparin and HS in decreasing atherosclerosis. We provide evidence for cross-talk between heparin through the TMEM184A heparin receptor and TNFα through inflammatory signaling. Our results demonstrate that heparin-induced DUSP1 expression in multiple EC types is similar to what has been reported previously in VSMCs and that this induction is necessary to decrease TNFα-induced JNK and p38 activation to prevent significant stress fiber formation. In vivo, HSPGs are likely natural ligands for TMEM184A, a possibility that requires further study. Additional investigation of the anti-inflammatory potential of heparin in vascular disease is important for improved exploitation of the effects of heparin on vascular cell physiology in a clinical setting.
Author Contributions
L .J. L. K. and M. D. C. conceived the study. L. J. L. K. coordinated the study and led the writing of the paper. S. L. N. F. conceived the nuclear studies, knockdown experiments, and stress fiber analysis; performed the experiments shown in all figures; and helped to write the paper. M. H. conceived and performed the experiments with whole-cell analysis for Figs. 1 and 2. D. K. carried out the experiments supporting whole-cell analysis for Figs. 1 and 2 and helped to conceive and carry out the initial studies shown in Figs. 3, 4, and 5. E. A. M. performed the studies shown in Figs. 1, 2, and 4. J. B. S. helped design the experiments in Figs. 1, 2, and 4, performed some of the experiments shown in Fig. 5, and helped to write the paper. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgment
We thank Yaqiu Li for purification of the HRmAbs used in this study.
This work was supported by National Institutes of Health Contract Grant HL54269 (to L. J. L. K.). It was initiated with funding from Lehigh Valley Hospital Department of Surgery and Division of Surgical Critical Care (to M. D. C. and L. J. L. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HS
- heparan sulfate(s)
- HSPG
- heparan sulfate proteoglycan
- VSMC
- vascular smooth muscle cell
- HRmAb
- mAb that blocks heparin binding to vascular cells
- EC
- endothelial cell
- NTD
- commercial antibody to the amino-terminal domain of TMEM184A
- TRITC
- tetramethylrhodamine isothiocyanate
- BAOEC
- bovine aortic endothelial cell
- RAOEC
- rat aortic endothelial cell
- HBMEC
- human brain microvascular endothelial cell
- TNFα
- tumor necrosis factor α.
References
- 1. Chiodelli P., Bugatti A., Urbinati C., and Rusnati M. (2015) Heparin/heparan sulfate proteoglycans glycomic interactome in angiogenesis: biological implications and therapeutical use. Molecules 20, 6342–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Engelberg H. (2001) Endogenous heparin activity deficiency: the “missing link” in atherogenesis? Atherosclerosis 159, 253–260 [DOI] [PubMed] [Google Scholar]
- 3. Vlodavsky I., Blich M., Li J.-P., Sanderson R. D., and Ilan N. (2013) Involvement of heparanase in atherosclerosis and other vessel wall pathologies. Matrix Biol. 32, 241–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koo A., Dewey C. F. Jr., and García-Cardeña G. (2013) Hemodynamic shear stress characteristic of atherosclerosis-resistant regions promotes glycocalyx formation in cultured endothelial cells. Am. J. Physiol. Cell Physiol. 304, C137–C146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xu D., and Esko J. D. (2014) Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 83, 129–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Couchman J. R., Gopal S., Lim H. C., Nørgaard S., and Multhaupt H. A. (2015) Syndecans: from peripheral coreceptors to mainstream regulators of cell behaviour. Int. J. Exp. Pathol. 96, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rapraeger A. C., Ell B. J., Roy M., Li X., Morrison O. R., Thomas G. M., and Beauvais D. M. (2013) Vascular endothelial-cadherin stimulates syndecan-1-coupled insulin-like growth factor-1 receptor and cross-talk between αVβ3 integrin and vascular endothelial growth factor receptor 2 at the onset of endothelial cell dissemination during angiogenesis. FEBS J. 280, 2194–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bârzu T., Molho P., Tobelem G., Petitou M., and Caen J. (1985) Binding and endocytosis of heparin by human endothelial cells in culture. Biochim. Biophys. Acta 845, 196–203 [DOI] [PubMed] [Google Scholar]
- 9. Castellot J. J. Jr., Wong K., Herman B., Hoover R. L., Albertini D. F., Wright T. C., Caleb B. L., and Karnovsky M. J. (1985) Binding and internalization of heparin by vascular smooth muscle cells. J. Cell. Physiol. 124, 13–20 [DOI] [PubMed] [Google Scholar]
- 10. Patton W. A. 2nd, Granzow C. A., Getts L. A., Thomas S. C., Zotter L. M., Gunzel K. A., and Lowe-Krentz L. J. (1995) Identification of a heparin-binding protein using monoclonal antibodies that block heparin binding to porcine aortic endothelial cells. Biochem. J. 311, 461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dougherty C. S., and Lowe-Krentz L. J. (1998) Heparin increases protein S levels in cultured endothelial cells by causing a block in degradation. J. Vasc. Res. 35, 437–448 [DOI] [PubMed] [Google Scholar]
- 12. Ettelaie C., Fountain D., Collier M. E., Elkeeb A. M., Xiao Y. P., and Maraveyas A. (2011) Low molecular weight heparin downregulates tissue factor expression and activity by modulating growth factor receptor-mediated induction of nuclear factor-κB. Biochim. Biophys. Acta 1812, 1591–1600 [DOI] [PubMed] [Google Scholar]
- 13. Thourani V. H., Brar S. S., Kennedy T. P., Thornton L. R., Watts J. A., Ronson R. S., Zhao Z. Q., Sturrock A. L., Hoidal J. R., and Vinten-Johansen J. (2000) Nonanticoagulant heparin inhibits NF-kB activation and attenuates myocardial reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 278, H2084–H2093 [DOI] [PubMed] [Google Scholar]
- 14. Wang L., Brown J. R., Varki A., and Esko J. D. (2002) Heparin's anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J. Clin. Invest. 110, 127–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han J., Ding R., Zhao D., Zhang Z., and Ma X. (2013) Unfractionated heparin attenuates lung vascular leak in a mouse model of sepsis: role of RhoA/Rho kinase pathway. Thromb. Res. 132, e42–e47 [DOI] [PubMed] [Google Scholar]
- 16. Gonzales J. N., Kim K. M., Zemskova M. A., Rafikov R., Heeke B., Varn M. N., Black S., Kennedy T. P., Verin A. D., and Zemskov E. A. (2014) Low anticoagulant heparin blocks thrombin-induced endothelial permeability in a PAR-dependent manner. Vascul. Pharmacol. 62, 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li X., Zheng Z., Li X., and Ma X. (2012) Unfractionated heparin inhibits lipopolysaccharide-induced inflammatory response through blocking p38 MAPK and NF-κB activation on endothelial cell. Cytokine 60, 114–121 [DOI] [PubMed] [Google Scholar]
- 18. Reilly C. F., Kindy M. S., Brown K. E., Rosenberg R. D., and Sonenshein G. E. (1989) Heparin prevents vascular smooth muscle cell progression through the G1 phase of the cell cycle. J. Biol. Chem. 264, 6990–6995 [PubMed] [Google Scholar]
- 19. Savage J. M., Gilotti A. C., Granzow C. A., Molina F., and Lowe-Krentz L. J. (2001) Antibodies against a heparin receptor slow cell proliferation and decrease MAPK activation in vascular smooth muscle cells. J. Cell. Physiol. 187, 283–293 [DOI] [PubMed] [Google Scholar]
- 20. Pukac L. A., Carter J. E., Ottlinger M. E., and Karnovsky M. J. (1997) Mechanisms of inhibition by heparin of PDGF stimulated MAP kinase activation in vascular smooth muscle cells. J. Cell. Physiol. 172, 69–78 [DOI] [PubMed] [Google Scholar]
- 21. Ottlinger M. E., Pukac L. A., and Karnovsky M. J. (1993) Heparin inhibits mitogen-activated protein kinase activation in intact rat vascular smooth muscle cells. J. Biol. Chem. 268, 19173–19176 [PubMed] [Google Scholar]
- 22. Gilotti A. C., Nimlamool W., Pugh R., Slee J. B., Barthol T. C., Miller E. A., and Lowe-Krentz L. J. (2014) Heparin responses in vascular smooth muscle cells involve cGMP dependent protein kinase. J. Cell. Physiol. 229, 2142–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pukac L. A., Castellot J. J. Jr., Wright T. C. Jr., Caleb B. L., and Karnovsky M. J. (1990) Heparin inhibits c-fos and c-myc mRNA expression in vascular smooth muscle cells. Cell Regul. 1, 435–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blaukovitch C. I., Pugh R., Gilotti A. C., Kanyi D., and Lowe-Krentz L. J. (2010) Heparin treatment of vascular smooth muscle cells results in the synthesis of the dual-specificity phosphatase MKP-1. J. Cell. Biochem. 110, 382–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kassel O., Sancono A., Krätzschmar J., Kreft B., Stassen M., and Cato A. C. (2001) Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 20, 7108–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lasa M., Abraham S. M., Boucheron C., Saklatvala J., and Clark A. R. (2002) Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol. Cell. Biol. 22, 7802–7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Manduteanu I., Dragomir E., Voinea M., Capraru M., and Simionescu M. (2007) Enoxaparin reduces H2O2-induced activation of human endothelial cells by a mechanism involving cell adhesion molecules and nuclear transcription factors. Pharmacology 79, 154–162 [DOI] [PubMed] [Google Scholar]
- 28. Pugh R., Slee J. B., Farwell S., Li Y., Barthol T., Pavio M., Patton W., and Lowe-Krentz L. (2016) TMEM184A is a receptor required for vascular smooth muscle cell responses to heparin. J. Biol. Chem. 291, 5326–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cuadrado A., and Nebreda A. (2010) Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429, 403–417 [DOI] [PubMed] [Google Scholar]
- 30. Bermudez O., Pagès G., and Gimond C. (2010) The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am. J. Physiol. Cell Physiol. 299, C189–C202 [DOI] [PubMed] [Google Scholar]
- 31. Sabio G., and Davis R. J. (2014) TNF and MAP kinase signalling pathways. Semin. Immunol. 26, 237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beyaert R., Cuenda A., Vanden Berghe W., Plaisance S., Lee J. C., Haegeman G., Cohen P., and Fiers W. (1996) The p38/RK mitogen-activated protein kinase pathway regulates interleukin-6 synthesis response to tumor necrosis factor. EMBO J. 15, 1914–1923 [PMC free article] [PubMed] [Google Scholar]
- 33. Damarla M., Hasan E., Boueiz A., Le A., Pae H. H., Montouchet C., Kolb T., Simms T., Myers A., Kayyali U. S., Gaestel M., Peng X., Reddy S. P., Damico R., and Hassoun P. M. (2009) Mitogen activated protein kinase activated protein kinase 2 regulates actin polymerization and vascular leak in ventilator associated lung injury. PLoS ONE 4, e4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chang E., Heo K. S., Woo C. H., Lee H., Le N. T., Thomas T. N., Fujiwara K., and Abe J. (2011) MK2 SUMOylation regulates actin filament remodeling and subsequent migration in endothelial cells by inhibiting MK2 kinase and HSP27 phosphorylation. Blood 117, 2527–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mengistu M., Slee J. B., and Lowe-Krentz L. (2012) in Actin: Structure, Functions and Disease (Consuelas V. A., ed.) pp. 177–205, Nova Sciences Publishers, Inc., Hauppauge, NY [Google Scholar]
- 36. Hotulainen P., and Lappalainen P. (2006) Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol. 173, 383–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsukita S., and Yonemura S. (1999) Cortical actin organization: lessons from ERM (ezrin/radixin/moesin) proteins. J. Biol. Chem. 274, 34507–34510 [DOI] [PubMed] [Google Scholar]
- 38. Sen A., Most P., and Peppel K. (2014) Induction of microRNA-138 by pro-inflammatory cytokines causes endothelial cell dysfunction. FEBS Lett. 588, 906–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang D., Xie P., Guo S., and Li H. (2010) Induction of MAPK phosphatase-1 by hypothermia inhibits TNF-α-induced endothelial barrier dysfunction and apoptosis. Cardiovasc. Res. 85, 520–529 [DOI] [PubMed] [Google Scholar]
- 40. Liu H. B., Zhang J., Xin S. Y., Liu C., Wang C. Y., Zhao D., and Zhang Z. R. (2013) Mechanosensitive properties in the endothelium and their roles in the regulation of endothelial function. J. Cardiovasc. Pharmacol. 61, 461–470 [DOI] [PubMed] [Google Scholar]
- 41. Borbiev T., Birukova A., Liu F., Nurmukhambetova S., Gerthoffer W. T., Garcia J. G., and Verin A. D. (2004) p38 MAP kinase-dependent regulation of endothelial cell permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 287, L911–L918 [DOI] [PubMed] [Google Scholar]
- 42. Brett J., Gerlach H., Nawroth P., Steinberg S., Godman G., and Stern D. (1989) Tumor necrosis factor/cachectin increases permeability of endothelial cell monolayers by a mechanism involving regulatory G proteins. J. Exp. Med. 169, 1977–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hamel M., Kanyi D., Cipolle M. D., and Lowe-Krentz L. (2006) Active stress kinases in proliferating endothelial cells associate with cytoskeletal structures. Endothelium 13, 157–170 [DOI] [PubMed] [Google Scholar]
- 44. Mengistu M., Brotzman H., Ghadiali S., and Lowe-Krentz L. (2011) fluid shear stress-induced JNK Activity leads to actin remodeling for cell alignment. J. Cell. Physiol. 226, 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Amini N., Boyle J. J., Moers B., Warboys C. M., Malik T. H., Zakkar M., Francis S. E., Mason J. C., Haskard D. O., and Evans P. C. (2014) Requirement of JNK1 for endothelial cell injury in atherogenesis. Atherosclerosis 235, 613–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chaudhury H., Zakkar M., Boyle J., Cuhlmann S., van der Heiden K., Luong le A., Davis J., Platt A., Mason J. C., Krams R., Haskard D. O., Clark A. R., and Evans P. C. (2010) c-Jun N-terminal kinase primes endothelial cells at atheroprone sites for apoptosis. Arterioscler. Thromb. Vasc. Biol. 30, 546–553 [DOI] [PubMed] [Google Scholar]
- 47. Kayyali U. S., Pennella C. M., Trujillo C., Villa O., Gaestel M., and Hassoun P. M. (2002) Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2. J. Biol. Chem. 277, 42596–42602 [DOI] [PubMed] [Google Scholar]
- 48. Kiemer A. K., Weber N. C., Fürst R., Bildner N., Kulhanek-Heinze S., and Vollmar A. M. (2002) Inhibition of p38 MAPK activation via induction of MKP-1: atrial natriuretic peptide reduces TNF-α-induced actin polymerization and endothelial permeability. Circ. Res. 90, 874–881 [DOI] [PubMed] [Google Scholar]
- 49. Azuma N., Akasaka N., Kito H., Ikeda M., Gahtan V., Sasajima T., and Sumpio B. E. (2001) Role of p38 MAP kinase in endothelial cell alignment induced by fluid shear stress. Am. J. Physiol. Heart Circ. Physiol. 280, H189–H197 [DOI] [PubMed] [Google Scholar]
- 50. Zakkar M., Chaudhury H., Sandvik G., Enesa K., Luong le A., Cuhlmann S., Mason J. C., Krams R., Clark A. R., Haskard D. O., and Evans P. C. (2008) Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ. Res. 103, 726–732 [DOI] [PubMed] [Google Scholar]
- 51. Ebong E. E., Lopez-Quintero S. V., Rizzo V., Spray D. C., and Tarbell J. M. (2014) Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr. Biol. (Camb.) 6, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baeyens N., Mulligan-Kehoe M. J., Corti F., Simon D. D., Ross T. D., Rhodes J. M., Wang T. Z., Mejean C. O., Simons M., Humphrey J., and Schwartz M. A. (2014) Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 17308–17313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Voyvodic P. L., Min D., Liu R., Williams E., Chitalia V., Dunn A. K., and Baker A. B. (2014) Loss of syndecan-1 induces a pro-inflammatory phenotype in endothelial cells with a dysregulated response to atheroprotective flow. J. Biol. Chem. 289, 9547–9559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sowa G. (2012) Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front. Physiol. 2, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rath G., Dessy C., and Feron O. (2009) Caveolae, caveolin and control of vascular tone: nitric oxide (NO) and endothelium derived hyperpolarizing factor (EDHF) regulation. J. Physiol. Pharmacol. 60, 105–109 [PubMed] [Google Scholar]
- 56. Chen K., and Williams K. J. (2013) Molecular mediators for raft-dependent endocytosis of syndecan-1, a highly conserved, multifunctional receptor. J. Biol. Chem. 288, 13988–13999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zeng Z., Li Y. C., Jiao Z. H., Yao J., and Xue Y. (2014) The cross talk between cGMP signal pathway and PKC in pulmonary endothelial cell angiogenesis. Int. J. Mol. Sci. 15, 10185–10198 [DOI] [PMC free article] [PubMed] [Google Scholar]