

Abstract
The epithelial-to-mesenchymal transition (EMT) is a process by which differentiated epithelial cells reprogram gene expression, lose their junctions and polarity, reorganize their cytoskeleton, increase cell motility and assume a mesenchymal morphology. Despite the critical functions of the microtubule (MT) in cytoskeletal organization, how it participates in EMT induction and maintenance remains poorly understood. Here we report that acetylated α-tubulin, which plays an important role in microtubule (MT) stabilization and cell morphology, can serve as a novel regulator and marker of EMT. A high level of acetylated α-tubulin was correlated with epithelial morphology and it profoundly decreased during TGF-β-induced EMT. We found that TGF-β increased the activity of HDAC6, a major deacetylase of α-tubulin, without affecting its expression levels. Treatment with HDAC6 inhibitor tubacin or TGF-β type I receptor inhibitor SB431542 restored the level of acetylated α-tubulin and consequently blocked EMT. Our results demonstrate that acetylated α-tubulin can serve as a marker of EMT and that HDAC6 represents an important regulator during EMT process.
Keywords: acetylation, epithelial-mesenchymal transition (EMT), histone deacetylase 6 (HDAC6), microtubule, transforming growth factor β (TGF-β)
Introduction
The epithelial-to-mesenchymal transition (EMT)2 describes a phenomenon in which epithelial cells lose polarity and intercellular contacts, and meanwhile gain mesenchymal properties such as increased migratory capacity and production of extracellular matrix protein (1). This transient and reversible process can be classified into three subtypes, depending on the biological and functional setting in which it occurs. Type 1 EMT underlies the generation of secondary epithelia and is critical during embryogenesis and organ development. The formation of fibroblasts during wound healing and fibrosis is considered a second distinct EMT subtype, while type 3 EMT is characterized by the transformation of epithelial cells into the invasive metastatic mesenchymal cells that underlie cancer progression (2). A hallmark of EMT is loss of tight junctions, desmosomes, adherent junctions and gap junctions (3). All this events trigger redistribution of key molecules, disruption of the polarity complex, and cytoskeletal reorganization.
EMT can be induced or regulated by various growth factors, among which TGF-β has been well documented as a major inducer of EMT during embryogenesis, cancer progression and fibrosis (4–6). Activation of the TGF-β signaling pathway begins from TGF-β binding to and activation of the receptor complex on the cell surface, which consists of two type I and two type II transmembrane serine/threonine kinase receptors. After the type II receptor phosphorylate the type I receptor, the type I receptor phosphorylates cytoplasmic Smad2/3 transcription factors. Phosphorylated Smad2/3, together with Smad4, translocate to the nucleus and interact with transcription factors, co-activators and/or co-repressors (7, 8). In addition to inducing growth inhibitory genes, Smad-dependent transcriptional program potently represses epithelial genes (e.g. E-cadherin and zonula occludens-1) and activates mesenchymal genes (e.g. N-cadherin and α-smooth muscle actin) (9). Furthermore, cross-talks among TGF-β, PDGF, Wnt, and Notch signaling pathways have been shown to contribute to EMT (2, 5, 6).
Microtubules (MTs) are the largest cytoskeletal components, which are not only involved in cell division, but that also contribute to many features of intracellular transport, organelle positioning, cell shape, and cell motility (10). α-Tubulin acetylation was first reported in 1983 (11) and later the N-terminal lysine-40 was identified as a acetylation site (12). Acetylation of α-tubulin is carried out by several reported histone acetyltransferases (13), and the major α-tubulin acetylase is MEC-17/aTAT (14). Like histone acetylation, tubulin acetylation is reversible. Two tubulin deacetylases HDAC6 and Sirtuin2 have been identified (15–17). Even though acetylation was one of the earliest tubulin post-translational modifications identified, its effects on cellular physiology have not been investigated clearly yet. A previous study reported that TGF-β-induced EMT is accompanied by HDAC6-dependent deacetylation of α-tubulin in lung adenocarcinoma A549 cells (18). Yet, it is unknown whether α-tubulin acetylation can serve as a marker in EMT in other types of epithelial cells and whether it plays an active role in EMT. Moreover, it is critical to identify the general mechanism underlying how TGF-β regulates HDAC6-dependent deacetylation of α-tubulin.
The current study sought to investigate the role of acetylated α-tubulin in TGF-β-induced EMT in mammary epithelial cells. We showed that acetylated α-tubulin occurs in immortalized mammary epithelial MCF-10A and NMuMG cells, but not in mesenchymal cell types such as fibroblasts or myoblasts. TGF-β induces a decrease in acetylated α-tubulin in mammary epithelial cells through activation of HDAC6, which could be blocked by inhibition of TGF-β receptor or directly tubacin or even paclitaxel. More interestingly, forced expression of acetylation-mimicking mutant of α-tubulin could block EMT, suggesting an active and direct role of α-tubulin in mediating EMT. Therefore, our results demonstrate that acetylated α-tubulin not only can serve as a marker for EMT, but also contributes to maintenance of epithelial cell shape.
Experimental Procedures
Antibodies, Reagents, and Plasmids
Antibodies against acetyl-α-tubulin (#T7451), α-tubulin (#T6793) and GAPDH (#G8795) were purchased from Sigma-Aldrich. Antibodies against E-cadherin (#3195), Vimentin (#5741), HDAC6 (#7558), p-Smad3 (#9520), Smad3 (#9523), acetyl-Histone H3 (#4499), acetyl-Histone H3 (Lys-9) (#9649), acetyl-Histone H3 (Lys-14) (#7627), acetyl-Histone H3 (Lys18) (#13998) and acetyl-Histone H3 (Lys-56) (#4243) were purchased from Cell Signaling Technology. Antibodies against ZO-1 (#21773, Invitrogen), N-cadherin (#1610920, BD Biosciences), MEC-17 (ab58742, Abcam), and Sirt2 (SC-28298, Santa Cruz Biotechnology) were also used.
Recombinant TGF-β (Stem RD), insulin (Invitrogen), human vascular endothelial growth factor (VEGF; BBI), human platelet-derived growth factor-BB (PDGF-BB; BBI), tumor necrosis factor-α (TNF-α; Sigma-Aldrich), and epidermal growth factor (EGF; Sigma-Aldrich) were utilized per manufacturers' instruction and described in the report. TβRI inhibitor SB431542, nicotiamide (Nico), and trichostatin A (TSA) were obtained from Sigma-Aldrich, while tubacin, nocodazole (NDL), and paclitaxel (PTX) were from Selleck Chem.
Cell Cultures and Transfection
MCF-10A cells were cultured in DMEM/F12 medium (Corning) supplemented with 5% horse serum (Invitrogen), insulin (10 μg/ml), EGF (20 ng/ml), cholera toxin (100 ng/ml) (Sigma-Aldrich), and hydrocortisone (0.5 μg/ml) (Sigma-Aldrich), and penicillin (50 units/ml) and streptomycin (50 μg/ml) at 37 °C in humidified incubator with 5% CO2. NMuMG cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) (Invitrogen), 10 μg/ml insulin, and antibiotics. L929, A549, and C3H10T1/2 cells were maintained in RPMI1640 (Corning) with 10% FBS. C2C12 cells were maintained with DMEM supplemented with 10% FBS. MCF-10A cells were transfected with X-tremeGENE (Roche Applied Science) and HEK293T cells with PEI (Polyscience).
Lentivirus Production and Stable Cell Line Generation
HDAC6 cDNA was subcloned into lentiviral vector pWPI-puro. For knocking down HDAC6, a shRNA against HDAC6 [sense: CCGGGCCTACGAGTTAACCCAGAACTCGAGTTCTGGGTTAAACTCGTAGGCTTTTTG; antisense: AATTCAAAAAGCCTACGAGTTTAACCCAGAACTCGAGTTCTGGGTTAAACTCGTAGGC] was cloned in lentiviral vector pLKO.1-shRNA-puro according to standard methods. Lentiviral vector plasmids were transfected into HEK293T cells together with packaging plasmid psPAX2 and envelope plasmid pMD2.G. After 48 h, lentiviral supernatants were collected and infected onto host cells. Stable cells were selected in the presence of 1 ng/ml of puromycin (Sigma).
Western Blotting
Cells were incubated with TGF-β (2 ng/ml) or chemical inhibitors for the indicated time periods, washed twice with ice-cold PBS and suspended in ice-cold lysis buffer (50 mm Tris/HCl, 1% Triton X-100 pH 7.4, 1% sodium deoxycholate, 0.1% SDS, 0.15% NaCl, 1 mm EDTA, 1 mm sodium orthovanadate) containing a protease inhibitor mixture (Sigma-Aldrich). The protein concentration was determined using the Bradford assay (Bio-Rad). Protein were solubilized in a sample buffer at 95 °C for 5 min and resolved by 10% SDS-PAGE. For immunoblotting, proteins were electro-transferred onto a PVDF membrane and blocked with 5% nonfat milk in TBS-0.10% Tween 20 (TBST) at room temperature for 1 h. Then, the membrane was incubated with primary antibody at 4 °C overnight. After washing (TBST) and subsequent blocking, the blot was incubated with HRP-conjugated secondary antibody (1:10000, Sigma-Aldrich) for 1 h at room temperature. After washing, antibody binding was detected with the ECL detection reagent (Thermo Fisher).
RT-PCR Analysis
Total RNAs were obtained by TRIzol method (Invitrogen). RNAs were reverse-transcribed to complementary DNA using the PrimeScript® RT reagent kit (TaKaRa). Quantitative reverse transcriptase (qRT)-PCR was performed on an ABI PRISM 7500 Sequence Detector System (Applied Biosystems) using gene-specific primers and SYBR Green Master Mix (Invitrogen). For the amplification the following primers were used (5′->3′): h-N-cadherin, ACCAGGTTTGGAATGGGACAG (forward) and GAGCATACAGCCCCATCACT (reverse); h-fibronectin, TGAAAGACCAGCAGAGGCATAAG (forward) and CTCATCTCCAACGGCATAATGG (reverse); h-SNAI1–1, TCGGAAGCCTAACTACAGCGA (forward) and AGATGAGCATTGGCAGCGAG; h-b-actin, CAAAGTTCACAATGTGGCCGAGGA (forward) and GGGACTTCCTGTAACAACGCATCT. (reverse).
Confocal Laser Scanning Microscopy
For immunostaining, cells were cultured on glass cover slips with or without 2 ng/ml of TGF-β for various time periods as indicated in the figure legends. After washing twice with PBS, cells were incubated with 4% PFA for 15 min and then incubated with 5% normal goat serum/1× PBS/0.3% Triton for 1 h at room temperature. Then, the cells were exposed to primary antibody at 4 °C overnight, rinsed three times with PBS and incubated with Alexa Fluor 488 or Alexa Fluor 546 goat anti-rabbit antibody (1:1000, Invitrogen) or donkey anti-mouse antibody (1:1000, Invitrogen) for 1.5 h at room temperature. For F-actin staining, cells were incubated with rhodamine-phalloidin (1:1000, Molecular Probes, Eugene, OR) for 40 min in the dark. After three washing steps, all the slides and coverslips were mounted with ProLong Gold antifade with DAPI reagent (Invitrogen).
Transwell Assays
The Transwell assay was performed using Transwell inserts (BD Bioscience). The inserts with 8.0 μm pore size were then washed with serum-free DMEM, and 100 μl of complete cell culture medium with 1 × 105 cells were seeded onto the insert. We then added 500 μl of complete cell culture medium into the well underside of the insert for 24 h at 37 °C and 5% CO2. After 8 h incubation, cells were fixed, stained with DAPI for 10 min and microscopically analyzed as described.
Wound Healing Assay
For wound healing assays, confluent cell cultures were uniformly scraped with a pipette tip across a 24-well plate. Following wounding, culture medium was replaced with fresh medium, and cells were exposed to 2 ng/ml of TGF-β for the indicated time periods.
HDAC6 Activity Assay
HDAC6 protein was immunoprecipitated from MCF-10A cells using specific antibody against HDAC6 and was incubated with a HDAC colorimetric substrate that comprises an acetylated lysine side chain to determine HDAC6 activity. For immunoprecipitation, cells (5 × 107) were incubated in 1 ml of lysis buffer (20 mm Tris, pH 7.5, 137 mm NaCl, 1.5 mm, 2 mm EDTA, 10% glycerol, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml aprotinin) on ice for 10 min. Cell lysates were sonicated and centrifuged at 13,000 × g for 10 min at 4 °C. Supernatants were incubated with antibodies against HDAC6 at 4 °C overnight. Immunocomplexes were recovered by incubation with 40 μl of 25% protein A/G-Sepharose. Immunocomplex beads were washed twice with lysis buffer, dissolved in 1% SDS lysis buffer, and subjected to HDAC6 enzymatic activity assay using a commercial kit (BioVision). According to the manufacturer's instruction, HDAC6 proteins precipitated from MCF-10A cells were incubated with a HDAC colorimetric substrate at 37 °C for 3 h to deacetylate the substrates. The assay was the stopped by the addition of 10 μl of lysine developer and was incubated at 37 °C for 30 min to produces a chromophore. Samples were read using an ELISA plate reader at a wavelength of 405 nm.
Statistical Analysis
Data are provided as means ± S.E., n represents the number of independent experiments. Data were tested for significance using unpaired Student's t test or ANOVA as appropriate. Differences were considered statistically significant when p values were < 0.05. Statistical analysis was performed with GraphPad InStat version 3.00 for Windows XP, GraphPad Software.
Results
Acetylated α-Tubulin Decreases during TGF-β-induced EMT
Acetylation of α-tubulin is known to be associated with microtubule stability (12, 19, 20). To investigate the role of acetylated α-tubulin in EMT, we compared the levels of acetylated α-tubulin in epithelial cells before and after TGF-β stimulation as well as fibroblasts. MCF-10A cells, an immortalized human mammary epithelial cell line, and NMuMG, an immortalized mouse mammary epithelial cell line, were employed. Both cell lines have been demonstrated to undergo EMT upon exposure to TGF-β. Upon 48 h of TGF-β stimulation, cells began to lose their polarized epithelial cell morphology and became scattered and spindle-like, resembling mesenchymal cell morphology (Fig. 1A). Measurement of acetylated α-tubulin by using Western blot analysis revealed that TGF-β treatment decreased the level of acetylated α-tubulin in both MCF-10A and NMuMG cells (Fig. 1B). The morphological and molecular changes could be reversed by SB431542, a specific inhibitor of TGF-β type I receptor. In contrast, neither morphology nor down-regulation of acetylated α-tubulin occurred after TGF-β treatment in various mesenchymal cell lines, including L929, C3H10T1/2 and myoblasts C2C12 (Fig. 1, C and D). Parallel comparison indicates that epithelial cells exhibited higher acetyl-α-tubulin level than mesenchymal cells. For instance, TGF-β treatment decreased acetyl-α-tubulin in MCF-10A cells to a level comparable to that in C2C12 cells (Fig. 1E). In accordance, confocal laser scanning microscopic analysis revealed that both acetyl-α-tubulin, while significantly reduced, and α-tubulin rearranged in MCF-10A cells upon TGF-β treatment, whereas no such TGF-β effects were observed in C2C12 cells (Fig. 1F). To ensure all the cell lines tested were normally responsive to TGF-β, we measured TGF-β-induced Smad3 phosphorylation and subsequent transcription of EMT markers. TGF-β could induce rapid activation of Smad3 in both MCF-10A and C2C12 cells. But the EMT marker expression and α-tubulin acetylation changed only in epithelial cell MCF-10A (Fig. 1G).
FIGURE 1.

Level of acetylated α-tubulin is decreased in TGF-β-induced EMT. A, TGF-β induces EMT in MCF-10A and NMuMG cells. Cells were treated with TGF-β (2 ng/ml) 48 h and/or SB431542 (5 μm), and then examined under microscope. B, TGF-β decreases the level of acetyl-α-tubulin in MCF-10A and NMuMG cells. Cells were treated with TGF-β (2 ng/ml) 48 h and/or SB431542 (5 μm), and then analyzed by using specific antibodies recognizing acetyl-α-tubulin, α-tubulin, and GAPDH. C, TGF-β exhibites no effect on morphology of L929, C3H10T1/2, and C2C12 cells. Cells were treated similarly as described in A. D, TGF-β does not decrease the level of acetyl-α-tubulin in L929, C3H10T1/2 and C2C12 cells. Cells were treated similarly as described in C and Western blotting as in B. E, TGF-β decreases the level of acetyl-α-tubulin in MCF-10A, but not C2C12 and NIH-3T3 cells. Immunoblotting analysis of acetyl-α-tubulin and GAPDH in MCF-10A, C2C12, and NIH-3T3 cells was done as described in B. F, TGF-β decreases the level of acetyl-α-tubulin in MCF-10A, but not C2C12 cells. Cells were treated with 2 ng/ml TGF-β (48 h), immunostained with specific antibodies for acetyl-α-tubulin or α-tubulin, and analyzed under confocal laser scanning microscopic as described under “Experimental Procedures.” Magnification 100×. G, immunoblotting analysis of E-cadherin, ZO-1, vimentin, N-cadherin, HDAC6, P-Smad3, Smad3, acetyl-α-tubulin, α-tubulin, and GAPDH. MCF-10A and C2C12 cells were stimulated with 2 ng/ml of TGF-β for the indicated time periods.
EMT is a reprogramming process. Is it possible that decreased acetylation of proteins is a general event during the process? We thus examined whether other acetylated proteins were subject to change in epithelial cells. Because histones are frequently acetylated in cells, we analyzed the global level of Histone H3 acetylation. As shown in Fig. 2, acetylated Histone H3 remained unchanged in MCF-10A, NMuMG, and A549 cells upon TGF-β treatment. Taken together, these results suggested that decrease in the global level of acetylated α-tubulin, but not histones, is associated with TGF-β-induced EMT.
FIGURE 2.

Global levels of Histone H3 acetylation are not decreased in TGF-β-induced EMT. MCF-10A, A549, and NMuMG cells were exposed to TGF-β (2 ng/ml, 48 h) in the presence of absence of SB431542 (5 μm, prior 2 h +additional 48 h). Acetyl-Histone H3 (lys9) or H3K9Ac, H3K14Ac, H3K18Ac, H3K56Ac, Histone H3, acetyl-α-tubulin, and GAPDH were examined by immunoblotting.
Loss of Acetylated α-Tubulin Is a Common Feature during EMT
EMT can be induced by many extracellular ligands (4). Thus, we sought to determine whether acetylated α-tubulin decreased during EMT induced by other cytokines. When stimulated by TNFα, PDGF-BB, VEGF, or EGF, cells underwent EMT and gained mesenchymal morphology to various degree (Fig. 3A). Meanwhile, the level of acetylated α-tubulin decreased as did the other epithelial markers such as E-cadherin and ZO-1, whereas N-cadherin and Vimentin were up-regulated (Fig. 3B). Interestingly, the degree of α-tubulin deacetylation was correlated with that of mesenchymal morphology. These data indicate that α-tubulin deacetylation may be as a general marker of EMT.
FIGURE 3.

Level of acetylated α-tubulin is decreased in EMT induced by multiple growth factors. A, immunoblotting analysis of E-cadherin, ZO-1, Vimentin, N-cadherin, acetyl-α-tubulin, and α-tubulin in MCF-10A cells. Cells treated with TGF-β (2 ng/ml), or TNFα (10 ng/ml), PDGF-bb (50 ng/ml), VEGF (50 ng/ml) or (EGF 100 ng/ml). B, morphology of MCF-10A cells. Culture conditions were the same as those in A.
High Levels of α-Tubulin Acetylation Block EMT
We next investigated whether decreased α-tubulin acetylation is essential for initiating/maintaining EMT. To this end, cells were treated with tubacin, which has been reported as α-tubulin acetylation inducer (21), prior to TGF-β treatment As shown in Fig. 4A, Western blotting analysis revealed that tubacin blocked TGF-β-induced down-regulation of E-cadherin and ZO-1 as well as up-regulation of Vimentin and N-cadherin. The effect of tubacin was similar as that of pretreatment with SB431542. PTX could also induce α-tubulin acetylation, blocked E-cadherin expression, but has detrimental effect on ZO-1 expression regardless of TGF-β stimulation (Fig. 4A). MT destabilizer NDL, the Sirtuin inhibitor nicotiamide (Nico) and the general HDAC inhibitor TSA did not exhibit any effect on EMT. These results were further supported by morphological assessment as shown in Fig. 4B. In the presence of tubacin or SB43154, MCF-10A cells retained typical epithelial morphology after TGF-β treatment, whereas PTX, NDL, and Nico did not prevent MCF-10A cells to gain TGF-β-induced spindle-like mesenchymal morphology.
FIGURE 4.

Inhibition of α-tubulin acetylation or disruption of its stability blocks EMT. A, immunoblotting analysis of E-cadherin, ZO-1, Vimentin, N-cadherin, acetyl-α-tubulin, and α-tubulin in MCF-10A cells. Cells were treated with SB431542 (5 μm), Tubacin (5 μm), Paclitaxol (PTX) (5 nm), Nocodazol (NDL) (5 nm), TSA (50 ng/liter), or Nicotiamide (Nico) (10 mm), followed by co-incubation with TGF-β (2 ng/ml). B, morphology of MCF-10A cells. Culture conditions were the same as those in A.
To distinguish whether it is the high level of acetylated α-tubulin, which is maintained by tubacin treatment, or nonspecific effect of tubacin that prevents EMT, we took advantage of tubulin-K40Q (18), a α-tubulin acetylation-mimicking mutant. Cells expressing tubulin-K40Q were unable to undergo EMT, as the levels of either epithelial markers E-cadherin and ZO-1 or mesenchymal markers Vimentin and N-cadherin remained unchanged upon TGF-β treatment. In sharp contrast, TGF-β-induced EMT took place in cells transfected with α-tubulin WT or an acetylation-resistant tubulin mutant (tubulin-K40R) (Fig. 5A). qRT-PCR analysis further supported that expression of tubulin-K40Q, but not tubulin-WT or tubulin K40R, significantly attenuated the TGF-β-induced up-regulation of mRNAs of N-cadherin, Fibronectin and Snail-1 (Fig. 5B). Expression of other EMT markers was also blocked in different degrees (data not shown). Furthermore, the effect of tubulin-K40Q on EMT was not related to cell cycle dysfunction (data not shown). Thus, excessive acetylation of α-tubulin prevented EMT. Taken together, these findings suggested that α-tubulin deacetylation probably ensures the onset of EMT.
FIGURE 5.

An acetylation-mimicking mutant of α-tubulin attenuates TGF-β-induced EMT. A, immunoblotting analysis of E-cadherin, ZO-1, Vimentin, N-cadherin, acetyl-α-tubulin, and α-tubulin in MCF-10A cells. MCF-10A cells were transfected to express wild type (WT), K40Q, or K40R mutant of α-tubulin. Cells were then treated with 2 ng/ml of TGF-β in the presence or absence of 5 μm of SB431542. GFP-tubulin and endogenous α-tubulin were indicated by arrows. B, K40Q mutant decreases TGF-β-induced expression of N-cadherin. MCF-10A cells infected with lentiviruses harboring α-tubulin WT, K40Q, or K40R were treated with TGF-β (2 ng/ml) 48 h. Total RNAs were isolated from treated cells and the N-cadherin mRNA levels were analyzed by using qRT-PCR. **, p < 0.01. C, K40Q mutant decreases TGF-β-induced expression of Fibronectin. qRT-PCR analysis was done as described in panel B. *, p < 0.05. D, K40Q mutant decreases TGF-β-induced expression of Snail-1. qRT-PCR analysis was done as described in panel B. *, p < 0.05.
TGF-β Increases the Activity of HDAC6
The balanced of α-tubulin acetylation is maintained by its acetylases and deacetylases. It has been well documented that HDAC6 and Sirtuin2 are tubulin deacetylases (15–17), whereas MEC-17/aTAT is the major α-tubulin acetylase (14). Having established that the α-tubulin acetylation level is reduced in TGF-β-induced EMT, we asked whether TGF-β achieved this through regulating α-tubulin acetylases or deacetylases. We found that neither the mRNA nor the protein levels of those enzymes were altered by TGF-β (Fig. 6A; data not shown). Interestingly, the HDAC6 activity was significantly increased after 24 h of TGF-β treatment (Fig. 6B). TGF-β-induced increase in the HDAC6 activity could be blocked by SB431542, suggesting that it was achieved through the TGF-β receptors (Fig. 6C). The increase was also similar to that in cells transfected with HDAC6 cDNA; meanwhile knockdown of HDAC6 expression led to an obvious decrease in its activity (Fig. 6D). These data might implicate that TGF-β increased HDAC6 activity to downregulate α-tubulin acetylation.
FIGURE 6.

TGF-β increases the activity of HDAC6. A, TGF-β and SB431542 have no effects on the mRNA levels of HDAC6 in MCF-10A cells. MCF-10A cells were treated with TGF-β (2 ng/ml) or SB431542 (5 μm) for 48 h. Total RNAs were isolated from treated cells and the HDAC6 mRNA levels in the treated cells were analyzed by using qRT-PCR. *, p < 0.05. B, TGF-β activates HDAC6 in MCF-10A cells. MCF-10A cells were treated with 2 ng/ml of TGF-β for the indicated time periods. Measurement of HDAC6 activity in cell lysates was carried out by using ELISA-based enzymatic assay as described under “Experimental Procedures.” C, TGF-β-induced HDAC6 activation is inhibited by SB431542 in MCF-10A cells. Measurement of HDAC6 activity was done as described in panel B. D, TGF-β activates HDAC6 in MCF-10A cells. MCF-10A cells stably expressing GFP (control), HDAC6 or shHDAC6 were treated with 2 ng/ml of TGF-β (48 h). Measurement of HDAC6 activity was done as described in panel B. E, TGF-β-induced HDAC6 activation is inhibited by SB431542, ActD and CHX in MCF-10A cells. MCF-10A cells were treated with SB431542, ActD and CHX for 48 h. HDAC6 activity was measured as described in panel B. F, TGF-β activates HDAC6 in HME cells. HME cells were treated with 2 ng/ml of TGF-β for the indicated time periods. HDAC6 activity was measured as described in panel B. G, TGF-β-induced HDAC6 activation is inhibited by Smad3 knockdown in HME cells. HME cells stably expressing an shRNA against Smad3 were treated with 2 ng/ml of TGF-β for the indicated time periods. HDAC6 activity was measured as described in panel B. Right, knockdown of Smad3 in HME was shown by Western blotting using indicated antibodies. H, TGF-β-induced HDAC6 activation is inhibited by Smad4 knock out in HaCaT cells. HaCaT cells with the Smad4 gene ablated were treated with 2 ng/ml of TGF-β for the indicated time periods. HDAC6 activity was measured as described in panel B. Right, knock-out of Smad4 in HaCaT was shown by Western blotting using indicated antibodies.
To further elucidate the mechanism how TGF-β increases HDAC6 activity, we sought to determine whether this increase requires new protein synthesis. As shown in Fig. 6E, treatment of actinomycin D (ActD) and cycloheximide (CHX), like SB431542, completely abolished TGF-β-increased HDAC6 activity. Because Smad proteins are the central effectors for TGF-β signaling, we determined the role of Smads in regulating HDAC6 activity. In our assays, we found that TGF-β could increase the HDAC6 activity in epithelial cell lines tested, including MCF-10A (Fig. 6, B–E), HME (Fig. 6F), and HaCaT (data not shown). Interestingly, knockdown of Smad3 rendered HME cells unresponsive to TGF-β-induced HDAC6 activity (Fig. 6G). Likewise, TGF-β no longer induced HDAC6 activity in Smad4−/− HaCaT cells (Fig. 6H). These data suggest that TGF-β increases HDAC6 activity in a manner dependent of the intact TGF-β signaling pathway.
Increased Expression of HDAC6 Induces EMT
To further examine the function of HDAC6 in EMT, we established MCF-10A cell lines that stably expressed HDAC6 or stably knocked down HDAC6. Forced expression of HDAC6 enabled the cells to lose polarity and intercellular contacts and gain typical mesenchymal morphology, while GFP-expressing control and shHDAC6 cells retained epithelial morphology (Fig. 7A). The molecular changes associated with EMT were also observed in HDAC6-expressing cells, which exhibited lower levels of E-cadherin and ZO-1 expression as well as higher levels of Vimentin and N-cadherin than GFP cells or shHDAC6 cells. In consistence, the level of acetylated α-tubulin decreased in HDAC6 cells, but increased in shHDAC6 cells (Fig. 7B). These results were further supported by confocal laser scanning microscopy showing that E-cadherin disappeared, Vimentin increased and actin cytoskeleton rearranged in HDAC6-expressing cells (Fig. 7C).
FIGURE 7.

Increased expression of HDAC6 induces EMT. A, morphology of MCF-10A cells stably expressing GFP (control), HDAC6, or shHDAC6. Cells were examined under a fluorescence microscope. Top, bright field; bottom, GFP fluorescence. B, HDAC6 induces EMT marker expression. MCF-10A cells stably expressing GFP (control), HDAC6 or shHDAC6 were subject to immunoblotting analysis using antibodies against E-cadherin, ZO-1, vimentin, N-cadherin, HDAC6, P-Smad3, Smad3, acetyl-α-tubulin, and α-tubulin. C, decreased acetyl-α-tubulin is correlated with EMT marker gene expression. Confocal laser scanning microscopy was used to analyze the levels of E-cadherin, vimentin, actin, acetyl-α-tubulin, and α-tubulin. Magnification 100×. D, HDAC6 promotes cell motility. Confluent monolayer of MCF-10A cells were scratched with a pipette tip to create a cell-free area. 24 h after addition of TGF-β (2 ng/ml), the wound closure was recorded by microphotography of the same region (Left). Quantification of cell motility (Right) was done by measuring the distance between the invading front of cells in three random selected microscopic fields for each condition and time point. The degree of motility is expressed as percentage of wound closure as compared with untransfected cells. E, HDAC6 promotes cell invasiveness. MCF-10A cells were cultured in the Transwell culture chambers with an 8-μm pore-size polycarbonate filter according to the manufacturer's instructions. 24 h later, invaded cells that were attached to the lower surface of the filter were stained with DAPI. The slices were imaged (Left). Quantification (Right) was by counting cell number in the lower surface. The degree of invasiveness is expressed as percentage of total cells seeded as compared with untransfected cells.
To this end, we assessed the migratory capacity of HDAC6-expressing cells by using wound healing assay and transwell assay. As shown in Fig. 7D, treatment with TGF-β resulted in increased cell motility in HDAC6-expressing MCF-10A cells, but had little or no effect in GFP control cells and shHDAC6 cells. In line with this finding, the transwell assay confirmed that increased invasive capacity was observed in HDAC6-expressing MCF-10A cells (Fig. 7E). We concluded that an appropriate expression level of HDAC6 is critical for EMT, cell motility, and cell invasiveness.
Discussion
EMT plays a critical role in development of many tissues and organs in the developing embryo. It is also a recognized process involved in wound healing, organ fibrosis and the initiation of metastasis for cancer progression. Important traits of EMT include loss of epithelial cell polarity, cytoskeletal rearrangement, and gain of mesenchymal characteristics (3). It has been well established that in TGF-β-induced EMT, Smads induce gene reprogramming through direct activation of EMT transcription factors and their subsequent cooperation in controlling transcription of target genes associated with EMT (6). There are many well-characterized gene markers associated with key events of EMT, including loss of epithelial or gain of mesenchymal morphology. Components of tight junctions, epithelial adhesions, and polarity proteins that are associated epithelial morphology are often downregulated during EMT. For instance, E-cadherin, ZO-1 is commonly indicative of epithelial phenotype and becomes remarkably lost. On the contrary, typical mesenchymal markers such as Vimentin, fibronectin, and transcription factors Twist, FoxC2, Snail1/2, and ZEB1/2 are often up-regulated during EMT. In addition to transcriptional regulation, EMT is regulated at post-transcriptional levels such as RNA splicing and non-coding RNAs. Recent studies have shown that microRNAs in the miR-200 family, miR-101 and miR-506 are markedly downregulated during EMT and also strong regulators of EMT through regulating transcription factors involved in EMT (22–24). Similarly, several lncRNAs are found to induce EMT, including HULC, MALAT-1, H19, and HOTAIR (25–28). These genes and RNAs are often recognized as markers for EMT.
Accompanied with reprogramming in gene expression, reorganization of the cytoskeletal architecture and its remodeling occurs in EMT, which enable cells to gain increased motility. Among non-Smad signaling pathways activated by TGF-β, the Rho-Rac-Cdc42 family GTPases are directly involved in the induced actin contractility, a reorganization of the cytoskeleton and dissolution of cell-cell contacts and the formation of stress fibers (29–31). Along with changes in actin contractility and stress fibers during EMT, the microtubule cytoskeleton plays an important role in epithelial basement membrane breakdown (32) and mediating TGF-β/SMAD2 signals to control E-cadherin gene expression (33). Microtubules, as an important component of the cytoskeleton, are mostly associated with cell adhesion, cell motility, and cell polarity (34). Tubulin acetylation is one kind of post-translational modifications on tubulin, which have been linked to MT stability (35). However, how tubulin acetylation participates in EMT is not clear. In TGF-β-induced EMT, knockdown or inhibition of HDAC6, a α-tubulin deacetylase, renders aberrant EMT marker gene expression in lung epithelial A549 cells (18). Reduced expression of HDAC6 also impairs the activation of SMAD3 in response to TGF-β (18). Thus, the Shan et al. study suggests that HDAC6 is an important regulator of TGF-β-induced EMT in lung epithelial A549 cells.
We undertook a small-scale screen for epigenetic genes involved in EMT. HDAC6 came out of the screen that could induce EMT. Our extensive study further extends and expands the finding by Shan (18) by revealing the following novel aspects. First, α-tubulin deacetylation is associated with EMT in mammalian epithelial cells and keratinocytes (data not shown) in addition to lung epithelial A549 cells reported. In MCF-10A and NMuMG cells, loss of α-tubulin acetylation is apparent upon TGF-β treatment. Fibroblasts or myoblasts has low levels of acetylated α-tubulin, which is not further altered by TGF-β treatment. Ectopic expression of HDAC6 in MCF-10A cells also triggers scattered and spindle-like mesenchymal cell morphology, up-regulation in mesenchymal markers Vimentin and N-cadherin, down-regulation in epithelial markers E-cadherin and ZO-1, actin and α-tubulin cytoskeleton rearrangement and increased cell motility. Similar increased cell motility is described in NIH-3T3 cells (16, 21). Secondly, loss of acetylated α-tubulin is not restricted to EMT where TGF-β is an inducer, but also associated with EMT induced by other growth factors, suggesting that α-tubulin deacetylation is a general marker for EMT. We reason that loss of α-tubulin acetylation can be attributed to either loss of acetylases or gain of HDAC6. Thirdly, we found that TGF-β does not change the mRNA or the protein levels of HDAC6 and the opposing acetylase MEC-17. Interestingly, α-tubulin deacetylation is attributed to the increased activity of HDAC6. But how by TGF-β increases the activity of HDAC6? We have found that HDAC6 activation requires new protein synthesis and the intact Smad pathway. Of course, the detailed mechanism underlying this TGF-β-induced Smad-dependent HDAC6 activation awaits further investigation. Finally, it is intriguing that an acetylation-mimicking mutant of tubulin-K40Q prevents EMT-associated decrease in epithelial markers and increase in mesenchymal markers (Fig. 5). This suggests that α-tubulin deacetylation is not just a marker for EMT, but also plays a direct role in controlling EMT, most likely through activation of Smad-mediated transcription.
It is previously reported that Smad2/3 bind with α-tubulin in the absence of TGF-β (36). Destabilization of MT dynamics can lead to Smad-MT dissociation and subsequently enhances TGF-β-induced Smad activation and transcriptional program (36), whereas PTX (a MT destabilizer) attenuates TGF-β signaling and lessens fibrosis that is often mediated by TGF-β (37). However, it is not known whether the Smad-MT dissociation plays a role in EMT. Since acetylated α-tubulin generally increase MT stability, is it possible that loss of MT stability due to reduced α-tubulin acetylation causes enhanced TGF-β signaling, leading to EMT? Indeed, treatment of tubacin inhibits the activity of HDAC6, which presumably increase MT stability, and blocks TGF-β-induced EMT. The efficacy of tubacin is similar to that of SB431542, an inhibitor of TGF-β type I receptor.
In summary, HDAC6 is activated to deacetylate α-tubulin in TGF-β-induced EMT in epithelial cells, thereby leading to decreased MT stability (Fig. 8). This results in more Smad-MT dissociation and thus Smad activation, suggesting that HDAC6 is a part of the MT-Smad regulatory loop. Indeed, the microtubule cytoskeleton plays an important role in regulating TGF-β/SMAD2 signals to control E-cadherin gene expression in MEE during palatal fusion (33). Meanwhile, decreased MT stability further facilitates cytoskeletal reorganization that accompanies increased cell motility during EMT. Finally, the current finding also suggest HDAC6 as a potential therapeutic target against cancer and fibrosis where pathological EMT represents a key step in the disease progression.
FIGURE 8.
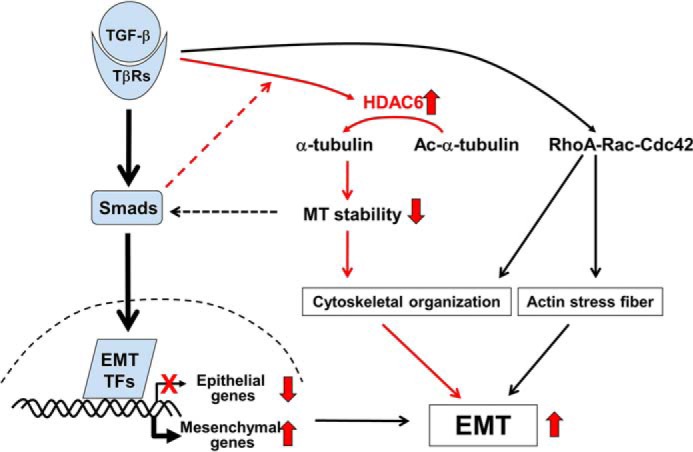
Working Model for TGF-β-induced EMT. In addition to the transcriptional reprogramming during TGF-β-induced EMT, TGF-β induces HDAC6 activity that converts acetyl-α-tubulin into α-tubulin. This deacetylation promotes cytoskeletal reorganization that favors EMT.
Author Contributions
X. H. F. conceived and coordinated the study. S. G., X. L., and X. H. F. wrote the paper. S. G., Y. L., B. Z., K. D., F. C. performed and analyzed the experiments. T. P. Y., L. Z., P. X., M. E., and T. L. provided key reagents, technical assistance and contributed to the preparation of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank the laboratory members for helpful discussion and technical assistance.
This research was supported by the Key Program of the Ministry of Sciences and Technology (2012CB966600), National Natural Science Foundation of China Grants (91540205, 31571447, 31401196), Major Program of the National Natural Science Foundation of China (31090360), and China Postdoctoral Science Foundation grant (2014T70571), the Fundamental Research Funds for the Central Universities, and National Institutes of Health grants (R01GM63773, R01AR053591, R01CA108454, and R01DK073932). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- EMT
- epithelial-to-mesenchymal transition
- MT
- microtubule
- Nico
- nicotiamide
- TSA
- trichostatin A
- NDL
- nocodazole
- PTX
- paclitaxel.
References
- 1. Hay E. D. (1995) An overview of epithelio-mesenchymal transformation. Acta Anatomica 154, 8–20 [DOI] [PubMed] [Google Scholar]
- 2. Kalluri R., and Weinberg R. A. (2009) The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zavadil J., and Böttinger E. P. (2005) TGF-β and epithelial-to-mesenchymal transitions. Oncogene 24, 5764–5774 [DOI] [PubMed] [Google Scholar]
- 4. Moustakas A., and Heldin C. H. (2007) Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Science 98, 1512–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xu J., Lamouille S., and Derynck R. (2009) TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lamouille S., Xu J., and Derynck R. (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Feng X. H., and Derynck R. (2005) Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 [DOI] [PubMed] [Google Scholar]
- 8. Massagué J. (2012) TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peinado H., Olmeda D., and Cano A. (2007) Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat. Rev. Cancer 7, 415–428 [DOI] [PubMed] [Google Scholar]
- 10. Gundersen G. G., and Cook T. A. (1999) Microtubules and signal transduction. Curr. Opin. Cell Biol. 11, 81–94 [DOI] [PubMed] [Google Scholar]
- 11. Dasgupta D., Rajgopalan R., and Gurnani S. (1983) Involvement of colchicine binding site of tubulin in the polymerisation process. FEBS Lett. 152, 101–104 [DOI] [PubMed] [Google Scholar]
- 12. Black M. M., and Keyser P. (1987) Acetylation of alpha-tubulin in cultured neurons and the induction of alpha-tubulin acetylation in PC12 cells by treatment with nerve growth factor. J. Neurosci. 7, 1833–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marmorstein R., and Trievel R. C. (2009) Histone modifying enzymes: structures, mechanisms, and specificities. Biochim. Biophys. Acta 1789, 58–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Akella J. S., Wloga D., Kim J., Starostina N. G., Lyons-Abbott S., Morrissette N. S., Dougan S. T., Kipreos E. T., and Gaertig J. (2010) MEC-17 is an alpha-tubulin acetyltransferase. Nature 467, 218–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsuyama A., Shimazu T., Sumida Y., Saito A., Yoshimatsu Y., Seigneurin-Berny D., Osada H., Komatsu Y., Nishino N., Khochbin S., Horinouchi S., and Yoshida M. (2002) In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 21, 6820–6831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X. F., and Yao T. P. (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 [DOI] [PubMed] [Google Scholar]
- 17. North B. J., Marshall B. L., Borra M. T., Denu J. M., and Verdin E. (2003) The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444 [DOI] [PubMed] [Google Scholar]
- 18. Shan B., Yao T. P., Nguyen H. T., Zhuo Y., Levy D. R., Klingsberg R. C., Tao H., Palmer M. L., Holder K. N., and Lasky J. A. (2008) Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J. Biol. Chem. 283, 21065–21073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cambray-Deakin M. A., and Burgoyne R. D. (1987) Acetylated and detyrosinated α-tubulins are co-localized in stable microtubules in rat meningeal fibroblasts. Cell Motil. Cytoskeleton 8, 284–291 [DOI] [PubMed] [Google Scholar]
- 20. Schulze E., Asai D. J., Bulinski J. C., and Kirschner M. (1987) Posttranslational modification and microtubule stability. J. Cell Biol. 105, 2167–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haggarty S. J., Koeller K. M., Wong J. C., Grozinger C. M., and Schreiber S. L. (2003) Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U.S.A. 100, 4389–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gregory P. A., Bert A. G., Paterson E. L., Barry S. C., Tsykin A., Farshid G., Vadas M. A., Khew-Goodall Y., and Goodall G. J. (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature Cell Biol. 10, 593–601 [DOI] [PubMed] [Google Scholar]
- 23. Kong D., Li Y., Wang Z., Banerjee S., Ahmad A., Kim H. R., and Sarkar F. H. (2009) miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells 27, 1712–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Korpal M., Lee E. S., Hu G., and Kang Y. (2008) The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 283, 14910–14914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao Y., Guo Q., Chen J., Hu J., Wang S., and Sun Y. (2014) Role of long non-coding RNA HULC in cell proliferation, apoptosis and tumor metastasis of gastric cancer: a clinical and in vitro investigation. Oncology Rep. 31, 358–364 [DOI] [PubMed] [Google Scholar]
- 26. Xu Z. Y., Yu Q. M., Du Y. A., Yang L. T., Dong R. Z., Huang L., Yu P. F., and Cheng X. D. (2013) Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int. J. Biol. Sci. 9, 587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo M., Li Z., Wang W., Zeng Y., Liu Z., and Qiu J. (2013) Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Letters 333, 213–221 [DOI] [PubMed] [Google Scholar]
- 28. Ying L., Chen Q., Wang Y., Zhou Z., Huang Y., and Qiu F. (2012) Upregulated MALAT-1 contributes to bladder cancer cell migration by inducing epithelial-to-mesenchymal transition. Mol. BioSys. 8, 2289–2294 [DOI] [PubMed] [Google Scholar]
- 29. Yilmaz M., and Christofori G. (2009) EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Reviews 28, 15–33 [DOI] [PubMed] [Google Scholar]
- 30. Schneider D., and Janshoff A. (2012) Inhibition of actin dynamics during epithelial-to-mesenchymal transition. Biochem. Biophys. Res. Commun. 419, 221–225 [DOI] [PubMed] [Google Scholar]
- 31. Hoj J. P., Davis J. A., Fullmer K. E., Morrell D. J., Saguibo N. E., Schuler J. T., Tuttle K. J., and Hansen M. D. (2014) Cellular contractility changes are sufficient to drive epithelial scattering. Exp. Cell Res. 326, 187–200 [DOI] [PubMed] [Google Scholar]
- 32. Nakaya Y., Sukowati E. W., Wu Y., and Sheng G. (2008) RhoA and microtubule dynamics control cell-basement membrane interaction in EMT during gastrulation. Nature Cell Biol. 10, 765–775 [DOI] [PubMed] [Google Scholar]
- 33. Kitase Y., and Shuler C. F. (2013) Microtubule disassembly prevents palatal fusion and alters regulation of the E-cadherin/catenin complex. Int. J. Dev. Biol. 57, 55–60 [DOI] [PubMed] [Google Scholar]
- 34. Westermann S., and Weber K. (2003) Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 4, 938–947 [DOI] [PubMed] [Google Scholar]
- 35. Sasse R., and Gull K. (1988) Tubulin post-translational modifications and the construction of microtubular organelles in Trypanosoma brucei. J. Cell Sci. 90, 577–589 [DOI] [PubMed] [Google Scholar]
- 36. Dong C., Li Z., Alvarez R. Jr., Feng X. H., and Goldschmidt-Clermont P. J. (2000) Microtubule binding to Smads may regulate TGF β activity. Mol. Cell 5, 27–34 [DOI] [PubMed] [Google Scholar]
- 37. Liu X., Zhu S., Wang T., Hummers L., Wigley F. M., Goldschmidt-Clermont P. J., and Dong C. (2005) Paclitaxel modulates TGFβ signaling in scleroderma skin grafts in immunodeficient mice. PLoS Medicine 2, e354. [DOI] [PMC free article] [PubMed] [Google Scholar]