

Abstract
Apolipoprotein (apo) A-I mediates many of the anti-atherogenic functions attributed to high density lipoprotein. Unfortunately, efforts toward a high resolution structure of full-length apoA-I have not been fruitful, although there have been successes with deletion mutants. Recently, a C-terminal truncation (apoA-IΔ185–243) was crystallized as a dimer. The structure showed two helical bundles connected by a long, curved pair of swapped helical domains. To compare this structure to that existing under solution conditions, we applied small angle x-ray scattering and isotope-assisted chemical cross-linking to apoA-IΔ185–243 in its dimeric and monomeric forms. For the dimer, we found evidence for the shared domains and aspects of the N-terminal bundles, but not the molecular curvature seen in the crystal. We also found that the N-terminal bundles equilibrate between open and closed states. Interestingly, this movement is one of the transitions proposed during lipid binding. The monomer was consistent with a model in which the long shared helix doubles back onto the helical bundle. Combined with the crystal structure, these data offer an important starting point to understand the molecular details of high density lipoprotein biogenesis.
Keywords: apolipoprotein, mass spectrometry (MS), oligomerization, small-angle x-ray scattering (SAXS), structural biology, structural model
Introduction
Apolipoprotein (apo)3 A-I is the most common protein constituent of human high density lipoprotein (HDL), comprising up to 70% of protein mass. As such, it is often credited with defining the functionality of HDL as related to its proposed cardioprotective benefits. For example, apoA-I dramatically stimulates lecithin:cholesterol acyl transferase, which esterifies cholesterol to create a concentration gradient that promotes free cholesterol movement from peripheral cells to HDL (1). apoA-I also interacts with the ATP binding cassette transporter A1 in the liver and periphery (2–5), an event that produces the bulk of circulating HDL. Given the recent revelations that HDL also contains greater than 90 “minor” proteins (HDL Proteome Watch, maintained by the Davidson Laboratory), apoA-I is likely an important scaffold that coordinates these factors to affect functions related to lipid metabolism, inflammation, innate immunity, and more (6).
Since the first descriptions of amphipathic helices inferred in the sequence of apoA-I in the 1970s (7, 8), considerable effort has been put into understanding apoA-I structure in its lipid-free form or when bound to lipid in HDL. With its flexibility and propensity to interact with lipid or itself (oligomerization), it has not yet been possible to apply traditional high resolution structural techniques such as nuclear magnetic resonance or x-ray crystallography to full-length, wild-type apoA-I. However, there has been success using deletion mutants. Borhani et al. (9) crystallized human apoA-I lacking the N-terminal 43 amino acids. The structure showed a ring-like tetramer with amphipathic helical domains coiled around each other. This was thought to reflect the lipid-bound form and became the basis for the double belt model of apoA-I (10). However, information about the monomeric, lipid-free form remained elusive but highly sought after, even prompting the publication of a falsified structure (11).
Recently, Mei and Atkinson (12) published a crystal structure of a mutant lacking the C-terminal 60 amino acids, apoA-IΔ185–243. It showed a curved dimer in which two four-helix bundles were connected by a pair of long antiparallel helices. This strongly resembled a structure of a dimeric apoA-IV mutant that we reported (13). These “helix-swapped” models are attractive in that they offer clear predictions of how: (i) a four-helix bundle can transition to the widely accepted double belt orientation upon lipid binding, and (ii) the dimer can transition to monomeric or trimeric forms (14).
Because apolipoproteins exhibit significant conformational flexibility, we investigated the structure of the truncated apoA-IΔ185–243 in both its dimeric and monomeric forms in solution to draw comparisons to the crystal structure. We used the lower resolution, but solution-based, techniques of chemical cross-linking and small angle x-ray scattering (SAXS). Our results confirm some of the general features of the crystal structure from Mei and Atkinson including the long antiparallel helices participating in the domain swap, but we note important differences in the flexibility of the N-terminal domain and the overall shape of the molecule.
Experimental Procedures
ApoA-IΔ185–243 Protein Expression and Purification
Our previously reported construct for recombinant, full-length apoA-I (15) was modified with a stop codon (TAG) after Asn184. The mutant apoA-IΔ185–243 was then expressed and purified as described (15). pET30 vectors (Novagen) containing mutant apoA-IΔ185–243 were transformed into BL-21 Escherichia coli cells. Cells were grown at 37 °C in Luria-Bertani culture medium containing kanamycin for selection of pET30 transformants. Protein expression was induced by addition of isopropyl β-d-thiogalactopyranoside (0.1 mm) followed by shaking at 225 RPM for 2 h at 37 °C. Cells were pelleted by centrifugation, supernatant was discarded, and cells were resuspended in binding buffer (5 mm imidazole, 500 mm NaCl, 20 mm Tris-HCl, pH 7.9) and lysed at 4 °C by probe sonication. The cell lysate was pelleted, and the supernatant was applied to His bind columns. Fractions containing apoA-IΔ185–243 were pooled, and the His tag was cleaved by tobacco etch virus protease at a mass ratio of 20 (apoA-I):1 (tobacco etch virus) for 2 h at room temperature. The sample was reapplied to the His bind resin to remove the His tag, and fractions containing apoA-IΔ185–243 were pooled, concentrated, and dialyzed into 10 mm NH4HCO3, pH 8.1, lyophilized to dryness, and stored at −80 °C until ready for use. The recombinant protein contained an additional glycine at the N terminus after cleavage of the His tag by tobacco etch virus. The sequence was confirmed by the Cincinnati Children's Hospital Sequencing Core. 15N-Labeled apoA-IΔ185–243 was generated as previously described (16).
14N and 15N versions of apoA-IΔ185–243 were solubilized individually in 3 m guanidine in Tris-HCl. The 14N and 15N apoA-IΔ185–243 were mixed at a 1:1 molar ratio at 37 °C for 1 h with intermediate vortexing. Protein was refolded with exhaustive dialysis against PBS, pH 7.4. Where indicated, apoA-IΔ185–243 was not subjected to denaturation (i.e. left in solution) to characterize any differences that may occur in the denaturation/reassembly process. All protein preparations were further purified using size exclusion chromatography using a single Superdex 200 gel filtration column (10/300 GL; GE Healthcare) on a Äkta FPLC system (GE Healthcare) in PBS to ensure removal of any bacterial protein contamination. Appropriate fractions were pooled and concentrated by ultrafiltration for cross-linking and SAXS. Protein concentration was determined by the Markwell modified Lowry assay (17). Purity was routinely >95% as determined by SDS-PAGE and mass spectrometry.
Cross-linking
All proteins were cross-linked with bis-(sulfosuccinimidyl) suberate (BS3) (Thermo Scientific) as previously described (16). Protein was cross-linked at a molar ratio of 50:1 cross-linker to protein for 12 h at 4 °C and quenched by excess Tris-HCl. Cross-linked monomeric and dimeric apoA-IΔ185–243 were separated by gel filtration chromatography on three Superdex 200 gel filtration columns (10/300 GL; GE Healthcare) in series on a Äkta FPLC system (GE Healthcare) in PBS at a flow rate of 0.3 ml/min. 0.25-ml fractions were collected and analyzed by SDS-PAGE. Fractions containing the pure monomeric and dimeric apoA-IΔ185–243 were pooled and concentrated. Samples undergoing MS analysis were dialyzed into 10 mm NH4HCO3, pH 8.1. 50-μg aliquots were digested with sequencing grade trypsin (Promega) at 1:20 mass ratio of trypsin to protein for 16 h at 37 °C. Peptides cross-linked with BS3 were lyophilized to dryness and stored at −20 °C until MS analysis. Cross-linking experiments were performed on two independent preparations of protein.
Mass Spectrometry and Identification of Cross-linked Peptides
Mass spectrometry analyses were performed as previously described (16). Nano-LC-MS/MS analyses were performed on a TripleTOF® 5600+ (AB Sciex, Toronto, Canada) coupled to an Eksigent (Dublin, CA) NanoLC-Ultra® nanoflow system. Dried samples were reconstituted in formic acid/H2O 0.1/99.9 (v/v), and 5 μl (∼1–2 μg of digest) was loaded onto C18 IntegraFritTM trap column (New Objective, Inc.) at 2 μl/min in FA/H2O 0.1/99.9 (v/v) for 15 min to desalt and concentrate the samples. For the chromatographic separation, the trap column was switched to align with the analytical column, Acclaim® PepMap100 (Dionex-Thermo Fisher Scientific). Peptides were eluted at 300 nl/min using a varying mobile phase gradient from 95% phase A (FA/H2O 0.1/99.9, v/v) to 40% phase B (FA/ACN 0.1/99.9 v/v) for 35 min (1% per min), then from 40% B to 85% B in 5 min with re-equilibration. Effluent was introduced to the mass spectrometer using a NANOSpray® III source (AB Sciex, Toronto, Canada). The instrument was operated in positive ion mode for 65 min, where each cycle consisted of one TOF-MS scan (0.25 s of accumulation time, in a 350–1500 m/z window) followed by 30 information-dependent acquisition mode MS/MS scans on the most intense candidate ions selected from initially performed TOF-MS scan during each cycle. Each product ion scan had an accumulation time of 0.075 s and CE of 43 with an 8-unit scan range. The .wiff files were converted to Mascot generic files using PeakView® v1.2.0.3 software (AB Sciex).
Mascot generic files were loaded into the SIM-XL search engine (18) for cross-linked peptides link version 1.1. Briefly, this latest version is optimized for characterizing interaction between homodimers by allowing the user to specify which proteins(s) in the sequence database have light (e.g. 14N) and heavy (e.g. 15N) versions. The carbamidomethylation of the cysteine and the BS3 cross-linker mass modification of 138.0681 at the N terminus and lysine was considered as fixed. A tolerance of 20 ppm was accepted at the MS1 and MS2 levels. All initial identification of cross-linked peptides required a SIM-XL primary score greater than 1.5. Because a single incorrect cross-link identification may lead to an erroneous model, a manual postvalidation of the search engine results, at the MS/MS level, was independently performed by two experienced analysts.
Small Angle X-ray Scattering
SAXS data were collected using the SIBYLS beamline (Berkeley, CA) (19). Cross-linked, monomeric, and dimeric apoA-IΔ185–243 were separated by gel filtration chromatography as described above. Purified samples were shipped overnight at 4 °C for SAXS data collection within 24 h of isolation to avoid aggregation artifacts. Three concentrations of the purified monomeric and dimeric apoA-IΔ185–243 in PBS were sampled at 10 °C with four exposure times; 0.5, 1.0, 2.0, and 5.0 s. Scattering profiles from samples suffering radiation damage were discarded. ScÅtter (SIBYLS) and ATSAS program suite (EMBL) were used for data analysis. 20 independent ab initio molecular envelope reconstructions were generated using the online DAMMIF server (EMBL-Hamburg) (20). The envelopes were superimposed and averaged using SUPCOMB and DAMAVER (ATSAS, EMBL-Hamburg). The averaged molecular envelope graphics were rendered using UCSF Chimera.
Model Generation and Evaluation
Three-dimensional composite models were generated based on the dimeric crystal structure of apoA-IΔ185–243 (Protein Data Bank entry 3R2P). The structure was manually manipulated in PyMOL guided by experimentally derived cross-links and SAXS data. Modeler v9.14 (21) was used to perform a sequence alignment and generate 100 iterations of each starting model. Models were constrained with identified cross-links with an upper bound of 26.0 ± 0.001 Å from C-α to C-α. The starting model for reported structures were chosen based on the best fit to experimental SAXS data using FoXS (22, 23). An energy minimization was performed on the initial structure using the AllosMod-FoXS web server (23, 24) to generate 3000 intermediate conformations consistent with the input structure using a temperature scan (300 K) (16). The final models were presented based on satisfaction of all cross-linking constraints and best fit to the experimental SAXS scattering profile.
Results
Concept of Isotope-assisted Cross-linking
A problem with using chemical cross-linking to understand the structure of a homodimer is the inability to distinguish between intra- and intermolecular cross-links. In previous work with apoA-IV, we solved this problem by isotopically labeling one polypeptide of the dimer with 15N, leaving the other with naturally occurring 14N (16). The two forms were expressed in bacteria grown with the appropriate nitrogen isotope, denatured in guanidine HCl, mixed together at a 1:1 molar ratio, and then allowed to refold into dimers with mixed isotopic species. We pursued the same strategy with apoA-IΔ185–243 (see “Experimental Procedures”). Fig. 1 illustrates the concept. Intermolecular cross-links between two peptides (A and B) result in four possible mass combinations depending on the isotopic makeup of the two species in a dimer: 14A to 14B, 15A to 15B, 14A to 15B, and 15A to 14B and a MS spectrum containing four sets of peaks (top panel). Alternatively, intramolecular cross-links only have two possibilities, 14A to 14B and 15A to 15B, resulting in two sets of peaks (bottom panel).
FIGURE 1.
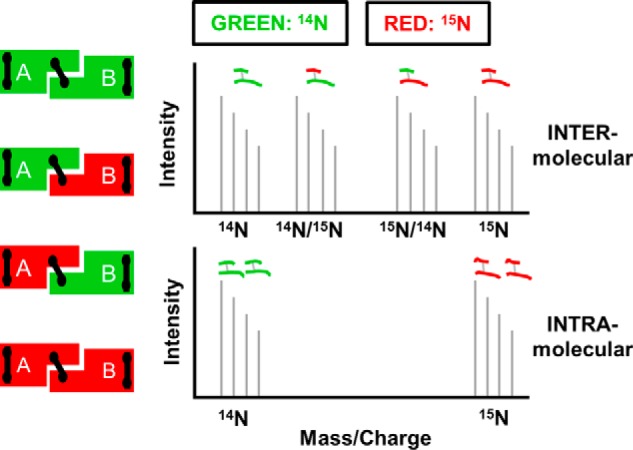
Principle behind isotope-assisted cross-linking. Recombinant proteins produced with either 14N amino acids (green) or 15N amino acids (red) are mixed at a 1:1 ratio under denaturing conditions and allowed to reassemble, resulting in the four combinations on the left. Proteins are locked into position by cross-linking and digested with trypsin. Intermolecular cross-links result in four mass peaks as shown in the top panel, whereas intramolecular cross-links result in two mass peaks shown in the bottom panel.
Oligomerization Properties of apoA-IΔ185–243
Fig. 2a contrasts WT apoA-I (28 kDa) with apoA-IΔ185–243 (22 kDa). The high purity of both preparations is apparent on the SDS gel and by direct injection MS of a mixture of both 14N apoA-IΔ185–243 and 15N apoA-IΔ185–243 (Fig. 2b). 14N apoA-IΔ185–243 exhibited an experimental molecular mass of 21,624 Da, nearly identical to its theoretical mass of 21,623.96 Da. 15N apoA-IΔ185–243 had an experimental molecular mass of 21,890 Da compared with a theoretical 21,894.25 Da. From both direct injection MS and peptide analyses, we determined that our labeling efficiency was >95%.
FIGURE 2.
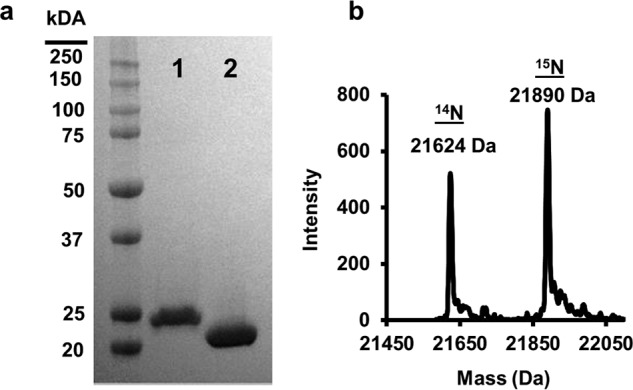
Expression and purification of lipid-free apoA-IΔ185–243. apoA-IΔ185–243 was expressed and purified from bacteria as described under “Experimental Procedures.” a, SDS-PAGE analysis of wild-type apoA-I (lane 1) and apoA-IΔ185–243 (lane 2). b, resolution and molecular weight determination of 14N and 15N apoA-IΔ185–243 using mass spectrometry.
We observed that unfolding the protein in guanidine HCl affected the relative monomer/dimer distribution of the truncation mutant (Fig. 3). When 14N apoA-IΔ185–243 was isolated directly from the bacteria without a refolding step, the preparation contained ∼60% monomer and 40% dimer by gel filtration chromatography at room temperature. However, denaturation in guanidine and refolding via dialysis resulted in a shift to the monomeric state (∼90%) with only ∼10% dimer. This agrees with a previous characterization of this mutant (25). This may result from concentration differences between compartments within the bacteria and bulk solution in vitro. Because it is not possible to perform the dual isotope technique with nonrefolded protein, we scaled up our separations to isolate enough dimer for the current cross-linking studies.
FIGURE 3.
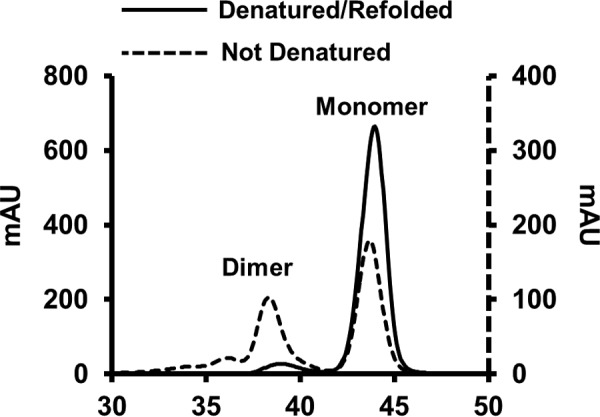
Redistribution of lipid-free apoA-IΔ185–243 after denaturation and reassembly. Distribution of dimeric and monomeric apoA-IΔ185–243 before (dotted line) and after denaturation and reassembly (solid line) using size exclusion chromatography.
Cross-linking
We used the homobifunctional cross-linker BS3, an NHS ester with a preference for lysines within its spacer arm length of 12 Å. However, it can also react with serine at a lower frequency (26, 27). Mixed 14N and 15N apoA-IΔ185–243 was cross-linked, yielding the monomeric and dimeric species shown in Fig. 4a (lane 1). Monomer and dimer were then isolated by gel filtration chromatography (Fig. 4b, lanes 1 and 2, respectively). The samples were subjected to tryptic digestion, and MS was used to identify the cross-links. In total, 29 cross-linked peptide pairs were identified in both monomeric and dimeric apoA-IΔ185–243; intramolecular cross-links are listed in Table 1 and intermolecular links are in Table 2. As expected, all cross-links in the monomer sample showed the two mass peak pattern described in Fig. 1 and exemplified in Fig. 4 (c and d, blue), indicating intramolecular span. The dimer sample (green) contained a mixture of intra- and intermolecular cross-links (the four-peak pattern). 13 of the identified cross-links were shared between monomer and dimer. Monomeric apoA-IΔ185–243 had 9 unique cross-links, whereas dimeric apoA-IΔ185–243 had 7 unique cross-links. This indicates that the dimer and monomer structures are related, but not identical. There were several examples of cross-links that were intramolecular in the monomer, but intermolecular in the dimer, despite linking the same Lys or Ser residues. This is a signature of a domain swap model of oligomerization.
FIGURE 4.

Separation and purification of monomeric and dimeric lipid-free apoA-IΔ185–243 by gel filtration and resulting MS spectra of intramolecular and intermolecular cross-links identified from each species. 14N and 15N apoA-IΔ185–243 were denatured, mixed 1:1, and reassembled as described under “Experimental Procedures.” Proteins were cross-linked and subjected to gel filtration chromatography. a, mixture of cross-linked monomeric and dimeric apoA-IΔ185–243. b, purified monomeric (lane 1) and dimeric (lane 2) samples after separation by gel filtration chromatography. The gels were stained with Coomassie Blue. c, mass spectra for the [M+4H]4+ ion of the cross-linked peptides spanning residues 107–116 and 117–123. Monomeric (blue) and dimeric (green) molecules exhibit a clear intramolecular span in both. d, mass spectra for the [M+4H]4+ ion of the cross-linked peptides spanning residues 117–123 and 132–140 exhibiting a clear intermolecular span in the dimer and intramolecular span in the monomer.
TABLE 1.
Identified INTRA-peptide BS3 cross-links in isolated lipid-free apoA-IΔ185–243 monomeric and dimeric samples derived from mixed 14N- and 15N-labeled proteins
| Cross-link | Peptides involveda | Mod.b | Sample | Massc |
|
|---|---|---|---|---|---|
| 14N | 15N | ||||
| Da | |||||
| Lys88–Lys94 | 84QEMSKDLEEVKAK96 | XL | M | 1671.86 | 1688.81 |
| XL | D | 1671.86 | 1688.81 | ||
| Lys12–Lys23 | 11VKDLATVYVDVLKDSGR27 | XL | M | 2015.13 | 2037.07 |
| XL | D | 2015.12 | 2037.06 | ||
| Lys23–Ser25 | 11VKDLATVYVDVLKDSGR27 | XL, H | M | 2171.22 | 2193.16 |
| XL, H | D | 2171.21 | 2193.15 | ||
| Lys133–Lys140 | 132QKLHELQEKLSPLGEEMR149 | XL | M | 2302.24 | 2329.16 |
| XL | D | 2302.23 | 2329.15 | ||
| Lys133–Ser142 | 132QKLHELQEKLSPLGEEMR149 | XL | M | 2302.24 | 2329.16 |
| D | NDd | ND | |||
| Lys140–Ser142 | 132QKLHELQEKLSPLGEEMR149 | XL, H | M | 2458.31 | 2485.24 |
| D | ND | ND | |||
| Lys106–Lys107 | 97VQPYLDDFQKKWQEEMELYR116 | XL | M | 2782.39 | 2811.32 |
| D | ND | ND | |||
a Lysines or serines involved in cross-links are in bold type.
b Mod., chemical modifications; XL, one complete cross-link (+138.068 Da); H, one hydrolyzed cross-linker (+156.079 Da).
c Experimentally derived monoisotopic mass for each peptide with each isotope and the combinations.
d ND, not detected. These ions were detectable in one sample (i.e. monomer or dimer) but not in the other.
TABLE 2.
Identified INTER-peptide BS3 cross-links in isolated lipid-free apoA-IΔ185–243 monomeric and dimeric samples derived from mixed 14N- and 15N-labeled proteins
| Cross-link | Peptides involveda | Mod.b | Samplec | Peptide massc |
Span | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 14N | 14N/15N | 15N/14N | 15N | |||||||
| Da | ||||||||||
| Lys118-Ser142 | 117QKVEPLR123 | XL | M | 2037.12 | NDd | ND | 2061.06 | Intra | ||
| 141LSPLGEEMR149 | D | ND | ND | ND | ND | ND | ||||
| Lys118–Lys133 | 117QKVEPLR123 | XL | M | 2158.25 | 2185.17 | Intra | ||||
| 132QKLHELQEK140 | D | 2158.24 | 2170.21 | 2173.20 | 2185.17 | Inter | ||||
| NT–Lys182 | −1GDEPPQSPWDR10 | XL | M | 2236.10 | ND | ND | 2261.03 | Intra | ||
| 178LEALKEN184 | D | ND | ND | ND | ND | ND | ||||
| NT–Lys118 | −1GDEPPQSPWDR10 | XL | M | ND | ND | ND | ND | ND | ||
| 117QKVEPLR123 | D | 2289.17 | 2301.13 | 2305.12 | 2317.07 | Inter | ||||
| Lys12–Lys182 | 11VKDLATVYVDVLK23 | XL | M | 2415.39 | ND | ND | 2439.32 | Intra | ||
| 178LEALKEN184 | D | ND | ND | ND | ND | ND | ||||
| Lys107–Lys118 | 107KWQEEMELYR116 | XL | M | 2417.28 | ND | ND | 2445.20 | Intra | ||
| 117QKVEPLR123 | D | 2417.27 | ND | ND | 2445.19 | Intra | ||||
| Lys12–Lys118 | 11VKDLATVYVDVLK23 | XL | M | ND | ND | ND | ND | ND | ||
| 117QKVEPLR123 | D | 2468.46 | 2480.42 | 2483.42 | 2495.38 | Inter | ||||
| NT–Lys107 | −1GDEPPQSPWDR10 | XL | M | ND | ND | ND | ND | ND | ||
| 107KWQEEMELYR116 | D | 2831.32 | 2847.28 | 2847.28 | 2863.25 | Inter | ||||
| NT–Lys12 | −1GDEPPQSPWDR10 | XL | M | 2882.52 | ND | ND | 2913.43 | Intra | ||
| 11VKDLATVYVDVLK23 | D | 2882.50 | ND | ND | 2913.42 | Intra | ||||
| Lys96–Ser167 | 95AKVQPYLDDFQK106 | XL | M | 2889.50 | 2921.41 | Intra | ||||
| 161THLAPYSDELR171 | D | 2889.49 | 2905.45 | 2905.45 | 2921.41 | Inter | ||||
| Lys118–Lys140 | 117QKVEPLR123 | XL | M | ND | ND | ND | ND | ND | ||
| 134LHELQEKLSPLGEEMR149 | D | 2914.60 | 2926.55 | 2937.53 | 2949.50 | Inter | ||||
| Lys118–Ser142 | 117QKVEPLR123 | XL | M | ND | ND | ND | ND | ND | ||
| 134LHELQEKLSPLGEEMR149 | D | 2914.60 | 2926.55 | 2937.53 | 2949.50 | Inter | ||||
| Lys88–Lys96 | 84QEMSKDLEEVK94 | XL | M | 2923.50 | ND | ND | 2953.41 | Intra | ||
| 95AKVQPYLDDFQK106 | D | 2923.49 | ND | ND | 2953.41 | Intra | ||||
| Lys40–Lys118 | 28DYVSQFEGSALGKQLNLK45 | XL | M | ND | ND | ND | ND | ND | ||
| 117QKVEPLR123 | D | 3002.65 | 3014.62 | 3025.55 | 3037.54 | Inter | ||||
| Lys118–Lys140 | 117QKVEPLR123 | XL, H | M | 3326.85 | ND | ND | 3365.74 | Intra | ||
| 132QKLHELQEKLSPLGEEMR149 | D | ND | ND | ND | ND | ND | ||||
| NT–Lys23 | −1GDEPPQSPWDR10 | XL, H | M | 3453.79 | ND | ND | 3491.68 | Intra | ||
| 11VKDLATVYVDVLKDSGR27 | D | 3453.77 | ND | ND | 3491.67 | Intra | ||||
| Lys77–Lys182 | 62EQLGPVTQEFWDNLEKETEGLR83 | XL | M | 3570.84 | ND | ND | 3609.73 | Intra | ||
| 178-LEALKEN-184 | D | ND | ND | ND | ND | ND | ||||
| NT–Lys140 | −1GDEPPQSPWDR10 | XL, H | M | ND | ND | ND | ND | ND | ||
| 132QKLHELQEKLSPLGEEMR149 | D | 3740.87 | 3756.79 | 3767.81 | 3783.76 | Inter | ||||
| NT–Lys77 | −1GDEPPQSPWDR10 | XL | M | 4037.97 | ND | ND | 4083.85 | Intra | ||
| 62EQLGPVTQEFWDNLEKETEGLR83 | D | 4037.95 | ND | ND | 4083.82 | Intra | ||||
| Lys23–Lys59 | 11VKDLATVYVDVLKDSGR27 | XL, H | M | 4052.22 | ND | ND | 4096.09 | Intra | ||
| 46LLDNWDSVTSTFSKLR61 | D | 4052.19 | ND | ND | 4096.07 | Intra | ||||
| Lys40–Lys140 | 28DYVSQFEGSALGKQLNLK45 | XL, H | M | 4454.38 | ND | ND | 4504.24 | Intra | ||
| 132QKLHELQEKLSPLGEEMR149 | D | 4454.35 | ND | ND | 4504.22 | Intra | ||||
| Lys88–Lys106 | 95AKVQPYLDDFQKKWQEEMELYR116 | XL, H, H | M | 4628.35 | ND | ND | 4674.21 | Intra | ||
| 84QEMSKDLEEVK94 | D | NDd | ND | ND | ND | ND | ||||
a Lysines or serines involved in cross-links are in bold type.
b Mod., chemical modifications; XL, one complete cross-link (+138.068 Da); H, one hydrolyzed cross-linker (+156.079 Da).
c Experimentally derived monoisotopic mass for each peptide with each isotope and the combinations.
d ND, not detected. These ions were detectable in one sample (i.e. monomer or dimer) but not in the other.
We also compared the cross-links observed in monomeric apoA-IΔ185–243 with those reported in the same region for WT apoA-I in solution. Including the current study, there have been 35 total cross-links reported in this region in 4 studies (28–30). This study reports 9 previously observed cross-links and 13 unique cross-links not previously found in WT apoA-I. This may suggest a general similarity in the structural fold of the two proteins, but we caution that the large deletion makes direct comparisons with full-length apoA-I difficult.
Small Angle X-ray Scattering
To assess the molecular shape of apoA-IΔ185–243, we performed SAXS experiments. Stability experiments demonstrated that the dimer slowly dissociated into monomer after purification by size exclusion chromatography (not shown). Therefore, to ensure the molecular homogeneity of the samples during SAXS data collection, we used BS3 to lock the dimer and monomers into their respective states and separated them by gel filtration as for the cross-linking/MS studies. The SAXS parameters for all samples are listed in Table 3. In all samples, the scattered intensity I(q) increased proportionally with sample concentration, and the Guinier range (used to calculate the radius of gyration, Rg) showed a linear response at low scattering angles (not shown), indicating high quality data with no evidence of protein aggregation.
TABLE 3.
Experimental parameters from SAXS sampling of apoA-IΔ185–243
| apoA-IΔ185–243 species | I(O) (Guinier) | Rg (Guinier) | Real space Rg | Dmax | Volume | DAMMIF NSDa | |
|---|---|---|---|---|---|---|---|
| cm−1 | Å | Å | Å | Å3 | Å | ||
| Monomer | |||||||
| 4.0 mg/ml | 1460 | 22.9 | 23.34 | 80 | 45,642 | 0.678 ± 0.059 | |
| 2.0 mg/ml | 859 | 23.4 | 23.75 | 78 | 49,575 | ||
| 1.0 mg/ml | 396 | 23.3 | 23.55 | 76 | 64,078 | ||
| Dimer (denatured/refolded) | |||||||
| 4.0 mg/ml | 2270 | 34.5 | 37.76 | 123 | 89,421 | 1.204 ± 0.529 | |
| 2.0 mg/ml | 1530 | 37.4 | 38.08 | 130 | 94,124 | ||
| 1.0 mg/ml | 890 | 40.2 | 38.41 | 125 | 103,611 | ||
| Dimer (not denatured) | |||||||
| 2.6 mg/ml | 1400 | 36.1 | 37.4 | 119 | 83,759 | 0.906 ± 0.324 | |
| 1.7 mg/ml | 925 | 36.7 | 37.7 | 122 | 83,373 | ||
| 1.0 mg/ml | 522 | 35.7 | 37.7 | 120 | 103,468 | ||
a NSD, normalized spatial discrepancy.
Dimer
We compared dimeric apoA-IΔ185–243 derived natively from E. coli (never denatured) or after denaturation/refolding. The SAXS profiles showed no difference in Rg (Guinier and real space) or Dmax, and the two samples were of comparable molecular volumes across all concentrations tested. Furthermore, scattering profiles and the pairwise distribution plots of both dimers were nearly identical (Fig. 5, a and b, respectively), indicating similar structures. Dimeric apoA-IΔ185–243 had an Rg value of 36.17 ± 0.50 Å, which, taken together with the pairwise distribution plot (Fig. 5b, violet), suggested an elongated rod-like structure. Transformation of the SAXS data exhibited a plateau in the q3·I(q) plot and lack of plateau in the q4·I(q) plot (Fig. 5, c and d, respectively), indicating that structures were folded yet contained a flexible domain (31, 32). Indeed, the low resolution envelope generated via ab initio reconstructions (Fig. 5e) confirmed the elongated, rod-like appearance. Interestingly, although a slight curvature was apparent, there was no evidence of the crescent shape observed in the crystal structure. The normalized spatial discrepancy, shown in Table 3, was reasonable suggesting high quantitative similarity between independent reconstructions.
FIGURE 5.

SAXS analysis of lipid-free apoA-IΔ185–243 dimer and monomer. a, intensity distribution of the SAXS scattering function of both dimer isolations (purple and green) and the monomer (tan). b, pairwise distribution function where AU equals arbitrary units. c, q3·I(q) versus q3(Å−1)3. d, q4·I(q) versus q4(Å−1)4. e and f, the SAXS ab initio reconstructions of the dimeric (e) and monomeric (f) apoA-IΔ185–243. The envelopes were generated using DAMMIF and DAMAVER. The averaged envelope was rendered and displayed using UCSF Chimera. The data were from three samples each at a different concentration. Each concentration was run at four different exposure times.
Monomer
Monomeric apoA-IΔ185–243 had Rg values of 23.20 ± 0.26 Å and a parabolic pairwise distribution plot (Fig. 5b, tan), consistent with a more globular structure than the dimer. Transformation of the monomer SAXS scattering profile showed a similar plateau in the q3·I(q) plot and lack of plateau in the q4·I(q) as compared with the dimer, indicating a rigid structure with a flexible domain. In agreement with the pairwise distance distribution profile, the ab initio reconstruction of the monomeric envelope (Fig. 5f) confirmed the globular appearance having similar width, but half the length of the dimer envelope. Indeed, the monomer envelope yielded a diameter of ∼78 Å with an average volume of 53,100 Å3, roughly half of the dimer (95,700 Å; Table 3).
Comparison to the Crystal Structure
We used the cross-linking and SAXS experimental data derived in solution to test the predictions from the crystal dimer of Mei and Atkinson. Fig. 6a shows a contact plot rendered from the crystal structure. Colored areas represent amino acids that fall within 26 Å of each other, i.e. within possible cross-linking distance. The cross-links in Tables 1 and 2 were placed on the contact plot showing that 13 of 20 were consistent. The majority of the violations were in the N-terminal region highlighted in Fig. 6b.
FIGURE 6.

Comparison of dimeric apoA-IΔ185–243 crystal structure with experimental cross-links. a, the cross-linking data from Tables 1 and 2 are superimposed on a molecular contact plot generated for the reported crystal structure of dimeric apoA-IΔ185–243(Protein Data Bank entry 3R2P). The x and y axes delineate the residue number of apoA-I from 1–243. Dark areas represent the truncated C-terminal segments of the protein. The diagonal line bisects the figure with the bottom right showing intramolecular interactions and the upper left showing intermolecular interactions. b, structural representation showing the cross-links (red dots circled in the contact plot) that deviate from predictions from the crystal structure.
Next, the crystal structure was compared with the SAXS data using FoXS (22, 23). Fig. 7 (a and b) shows significant deviation between the theoretical scattering profile and the experimental data with a poor χ of 3.25. We solvated the crystal structure and generated a theoretical pairwise distribution curve using Scatter (Fig. 7c, red). The biphasic pattern for the crystal structure clearly deviates from the experimental pairwise distribution curve. Lastly, we superimposed the crystal structure on the molecular envelopes generated from the SAXS experimental data (Fig. 7d), revealing large inconsistencies. Taking the cross-link violations with the SAXS discrepancies, we concluded that apoA-IΔ185–243 adopts an alternate configuration in solution. Thus, we set out to generate a solution-state model of apoA-IΔ185–243.
FIGURE 7.

Comparison of dimeric apoA-IΔ185–243 with experimental SAXS data. a, the theoretical scattering profile of the crystal dimer generated by FoXS superimposed onto the experimental scattering profile from the SAXS analysis with the chi distribution values in parenthesis. b, the residuals normalized to the experimental scattering profile (black dotted line). c, the pairwise distribution function of the experimental dimer (black) and the theoretical distribution generated after solvation using ScÅtter where AU equals arbitrary units. d, the crystal structure superimposed onto the molecular envelopes generated from the SAXS analysis.
Derivation of a Dimeric apoA-IΔ185–243 Solution Model
Because many of the experimental cross-links were consistent with the crystal structure, we used it as a starting model. We used as few manipulations as possible to bring it in line with the cross-linking and SAXS data. Previous studies on discoidal HDL suggest the antiparallel 5/5 helices can form a hairpin (10, 33). Implementation of hairpins and juxtaposition to residues 106–116 resolved some of the problematic cross-links and resulted in the overall shortening of the structure imposed by the SAXS data. Focusing on the N termini, we observed cross-links consistent with the crystal structure (Fig. 8, top inset) but also links that strongly suggested an interaction between the N terminus and middle of the molecule (Fig. 8, bottom inset). We concluded that (i) satisfying all cross-links in a single dimer model was only feasible by collapsing the structure into a compact, spherical globular protein or (ii) the N terminus is dynamic, and moving back and forth from the end to the middle of the molecule and the cross-linker was capturing it in both positions. The elongated structure implied by the SAXS data ruled out the first possibility. Therefore, we generated two initial models differing in the placement of the N terminus (residues 1–37). The “open” conformation has the N terminus folded across the ends of the dimer to complete four-helix bundles on each end (like the crystal structure). A second “closed” conformation uses the N-terminal minor helix (residues 37–42) as a hinge so the N terminus can flip over and interact with the middle of the molecule.
FIGURE 8.

Derivation of the starting model(s) of dimeric apoA-IΔ185–243. The open model of apoA-IΔ185–243 was generated by applying a hairpin in helix 5 of each apoA-I molecule. The open model consists of cross-links consistent with the placement of the N terminus (red) in the crystal structure (top inset). The closed initial model was generated by folding the N-terminal segments (red) to the middle of the molecule guided by remaining intermolecular cross-links observed in that region (bottom inset).
Modeler v9.14 was used to generate 100 iterations of each initial structure constrained by experimental cross-links respective to the open and closed conformations. The best initial model for each conformation was determined using FoXS, which gave improved fits (χ = 1.32 and χ = 0.95 for the open and closed, respectively) compared with the crystal structure (χ = 3.25). We performed an energy minimization on each best fit conformation and then used AllosMod-FoXS (23, 24) to generate 3000 alternate structures of each model. These were constrained to their respective cross-link sets and simulated at 300 K to determine whether an alternate model at a physiological temperature better represents the SAXS profile. Fig. 9 (a and b) shows the final open and closed models superimposed onto the SAXS molecular envelope. Both show good visual fit to the molecular envelopes and excellent agreement with the experimental SAXS profile shown in Fig. 9c (χ = 0.73 and 0.83 for the open and closed, respectively). Additionally, MultiFoXS was used on a pool of all 6000 independent models (3000 each of the open and closed) to determine whether a two-state model better fit the SAXS profile. The crystal structure was included in the pool as a control. Indeed, MultiFoXS picked a single model from each conformation for an improved fit of χ = 0.67 weighted at 64% open and 36% closed (Fig. 9c, orange). Lastly, contact plots were generated and superimposed for both conformations in Fig. 9d, showing that 100% of the observed cross-links are accounted for between the two models.
FIGURE 9.

AllosMod-FoXS modeling of the open and closed conformations of dimeric apoA-IΔ185–243. a and b, the best fit open and closed conformations are superimposed onto the ab initio dimeric apoA-IΔ185–243 molecular envelope in a and b, respectively. c, comparison of the experimental dimeric apoA-IΔ185–243 x-ray scattering profile (black line) to the best fit open (green line), closed (purple line), and two-state (orange line) theoretical profiles with the chi distribution in parenthesis. d, the residual scatter plot normalized to the experimental scatter profile (black dotted line). e, cross-linking data from Tables 1 and 2 superimposed on a molecular contact blot generated from the best fit of the combined open (dark) and closed (light) models.
Derivation of a Monomeric apoA-IΔ185–243 Solution Model
Mei and Atkinson (12) postulated that monomeric apoA-IΔ185–243 occurs when the long helix (residues 118–184), which participates in the dimer domain swap, doubles back onto the four-helix bundle in the exact same position that the same sequence from the other monomer would sit in the dimer. We generated a starting model of this concept based on the crystal structure (Fig. 10a). Interestingly, Fig. 10b shows that the cross-linking pattern was 100% compatible, even on the unoptimized model. Using Modeler 9.14, 100 iterations of the model were generated with the imposed cross-link constraints. The best fit model was then run through AllosMod-FoXS to generate 3000 alternate structures at 300 K. The final best fit model is shown superimposed on the molecular SAXS envelope for monomeric apoA-IΔ185–243 (Fig. 10c) with an excellent fit of χ = 0.78 (Fig. 10d).
FIGURE 10.

AllosMod-FoXS modeling of the monomeric apoA-IΔ185–243. a, derivation of the initial model as postulated by Mei and Atkinson (12). b, cross-linking data from Tables 1 and 2 superimposed on a molecular contact blot generated for the theoretical model. c, single best fit conformation from AllosMod-FoXS output superimposed on the molecular envelope. d, comparison of the experimental monomeric apoA-IΔ185–243 x-ray scattering profile (black line) to the single best fit initial (green line) and final (purple line) profiles generated by AllosMod-FoXS. e, regions of α-helicity in our final model of monomeric apoA-I Δ185–243 (magenta) versus that determined in monomeric full-length apoA-I by Chetty et al. (44) (black) and the crystal structure (green). The boxes represent α-helical segments.
Discussion
Deletion mutants have become a valuable work-around to the notorious problem of getting full-length exchangeable apolipoproteins to crystallize for high resolution structural studies. Typically, apolipoproteins have low thermodynamic stability with highly dynamic domains that are poised to interact with lipid (34, 35). Removing these domains may make crystallization more tractable, but there are drawbacks to this approach. First, it is never clear how the structure of the deletion mutant relates to that of the full-length protein. Even though the missing domain is flexible, it may participate in interactions that are key to the global structure of the WT protein. Second, apolipoproteins are likely more sensitive than most proteins to high concentrations and nonphysiological precipitants required for crystallization. Therefore, we believe that it is critical to evaluate predictions from crystal structures on proteins that are in solution.
We previously reported the crystal structure of a deletion mutant of apoA-IV, which showed a remarkably similar helical “swap” as that reported by Mei and Atkinson (12) for apoA-IΔ185–243. SAXS and cross-linking analyses of dimeric apoA-IV64–333 in solution agreed quite well with the crystal structure (13), and we went on to propose a structure for full-length apoA-IV using the crystal structure as a template. In the case of apoA-IΔ185–243, however, it was immediately clear that both the SAXS analysis and the cross-linking patterns were not fully compatible with the crystal structure, at least in the dimer. Despite these differences, important predictions of the crystal structure were clearly borne out in solution. For example, the long swapped helix between the dimers was confirmed. Intermolecular cross-links observed between Lys96 and Ser167, Lys118 and Lys140, Lys118 and Ser142, and Lys118 and Lys133 are consistent with the extended helix swap. There was also confirmatory evidence of the folded helical hairpins at each end of the dimer with the intramolecular cross-link between Lys23 and Lys59, for example.
However, we reproducibly noted interactions that could not be reconciled in the rigid crystal structure. Implementation of the hairpins in helix 5 and their subsequent juxtaposition to residues 106–116 satisfied critical cross-links constraints while also improving the fit to the SAXS data. Several studies have postulated the existence of a hairpin in helix 5 despite its absence in either crystal structure (9, 12). In the double belt disc model, Li et al. (10) hypothesized that helix 5 hairpins could reduce the diameter of discoidal HDL. Applying to the crystal structure of Mei and Atkinson (12), helix 5 flexibility might allow the formation of a “presentation tunnel” for the docking of lecithin:cholesterol acyl transferase and the subsequent influx of cholesterol ester, an idea supported by molecular dynamics studies (10, 36). We also observed interactions between the N terminus and the central domain of the dimer. For example, the intermolecular cross-link between the N terminus and Lys118 can only occur if the N-terminal major helix (residues 7–34) swings back across the dimer as illustrated in our closed model. However, we also saw the N terminus interacting intramolecularly with Lys77, which is consistent with the crystal structure. Because the N terminus cannot be in two places at once, we were forced to postulate at least two models in equilibrium.
This result is intriguing when considering the transition of apoA-I to a lipid-bound species. Mei and Atkinson (12) postulated that the crystal structure could form a discoidal HDL particle through a “sequential unhinging of the N-terminal bundle.” They proposed that the hairpin at each end of the dimer can swing away from the AB-repeating backbone and then unfurl to form a ring that approximates the double belt model of lipid-bound apoA-I (10). This movement is quite analogous to our closed (i.e. closed ring) conformation of the lipid-free dimer. Both transitions are predicted to occur using the N-terminal minor helix as a hinge.
Mei and Atkinson (12) proposed that the N-terminal bundles are stabilized by two hydrophobic clusters at each end of the bundle (Fig. 11a). The N-terminal aromatic cluster holds the first helix, the second helix B of H1, and the helix of the first A unit of H2 together. The C aromatic cluster holds the N-terminal helix and H4(AB2) together through π-π interactions. Influx of lipid was suggested to open the N-terminal helix bundle by disrupting one or both staple domains. Our data suggests that, in solution, these helical bundles may be more dynamic (Fig. 11, b and c). Interestingly, we found similar aromatic clusters in the closed model that might contribute to its stability (Fig. 11c). Trp8 is in close proximity to Trp108 and Phe104 in the long swapped helix. Additionally, Phe33 from both N-terminal major helices are in close proximity in the middle of the molecule. Thus, hydrophobic clusters could be pseudostabilizing features in both conformers in solution.
FIGURE 11.

Disruption of hydrophobic “staples” that allow mobility of the N terminus in solution. a, one monomer of the Mei crystal structure of apoA-I showing the C aromatic cluster (comprised of residues Phe33, Phe104, and Trp108) and an N aromatic cluster (Trp8, Phe71, and Trp72) that is proposed to stabilize the four-helix bundles on each end of the dimer structure. b, the open form of the solution model reported here showing the plausibility of both interactions. c, the closed form of the solution model showing disruptions of the N aromatic cluster allowing the N-terminal major helix, using the N-terminal minor helix (blue) as a hinge, to swing toward the middle of the dimer. Stabilization of the N-terminal helix is postulated to occur with two alternative aromatic clusters: (i) between Phe33 on the N-terminal major helices of each molecule and (ii) Trp8 at the end of the N-terminal major helix with Trp108 and Phe104 on the domain swap.
We caution that this proposed structural equilibrium is only relevant in the context of this particular deletion mutant and its role with respect to lipid binding or other functions in the full-length protein is less clear. The missing C terminus has been shown repeatedly to play a major role in lipid binding (34, 37–40). It is possible that, when present, the C terminus stabilizes the N-terminal bundles in much the same way they appear to be under crystallization conditions (30). Indeed, in apoA-IV, there are extensive stabilizing interactions between the N and C termini at both ends of its dimer (41). Engagement of the apoA-I C terminus, by lipid or perhaps ABCA1, may free up the N-terminal helix to swing away from the helical bundle as part of the particle assembly process. Nevertheless, the absence of the C terminus may have allowed the fortuitous visualization of a transition step (the opening of the N-terminal bundles) during lipid binding that would otherwise not be apparent in a static full-length structure.
Based on our SAXS data, the half circle curvature likely manifests as lipid accumulates. The curvature in the crystal structure may have arisen from a pseudolipid-like environment contributed by PEG or other additives during crystallization. Indeed, additives like isopropanol can induce a lipid-bound-like structure to otherwise lipid-free apoA-I (42). Crystal packing and other factors could also be responsible (43).
With regard to monomeric apoA-IΔ185–243, our results are highly compatible with the monomer scheme proposed by Mei and Atkinson (12). The idea is quite similar to the “pocket knife closing” model that we proposed for apoA-IV (13). The overall α-helicity of our model is 61%, which matches nicely with circular dichroism data estimating 59% helicity for this mutant in solution (12). Although it is dangerous to make direct comparisons from a deletion mutant to the monomeric full-length version of apoA-I, we did find it interesting that our final model exhibited α-helical character in many of the same regions assigned by the hydrogen-deuterium exchange experiments of Chetty et al. (44) in WT apoA-I; there was 65% overlap of helical residues (Fig. 10e). There was also some cross-link overlap between apoA-IΔ185–243 versus those reported for full-length apoA-I. These data imply that some of the structural features of apoA-IΔ185–243 could apply to WT apoA-I. Confirmation awaits more detailed studies on WT apoA-I.
Finally, we acknowledge the strong experimental evidence showing that apoA-I exhibits molten globule characteristics in solution (29, 30, 35, 45). apoA-I has a free energy of denaturation that is well below that of most soluble proteins (45, 46) with helical segments that are constantly folding and unfolding on a time scale of seconds (44). Although the molecules must have a distinct shape as evidenced by the SAXS data, the two dimeric structures reported here are probably best thought of as two general conformational classes, each representing a set dynamically related structures which coexist at any given time.
In summary, we report two related models for soluble dimeric apoA-IΔ185–243 that differ by the location of the N terminus. We also provided strong evidence supporting the postulated monomeric structure of apoA-IΔ185–243. This work emphasizes that high resolution structural studies should be coupled with innovative in-solution experiments to understand the dynamics of the exchangeable apolipoproteins. This will allow us to better understand how they transition in response to lipid. Current work is focused on deriving a structure for full-length apoA-I using these models as a foundation.
Author Contributions
J. T. M. conducted experiments, derived the models, and wrote the paper. J. M. and M. C. conducted experiments. R. G. W., T. B. T., M. K. J., J. P. S., and W. S. D. analyzed data, contributed to model building, and assisted in manuscript writing. D. B. L., P. C. C., and F. C. G. analyzed data.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM098458 (to W. S. D. and T. B. T.) and HL67093 (to W. S. D.), National Institute of Health Project MINOS R01GM105404, and the Department of Energy Office of Basic Energy Sciences through the Integrated Diffraction Analysis Technologies program, supported by Department of Energy Office of Biological and Environmental Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- apo
- apolipoprotein
- HDL
- high density lipoprotein
- SAXS
- small angle x-ray scattering
- BS3
- bis-(sulfosuccinimidyl) suberate.
References
- 1. Soutar A. K., Garner C. W., Baker H. N., Sparrow J. T., Jackson R. L., Gotto A. M., and Smith L. C. (1975) Effect of the human plasma apolipoproteins and phosphatidylcholine acyl donor on the activity of lecithin: cholesterol acyltransferase. Biochemistry 14, 3057–3064 [DOI] [PubMed] [Google Scholar]
- 2. Bodzioch M., Orsó E., Klucken J., Langmann T., Böttcher A., Diederich W., Drobnik W., Barlage S., Büchler C., Porsch-Ozcürümez M., Kaminski W. E., Hahmann H. W., Oette K., Rothe G., Aslanidis C., Lackner K. J., and Schmitz G. (1999) The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 22, 347–351 [DOI] [PubMed] [Google Scholar]
- 3. Brooks-Wilson A., Marcil M., Clee S. M., Zhang L. H., Roomp K., van Dam M., Yu L., Brewer C., Collins J. A., Molhuizen H. O., Loubser O., Ouelette B. F., Fichter K., Ashbourne-Excoffon K. J., Sensen C. W., Scherer S., Mott S., Denis M., Martindale D., Frohlich J., Morgan K., Koop B., Pimstone S., Kastelein J. J., Genest J. Jr., and Hayden M. R. (1999) Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 22, 336–345 [DOI] [PubMed] [Google Scholar]
- 4. Rust S., Rosier M., Funke H., Real J., Amoura Z., Piette J. C., Deleuze J. F., Brewer H. B., Duverger N., Denèfle P., and Assmann G. (1999) Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 22, 352–355 [DOI] [PubMed] [Google Scholar]
- 5. Oram J. F., Lawn R. M., Garvin M. R., and Wade D. P. (2000) ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J. Biol. Chem. 275, 34508–34511 [DOI] [PubMed] [Google Scholar]
- 6. Shah A. S., Tan L., Long J. L., and Davidson W. S. (2013) Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 54, 2575–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Segrest J. P. (1977) Amphipathic helixes and plasma lipoproteins: thermodynamic and geometric considerations. Chem. Phys. Lipids 18, 7–22 [DOI] [PubMed] [Google Scholar]
- 8. Segrest J. P., and Feldmann R. J. (1977) Amphipathic helixes and plasma lipoproteins: a computer study. Biopolymers 16, 2053–2065 [DOI] [PubMed] [Google Scholar]
- 9. Borhani D. W., Rogers D. P., Engler J. A., and Brouillette C. G. (1997) Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc. Natl. Acad. Sci. U.S.A. 94, 12291–12296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li L., Chen J., Mishra V. K., Kurtz J. A., Cao D., Klon A. E., Harvey S. C., Anantharamaiah G. M., and Segrest J. P. (2004) Double belt structure of discoidal high density lipoproteins: molecular basis for size heterogeneity. J. Mol. Biol. 343, 1293–1311 [DOI] [PubMed] [Google Scholar]
- 11. Borrell B. (2009) Fraud Rocks Protein Community. Nature 462, 970. [DOI] [PubMed] [Google Scholar]
- 12. Mei X., and Atkinson D. (2011) Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J. Biol. Chem. 286, 38570–38582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deng X., Morris J., Dressmen J., Tubb M. R., Tso P., Jerome W. G., Davidson W. S., and Thompson T. B. (2012) The structure of dimeric apolipoprotein A-IV and its mechanism of self-association. Structure 20, 767–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deng X., Walker R. G., Morris J., Davidson W. S., and Thompson T. B. (2015) Role of conserved proline residues in human apolipoprotein A-IV structure and function. J. Biol. Chem. 290, 10689–10702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tubb M. R., Smith L. E., and Davidson W. S. (2009) Purification of recombinant apolipoproteins A-I and A-IV and efficient affinity tag cleavage by tobacco etch virus protease. J. Lipid Res. 50, 1497–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walker R. G., Deng X., Melchior J. T., Morris J., Tso P., Jones M. K., Segrest J. P., Thompson T. B., and Davidson W. S. (2014) The structure of human apolipoprotein A-IV as revealed by stable isotope-assisted cross-linking, molecular dynamics and small angle x-ray scattering. J. Biol. Chem. 289, 5596–5608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 18. Lima D. B., de Lima T. B., Balbuena T. S., Neves-Ferreira A. G., Barbosa V. C., Gozzo F. C., and Carvalho P. C. (2015) SIM-XL: A powerful and user-friendly tool for peptide cross-linking analysis. J. Proteomics 129, 51–55 [DOI] [PubMed] [Google Scholar]
- 19. Dyer K. N., Hammel M., Rambo R. P., Tsutakawa S. E., Rodic I., Classen S., Tainer J. A., and Hura G. L. (2014) High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods Mol. Biol. 1091, 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Franke S. D. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 22. Schneidman-Duhovny D., Hammel M., Tainer J. A., and Sali A. (2013) Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 105, 962–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schneidman-Duhovny D., Hammel M., and Sali A. (2010) FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38, W540–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weinkam P., Pons J., and Sali A. (2012) Structure-based model of allostery predicts coupling between distant sites. Proc. Natl. Acad. Sci. U.S.A. 109, 4875–4880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laccotripe M., Makrides S. C., Jonas A., and Zannis V. I. (1997) The carboxyl-terminal hydrophobic residues of apolipoprotein A-I affect its rate of phospholipid binding and its association with high density lipoprotein. J. Biol. Chem. 272, 17511–17522 [DOI] [PubMed] [Google Scholar]
- 26. Swaim C. L., Smith J. B., and Smith D. L. (2004) Unexpected products from the reaction of the synthetic cross-linker 3,3′-dithiobis(sulfosuccinimidyl propionate), DTSSP with peptides. J. Am. Soc. Mass Spectrom. 15, 736–749 [DOI] [PubMed] [Google Scholar]
- 27. Leavell M. D., Novak P., Behrens C. R., Schoeniger J. S., and Kruppa G. H. (2004) Strategy for selective chemical cross-linking of tyrosine and lysine residues. J. Am. Soc. Mass Spectrom 15, 1604–1611 [DOI] [PubMed] [Google Scholar]
- 28. Silva R. A., Hilliard G. M., Fang J., Macha S., and Davidson W. S. (2005) A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry 44, 2759–2769 [DOI] [PubMed] [Google Scholar]
- 29. Pollard R. D., Fulp B., Samuel M. P., Sorci-Thomas M. G., and Thomas M. J. (2013) The conformation of lipid-free human apolipoprotein A-I in solution. Biochemistry 52, 9470–9481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Segrest J. P., Jones M. K., Shao B., and Heinecke J. W. (2014) An experimentally robust model of monomeric apolipoprotein A-I created from a chimera of two X-ray structures and molecular dynamics simulations. Biochemistry 53, 7625–7640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rambo R. P., and Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hammel M. (2012) Validation of macromolecular flexibility in solution by small-angle x-ray scattering (SAXS). Eur. Biophys. J. 41, 789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones M. K., Catte A., Li L., and Segrest J. P. (2009) Dynamics of activation of lecithin:cholesterol acyltransferase by apolipoprotein A-I. Biochemistry 48, 11196–11210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davidson W. S., Hazlett T., Mantulin W. W., and Jonas A. (1996) The role of apolipoprotein AI domains in lipid binding. Proc. Natl. Acad. Sci. U.S.A. 93, 13605–13610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saito H., Dhanasekaran P., Nguyen D., Holvoet P., Lund-Katz S., and Phillips M. C. (2003) Domain structure and lipid interaction in human apolipoproteins A-I and E, a general model. J. Biol. Chem. 278, 23227–23232 [DOI] [PubMed] [Google Scholar]
- 36. Segrest J. P., Jones M. K., Catte A., and Thirumuruganandham S. P. (2012) Validation of previous computer models and MD simulations of discoidal HDL by a recent crystal structure of apoA-I. J. Lipid Res. 53, 1851–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Palgunachari M. N., Mishra V. K., Lund-Katz S., Phillips M. C., Adeyeye S. O., Alluri S., Anantharamaiah G. M., and Segrest J. P. (1996) Only the two end helixes of eight tandem amphipathic helical domains of human apo A-I have significant lipid affinity: implications for HDL assembly. Arterioscl. Thromb. Vasc. Biol. 16, 328–338 [DOI] [PubMed] [Google Scholar]
- 38. Holvoet P., Zhao J. Z., Vanloo B., Vos R., Deridder E., Dhoest A., Taveirne J., Brouwers E., Demarsin E., Engelborghs Y., Rosseneu M., Collen D., and Brasseur R. (1995) Phospholipid binding and lecithin-cholesterol acyltransferase activation properties of apolipoprotein A-I mutants. Biochemistry 34, 13334–13342 [DOI] [PubMed] [Google Scholar]
- 39. Minnich A., Collet X., Roghani A., Cladaras C., Hamilton R. L., Fielding C. J., and Zannis V. I. (1992) Site-directed mutagenesis and structure-function analysis of the human apolipoprotein A-I. Relation between lecithin-cholesterol acyltransferase activation and lipid binding. J. Biol. Chem. 267, 16553–16560 [PubMed] [Google Scholar]
- 40. Panagotopulos S. E., Witting S. R., Horace E. M., Hui D. Y., Maiorano J. N., and Davidson W. S. (2002) The role of apolipoprotein A-I helix 10 in apolipoprotein-mediated cholesterol efflux via the ATP-binding cassette transporter ABCA1. J. Biol. Chem. 277, 39477–39484 [DOI] [PubMed] [Google Scholar]
- 41. Deng X., Morris J., Chaton C., Schröder G. F., Davidson W. S., and Thompson T. B. (2013) Small-angle x-ray scattering of apolipoprotein A-IV reveals the importance of its termini for structural stability. J. Biol. Chem. 288, 4854–4866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leroy A., and Jonas A. (1994) Native-like structure and self-association behavior of apolipoprotein A-I in a water/n-propanol solution. Biochim. Biophys. Acta 1212, 285–294 [DOI] [PubMed] [Google Scholar]
- 43. Segrest J. P., Jones M. K., Catte A., Manchekar M., Datta G., Zhang L., Zhang R., Li L., Patterson J. C., Palgunachari M. N., Oram J. F., and Ren G. (2015) Surface density-induced pleating of a lipid monolayer drives nascent high-density lipoprotein assembly. Structure 23, 1214–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chetty P. S., Mayne L., Lund-Katz S., Stranz D., Englander S. W., and Phillips M. C. (2009) Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 106, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gursky O., and Atkinson D. (1996) Thermal unfolding of human high-density apolipoprotein A-1: implications for a lipid-free molten globular state. Proc. Natl. Acad. Sci. U.S.A. 93, 2991–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reijngoud D. J., and Phillips M. C. (1982) Mechanism of dissociation of human apolipoprotein A-I from complexes with dimyristoylphosphatidylcholine as studied by guanidine-hydrochloride denaturation. Biochemistry 21, 2969–2976 [DOI] [PubMed] [Google Scholar]