

Abstract
CaMKII is a serine–threonine protein kinase that is abundant in myocardium. Emergent evidence suggests that CaMKII may play an important role in promoting atrial fibrillation (AF) by targeting a diverse array of proteins involved in membrane excitability, cell survival, calcium homeostasis, matrix remodelling, inflammation, and metabolism. Furthermore, CaMKII inhibition appears to protect against AF in animal models and correct proarrhythmic, defective intracellular Ca2+ homeostasis in fibrillating human atrial cells. This review considers current concepts and evidence from animal and human studies on the role of CaMKII in AF.
Keywords: CaMKII, Atrial fibrillation, Atrial remodelling, Arrhythmias
1. Introduction
Atrial fibrillation (AF) is the most common sustained clinical arrhythmia, affecting approximately 33.5 million people annually worldwide.1 Age is a major risk factor for AF, and the disease incidence and prevalence have increased with the ageing of the general population.2,3 AF is associated with increased morbidity, primarily from stroke and heart failure, and increased mortality.4,5
AF is a progressive disease, and the current clinical classification of AF is based on its pattern and duration.6 AF is classified as paroxysmal AF (episodes that last <7 days with or without intervention), persistent AF (episodes lasting ≥7 days), long-standing persistent AF (continuous AF >12 months in duration), or permanent AF (when no further attempts are made to achieve normal sinus rhythm). Affected individuals do not necessarily progress through these various forms of AF in a linear or predictable fashion. The pathophysiology of AF consists of mechanisms involved in the initiation of the arrhythmia, maintenance of the arrhythmia, and progression to long-lasting forms of AF.7 The progression of AF is associated with the concept of ‘AF begets AF’, in which changes in atrial structure and function promote and perpetuate atrial arrhythmias,8 and these changes are referred to as atrial arrhythmogenic remodelling.9
An understanding of the molecular basis for these AF-related changes and how they pertain to the initiation and progression of the disease is essential to the development of effective therapeutic strategies. A conceptual framework for a mechanistic understanding of these mechanisms can be divided into three components.10 The first component is the basic arrhythmia mechanism responsible for the initiation and maintenance of AF. This refers to triggered (ectopic) and reentrant activities. AF initiation fundamentally requires a trigger in the form of either triggered (ectopic) activity or reentrant activity usually in the presence of a vulnerable substrate that may be anatomic or functional. An exception will be in lone AF in which AF occurs in the absence of structural heart disease. Both focal ectopic firing and reentrant mechanisms can maintain AF; however, the latter is the likely mechanism in the majority of cases. The second mechanistic component is atrial arrhythmogenic remodelling that promotes AF and comprises electrical, structural, and autonomic nervous system and Ca2+-handling changes observed in AF.9 The last component consists of the aetiological factors that predispose to AF. These include concurrent cardiovascular diseases such as heart failure, sinus node dysfunction (SND), valvular heart disease, hypertension, and ageing; genetic factors such as single gene mutations, gene polymorphisms, and gene variants; and other extrinsic factors such as sleep apnoea, obesity, and thyroid disease. Atrial remodelling can occur either as a consequence of these aetiological factors or as a direct consequence of AF itself.
The Ca2+- and calmodulin-dependent protein kinase II (CaMKII) is a multifunctional serine–threonine kinase that is now recognized as a critical Ca2+ and reactive oxygen species (ROS) sensor that mechanistically links several of the observed cellular perturbations and mechanisms in AF with proarrhythmic atrial remodelling.
The goal of this review is to highlight new and emerging evidence highlighting the role of CaMKII in AF and how CaMKII participates in the complex interplay of the various mechanistic components of AF. We start with an overview of CaMKII and relevant downstream targets and then proceed to discuss known and potential effects of CaMKII in the spectrum of AF disease. We conclude with a discussion of potential therapeutic implications. Most of our knowledge regarding CaMKII comes from studies performed in ventricular cardiomyocytes. Although discussing a wide range of studies, we have tried to emphasize a growing body of work in humans and animals using atrial myocardium.
2. CaMKII—biology, activation, and downstream targets
2.1. Biology and structure
CaMKII is a multifunctional serine–threonine protein kinase that is abundantly expressed in various tissues, including the heart. There are four CaMKII gene products resulting in four homologous isoforms of CaMKII—α, β, δ, and γ, and these share activation mechanisms.11–14 These isoforms are thought to co-assemble as heteromultimeric holoenzymes and are known to have varied expression levels in different tissues12 (Figure 1). For instance, CaMKIIα and CaMKIIβ are preferentially enriched in neuronal tissues, whereas CaMKIIδ and CaMKIIγ are the predominant isoforms in the heart. There is a hypervariable region in CaMKII located between the association domain and the C-terminus of the regulatory domain that gives rise to various splice variants. There is a CaMKIIδ splice variant with a nuclear localization signal (NLS; CaMKIIδB) and another lacking an NLS sequence (CaMKIIδC).15 The presence or absence of the NLS does not absolutely determine localization, perhaps because these isoforms co-assemble as heteromultimers and the ratio of the NLS-containing isoforms biases cellular targeting.15 Nevertheless, the specific role of these splice variants remains unclear, and both CaMKIIδB and CaMKIIδC are present in nuclear and cytosolic compartments.16
Figure 1.

Structure, activation, and post-translational modifications of CaMKII. CaMKII monomers consist of a catalytic domain, a regulatory domain, and an association domain. The regulatory domain has an autoinhibitory binding region with several sites for post-translational modification and a CaM binding region. Co-assembly of monomers through the association domain forms the CaMKII holoenzyme. In the presence of Ca2+, Ca2+/CaM binds to the regulatory site, which causes a conformational change and activation of the autoinhibited inactive enzyme with exposure of the catalytic domain. Post-translational modification of the autoinhibitory region at any of the sites highlighted results in constitutive activity of CaMKII that is autonomous of Ca2+/CaM. CaM, calmodulin.
Structurally, CaMKII exists as a holoenzyme that consists of two stacked ring-shaped hexamers.14 Each monomer consists of an N-terminal catalytic domain, a central regulatory domain, and a C-terminal association domain.17 The catalytic domain harbours the adenosine triphosphate (ATP) and substrate binding sites and is responsible for the catalytic activity of the protein. The regulatory domain contains an inhibitory pseudosubstrate sequence, several amino acid residues susceptible to post-translational modification, and the calmodulin (CaM) binding region. The association domain is responsible for binding with other subunits to form the holoenzyme (Figure 1).14
2.2. Activation
In the inactive state of CaMKII, the pseudosubstrate region of the regulatory domain inhibits the catalytic domain of each CaMKII subunit by sterically blocking the substrate and ATP-binding pocket.18,19 With increases in intracellular [Ca2+], Ca2+ binds to CaM and the calcified CaM (Ca2+/CaM) activates CaMKII by binding to the C-terminal region of the regulatory domain. This causes a conformational change with allosteric displacement of the pseudosubstrate region, allowing the substrate and ATP access to the catalytic domain.20 Sustained increases in Ca2+/CaM in the presence of ATP result in autophosphorylation at Thr 287 in the autoinhibitory region of the regulatory domain. Autophosphorylation promotes a conformational change that prevents the association of the catalytic and regulatory domains, resulting in sustained Ca2+-independent activation.21–23
Our group discovered that oxidation of a pair of methionine residues at positions 281/282 results in a similar form of Ca2+/CaM-independent persistent activation compared with autophosphorylation.24 Other modes of post-translational modification that result in autonomous CaMKII activity include O-linked glycosylation of serine at position 280 by O-linked N-acetylgluocasmine25 and NO-dependent nitrosylation of cysteine residues at putative positions 116, 273, or 290.26 Interestingly, phosphorylation of Thr 306/307 causes CaMKII inactivation by decreasing the binding to Ca/CaM complexes.27 Very recently, it was discovered that NO modification of Ser 273, a position outside of the autoinhibitory domain, inactivates CaMKII.28 Thus, CaMKII may be activated or inactivated by post-translational modifications, but post-translational modifications to the CaMKII autoinhibitory domain provide a path for CaMKII to become persistently active under the influence of upstream signals that increase intracellular [Ca2+] and ROS.24,29
The capacity of CaMKII to transduce the effects of a multitude of upstream messengers raises the question of the existence of a central mechanism that regulates the activation of CaMKII similar to the protein kinase A (PKA) signalling pathway. Such a central mechanism does not appear to exist; rather what is clear is that initiation of CaMKII activation requires Ca2+/CaM binding, and sustained elevation in intracellular [Ca2+] can promote Ca2+/CaM autonomous activity by ‘autophosphorylation’ at Thr 287. However, Ca2+/CaM-independent activity can also follow increased ROS, even in the presence of physiological (diastolic) intracellular [Ca2+].30 Additionally, similar to PKA, β-adrenergic stimulation can activate CaMKII through the cAMP-Epac pathway (discussed subsequently). Changes in global cystolic [Ca2+], such as those occurring in excitation–contraction coupling (ECC) and local [Ca2+], for example, in the dyadic space can activate CaMKII. CaMKII achieves signal specificity through a variety of mechanisms, as discussed subsequently.
2.3. Localization and intracellular targeting
Cellular signalling systems generally have mechanisms that facilitate the interaction of effector molecules with their targets often through close proximity to achieve signal specificity and efficiency. CaMKII achieves subcellular targeting in a heterogeneous fashion such as compartmentalization, employing anchoring proteins and also identifying target protein motifs with homology to the CaMKII regulatory domain autoinhibitory region.11,31 CaMKII is enriched in specific subcellular domains such as those in the transverse tubules adjacent to l-type Ca2+ channels (LTCC) and ryanodine receptors.32 Earlier we briefly mentioned the existence of CaMKIIδ splice variants—CaMKIIδB and δC. It was initially thought that CaMKIIδB preferentially localized to the nucleus and CaMKIIδC to the cytosol; however, it has subsequently been shown that these isoforms are not exclusively limited to these cellular compartments.16 In fact, these and other isoforms of CaMKII co-assemble as heteromultimers, and the relative composition of the heteromultimer biases cellular targeting. The study of Mishra et al.16 showed that there is compartmentalized activation of CaMKIIδ independent of the isoform with the location of activation determined by the stimulus and the Ca2+ release site. Phenylephrine preferentially activated both CaMKIIδ isoforms in the nuclear compartment, whereas caffeine preferentially activated these isoforms in the sarcoplasmic reticulum (SR). Compartmentalization also had functional significance, with phenylephrine selectively phosphorylating nuclear transcriptional regulator HDAC5 at a specific CaMKII site (Ser 498) and caffeine selectively phosphorylating phospholamban (PLN) at the CaMKII-specific phosphorylation site (Thr 17).
Unlike other signalling molecules such as PKA that have extensively validated anchoring proteins that act as adaptors, there is less known about how CaMKII achieves target specificity. However, a few anchoring proteins have been described that regulate coupling of CaMKII to some target proteins, thus enhancing signalling specificity. The first of these was the N-methyl-d-aspartate subunit NR2B in the nervous system, which caused activation-dependent subcellular targeting through translocation.33 Subsequently, other anchoring proteins have been described in the heart. Alpha-kinase anchoring protein is an example of a protein that anchors sarcoplasmic endoplasmic reticulum ATPase (SERCA2a) and CaMKII, and through this interaction modulates CaMKII-dependent PLN phosphorylation.34 Another is β-arrestin that interacts with CaMKIIδ35 and functions as an anchoring protein that links β1-adrenergic receptors to the cAMP-Epac-CaMKII signalling pathway.36 It forms a multimeric complex with CaMKII and Epac in the cytoplasm, and this complex then translocates to the plasma membrane after β-adrenergic stimulation to bring CaMKII and Epac in close proximity with cAMP. A membrane-associated guanylate kinase protein, SAP97, has also been shown in rat atrial myocytes to anchor CaMKII to the C-terminus of Kv4.3 orchestrating CaMKII-mediated increases in the transient outward current (Ito).37
CaMKII uses recognition and targeting of CaMKII-binding motifs in its interaction with LTCC by binding to a CaMKII binding site on the β2a subunit of LTCC.38 Similarly, CaMKII uses this mechanism to bind to βIV-spectrin in targeting voltage-gated Na+ channels.39 Taken together, CaMKII employs different mechanisms as described to achieve target specificity and efficiency in its role as a master regulator of several cellular processes.
2.4. Downstream targets
CaMKII acts on diverse downstream targets in cardiomyocytes by lowering the free energy required to phosphorylate a target serine or threonine residue. Phosphorylation can lead to conformational changes that affect protein function. A full discussion of CaMKII target proteins is beyond the scope of this review, but CaMKII targets related to myocardial ECC may be particularly important for understanding the role of CaMKII in AF. CaMKII appears to affect myocardial cell membrane excitability through its action on most voltage-gated ion channels such as LTCCs,40–42 K+ channels,37,43,44 and Na+ channels.45–47 Other CaMKII targets include Ca2+ cycling proteins such as the type 2 ryanodine receptor (RyR2)48 and phospholamban (PLN).49 The net effect of CaMKII phosphorylation on these targets, in the context of pathological stress, is to promote a proarrhythmic circumstance by favouring cell membrane hyperexcitability and afterdepolarizations. Collectively, early afterdepolarizations (EADs) and delayed afterdepolarizations (DADs) are thought to be triggers for initiating arrhythmias, including AF (see the section on Mechanisms and experimental models of AF).
2.4.1. l-type Ca2+ channels
The LTCC is the major portal for extracellular Ca2+ to enter the cytoplasm.50 CaMKII-catalysed phosphorylation of LTCCs produces high-activity mode-2 gating, resulting in increased open probability of ICa.L channels.38 This increases the influx of Ca2+ into atrial myocytes. CaMKII also results in ‘Ca2+-dependent ICa.L-facilitation’40 in which there is increased peak ICa.L and slowed ICa.L inactivation,51 a signature LTCC response to repeated depolarizing pulses.11 Mode-2 gating, and ICa facilitation are thought to be proarrhythmic by predisposing to action potential (AP) prolongation, EADs, SR Ca2+-leak, inward Na+/Ca2+ exchanger (NCX) current, and DADs.32,38,52,53
2.4.2. Voltage-gated Na+ channels
The α isoform of the cardiac sodium (Na) channel (Nav1.5) is phosphorylated by CaMKII.45 This leads to Na+ gating modification with increased steady-state inactivation of the Na2+ channel with high heart rates. CaMKII-dependent phosphorylation of Nav1.5 also slows Na2+ current (INa) inactivation and augments the non-inactivating ‘late’ component of INa.39,54 Sustained late INa increases subsarcolemmal [Na+] and indirectly increases intracellular [Ca2+] by prolonging AP duration (APD) and by reducing the efficacy of the NCX to extrude Ca2+ from the cytoplasm to the extracellular space.55 These effects contribute to EADs and DADs. CaMKII inhibition by autocamide-2-related inhibitory peptide in murine and human atrial myocytes or genetic knockout of CaMKIIδc (CaMKIIδc-knockout mice) prevents late INa-dependent SR Ca2+-leak.56
2.4.3. Voltage-gated K+ channels
Voltage-gated K+ (IK) currents are primarily responsible for membrane repolarization. IK is a composite of the effect of several ion channels. Here, we discuss the IK channels that are best understood to be directly affected by CaMKII. The transient outward K+ current (Ito) is responsible for early repolarization and termination of the plateau phase of the AP, and CaMKII activates Ito in atrial myocytes.43,57 This results in slowing of Ito inactivation and acceleration of its recovery, with the overall effect of shortening the AP and decreasing the refractory period. The KV4.3 pore-forming subunit of Ito is regulated by CaMKII-dependent phosphorylation through the accessory protein SAP97, and this also results in increased Ito.37 KV1.4 is postulated to have a CaMKII phosphorylation target motif.58 The inward rectifier current (IK1) is another component of IK that is activated by CaMKII.46 This is the primary cardiac inward rectifier current, and the protein Kir2.1 subunit is the essential pore-forming unit for this channel. IK1 is important for maintaining the resting cell membrane potential and sculpting terminal phase 3 repolarization. IK1 is increased in chronic AF,59,60 and this helps sustain high-frequency reentrant sources called rotors that are characteristic of reentrant arrhythmias.61 IK1 and the ultrarapid delayed rectifier K+ current (IKUR) both appear to be augmented by CaMKII,43,57 which offsets the APD-prolonging effects of CaMKII-dependent ICa.L and INa phosphorylation. Contrary to earlier studies, a study of atrial myocytes from human subjects in SR and AF showed that CaMKII inhibition with KN-93 had no effect on IK1 current in sinus rhythm or chronic AF patients, whereas protein kinase C (PKC) decreased IK1 current in chronic AF.62 Further, PKC-dependent phosphorylation also increased constitutive acetylcholine-sensitive K+ current (IK.Ach) activity in chronic AF.62 Thus, complex actions at multiple voltage-gated ion channels can contribute to arrhythmias by promoting EADs and DADs (i.e. arrhythmia triggers) and by augmenting dispersion of repolarization that may be a substrate for reentry.
2.4.4. Type 2 ryanodine receptor
The RyR2 is an intracellular Ca2+ channel that controls release of myofilament activating Ca2+ from the SR lumen to the cytoplasm. This Ca2+ initiates the mechanical phase of ECC, but can also influence Ca2+-sensitive cell membrane conductances (i.e. by actions on ion channels, exchangers, and transporters), transcription, and Ca2+-regulated mitochondrial processes. A mostly LTCC-mediated trigger of intracellular Ca2+ is the fundamental signal for activating RyR2, but its opening probability is biased by phosphorylation. CaMKII-dependent hyperphosphorylation of Ser 2814 on RyR2 increases open probability, augmenting dyssynchronous SR Ca2+ release48 and proarrhythmic DADs,63 which are mostly due to inward NCX current (INCX). There is increased NCX expression in persistent AF, which magnifies the size of DAD-generating inward INCX for any given amount of aberrant Ca2+ release. RyR2 dysfunction that destabilizes the closed state increases AF susceptibility in genetic mouse models.64–66 Inhibition of CaMKII-dependent RyR2 phosphorylation reduces the incidence of rapid pacing-induced AF in mice.66,67
2.4.5. Phospholamban
Phospholaman (PLN) is a negative regulator of the SERCA2a.68,69 Phosphorylation of PLN by PKA (at Ser 16) or CaMKII (Thr 17) promotes PLN aggregation and disengagement from SERCA2a and SERCA2a disinhibition, resulting in increased SR Ca2+-reuptake from the cytoplasm.70,71
Activation of these target proteins, by CaMKII-catalysed phosphorylation, can explain the acute proarrhythmic role of CaMKII in AF and other arrhythmias, by enhancing the probability of afterdepolarizations and reentry.72,73 Chronic CaMKII activation favours cardiomyocyte death and fibrosis, myocardial hypertrophy, and extracellular matrix remodelling—processes that contribute to proarrhythmic remodelling of the tissue substrate. In the case of AF, tissue substrate remodelling is an important factor in the transition of AF from sporadic or paroxysmal forms to chronic and permanent forms.72,74
Regulation of phosphorylation, and thus activity of the various Ca2+-handling proteins discussed earlier, does not depend solely on the actions of CaMKII. Other protein kinases such as PKA and PKC also play significant roles in the regulation of these proteins. For the purposes of this review, we briefly highlight the PKA signalling pathway and how this interacts with CaMKII signalling in the context of AF. Both PKA and CaMKII act at different sites on these proteins with similar consequences in most, but not all, cases. Some of the known PKA phosphorylation sites for these proteins include Ser 1700 for LTCC, Ser 2030 and Ser 2808 for RyR2, and Ser 16 for PLN. Classic PKA signalling involves cyclic adenosine monophosphate (cAMP) activation of PKA in response to β-adrenergic signalling. PKA activation under physiological conditions occurs through β-adrenergic signalling and causes intracellular influx of Ca2+ and increased SR Ca2+ uptake and storage that culminates in increased systolic Ca2+ transients.75 CaMKII activation occurs either directly through increased intracellular Ca2+ (as discussed earlier) or alternatively through a cAMP-dependent mechanism. The latter occurs via activation of adenylyl cyclases with subsequent increase in cytosolic cAMP that then binds and activates a guanine nucleotide exchange protein activated by cAMP (Epac). Epac modulates cardiac Ca2+-handling in various ways such as increasing the frequency of Ca2+ oscillations and inducing SR Ca2+ release independent of PKA through effectors such as phospholipase C and CaMKII.76–78 PKC also may affect CaMKII through this mechanism.78 Epac increases CaMKII-mediated diastolic Ca2+-leak through RyR2 activation.77 This is considered the cAMP- and Epac-induced SR Ca2+-leak pathway. RyR2 PKA hyperphosphorylation is present in atrial tissue from a canine model of atrial pacing-induced AF and in atrial tissue from humans with AF when compared with sinus rhythm,79 and this results in increased open probability of the channel.80
β-adrenergic stimulation also induces PKA-dependent and PKA-independent CaMKII-mediated diastolic SR Ca2+-leak.80,81 Late INa also increases diastolic SR Ca2+-leak in atrial myocytes/myocardium from mice and humans with permanent AF by CaMKII- and PKA-dependent phosphorylation of PLN (Thr 17 and Ser 16, respectively) and RyR2 (Ser 2814 and Ser 2808, respectively).56 Both kinases appeared to be necessary for late INa-dependent SR Ca2+-leak induction.
It is clear that several signalling pathways including cAMP/PKA, cAMP/Epac, and direct CaMKII activation are involved in AF; however, the relative contribution of PKA and CaMKII and the relevant signalling pathways to the initiation, maintenance, or progression of AF are unclear. Previous studies suggest differential contribution of both kinases. For instance, PKA activity significantly activated ICa and SR Ca2+ uptake with modest effects on RyR2.82,83 CaMKII, in contrast, had more robust RyR2 effects with modest ICa activation and SR Ca2+ uptake.84 Historically, there has been some controversy on the effects of PKA and CaMKII on RyR2, with CaMKII considered to be downstream of PKA. However, more recent studies demonstrate that these kinases act independently but synergistically on β-adrenergic stimulation.56,81,85 There is ample evidence of crosstalk between PKA and CaMKII-dependent signalling at different levels. With regard to Ca2+-signalling, CaMKII affects cAMP levels in a negative-feedback loop fashion through its action on phosphodiesterase 4D, thus indirectly affecting PKA activity.86 PKA also facilitates CaMKII-dependent SR Ca2+-leak induction by maintaining SR Ca2+-load.56
3. CaMKII in AF
CaMKII appears to be a myocardial stress response modulator because its activity and expression are increased in diverse forms of structural heart disease. Furthermore, CaMKII activity seems to augment classical fight or flight heart rate responses.87 Tables 1 and 2 show a summary of various studies evaluating CaMKII protein expression and activity in animal models (Table 1) and human tissue (Table 2) with atrial tachyarrhythmias and AF.
Table 1.
CaMKII in AF: summary of animal studies
| Author | Year | Species | Arrhythmia model | CaMKII | Effect |
|---|---|---|---|---|---|
| Greiser et al.88 | 2009 | Goat | Rapid atrial pacing | CaMKII-dependent hyperphosphorylation of RyR2 | Atrial hypocontractility Decreased SR Ca2+ load |
| Chelu et al.89 | 2009 | Mouse | Genetic mouse model (CREM-IbΔC-X transgenic mice) | Increased expression and autophosphorylation of CaMKIIδ | Hyperphosphorylation of RyR2, arrhythmia suppression by CaMKII inhibition. |
| Chelu et al.89 | 2009 | Goat | Rapid atrial pacing | Increased expression and autophosphorylation of CaMKIIδ | Hyperphosphorylation of RyR2 |
| Wakili et al.90 | 2010 | Canine | Rapid atrial pacing | Increased expression and autophosphorylation of CaMKIIδ | Atrial hypocontractility Decreased APD Decreased Ca2+ transient |
| Purohit et al.66 | 2013 | Mouse | Angiotensin-II | Increased ox-CaMKII | Increased AF susceptibility dependent on ox-CaMKIIδ and RyR2 S2814 phosphorylation, arrhythmia suppression by CaMKII inhibition |
| Greiser et al.91 | 2014 | Rabbit | Atrial myocytes | No change in CaMKII-dependent RyR2 phosphorylation or CaMKII activity | Ca2+ signalling silencing with increased intracellular Ca2+ and no changes in Ca2+ sparks, Ca2+ waves, Ca2+ release, and Ca2+ release instability |
| Fischer et al.56 | 2015 | Mouse | Atrial cardiomyocytes treated with ATX-II | Increased p-CaMKII | Enhanced late INa Increased SR Ca2+-leak Hyperphosphorylation of CaMKII target sites: RyR2 (Ser2815) and PLN (Thr17) |
| Lenski et al.92 | 2015 | NVRM | Electrical stimulation | Increased p-CaMKII/CaMKII | Increased p-AMPK/AMPK Increased FA uptake Decreased glucose uptake |
AP, action potential; AMPK, AMP-activated protein kinase; ATX-II, Anemonia-sulcata toxin-II; FA, fatty acid; NVRM, neonatal ventricular rat myocytes; ox-CaMKII, oxidized CaMKII; p-CaMKII, phosphorylated CaMKII; p-AMPK, phosphorylated AMPK; PLN, phospholamban; SR, sarcoplasmic reticulum; RyR2, ryanodine type 2 receptor.
Table 2.
CaMKII in AF: summary of human studies
| Author | Year | Species | Type of AF | Tissue | CaMKII | Effect |
|---|---|---|---|---|---|---|
| Tessier et al.43 | 1999 | Human | Chronic AF | RA | Increased CaMKIIδ expression | CaMKII slowed Ito inactivation; CaMKII inhibition hastened Ito inactivation |
| Christ et al.93 | 2004 | Human | Chronic AF | RAA | Increased protein expression of the catalytic subunit of protein phosphatase 2A and phosphatase activity | Decreased basal ICaL density in chronic AF |
| Chelu et al.89 | 2009 | Human | Chronic AF | RAA | Increased CaMKIIδ expression and autophosphorylation | Hyperphosphorylation of RyR2 |
| Neef et al.94 | 2010 | Human | Unclassified | RA | Increased CaMKII expression | Increased SR Ca2+-leak and diastolic Ca Increased RyR2 phosphorylation, reversal of SR Ca2+-leak with CaMKII inhibition |
| Voigt et al.95 | 2012 | Human | Chronic AF | RAA | Increased autophosphorylation of CaMKII | Hyperphosphorylation of RyR2 Increased SR Ca2+-leak, diastolic Ca2+, frequency of spontaneous Ca2+ release events Reversal of SR Ca2+-leak and afterdepolarizations with CaMKII inhibition |
| Purohit et al.66 | 2013 | Human | Mixed (paroxysmal, persistent/permanent, and unclassified) | RA | Increased ox-CaMKII | AF patients had more ox-CaMKII compared with patients in sinus rhythm |
| Greiser et al.91 | 2014 | Human | Persistent AF | RA | No change in CaMKII-dependent RyR2 phosphorylation or CaMKII activity | Ca2+ signalling silencing with increased intracellular Ca2+ and no changes in Ca2+ sparks, Ca2+ waves, Ca2+ release, and Ca2+ release instability |
| Christ et al.96 | 2014 | Human | Chronic AF | RAA | Reduced CaMKII-dependent phosphorylation of PLN but not RyR2 | Blunting of agonist-evoked arrhythmias in atrial myocytes from patients with AF compared with those in sinus rhythm |
| Lenski et al.92 | 2015 | Human | Permanent AF | LAA | Increased p-CaMKII/CaMKII | Increased FA uptake Decreased glucose uptake |
Ito, transient outward K+ current; RA, right atrium; RAA, right atrial appendage; LAA, left atrial appendage; FA, fatty acid; ox-CaMKII, oxidized CaMKII; p-CaMKII, phosphorylated CaMKII; SR, sarcoplasmic reticulum; PLN, phospholamban; RyR2, ryanodine type 2 receptor.
4. Mechanisms and experimental models of AF
AF and atrial tachycardia-related remodelling are the consequences of a complex interplay of pathophysiological mechanisms. An understanding of the fundamental processes that constitute the molecular basis of AF is necessary to highlight the particular steps now thought to be affected by CaMKII. The primary mechanisms of AF are focal ectopic or triggered activity and reentrant activity.97 Triggered activity can occur after full repolarization of the AP and these are called DADs. Alternatively, EADs are triggered activities that occur during phase 2 or 3 of the AP. Ca2+ overload with associated shortening of the APD favours generation of DADs and these are rate-dependent, whereas EADs are favoured by APD prolongation, persistent inward ICa.L,32 INa,39 and spontaneous SR Ca2+ release.98–100 EADs and DADs can also be driven by adrenergic stimulation, at least in part because of their action to activate CaMKII and its downstream targets. Reentrant activity occurs in the presence of suitable substrate, which can be either anatomic (e.g. fibrosis) or functional (e.g. electrical refractoriness) that allows for the perpetuation of an excitable circuit. The substrate can be the consequence of AF-related atrial remodelling or the result of other conditions such as genetic mutations, ageing, structural heart disease, or other systemic disease.7 Reentrant activity is the primary mode for AF maintenance. Persistent AF is likely maintained by the combination of complex multiple circuit reentry and ectopic activity.101
Importantly, as discussed in the previous section, excessive CaMKII activation has been implicated in AF triggering and reentry mechanisms through its downstream effects on ion channels and proarrhythmic remodelling that results from cell death, fibrosis, and hypertrophy.
Various experimental paradigms have been developed to study the mechanisms of AF.97 Clinical AF is a heterogeneous mix of phenotypes. AF often exists in association with structural heart disease such as congestive heart failure, valvular disease, or hypertension; however, there are times that AF occurs in the absence of any detectable structural heart disease (‘lone AF’). Also there are different clinical forms of AF: paroxysmal, persistent, and permanent AF. The dominant paradigm for the natural progression of the disease is from paroxysmal to permanent AF. The rapid atrial pacing (RAP) model8,102 of experimental AF aims to mimic ‘lone AF’. Of note, earlier studies used a single atrial burst pacemaker,103 and these were associated with high ventricular rates that likely caused mild tachycardia-mediated cardiomyopathy that potentially complicated the observations in these studies. More recent studies combine RAP with complete atrioventricular block104,105 to avoid or minimize this effect. Both models are relevant to human disease as the former simulates AF with no rate control and the latter AF with adequate rate control.97 The RAP model is also of clinical significance as a model for studying the transition from paroxysmal to chronic AF. Another AF experimental model to study the relationship between heart failure and AF exists, in which rapid ventricular pacing induces heart failure.104 There are differences in the electrical and structural remodelling observed in RAP model when compared with the heart failure model. Other forms of experimental AF in animals include AF combined with structural heart disease,106 atrial dilatation without heart failure,107 and sterile pericarditis to mimic post-operative AF.108 Increasingly, transgenic mice are being used as AF experimental models.66,89,109,110 The main advantage of mouse models is their genetic tractability and ability to generate mutant genes to mimic human diseases and to dissect candidate AF molecular mechanisms. For the purposes of this review, we will focus primarily on the RAP model in ‘large’ animals, as a model to study AF-related atrial remodelling and AF progression, and on various genetically modified mouse models with the power to dissect defined molecular components of AF pathways.
The transition from paroxysmal to the more chronic forms of AF is poorly understood, and the role that CaMKII plays in this process is an area of on-going interest in the field. The most established role for CaMKII in AF in in vivo (animal) systems relates to mechanisms of arrhythmia triggering, by contributing to RyR2 hyperphosphorylation and Ca2+-leak, which triggers afterdepolarizations.56,67,89,111 Importantly, this mechanism pertains to fibrillating human atrium.94,95 Although triggering may be important for paroxysmal or more permanent forms of AF,95 it appears to be required for paroxysmal AF in the absence of structural heart disease (i.e. lone AF). This is consistent with reports in which CaMKII contributes to triggering in pulmonary vein cardiomyocytes.112 Taken together, these data align with the idea that CaMKII contributes to early stage AF. However, CaMKII could also contribute to the transition from paroxysmal to more permanent forms of AF by several mechanisms: loss of normal intracellular [Ca2+] homeostasis, increased cardiomyocyte death and fibrosis, matrix remodelling by elaboration of matrix metalloproteinase-9 (MMP-9), and inflammatory gene transcription. Although intriguing and of potential clinical relevance, the role of CaMKII in late stage AF pathophysiology is less completely understood.
5. Components of atrial remodelling in AF
Atrial remodelling is characterized by several atrial structural and functional alterations that occur either as a result of AF or underlying structural heart disease that predates or coexists with AF. These changes can be classified as electrical remodelling, tissue (structural) remodelling, Ca2+-handling abnormalities, and neurohormonal.9 Although tissue, Ca2+, and electrical remodelling are complex and involve diverse signalling pathways, CaMKII has emerged as a nodal signal with the potential to orchestrate many of these changes. Furthermore, CaMKII activity is activated downstream to neurohumoral agonists, suggesting that CaMKII is a transduction signal linking neurohumoral signalling and ROS with proarrhythmic mechanisms in AF. Here, we will focus on the components that are known or postulated to be directly modulated by CaMKII and also discuss several CaMKII-dependent molecular changes that occur in AF.
6. Electrical remodelling
Electrical remodelling entails changes in the electrophysiological properties of the atria. This is a composite of the alterations that occur in ion channels, pumps, and exchangers in atrial cells.
APD shortening and enhanced variability of APD are central features of AF-related electrical remodelling and are accompanied by loss of rate adaptation of the atrial effective refractory period (AERP), which facilitates the maintenance of reentrant circuits in both animal AF models and human AF.8,113–115 Reduced AP duration is a result of reduced ICa,L.113,116,117 The reduced ICa,L measured in atrial myocytes isolated from chronic AF patients may indicate an adaptive response to arrhythmia-induced intracellular Ca2+ overload.117 The reduction in ICa,L is due to decreased mRNA and protein expression of the α subunit of ICa,L(CaV1.2) in AF through the negative feedback action of Ca2+-dependent phosphatase calcineurin/nuclear factor of activated T-cells (NFAT).118 Translocation of NFAT into the nucleus results in transcriptional downregulation of the CaV1.2 gene with consequent AP duration shortening.118 Although most of the available data show increased mRNA and protein levels of CaV1.2, others have found no alteration in protein levels in AF.93,119 An intriguing finding in one of these studies was that CaMKII inhibition with KN-93 resulted in about a 40% reduction in basal ICa,L in patients with sinus rhythm, but had no effect on basal ICa,L in patients with chronic AF.93 This was associated with increased phosphatase activity (especially protein phosphatase 2A) in chronic AF, suggesting that an imbalance in the kinase-phosphatase activity in favour of increased phosphatase activity is responsible for the reduced basal ICa,L in patients with chronic AF. The local CaMKII activity was not measured, but hypothetically there could be a local reduction in CaMKII activity due to several factors, such as cytoskeletal protein abnormalities due to AF that affect CaMKII activity, medications, or differences in experimental models.93,119 Also, increased activation of the Ca2+-dependent protease calpain promotes breakdown of ICa,L channels.120 Taken together, this suggests that the net effect on ICa,L may depend more on post-translational modification of the channel by phosphorylation rather than by expression.
A recent study by Greiser et al.91 challenges the current paradigm in the field that atrial remodelling results from dysfunctional Ca2+ handling that results in increased triggered activity and the role that CaMKII plays in tachycardia-mediated atrial remodelling. They observed a paradoxical suppression of central Ca2+ release with no changes in Ca2+ sparks, Ca2+ waves, SR Ca2+ release, and Ca2+ release instability in atrial myocytes from rabbits exposed to RAP for 5 days and in atrial myocytes from chronic AF patients. This was termed ‘Ca2+ signalling silencing’. It was attributed to increased Ca2+ buffering capacity of the atrial myocytes through binding to myofilaments and reduction in RyR2 expression with smaller RyR2 clusters in the SR. Additionally, there was hyperphosphorylation of RyR2 at the PKA phosphorylation site (Ser 2808), but reduced CaMKII-dependent RyR2 phosphorylation (Ser 2815) with no changes in CaMKII and PLN activity. It should be noted that the animal model used was a short-lived duration (5 days) of atrial pacing, with no evidence of AF in the rabbits. Further studies are needed to determine whether Ca2+ signalling silencing is a transient response to tachycardia or a more sustained response to chronic rapid activation with potential relevance to the pathophysiology of AF. Another group has reported a marked reduction in agonist-induced arrhythmias in the atria of patients with AF when compared with those in sinus rhythm with a corresponding reduction in maximal ICaL responses.96 The agonists used were norepinephrine, epinephrine, serotonin, and forskolin (a cAMP activator). CaMKII inhibition with KN-93 reduced norepinephrine-evoked ICaL responses in the atria from myocytes from patients with sinus rhythm but not in atrial myocytes from patients with AF. Also, adrenergic stimulation evoked reduced CaMKII-dependent phosphorylation of PLN (Thr 17), but no changes in PKA-dependent PLN phosphorylation (Ser 16) and no changes in RyR2 phosphorylation either by CaMKII or by PKA. Taken together, this was suggestive of loss of CaMKII-dependent phosphorylation of LTCC and PLN in response to adrenergic stimulation in chronic AF. It is not clear to us how to reconcile these results from Christ with the report by Voigt et al.,95 showing that CaMKII is critical for agonist-evoked RyR2 Ca2+-leak and arrhythmia triggering. These studies highlight the complexity of the mechanisms involved in AF and more specifically raise a question about the role of CaMKII in chronic AF. Further investigations to definitively understand these issues better are needed.
Decreased atrial conduction velocity may contribute to reentry and maintenance of AF. This occurs via a reduction in INa or direct inhibition of gap-junction channels in atrial cardiomyocytes.121 In murine experiments, decreased INa was shown to be due to an acute Ca2+-dependent inhibition of INa and reduced Nav1.5 subunit expression under chronic conditions.122 This Ca2+-dependent reduction in INa promotes the stability of AF primarily by a reentrant mechanism, but may also reduce ectopic activity.123 Oxidized CaMKII (ox-CaMKII) slows conduction in the infarct border zone of ventricular myocardium,124 but it is currently unknown whether CaMKII modulates atrial conduction velocity. However, it has been suggested that CaMKII-dependent phosphorylation could also reduce INa, especially at fast heart rates as seen in AF.45
7. Structural remodelling
The hallmark of structural atrial remodelling is atrial fibrosis.104,125 It is a common final pathway for AF promoting conditions and a negative predictor of treatment outcomes.9,126 Atrial fibrosis results from interaction between atrial cardiomyocytes and fibroblasts. Ca2+ influx into atrial fibroblasts induces proliferation and differentiation into collagen-secreting myofibroblasts, resulting in increased fibrosis and consequently heterogeneous conduction slowing and reentry.127 These changes are brought about by influx of Ca2+ into atrial fibroblasts via multiple ion channels. In human atrial fibroblasts, transient-receptor potential (TRP) channels are major participants in this process, especially TRP melastatin-related-7 (TRPM7) and canonical-3 (TRPC3) channels.128,129 CaMKII putatively can act both upstream and downstream of these TRP channels.130 AF patients have larger TRPM7 currents in their atrial fibroblasts, and knockdown of TRPM7 expression reduces basal differentiation of fibroblasts from chronic AF patients.128 It is unknown whether CaMKII interacts with TRPM7 in atrial myocytes, although CaMKII was shown to inhibit TRPM7-like channels in hepactocytes.131 Influx of Ca2+ through TRPC3 channel promotes CaMKII activation and NADPH-oxidase-mediated production of ROS in a genetic mouse model of dilated cardiomyopathy.132
CaMKII is involved in profibrotic signalling in the myocardium. Aldosterone stimulation leads to increased ox-CaMKII and activation of myocyte enhancer factor 2 that causes increased myocardial MMP-9 synthesis.29 Angiotensin II increases ox-CaMKII that causes sinoatrial cell death, SND, and atrial fibrosis, whereas CaMKII inhibition is protective against death and fibrosis in sinoatrial cells.133 Further investigation is needed to determine whether CaMKII contributes to atrial fibrosis in AF by these or other pathways, but CaMKII inhibition does reduce fibrosis in ventricular myocardium.29
Excessive CaMKII activity triggers cell death, and this results in reparative fibrosis and adverse remodelling in ventricular myocytes in a pressure overload model of heart failure,134 ventricular myocytes, and sinoatrial cells following Ang II infusion.24,133 CaMKII inhibition is also protective against beta-adrenergic-receptor agonist-induced apoptosis, and this anti-apoptotic mechanism involves PLN regulation of SR Ca2+ content135,136 and mitochondria.137 Mitochondrial-targeted CaMKII inhibition prevents myocyte death following myocardial infarction and catecholamine infusion137 (discussed subsequently). These various CaMKII-mediated mechanisms of cell death have not been directly tested in AF.
There is an established link between inflammation and AF,138,139 and several inflammatory markers such as cytokines, C-reactive protein, and complementary factors are elevated in AF. Inflammation leads to structural remodelling, primarily in the form of fibrosis. We previously showed that CaMKII induces local inflammatory response through the nuclear factor-κB pathway in cardiomyocytes when exposed to bacterial endotoxin or myocardial infarction and CaMKII inhibition suppressed this response.140 CaMKII is also oxidatively activated during myocardial infarction or by endotoxin as part of a toll-like receptor/myeloid differentiation protein 88 pathway.141 All these findings suggest that CaMKII may play an important role in proarrhythmic atrial remodelling favouring AF.
Atrial enlargement is another component of structural remodelling. Mice with transgenic overexpression of the transcriptional repressor CREM-IbΔC-X in cardiomyocytes (CREM mice) develop age-dependent progression from spontaneous atrial ectopy to paroxysmal and long-lasting AF episodes.142 Ca2+-handling abnormalities and atrial enlargement precede the spontaneous development of AF. Genetic inhibition of CaMKII-dependent RyR2 phosphorylation, by knockin replacement of a validated CaMKII-targeted phosphorylation site (RyR2-Ser 2814), in CREM mice prevents Ca2+-handling abnormalities and spontaneous AF, atrial dilatation, and conduction abnormalities.142 Thus CaMKII-dependent RyR2 hyperphosphorylation leads to both triggered activity and progressive development of an AF substrate, promoting atrial hypertrophy and dilatation, and AF progression. These mechanistic insights suggest that targeted treatment of CaMKII or SR Ca2+-leak via RyR2 has the potential to slow or arrest the progression of AF.
8. Role of CaMKII in ROS/redox sensing and signalling
ROS are involved in modulating the action of various cellular targets with functional consequences under physiological and pathological conditions in the cardiovascular system. AF is associated with increased oxidative stress, and this has been linked to AF-related atrial remodelling.143–145 There is increasing interests in the role of ROS and CaMKII in cardiac disease, with significant progress made in recent years in our understanding of links between ROS and CaMKII in the pathophysiological mechanisms of AF.146
ROS directly activates CaMKII by oxidation with subsequent persistent constitutive CaMKII activity, which is proarrhythmic in its effect on downstream targets.24,66,147 The acute proarrhythmic actions of ox-CaMKII are due to increased EADs and DADs54,148 and reentry.124
NADPH oxidases (Nox) are important sources of ROS in AF.149 Increased Nox4 expression in cardiomyocytes is associated with mitochondrial damage and apoptosis.150 In a zebrafish model, overexpression of Nox is associated with an arrhythmogenic phenotype, and this effect is mediated by ROS-induced activation of CaMKII.151 Mice that lack functional Nox (p47−/− mice) are resistant to angiotensin II-dependent increases in ROS and oxidation of CaMKII and are also resistant to angiotensin-II-mediated AF.66 Inhibition of Nox4 by apocynin attenuates electrical remodelling and AF inducibility in a rabbit model of RAP.152 As the duration of AF increases, sources of intracellular ROS shift from Nox to other cellular sources, including mitochondrial oxidases (xanthine oxidase), monoamine oxidase, and uncoupled eNOS.153
In a computational model,154 the arrhythmogenic pattern of EADs observed during oxidative stress was dependent on ox-CaMKII activation and pacing-cycle length. Oxidative stress is also associated with reduced AERP.143
Despite the link between oxidative stress and AF, attempts at utilizing antioxidants to prevent AF and AF-related atrial remodelling have been largely disappointing.155 One idea is that general antioxidants lack potency, pharmacokinetic attributes, or access to key subcellular domains necessary to exert beneficial actions. The shift from Nox as a source of cellular ROS to other cellular sources may explain why statins, which decrease ROS production by Nox via inhibition of Rac1, are only effective in acute AF models such as post-operative AF but ineffective in ameliorating ROS production in chronic AF models such as permanent AF.153 There was a recent comprehensive review of this topic.153 The finding that genetically modified mice with ROS-resistant CaMKII were protected from AF triggered by rapid pacing and primed by angiotensin II infusion66 suggests that CaMKII could be part of a concise ROS-responsive molecular pathway and a candidate therapeutic antioxidant target for AF.
9. CaMKII and mitochondria, energy expenditure, and its implications for AF mechanism
Mitochondria are essential for ATP production, the primary form of myocardial cellular energy, and they are the primary sources of ROS. They are an integral component of intracellular Ca2+ and ROS signalling. In cardiomyocytes, excessive mitochondrial Ca2+ and ROS cause opening of the mitochondrial permeability transition pore (mPTP), which leads to dissipation of the mitochondrial inner membrane potential and decreased ATP synthesis and this results in myocardial cell death via apoptotic mechanisms.156–158 CaMKII through its dual action of increasing ROS production and intracellular Ca2+ via SR Ca2+-leak results in mPTP opening.159 We previously showed that CaMKII is present in the mitochondria and that cardiomyocytes from transgenic mice with mitochondrial-targeted CaMKII inhibitory protein137 had a blunted mPTP response to Ca2+. These mice were protected from programmed cell death with exposure to different myocardial stressors such as ischaemia/reperfusion, myocardial infarction, and adrenergic stimulation.137 At this time, the constituents of mPTP are unknown and this pathway has not been directly tested in AF models. CaMKII also increases mitochondrial Ca2+ by phosphorylating the mitochondrial Ca2+ uniporter,137 although this is controversial.160 This Ca2+-sensitive channel is a major Ca2+ entry portal through the mitochondrial inner membrane into the mitochondria.161–163 Thus, mitochondrial CaMKII may contribute to a feed-forward mechanism by which Ca2+ overload triggers mPTP opening and increases ROS production, thereby causing more CaMKII activation and activity.
Increased oxidative stress and increased ox-CaMKII have been demonstrated in the atria of AF patients,66,145,164 and this was associated with mitochondrial DNA damage and reduction in ATP synthesis in these patients.164 Additionally, mitochondria undergo structural and morphological changes such as swelling and disturbance of cristae structure during RAP and AF.164–166 Functionally, impaired oxidative phosphorylation and respiration are observed in atrial tissue in response to RAP.166,167 These changes were Ca2+-dependent and were prevented by blocking LTCCs.
Ca2+ thus functions as a second messenger that links AF to structural and functional mitochondrial changes. Also, CaMKII plays an important role in the pathological mitochondrial changes that result in mitochondrial dysfunction and myocardial cell death in AF and likely contributes to atrial remodelling. The potential role of mitochondrial CaMKII represents an interesting and potentially important frontier in CaMKII research in AF.
10. CaMKII and atrial metabolic remodelling in AF
There is interplay between CaMKII and AMP-activated protein kinase (AMPK). AMPK is a serine/threonine kinase that regulates cellular energy metabolism and protein synthesis.168 CaMKII, Ca2+/calmodulin-dependent protein kinase kinase (CaMKKII), or Ca2+ alone can modulate the activity of AMPK in its role as a cellular energy sensor.169,170 The net effect of AMPK activation is enhancing energy-generating pathways and reducing energy demand processes.168,171,172 Lenski et al.92 recently demonstrated both in vitro and in vivo that arrhythmia induces metabolic changes in atrial myocardium characterized by lipid accumulation and reduced glucose uptake. They postulated that activation of AMPK causes these changes and CaMKII can activate AMPK. Inhibition of CaMKII and AMPK independently prevented these changes in vitro. This pathway involves elevated fatty acid translocase (FAT/CD36) membrane expression with resultant increased fatty acid uptake and lipid accumulation. Also, there is decreased membrane expression of synaptosomal-associated protein 23 in the plasma membrane, which results in reduced AMPK-induced glucose transporter subtype 4 membrane translocation and glucose uptake.92 These changes correlated with activation of proapoptotic pathways and atrial remodelling.
AMPK activation in AF was recently shown in canine and human atrial myocytes to be protective against AF-induced metabolic stress.173 High atrial rate and associated metabolic stress resulted in decreased ICa.L, intracellular Ca2+ transients, and cell shortening. The increased energy demand caused AMPK phosphorylation and subsequent regulation of Ca2+ handling through activation of and interaction with intracellular targets such as Cav1.2 and NCX, to counteract the effects of the metabolic stress. There was no evidence of interaction with other Ca2+-handling proteins such as RyR2, PLN, and SERCA2a. AMPK phosphorylation was increased in canine left atrial tissue after 1 week of tachypacing model of AF. Similarly, fractional AMPK phosphorylation was increased in human right atrial appendage tissue from subjects with paroxysmal AF compared with SR; however, there was a significant decrease in chronic AF subjects. This finding is interesting as it may be an indication that the AMPK system protects against paroxysmal or short bouts of AF, but does not necessarily play a role in persistent AF. It also buttresses the idea that there are likely different mechanisms underlying the different clinical forms of AF.
11. Atrial mechanical dysfunction
Atrial mechanical dysfunction, which manifests as atrial hypocontractility, is another feature of AF-related atrial remodelling and is a major cause of stroke and thromboembolism associated with AF.174,175 It coexists with on-going AF and can persist for variable periods after conversion to sinus rhythm in patients with long-standing AF.176 The latter is known as ‘atrial stunning’, and the degree of hypocontractility correlates with the duration of AF.176 In a canine atrial tachycardia model, there was reduced fractional cell shortening in atrial myocytes, and this was explained, in part, by abnormal intracellular Ca2+ handling and decreased systolic Ca2+ transients.175,177 In humans with prolonged AF, atrial contractility is reduced by as much as 75%.177 Others have shown that AP duration shortening could be responsible for the observed atrial hypocontractility.178 Additionally, in a canine model of tachypacing, there is altered myofilament function due to abnormal phosphorylation of myosin and myosin-binding protein C (MyBP-C).90 In this study, there was increased autophosphorylation and expression of CaMKIIδ and protein phosphatase I activity with downregulation of PKA. However, a direct link between CaMKII and altered myofilament function was not shown. Several studies show that MyBP-C is phosphorylated by CaMKII, which leads to hypocontractility and impaired relaxation in heart failure.179–181 Acute β-adrenergic stimulation leads to CaMKII phosphorylation of MyBP-C,179 but the effect of chronic β-adrenergic stimulation on this and the functional significance is unclear.182 Titin, which is a large myofilament protein, contributes to myocardial diastolic stiffness, and its mechanical properties are changed in heart failure.183–185 Specific CaMKII phosphorylation sites were recently identified on titin.186 CaMKII-dependent titin phosphorylation resulted in decreased myocardial passive force and altered diastolic stress in murine and human failing hearts,186 potentially linking diastolic dysfunction and AF via CaMKII. Thus, CaMKII acts through diverse mechanisms with the potential to reduce mechanical function. However, more definitive studies and more thorough identification of key CaMKII target sites will be necessary to test these concepts definitively.
12. AF and heart failure
AF commonly coexists with structural heart disease such as heart failure in patients.187,188 Atrial CaMKII activity is increased in a ventricular tachypacing-induced canine model of heart failure189 and in goats with atrial dilatation.88 In heart failure, there is chronic activation of neurohormonal agonist signalling, which begins as an adaptive response but subsequently becomes pathological and contributes to disease progression. This chronic pathological response results from increased intracellular Ca2+ and ROS.11 The agonists involved in this pathway are β-adrenergic receptors agonists, angiotensin II, and aldosterone, and all activate CaMKII.24,29,190 Indeed, CaMKII transcription, protein expression, and activity are increased in the failing heart.29,191–193 In addition to the neurohormonal system activation, other mechanisms by which CaMKII is increased in heart failure include increased calcineurin activity.194,195 The mechanism of increased CaMKII activity in heart failure appears to hinge on autophosphorylation of Thr 287 and/or oxidation of methionines 281 and 282.11 We found that ox-CaMKII was increased after myocardial infarction and was further enhanced by angiotensin II,24 aldosterone,29 or hyperglycaemia.196 Pathological β-adrenergic receptor stimulation in heart failure could activate CaMKII by cAMP-EPAC-dependent and cAMP-independent mechanisms to hyperphosphorylate RyR, causing SR Ca2+-leak and arrhythmias.77,78,85 As described earlier, the downstream effects of CaMKII activation likely result in AF-related atrial remodelling changes that promote AF initiation and progression. This may explain, in part, the association between heart failure and AF.
13. SND and AF
AF frequently coexists with SND.197 In the MOST trial, among patients with SND who required pacemakers, AF was the most frequent arrhythmia. About 45–50% of the subjects had a history of AF at the time of pacemaker implantation.198 SND is associated with changes in the sinoatrial pacemaker complex that are similar to AF-related atrial remodelling.199,200 AF has been shown to cause SND201 and conversely, SND predisposes to AF.199,202,203 The mechanisms for these observations are due to atrial remodelling—structural remodelling especially fibrosis,204 electrical remodelling, and neurohormonal stimulation, as described earlier in this review.
CaMKII through its effect on intracellular Ca2+ dynamics plays a key role in pacemaker activity.205 In this study, inhibition of CaMKII either by autocamide-2-inhibitory peptide (AIP), a specific peptide inhibitor, or by KN-93 resulted in decreased ICa.L amplitude and complete arrest of sinoatrial cells. Work from our laboratory indicated that CaMKII contributes approximately half of the dynamic range of fight or flight heart rate increases by actions in sinoatrial cells32 and that angiotensin II infusion in mice caused increased atrial and sinoatrial node ox-CaMKII. Ox-CaMKII triggered a critical threshold of sinoatrial cell apoptosis, atrial fibrosis, and conduction slowing that contributed to SND.133 Targeted CaMKII inhibition in the heart or sinoatrial node of mice protected against the deleterious effects of angiotensin II: sinoatrial cell apoptosis, fibrosis, and SND. We also found elevated levels of ox-CaMKII in the right atria from human patients with SND133 and AF when compared with those in sinus rhythm and also in the atria of Ang-II-infused mice.66 These mice had increased ROS, SR Ca2+ sparks, DADs, and heightened AF susceptibility compared with saline-infused mice. Mice with genetically engineered resistance to CaMKII oxidation and mice lacking functional NADPH oxidase (p47−/−) were resistant to angiotensin-II-mediated AF. These data show that ox-CaMKII is involved in the pathophysiology of both AF and SND and may be important in the clinical coexistence and synergy between both diseases.
14. Genetics and CaMKII in AF
It is recognized that there is a genetic contribution to AF, and certain familial and inheritable forms of AF have been described. Additionally, common genetic variants and single nucleotide polymorphisms have been identified in relation to AF. This was recently comprehensively reviewed.206 Although CaMKII has been implicated in some inherited arrhythmias such as catecholaminergic polymorphic ventricular tachycardia (CPVT), long QT type 3, ankyrin-B syndrome (long QT type 4),207,208 and Timothy syndrome (long QT type 8),209 we are unaware of a specific CaMKII genetic mutation or variation that has been identified in relation to AF. A loss-of-function mutation in ANK2 (a gene that encodes for ankyrin-B) was noted in some patients with early onset AF.210 In this study, mice with ankyrin-B deficiency manifested electrophysiological properties consistent with human AF, and DeGrande et al.207 showed that CaMKII inhibition rescued the proarrhythmic features of ankyrin-B deficiency in models of human ankyrin-B syndrome. Also, gain-of-function mutations in RyR2 have been shown to predispose to CPVT and AF.65,89,211,212
15. Potential as a therapeutic target in AF
Current treatment strategies for the management of AF include anti-arrhythmic drugs, ablation, and surgical procedures. Although useful, these oftentimes have suboptimal efficacy, and the anti-arrhythmic drugs have potential adverse effects and toxicities, including life-threatening proarrhythmia.213 The central role of Ca2+-dysregulation and CaMKII in the pathophysiology of AF makes CaMKII an attractive therapeutic target.
15.1. Pharmacological approaches to modulation of CaMKII activity
Several CaMKII-specific inhibitors exist, but none possesses drug-like properties necessary for therapeutic purposes. This topic was recently reviewed in detail.214 Here we discuss the use of relevant CaMKII inhibitors and highlight their use in relation to AF. KN-93 is a relatively specific CaMKII inhibitor215 that is widely employed for experimental purposes214 and is the best CaMKII inhibitor available. It competitively and selectively binds to the CaM-binding site of CaMKII, allosterically blocking activation. It does not compete with ATP and so does not have an effect on the activity of autophosphorylated CaMKII. Thus, once CaMKII is activated and autophosphorylated, KN-93 is ineffective in CaMKII inhibition, irrespective of the mode of activation. It selectively inhibits CaMKII relative to other kinases such as PKA and PKC, but has been shown to also effectively inhibit CaMKI and CaMKIV,216,217 and a few other kinase targets.218 A drawback to the use of KN-93 is its off-target effects. It affects LTCC52,219 and several voltage-gated potassium channels.220 Unfortunately, these off-target actions occur at concentrations necessary for CaMKII inhibition (e.g. IC50 in myocardial lysates of ∼2.5 mM)52 and are not shared by the control drug, KN-92. Thus, it is our view that it is best to use a variety of complementary approaches to CaMKII inhibition, such as gene deletion and inhibitory peptides, in order to be confident that experimental outcomes are not due to off-target actions. This is particularly true for studies of arrhythmias, in which ion channel antagonist actions of KN-93 have the potential to achieve anti-arrhythmic actions. However, these compounds have been useful and remain a key tool in our understanding of CaMKII signalling. KN-93 has been shown in both in vitro and in vivo mouse models to prevent pacing-induced AF.89 It also decreases SR Ca2+-leak in atrial myocytes from AF patients.94,95 KN-62 is another CaMKII inhibitor that is structurally similar to KN-93 and has the same mechanism of action, but has largely been replaced by KN-93.
CaMKII inhibitory peptides, which mimic the autoinhibitory region of the CaMKII regulatory domain, include AIP and autocamide-3 inhibitor (AC3-I). These are highly selective inhibitors of CaMKII relative to PKA, PKC, and CaMKIV221,222 and have been used experimentally to study CaMKII function in cardiovascular physiology and disease. In vivo murine studies have used internalization sequences or transgenic expression. Mice with transgenic myocardial expression of AC3-I were protected from pacing-induced AF, following angiotensin II infusion.66 There is the potential for off-target effects and altered selectivity with modification to these peptides either from fusion with green fluorescent protein, myristoylation or from changes to internalization sequences to enhance specific attributes of the peptide, such as increased expression, metabolic stability, or cell permeation.87,214,223 AC3-I has the potential to inhibit protein kinase D,223 but this potential has not been validated in other studies.224
CaMKIIN is a small protein endogenous to neurons but not to myocardium that inhibits CaMKII with high specificity and potency. We have used CaMKIIN as an experimental tool that can be delivered to specific target sites through local application of adenoviral constructs, targeted transgenic expression to specific tissues, and/or intracellular compartments.87,133,137 Sinoatrial node-targeted CaMKII inhibition by local application of adenoviral construct of CaMKIIN was protective against Ang-II-induced SND in mice.133 There are no studies of these peptides in AF models to our knowledge.
The CaMKII inhibitors described earlier have all been in the context of experimental models, but highlight the potential for CaMKII-targeted therapeutics. Although there are currently no CaMKII inhibitors available clinically, several potential small molecule inhibitors for cardiovascular application are in early stages of development.
15.2. Potential therapeutic application of CaMKII inhibition
CaMKII inhibition or elimination of CaMKII-dependent RyR2 phosphorylation has been shown to protect from AF in mouse models and is also beneficial in atrial cardiomyocytes of chronic AF patients.66,67,95 Furthermore, CaMKII is a truly nodal signal, so that CaMKII inhibition has the potential to counter AF by actions relevant to its progression from paroxysmal to permanent. However, given the ubiquitous nature of CaMKII and its multifunctional role in several physiological processes, it will be important to prove that systemic CaMKII inhibition does not contribute to undesirable side effects such as reduced fertility and impaired memory.225,226 Despite these potential issues, CaMKII is dispensable, as demonstrated by the viability of CaMKII knockout mice, and is likely present in heart to maximize heart rate and mechanical performance as part of the fight or flight response. Arguably, these attributes are not of central importance to most AF patients. A clinically useful CaMKII inhibitor likely should not cross the blood–brain barrier and should possess a sufficiently wide therapeutic window to avoid potential toxicities. Allosteric or isoform-specific CaMKII inhibitors could also enhance selectivity and reduce the potential for off-target effects.227
MicroRNAs have emerged as another potential therapeutic tool in the management of human disease.228 Some microRNAs have been identified that are involved in the regulation of CaMKIIδ expression such as microRNA-145229 and microRNA-30b-5p,230 whereas several others have been identified in the transcriptional changes that occur in AF.231 Targeting these microRNAs in the heart may emerge as a unique tool managing arrhythmias and other cardiac diseases. The pathophysiological mechanisms and importance of CaMKII may vary based on experimental AF models and in different forms of clinical AF, and the effect of CaMKII inhibition in these various conditions is unclear. Pacing-induced AF in the setting of angiotensin II infusion66 may mirror post-operative AF because of the shared circumstance of neurohumoral activation and elevated ROS. Based on the link between CaMKII activation and tissue remodelling, it is possible that a CaMKII inhibitor drug could reduce progression of AF from paroxysmal to chronic or permanent forms.
Given the landscape of potential therapeutic targets, our view is that CaMKII inhibition is a promising approach.
16. Conclusion
AF and atrial remodelling in the context of AF occur through a complex interplay of several molecular and cellular mechanisms, and these are implicated in the mechanisms for the initiation, maintenance, and progression of AF. Available evidence shows that there is excessive CaMKII activity in the setting of AF. CaMKII by its actions as a Ca2+ and ROS sensor modulates several of the pathways involved in AF and atrial remodelling to promote a proarrhythmic milieu in the atrium (Figures 2 and 3). Animal studies show that inhibition of CaMKII is protective against AF. This suggests that CaMKII inhibition is a potential therapeutic target for the management of AF. Further studies are however needed to understand the mechanistic role of CaMKII in atrial remodelling and AF.
Figure 2.
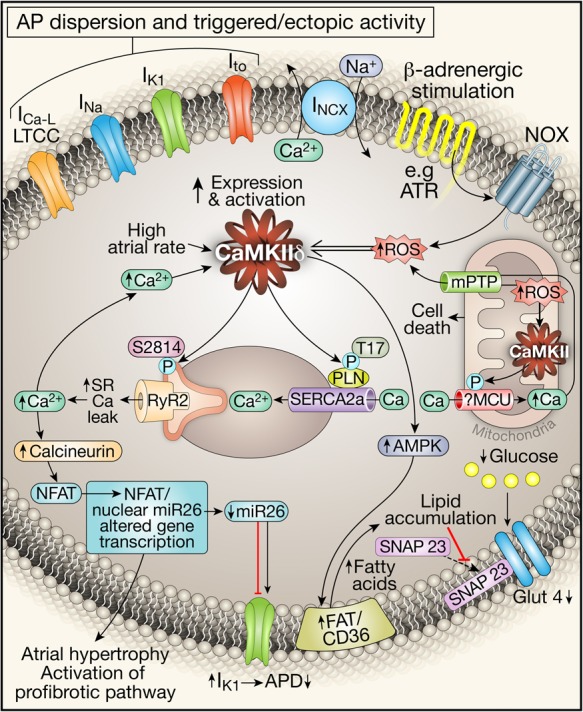
CaMKII-mediated intracellular mechanisms of AF-related atrial remodelling. Stress signals result in increased intracellular Ca2+ and ROS that lead to increased CaMKII expression and activity. CaMKII has diverse downstream targets through which it promotes arrhythmogenesis. Its cumulative effect on ionic channels (ICa.L, INa, IK1, Ito, and INCX) is inhomogeneous AP propagation and repolarization dispersion that leads to triggered activity. It also results in increased SR Ca2+, further increasing intracellular [Ca2+]. This acts via the calcineurin/NFAT pathway to activate profibrotic pathways and induce atrial hypertrophy. Increased intracellular [Ca2+] also leads to increased mitochondrial [Ca2+], which through its action on mPTP opening causes increased ROS and CaMKII activation in the mitochondria that can cause cell death. CaMKII also acts through AMPK activation to increase lipid accumulation and to decrease glucose uptake. β-adrenergic stimulation acts through angiotensin receptors to activate NADPH oxidase (NOX) to increase intracellular ROS and to activate CaMKI via oxidation.
Figure 3.
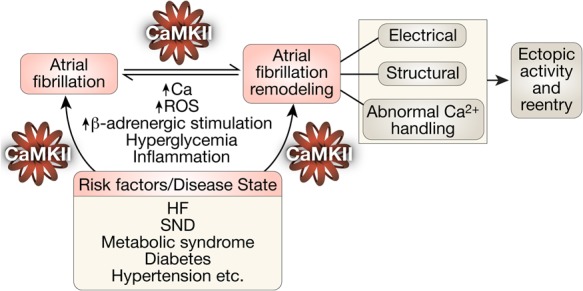
CaMKII transduces proarrhythmic signalling in atrial remodelling and AF in a feed-forward manner. Increased intracellular Ca2+ and ROS induce autonomous CaMKII activity. Excessive CaMKII activity through its action on multiple downstream targets (ion channels, proteins, etc.) results in triggered and reentrant activities that can initiate and perpetuate AF. It also results in several changes that are associated with atrial remodelling in AF. Underlying disease states or risk factors also cause excessive CaMKII activity with similar proarrhythmic downstream consequences. HF, heart failure; SND, sinus node dysfunction.
Funding
This work was supported in part by the US National Institutes of Health (NIH) grants R01-HL079031, R01-HL096652, R01-HL070250, and R01-HL113001. O.O.M. was supported by an institutional training grant from the US NIH—T32-HL007227.
Acknowledgement
We are grateful to Mr Shawn Roach who produced the artwork for the figures.
Conflict of interest: M.E.A. is a named inventor on intellectual property claiming to treat arrhythmias by CaMKII inhibition and is a cofounder of Allosteros Therapeutics, a company aiming to develop CaMKII inhibitor therapeutics. O.O.M. reports no conflict of interest.
References
- 1.Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH Jr, Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, Ezzati M, Murray CJ. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation 2014;129:837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kannel WB, Wolf PA, Benjamin EJ, Levy D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol 1998;82:2N–9N. [DOI] [PubMed] [Google Scholar]
- 3.Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol 2009;104:1534–1539. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin EJ, Wolf PA, D'Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation 1998;98:946–952. [DOI] [PubMed] [Google Scholar]
- 5.Thrall G, Lane D, Carroll D, Lip GY. Quality of life in patients with atrial fibrillation: a systematic review. Am J Med 2006;119:448.e1–448.e19. [DOI] [PubMed] [Google Scholar]
- 6.January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC Jr, Conti JB, Ellinor PT, Ezekowitz MD, Field ME, Murray KT, Sacco RL, Stevenson WG, Tchou PJ, Tracy CM, Yancy CW, American College of Cardiology/American Heart Association Task Force on Practice G. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2014;64:e1–e76. [DOI] [PubMed] [Google Scholar]
- 7.Wakili R, Voigt N, Kaab S, Dobrev D, Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest 2011;121:2955–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995;92:1954–1968. [DOI] [PubMed] [Google Scholar]
- 9.Nattel S, Harada M. Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J Am Coll Cardiol 2014;63:2335–2345. [DOI] [PubMed] [Google Scholar]
- 10.Camm AJ, Al-Khatib SM, Calkins H, Halperin JL, Kirchhof P, Lip GY, Nattel S, Ruskin J, Banerjee A, Blendea D, Guasch E, Needleman M, Savelieva I, Viles-Gonzalez J, Williams ES. A proposal for new clinical concepts in the management of atrial fibrillation. Am Heart J 2012;164:292–302.e1. [DOI] [PubMed] [Google Scholar]
- 11.Swaminathan PD, Purohit A, Hund TJ, Anderson ME. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res 2012;110:1661–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tobimatsu T, Fujisawa H. Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J Biol Chem 1989;264:17907–17912. [PubMed] [Google Scholar]
- 13.Shenolikar S, Lickteig R, Hardie DG, Soderling TR, Hanley RM, Kelly PT. Calmodulin-dependent multifunctional protein kinase. Evidence for isoenzyme forms in mammalian tissues. Eur J Biochem 1986;161:739–747. [DOI] [PubMed] [Google Scholar]
- 14.Hudmon A, Schulman H. Structure–function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J 2002;364:593–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srinivasan M, Edman CF, Schulman H. Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J Cell Biol 1994;126:839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mishra S, Gray CB, Miyamoto S, Bers DM, Brown JH. Location matters: clarifying the concept of nuclear and cytosolic CaMKII subtypes. Circ Res 2011;109:1354–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell 2003;11:1241–1251. [DOI] [PubMed] [Google Scholar]
- 18.Yang E, Schulman H. Structural examination of autoregulation of multifunctional calcium/calmodulin-dependent protein kinase II. J Biol Chem 1999;274:26199–26208. [DOI] [PubMed] [Google Scholar]
- 19.Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell 2005;123:849–860. [DOI] [PubMed] [Google Scholar]
- 20.Saitoh T, Schwartz JH. Phosphorylation-dependent subcellular translocation of a Ca2+/calmodulin-dependent protein kinase produces an autonomous enzyme in Aplysia neurons. J Cell Biol 1985;100:835–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lou LL, Lloyd SJ, Schulman H. Activation of the multifunctional Ca2+/calmodulin-dependent protein kinase by autophosphorylation: ATP modulates production of an autonomous enzyme. Proc Natl Acad Sci USA 1986;83:9497–9501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schworer CM, Colbran RJ, Soderling TR. Reversible generation of a Ca2+-independent form of Ca2+(calmodulin)-dependent protein kinase II by an autophosphorylation mechanism. J Biol Chem 1986;261:8581–8584. [PubMed] [Google Scholar]
- 23.Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science 1992;256:1199–1202. [DOI] [PubMed] [Google Scholar]
- 24.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008;133:462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013;502:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gutierrez DA, Fernandez-Tenorio M, Ogrodnik J, Niggli E. NO-dependent CaMKII activation during beta-adrenergic stimulation of cardiac muscle. Cardiovasc Res 2013;100:392–401. [DOI] [PubMed] [Google Scholar]
- 27.Colbran RJ. Inactivation of Ca2+/calmodulin-dependent protein kinase II by basal autophosphorylation. J Biol Chem 1993;268:7163–7170. [PubMed] [Google Scholar]
- 28.Erickson JR, Nichols CB, Uchinoumi H, Stein ML, Bossuyt J, Bers DM. S-nitrosylation induces both autonomous activation and inhibition of calcium/calmodulin-dependent protein kinase II delta. J Biol Chem 2015;290:25646–25656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He BJ, Joiner ML, Singh MV, Luczak ED, Swaminathan PD, Koval OM, Kutschke W, Allamargot C, Yang J, Guan X, Zimmerman K, Grumbach IM, Weiss RM, Spitz DR, Sigmund CD, Blankesteijn WM, Heymans S, Mohler PJ, Anderson ME. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med 2011;17:1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palomeque J, Rueda OV, Sapia L, Valverde CA, Salas M, Petroff MV, Mattiazzi A. Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ Res 2009;105:1204–1212. [DOI] [PubMed] [Google Scholar]
- 31.Hudmon A, Schulman H. Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem 2002;71:473–510. [DOI] [PubMed] [Google Scholar]
- 32.Wu Y, MacMillan LB, McNeill RB, Colbran RJ, Anderson ME. CaM kinase augments cardiac l-type Ca2+ current: a cellular mechanism for long Q-T arrhythmias. Am J Physiol 1999;276:H2168–H2178. [DOI] [PubMed] [Google Scholar]
- 33.Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 2001;411:801–805. [DOI] [PubMed] [Google Scholar]
- 34.Singh P, Salih M, Tuana BS. Alpha-kinase anchoring protein alphaKAP interacts with SERCA2A to spatially position Ca2+/calmodulin-dependent protein kinase II and modulate phospholamban phosphorylation. J Biol Chem 2009;284:28212–28221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR III, Lefkowitz RJ. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci USA 2007;104:12011–12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mangmool S, Shukla AK, Rockman HA. beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J Cell Biol 2010;189:573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El-Haou S, Balse E, Neyroud N, Dilanian G, Gavillet B, Abriel H, Coulombe A, Jeromin A, Hatem SN. Kv4 potassium channels form a tripartite complex with the anchoring protein SAP97 and CaMKII in cardiac myocytes. Circ Res 2009;104:758–769. [DOI] [PubMed] [Google Scholar]
- 38.Koval OM, Guan X, Wu Y, Joiner ML, Gao Z, Chen B, Grumbach IM, Luczak ED, Colbran RJ, Song LS, Hund TJ, Mohler PJ, Anderson ME. CaV1.2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci USA 2010;107:4996–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest 2010;120:3508–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson ME, Braun AP, Schulman H, Premack BA. Multifunctional Ca2+/calmodulin-dependent protein kinase mediates Ca(2+)-induced enhancement of the l-type Ca2+ current in rabbit ventricular myocytes. Circ Res 1994;75:854–861. [DOI] [PubMed] [Google Scholar]
- 41.Yuan W, Bers DM. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am J Physiol 1994;267:H982–H993. [DOI] [PubMed] [Google Scholar]
- 42.Xiao RP, Cheng H, Lederer WJ, Suzuki T, Lakatta EG. Dual regulation of Ca2+/calmodulin-dependent kinase II activity by membrane voltage and by calcium influx. Proc Natl Acad Sci USA 1994;91:9659–9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tessier S, Karczewski P, Krause EG, Pansard Y, Acar C, Lang-Lazdunski M, Mercadier JJ, Hatem SN. Regulation of the transient outward K(+) current by Ca(2+)/calmodulin-dependent protein kinases II in human atrial myocytes. Circ Res 1999;85:810–819. [DOI] [PubMed] [Google Scholar]
- 44.Xiao L, Coutu P, Villeneuve LR, Tadevosyan A, Maguy A, Le Bouter S, Allen BG, Nattel S. Mechanisms underlying rate-dependent remodeling of transient outward potassium current in canine ventricular myocytes. Circ Res 2008;103:733–742. [DOI] [PubMed] [Google Scholar]
- 45.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 2006;116:3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, Marionneau C, Zhang R, Shah V, Hell JW, Nerbonne JM, Anderson ME. Calmodulin kinase II inhibition shortens action potential duration by upregulation of K+ currents. Circ Res 2006;99:1092–1099. [DOI] [PubMed] [Google Scholar]
- 47.Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, Bers DM, Hudmon A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem 2012;287:19856–19869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res 2004;94:e61–e70. [DOI] [PubMed] [Google Scholar]
- 49.Kranias EG. Regulation of Ca2+ transport by cyclic 3′,5′-AMP-dependent and calcium-calmodulin-dependent phosphorylation of cardiac sarcoplasmic reticulum. Biochim Biophys Acta 1985;844:193–199. [DOI] [PubMed] [Google Scholar]
- 50.Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci USA 2002;99:17185–17190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hashambhoy YL, Winslow RL, Greenstein JL. CaMKII-induced shift in modal gating explains l-type Ca(2+) current facilitation: a modeling study. Biophys J 2009;96:1770–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson ME, Braun AP, Wu Y, Lu T, Wu Y, Schulman H, Sung RJ. KN-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther 1998;287:996–1006. [PubMed] [Google Scholar]
- 53.Curran J, Brown KH, Santiago DJ, Pogwizd S, Bers DM, Shannon TR. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca(2+)-calmodulin-dependent protein kinase II. J Mol Cell Cardiol 2010;49:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res 2011;108:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song Y, Shryock JC, Belardinelli L. A slowly inactivating sodium current contributes to spontaneous diastolic depolarization of atrial myocytes. Am J Physiol Heart Circ Physiol 2009;297:H1254–H1262. [DOI] [PubMed] [Google Scholar]
- 56.Fischer TH, Herting J, Mason FE, Hartmann N, Watanabe S, Nikolaev VO, Sprenger JU, Fan P, Yao L, Popov AF, Danner BC, Schondube F, Belardinelli L, Hasenfuss G, Maier LS, Sossalla S. Late INa increases diastolic SR-Ca2+-leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc Res 2015;107:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wagner S, Hacker E, Grandi E, Weber SL, Dybkova N, Sossalla S, Sowa T, Fabritz L, Kirchhof P, Bers DM, Maier LS. Ca/calmodulin kinase II differentially modulates potassium currents. Circ Arrhythm Electrophysiol 2009;2:285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hagiwara K, Nunoki K, Ishii K, Abe T, Yanagisawa T. Differential inhibition of transient outward currents of Kv1.4 and Kv4.3 by endothelin. Biochem Biophys Res Commun 2003;310:634–640. [DOI] [PubMed] [Google Scholar]
- 59.Gaborit N, Steenman M, Lamirault G, Le Meur N, Le Bouter S, Lande G, Leger J, Charpentier F, Christ T, Dobrev D, Escande D, Nattel S, Demolombe S. Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation 2005;112:471–481. [DOI] [PubMed] [Google Scholar]
- 60.Dobrev D, Graf E, Wettwer E, Himmel HM, Hala O, Doerfel C, Christ T, Schuler S, Ravens U. Molecular basis of downregulation of G-protein-coupled inward rectifying K(+) current (I(K,ACh) in chronic human atrial fibrillation: decrease in GIRK4 mRNA correlates with reduced I(K,ACh) and muscarinic receptor-mediated shortening of action potentials. Circulation 2001;104:2551–2557. [DOI] [PubMed] [Google Scholar]
- 61.Pandit SV, Berenfeld O, Anumonwo JM, Zaritski RM, Kneller J, Nattel S, Jalife J. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J 2005;88:3806–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Voigt N, Friedrich A, Bock M, Wettwer E, Christ T, Knaut M, Strasser RH, Ravens U, Dobrev D. Differential phosphorylation-dependent regulation of constitutively active and muscarinic receptor-activated IK,ACh channels in patients with chronic atrial fibrillation. Cardiovasc Res 2007;74:426–437. [DOI] [PubMed] [Google Scholar]
- 63.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 2005;97:1314–1322. [DOI] [PubMed] [Google Scholar]
- 64.Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, Wehrens XH. Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm 2008;5:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shan J, Xie W, Betzenhauser M, Reiken S, Chen BX, Wronska A, Marks AR. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res 2012;111:708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, Stefansdottir H, Behunin AC, Li N, El-Accaoui RN, Yang B, Swaminathan PD, Weiss RM, Wehrens XH, Song LS, Dobrev D, Maier LS, Anderson ME. Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 2013;128:1748–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li N, Wang T, Wang W, Cutler MJ, Wang Q, Voigt N, Rosenbaum DS, Dobrev D, Wehrens XH. Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ Res 2012;110:465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tada M, Kirchberger MA, Repke DI, Katz AM. The stimulation of calcium transport in cardiac sarcoplasmic reticulum by adenosine 3′:5′-monophosphate-dependent protein kinase. J Biol Chem 1974;249:6174–6180. [PubMed] [Google Scholar]
- 69.Kim HW, Steenaart NA, Ferguson DG, Kranias EG. Functional reconstitution of the cardiac sarcoplasmic reticulum Ca2(+)-ATPase with phospholamban in phospholipid vesicles. J Biol Chem 1990;265:1702–1709. [PubMed] [Google Scholar]
- 70.Kranias EG, Mandel F, Wang T, Schwartz A. Mechanism of the stimulation of calcium ion dependent adenosine triphosphatase of cardiac sarcoplasmic reticulum by adenosine 3′,5′-monophosphate dependent protein kinase. Biochemistry 1980;19:5434–5439. [DOI] [PubMed] [Google Scholar]
- 71.Kranias EG, Bilezikjian LM, Potter JD, Piascik MT, Schwartz A. The role of calmodulin in regulation of cardiac sarcoplasmic reticulum phosphorylation. Ann N Y Acad Sci 1980;356:279–291. [DOI] [PubMed] [Google Scholar]
- 72.Rokita AG, Anderson ME. New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation 2012;126:2125–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Onal B, Unudurthi SD, Hund TJ. Modeling CaMKII in cardiac physiology: from molecule to tissue. Front Pharmacol 2014;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol 2008;51:802–809. [DOI] [PubMed] [Google Scholar]
- 75.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 2008;70:23–49. [DOI] [PubMed] [Google Scholar]
- 76.Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc'h F. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ Res 2005;97:1296–1304. [DOI] [PubMed] [Google Scholar]
- 77.Pereira L, Metrich M, Fernandez-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Benitah JP, Lezoualc'h F, Gomez AM. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol 2007;583:685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J Biol Chem 2009;284:1514–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vest JA, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR, Marks AR. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 2005;111:2025–2032. [DOI] [PubMed] [Google Scholar]
- 80.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 2000;101:365–376. [DOI] [PubMed] [Google Scholar]
- 81.Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XH, Chen J, Bers DM. Epac2 mediates cardiac beta1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 2013;127:913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res 2002;90:309–316. [DOI] [PubMed] [Google Scholar]
- 83.Ginsburg KS, Bers DM. Modulation of excitation–contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol 2004;556:463–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huke S, Bers DM. Temporal dissociation of frequency-dependent acceleration of relaxation and protein phosphorylation by CaMKII. J Mol Cell Cardiol 2007;42:590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res 2007;100:391–398. [DOI] [PubMed] [Google Scholar]
- 86.Mika D, Richter W, Conti M. A CaMKII/PDE4D negative feedback regulates cAMP signaling. Proc Natl Acad Sci USA 2015;112:2023–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu Y, Gao Z, Chen B, Koval OM, Singh MV, Guan X, Hund TJ, Kutschke W, Sarma S, Grumbach IM, Wehrens XH, Mohler PJ, Song LS, Anderson ME. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc Natl Acad Sci USA 2009;106:5972–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Greiser M, Neuberger HR, Harks E, El-Armouche A, Boknik P, de Haan S, Verheyen F, Verheule S, Schmitz W, Ravens U, Nattel S, Allessie MA, Dobrev D, Schotten U. Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block-induced atrial dilatation and atrial fibrillation. J Mol Cell Cardiol 2009;46:385–394. [DOI] [PubMed] [Google Scholar]
- 89.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest 2009;119:1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wakili R, Yeh YH, Yan Qi X, Greiser M, Chartier D, Nishida K, Maguy A, Villeneuve LR, Boknik P, Voigt N, Krysiak J, Kaab S, Ravens U, Linke WA, Stienen GJ, Shi Y, Tardif JC, Schotten U, Dobrev D, Nattel S. Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ Arrhythm Electrophysiol 2010;3:530–541. [DOI] [PubMed] [Google Scholar]
- 91.Greiser M, Kerfant BG, Williams GS, Voigt N, Harks E, Dibb KM, Giese A, Meszaros J, Verheule S, Ravens U, Allessie MA, Gammie JS, van der Velden J, Lederer WJ, Dobrev D, Schotten U. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J Clin Invest 2014;124:4759–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lenski M, Schleider G, Kohlhaas M, Adrian L, Adam O, Tian Q, Kaestner L, Lipp P, Lehrke M, Maack C, Bohm M, Laufs U. Arrhythmia causes lipid accumulation and reduced glucose uptake. Basic Res Cardiol 2015;110:40. [DOI] [PubMed] [Google Scholar]
- 93.Christ T, Boknik P, Wohrl S, Wettwer E, Graf EM, Bosch RF, Knaut M, Schmitz W, Ravens U, Dobrev D. l-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 2004;110:2651–2657. [DOI] [PubMed] [Google Scholar]
- 94.Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G, Maier LS. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res 2010;106:1134–1144. [DOI] [PubMed] [Google Scholar]
- 95.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012;125:2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Christ T, Rozmaritsa N, Engel A, Berk E, Knaut M, Metzner K, Canteras M, Ravens U, Kaumann A. Arrhythmias, elicited by catecholamines and serotonin, vanish in human chronic atrial fibrillation. Proc Natl Acad Sci USA 2014;111:11193–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev 2011;91:265–325. [DOI] [PubMed] [Google Scholar]
- 98.Katra RP, Laurita KR. Cellular mechanism of calcium-mediated triggered activity in the heart. Circ Res 2005;96:535–542. [DOI] [PubMed] [Google Scholar]
- 99.Ming Z, Nordin C, Aronson RS. Role of l-type calcium channel window current in generating current-induced early afterdepolarizations. J Cardiovasc Electrophysiol 1994;5:323–334. [DOI] [PubMed] [Google Scholar]
- 100.Volders PG, Kulcsar A, Vos MA, Sipido KR, Wellens HJ, Lazzara R, Szabo B. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovasc Res 1997;34:348–359. [DOI] [PubMed] [Google Scholar]
- 101.Allessie MA, de Groot NM, Houben RP, Schotten U, Boersma E, Smeets JL, Crijns HJ. Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: longitudinal dissociation. Circ Arrhythm Electrophysiol 2010;3:606–615. [DOI] [PubMed] [Google Scholar]
- 102.Morillo CA, Klein GJ, Jones DL, Guiraudon CM. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation 1995;91:1588–1595. [DOI] [PubMed] [Google Scholar]
- 103.Gaspo R, Bosch RF, Bou-Abboud E, Nattel S. Tachycardia-induced changes in Na+ current in a chronic dog model of atrial fibrillation. Circ Res 1997;81:1045–1052. [DOI] [PubMed] [Google Scholar]
- 104.Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation 1999;100:87–95. [DOI] [PubMed] [Google Scholar]
- 105.Schoonderwoerd BA, Ausma J, Crijns HJ, Van Veldhuisen DJ, Blaauw EH, Van Gelder IC. Atrial ultrastructural changes during experimental atrial tachycardia depend on high ventricular rate. J Cardiovasc Electrophysiol 2004;15:1167–1174. [DOI] [PubMed] [Google Scholar]
- 106.Neuberger HR, Schotten U, Blaauw Y, Vollmann D, Eijsbouts S, van Hunnik A, Allessie M. Chronic atrial dilation, electrical remodeling, and atrial fibrillation in the goat. J Am Coll Cardiol 2006;47:644–653. [DOI] [PubMed] [Google Scholar]
- 107.Boyden PA, Hoffman BF. The effects on atrial electrophysiology and structure of surgically induced right atrial enlargement in dogs. Circ Res 1981;49:1319–1331. [DOI] [PubMed] [Google Scholar]
- 108.Page PL, Plumb VJ, Okumura K, Waldo AL. A new animal model of atrial flutter. J Am Coll Cardiol 1986;8:872–879. [DOI] [PubMed] [Google Scholar]
- 109.Hagendorff A, Schumacher B, Kirchhoff S, Luderitz B, Willecke K. Conduction disturbances and increased atrial vulnerability in Connexin40-deficient mice analyzed by transesophageal stimulation. Circulation 1999;99:1508–1515. [DOI] [PubMed] [Google Scholar]
- 110.Verheule S, Sato T, Everett T, Engle SK, Otten D, von der Lohe MR, Nakajima HO, Nakajima H, Field LJ, Olgin JE. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta 1. Circ Res 2004;94:1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guo X, Yuan S, Liu Z, Fang Q. Oxidation- and CaMKII-mediated sarcoplasmic reticulum Ca(2+) leak triggers atrial fibrillation in aging. J Cardiovasc Electrophysiol 2014;25:645–652. [DOI] [PubMed] [Google Scholar]
- 112.Lo LW, Chen YC, Chen YJ, Wongcharoen W, Lin CI, Chen SA. Calmodulin kinase II inhibition prevents arrhythmic activity induced by alpha and beta adrenergic agonists in rabbit pulmonary veins. Eur J Pharmacol 2007;571:197–208. [DOI] [PubMed] [Google Scholar]
- 113.Yue L, Feng J, Gaspo R, Li GR, Wang Z, Nattel S. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res 1997;81:512–525. [DOI] [PubMed] [Google Scholar]
- 114.Daoud EG, Bogun F, Goyal R, Harvey M, Man KC, Strickberger SA, Morady F. Effect of atrial fibrillation on atrial refractoriness in humans. Circulation 1996;94:1600–1606. [DOI] [PubMed] [Google Scholar]
- 115.Franz MR, Karasik PL, Li C, Moubarak J, Chavez M. Electrical remodeling of the human atrium: similar effects in patients with chronic atrial fibrillation and atrial flutter. J Am Coll Cardiol 1997;30:1785–1792. [DOI] [PubMed] [Google Scholar]
- 116.Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C, Kuhlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res 1999;44:121–131. [DOI] [PubMed] [Google Scholar]
- 117.Van Wagoner DR, Pond AL, Lamorgese M, Rossie SS, McCarthy PM, Nerbonne JM. Atrial l-type Ca2+ currents and human atrial fibrillation. Circ Res 1999;85:428–436. [DOI] [PubMed] [Google Scholar]
- 118.Qi XY, Yeh YH, Xiao L, Burstein B, Maguy A, Chartier D, Villeneuve LR, Brundel BJ, Dobrev D, Nattel S. Cellular signaling underlying atrial tachycardia remodeling of l-type calcium current. Circ Res 2008;103:845–854. [DOI] [PubMed] [Google Scholar]
- 119.Schotten U, Haase H, Frechen D, Greiser M, Stellbrink C, Vazquez-Jimenez JF, Morano I, Allessie MA, Hanrath P. The l-type Ca2+-channel subunits alpha1C and beta2 are not downregulated in atrial myocardium of patients with chronic atrial fibrillation. J Mol Cell Cardiol 2003;35:437–443. [DOI] [PubMed] [Google Scholar]
- 120.Brundel BJ, Kampinga HH, Henning RH. Calpain inhibition prevents pacing-induced cellular remodeling in a HL-1 myocyte model for atrial fibrillation. Cardiovasc Res 2004;62:521–528. [DOI] [PubMed] [Google Scholar]
- 121.King JH, Zhang Y, Lei M, Grace AA, Huang CL, Fraser JA. Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol (Oxf) 2013;207:308–323. [DOI] [PubMed] [Google Scholar]
- 122.King JH, Wickramarachchi C, Kua K, Du Y, Jeevaratnam K, Matthews HR, Grace AA, Huang CL, Fraser JA. Loss of Nav1.5 expression and function in murine atria containing the RyR2-P2328S gain-of-function mutation. Cardiovasc Res 2013;99:751–759. [DOI] [PubMed] [Google Scholar]
- 123.Heijman J, Wehrens XH, Dobrev D. Atrial arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia—is there a mechanistic link between sarcoplasmic reticulum Ca(2+) leak and re-entry? Acta Physiol (Oxf) 2013;207:208–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Christensen MD, Dun W, Boyden PA, Anderson ME, Mohler PJ, Hund TJ. Oxidized calmodulin kinase II regulates conduction following myocardial infarction: a computational analysis. PLoS Comput Biol 2009;5:e1000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res 2002;54:230–246. [DOI] [PubMed] [Google Scholar]
- 126.Oakes RS, Badger TJ, Kholmovski EG, Akoum N, Burgon NS, Fish EN, Blauer JJ, Rao SN, DiBella EV, Segerson NM, Daccarett M, Windfelder J, McGann CJ, Parker D, MacLeod RS, Marrouche NF. Detection and quantification of left atrial structural remodeling with delayed-enhancement magnetic resonance imaging in patients with atrial fibrillation. Circulation 2009;119:1758–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yue L, Xie J, Nattel S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc Res 2011;89:744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, Yue L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res 2010;106:992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, Shi Y, Kamiya K, Murohara T, Kodama I, Tardif JC, Schotten U, Van Wagoner DR, Dobrev D, Nattel S. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 2012;126:2051–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Heijman J, Voigt N, Wehrens XH, Dobrev D. Calcium dysregulation in atrial fibrillation: the role of CaMKII. Front Pharmacol 2014;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mishra R, Rao V, Ta R, Shobeiri N, Hill CE. Mg2+- and MgATP-inhibited and Ca2+/calmodulin-sensitive TRPM7-like current in hepatoma and hepatocytes. Am J Physiol Gastrointest Liver Physiol 2009;297:G687–G694. [DOI] [PubMed] [Google Scholar]
- 132.Kitajima N, Watanabe K, Morimoto S, Sato Y, Kiyonaka S, Hoshijima M, Ikeda Y, Nakaya M, Ide T, Mori Y, Kurose H, Nishida M. TRPC3-mediated Ca2+ influx contributes to Rac1-mediated production of reactive oxygen species in MLP-deficient mouse hearts. Biochem Biophys Res Commun 2011;409:108–113. [DOI] [PubMed] [Google Scholar]
- 133.Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, Kutschke W, Yang J, Donahue JK, Weiss RM, Grumbach IM, Ogawa M, Chen PS, Efimov I, Dobrev D, Mohler PJ, Hund TJ, Anderson ME. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest 2011;121:3277–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J Jr, Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res 2003;92:912–919. [DOI] [PubMed] [Google Scholar]
- 135.Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, Yang J, Price EE, Gleaves L, Eren M, Ni G, Vaughan DE, Xiao RP, Anderson ME. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol 2006;291:H3065–H3075. [DOI] [PubMed] [Google Scholar]
- 136.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, Devic E, Kobilka BK, Cheng H, Xiao RP. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest 2003;111:617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature 2012;491:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Issac TT, Dokainish H, Lakkis NM. Role of inflammation in initiation and perpetuation of atrial fibrillation: a systematic review of the published data. J Am Coll Cardiol 2007;50:2021–2028. [DOI] [PubMed] [Google Scholar]
- 139.Rudolph V, Andrie RP, Rudolph TK, Friedrichs K, Klinke A, Hirsch-Hoffmann B, Schwoerer AP, Lau D, Fu X, Klingel K, Sydow K, Didie M, Seniuk A, von Leitner EC, Szoecs K, Schrickel JW, Treede H, Wenzel U, Lewalter T, Nickenig G, Zimmermann WH, Meinertz T, Boger RH, Reichenspurner H, Freeman BA, Eschenhagen T, Ehmke H, Hazen SL, Willems S, Baldus S. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat Med 2010;16:470–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Singh MV, Kapoun A, Higgins L, Kutschke W, Thurman JM, Zhang R, Singh M, Yang J, Guan X, Lowe JS, Weiss RM, Zimmermann K, Yull FE, Blackwell TS, Mohler PJ, Anderson ME. Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest 2009;119:986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol 2012;52:1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li N, Chiang DY, Wang S, Wang Q, Sun L, Voigt N, Respress JL, Ather S, Skapura DG, Jordan VK, Horrigan FT, Schmitz W, Muller FU, Valderrabano M, Nattel S, Dobrev D, Wehrens XH. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 2014;129:1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Carnes CA, Chung MK, Nakayama T, Nakayama H, Baliga RS, Piao S, Kanderian A, Pavia S, Hamlin RL, McCarthy PM, Bauer JA, Van Wagoner DR. Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ Res 2001;89:E32–E38. [DOI] [PubMed] [Google Scholar]
- 144.Yang KC, Dudley SC Jr. Oxidative stress and atrial fibrillation: finding a missing piece to the puzzle. Circulation 2013;128:1724–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mihm MJ, Yu F, Carnes CA, Reiser PJ, McCarthy PM, Van Wagoner DR, Bauer JA. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 2001;104:174–180. [DOI] [PubMed] [Google Scholar]
- 146.Anderson ME. Oxidant stress promotes disease by activating CaMKII. J Mol Cell Cardiol 2015;89:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Youn JY, Zhang J, Zhang Y, Chen H, Liu D, Ping P, Weiss JN, Cai H. Oxidative stress in atrial fibrillation: an emerging role of NADPH oxidase. J Mol Cell Cardiol 2013;62:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res 2009;104:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Afanas'ev I. ROS and RNS signaling in heart disorders: could antioxidant treatment be successful? Oxid Med Cell Longev 2011;2011:293769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 2010;106:1253–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhang Y, Shimizu H, Siu KL, Mahajan A, Chen JN, Cai H. NADPH oxidase 4 induces cardiac arrhythmic phenotype in zebrafish. J Biol Chem 2014;289:23200–23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang DX, Ren K, Guan Y, Wang YT, Shan ZL. Protective effects of apocynin on atrial electrical remodeling and oxidative stress in a rabbit rapid atrial pacing model. Chin J Physiol 2014;57:76–82. [DOI] [PubMed] [Google Scholar]
- 153.Wolke C, Bukowska A, Goette A, Lendeckel U. Redox control of cardiac remodeling in atrial fibrillation. Biochim Biophys Acta 2015;1850:1555–1565. [DOI] [PubMed] [Google Scholar]
- 154.Foteinou PT, Greenstein JL, Winslow RL. Mechanistic investigation of the arrhythmogenic role of oxidized CaMKII in the heart. Biophys J 2015;109:838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Violi F, Pastori D, Pignatelli P, Loffredo L. Antioxidants for prevention of atrial fibrillation: a potentially useful future therapeutic approach? A review of the literature and meta-analysis. Europace 2014;16:1107–1116. [DOI] [PubMed] [Google Scholar]
- 156.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med 2000;6:513–519. [DOI] [PubMed] [Google Scholar]
- 157.Clapham DE. Calcium signaling. Cell 2007;131:1047–1058. [DOI] [PubMed] [Google Scholar]
- 158.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell 2008;134:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Odagiri K, Katoh H, Kawashima H, Tanaka T, Ohtani H, Saotome M, Urushida T, Satoh H, Hayashi H. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J Mol Cell Cardiol 2009;46:989–997. [DOI] [PubMed] [Google Scholar]
- 160.Fieni F, Johnson DE, Hudmon A, Kirichok Y. Mitochondrial Ca2+ uniporter and CaMKII in heart. Nature 2014;513:E1–E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004;427:360–364. [DOI] [PubMed] [Google Scholar]
- 162.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011;476:341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011;476:336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lin PH, Lee SH, Su CP, Wei YH. Oxidative damage to mitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic Biol Med 2003;35:1310–1318. [DOI] [PubMed] [Google Scholar]
- 165.Lendeckel U, Muller C, Rocken C, Laube B, Tager M, Huth C, Klein HU, Goette A. Expression of opioid receptor subtypes and their ligands in fibrillating human atria. Pacing Clin Electrophysiol 2005;28(Suppl 1):S275–S279. [DOI] [PubMed] [Google Scholar]
- 166.Schild L, Bukowska A, Gardemann A, Polczyk P, Keilhoff G, Tager M, Dudley SC, Klein HU, Goette A, Lendeckel U. Rapid pacing of embryoid bodies impairs mitochondrial ATP synthesis by a calcium-dependent mechanism—a model of in vitro differentiated cardiomyocytes to study molecular effects of tachycardia. Biochim Biophys Acta 2006;1762:608–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bukowska A, Schild L, Keilhoff G, Hirte D, Neumann M, Gardemann A, Neumann KH, Rohl FW, Huth C, Goette A, Lendeckel U. Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp Biol Med (Maywood) 2008;233:558–574. [DOI] [PubMed] [Google Scholar]
- 168.Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res 2007;100:474–488. [DOI] [PubMed] [Google Scholar]
- 169.Frederich M, Balschi JA. The relationship between AMP-activated protein kinase activity and AMP concentration in the isolated perfused rat heart. J Biol Chem 2002;277:1928–1932. [DOI] [PubMed] [Google Scholar]
- 170.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 2007;100:328–341. [DOI] [PubMed] [Google Scholar]
- 171.Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol 2006;574:95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, Giri S, Andreelli F. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol 2010;45:276–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Harada M, Tadevosyan A, Qi X, Xiao J, Liu T, Voigt N, Karck M, Kamler M, Kodama I, Murohara T, Dobrev D, Nattel S. Atrial fibrillation activates AMP-dependent protein kinase and its regulation of cellular calcium handling: potential role in metabolic adaptation and prevention of progression. J Am Coll Cardiol 2015;66:47–58. [DOI] [PubMed] [Google Scholar]
- 174.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991;22:983–988. [DOI] [PubMed] [Google Scholar]
- 175.Sun H, Gaspo R, Leblanc N, Nattel S. Cellular mechanisms of atrial contractile dysfunction caused by sustained atrial tachycardia. Circulation 1998;98:719–727. [DOI] [PubMed] [Google Scholar]
- 176.Manning WJ, Silverman DI, Katz SE, Riley MF, Come PC, Doherty RM, Munson JT, Douglas PS. Impaired left atrial mechanical function after cardioversion: relation to the duration of atrial fibrillation. J Am Coll Cardiol 1994;23:1535–1540. [DOI] [PubMed] [Google Scholar]
- 177.Schotten U, Ausma J, Stellbrink C, Sabatschus I, Vogel M, Frechen D, Schoendube F, Hanrath P, Allessie MA. Cellular mechanisms of depressed atrial contractility in patients with chronic atrial fibrillation. Circulation 2001;103:691–698. [DOI] [PubMed] [Google Scholar]
- 178.de Haan S, Greiser M, Harks E, Blaauw Y, van Hunnik A, Verheule S, Allessie M, Schotten U. AVE0118, blocker of the transient outward current (I(to)) and ultrarapid delayed rectifier current (I(Kur)), fully restores atrial contractility after cardioversion of atrial fibrillation in the goat. Circulation 2006;114:1234–1242. [DOI] [PubMed] [Google Scholar]
- 179.Hartzell HC, Glass DB. Phosphorylation of purified cardiac muscle C-protein by purified cAMP-dependent and endogenous Ca2+-calmodulin-dependent protein kinases. J Biol Chem 1984;259:15587–15596. [PubMed] [Google Scholar]
- 180.Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J 1995;14:1952–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cheng J, Xu L, Lai D, Guilbert A, Lim HJ, Keskanokwong T, Wang Y. CaMKII inhibition in heart failure, beneficial, harmful, or both. Am J Physiol Heart Circ Physiol 2012;302:H1454–H1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J Mol Cell Cardiol 2010;48:322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Linke WA, Popov VI, Pollack GH. Passive and active tension in single cardiac myofibrils. Biophys J 1994;67:782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation 2006;113:1966–1973. [DOI] [PubMed] [Google Scholar]
- 185.Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA. Titin isoform switch in ischemic human heart disease. Circulation 2002;106:1333–1341. [DOI] [PubMed] [Google Scholar]
- 186.Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Kruger M, Backs J, Linke WA. Crucial role for Ca2(+)/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res 2013;112:664–674. [DOI] [PubMed] [Google Scholar]
- 187.Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med 1982;306:1018–1022. [DOI] [PubMed] [Google Scholar]
- 188.Maisel WH, Stevenson LW. Atrial fibrillation in heart failure: epidemiology, pathophysiology, and rationale for therapy. Am J Cardiol 2003;91:2D–8D. [DOI] [PubMed] [Google Scholar]
- 189.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol 2008;1:93–102. [DOI] [PubMed] [Google Scholar]
- 190.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med 2005;11:409–417. [DOI] [PubMed] [Google Scholar]
- 191.Anderson ME. Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol Ther 2005;106:39–55. [DOI] [PubMed] [Google Scholar]
- 192.Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittkopper K, Renner A, Schmitto JD, Gummert J, El-Armouche A, Hasenfuss G, Maier LS. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res 2010;107:1150–1161. [DOI] [PubMed] [Google Scholar]
- 193.Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res 1999;84:713–721. [DOI] [PubMed] [Google Scholar]
- 194.Khoo MS, Li J, Singh MV, Yang Y, Kannankeril P, Wu Y, Grueter CE, Guan X, Oddis CV, Zhang R, Mendes L, Ni G, Madu EC, Yang J, Bass M, Gomez RJ, Wadzinski BE, Olson EN, Colbran RJ, Anderson ME. Death, cardiac dysfunction, and arrhythmias are increased by calmodulin kinase II in calcineurin cardiomyopathy. Circulation 2006;114:1352–1359. [DOI] [PubMed] [Google Scholar]
- 195.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998;93:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ, Anderson ME. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest 2013;123:1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gomes JA, Kang PS, Matheson M, Gough WB Jr, El-Sherif N. Coexistence of sick sinus rhythm and atrial flutter-fibrillation. Circulation 1981;63:80–86. [DOI] [PubMed] [Google Scholar]
- 198.Lamas GA, Lee KL, Sweeney MO, Silverman R, Leon A, Yee R, Marinchak RA, Flaker G, Schron E, Orav EJ, Hellkamp AS, Greer S, McAnulty J, Ellenbogen K, Ehlert F, Freedman RA, Estes NA III, Greenspon A, Goldman L, Mode Selection Trial in Sinus-Node D. Ventricular pacing or dual-chamber pacing for sinus-node dysfunction. N Engl J Med 2002;346:1854–1862. [DOI] [PubMed] [Google Scholar]
- 199.Sanders P, Morton JB, Kistler PM, Spence SJ, Davidson NC, Hussin A, Vohra JK, Sparks PB, Kalman JM. Electrophysiological and electroanatomic characterization of the atria in sinus node disease: evidence of diffuse atrial remodeling. Circulation 2004;109:1514–1522. [DOI] [PubMed] [Google Scholar]
- 200.Sparks PB, Jayaprakash S, Vohra JK, Kalman JM. Electrical remodeling of the atria associated with paroxysmal and chronic atrial flutter. Circulation 2000;102:1807–1813. [DOI] [PubMed] [Google Scholar]
- 201.Elvan A, Wylie K, Zipes DP. Pacing-induced chronic atrial fibrillation impairs sinus node function in dogs. Electrophysiological remodeling. Circulation 1996;94:2953–2960. [DOI] [PubMed] [Google Scholar]
- 202.Luck JC, Engel TR. Dispersion of atrial refractoriness in patients with sinus node dysfunction. Circulation 1979;60:404–412. [DOI] [PubMed] [Google Scholar]
- 203.De Sisti A, Attuel P, Manot S, Fiorello P, Halimi F, Leclercq JF. Electrophysiological determinants of atrial fibrillation in sinus node dysfunction despite atrial pacing. Europace 2000;2:304–311. [DOI] [PubMed] [Google Scholar]
- 204.Akoum N, McGann C, Vergara G, Badger T, Ranjan R, Mahnkopf C, Kholmovski E, MacLeod R, Marrouche N. Atrial fibrosis quantified using late gadolinium enhancement MRI is associated with sinus node dysfunction requiring pacemaker implant. J Cardiovasc Electrophysiol 2012;23:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Vinogradova TM, Zhou YY, Bogdanov KY, Yang D, Kuschel M, Cheng H, Xiao RP. Sinoatrial node pacemaker activity requires Ca(2+)/calmodulin-dependent protein kinase II activation. Circ Res 2000;87:760–767. [DOI] [PubMed] [Google Scholar]
- 206.Tucker NR, Ellinor PT. Emerging directions in the genetics of atrial fibrillation. Circ Res 2014;114:1469–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.DeGrande S, Nixon D, Koval O, Curran JW, Wright P, Wang Q, Kashef F, Chiang D, Li N, Wehrens XH, Anderson ME, Hund TJ, Mohler PJ. CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome. Heart Rhythm 2012;9:2034–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 2003;421:634–639. [DOI] [PubMed] [Google Scholar]
- 209.Thiel WH, Chen B, Hund TJ, Koval OM, Purohit A, Song LS, Mohler PJ, Anderson ME. Proarrhythmic defects in Timothy syndrome require calmodulin kinase II. Circulation 2008;118:2225–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Cunha SR, Hund TJ, Hashemi S, Voigt N, Li N, Wright P, Koval O, Li J, Gudmundsson H, Gumina RJ, Karck M, Schott JJ, Probst V, Le Marec H, Anderson ME, Dobrev D, Wehrens XH, Mohler PJ. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 2011;124:1212–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sumitomo N, Sakurada H, Taniguchi K, Matsumura M, Abe O, Miyashita M, Kanamaru H, Karasawa K, Ayusawa M, Fukamizu S, Nagaoka I, Horie M, Harada K, Hiraoka M. Association of atrial arrhythmia and sinus node dysfunction in patients with catecholaminergic polymorphic ventricular tachycardia. Circ J 2007;71:1606–1609. [DOI] [PubMed] [Google Scholar]
- 212.Zhabyeyev P, Hiess F, Wang R, Liu Y, Wayne Chen SR, Oudit GY. S4153R is a gain-of-function mutation in the cardiac Ca(2+) release channel ryanodine receptor associated with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. Can J Cardiol 2013;29:993–996. [DOI] [PubMed] [Google Scholar]
- 213.Camm J. Antiarrhythmic drugs for the maintenance of sinus rhythm: risks and benefits. Int J Cardiol 2012;155:362–371. [DOI] [PubMed] [Google Scholar]
- 214.Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol 2014;5:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun 1991;181:968–975. [DOI] [PubMed] [Google Scholar]
- 216.Mochizuki H, Ito T, Hidaka H. Purification and characterization of Ca2+/calmodulin-dependent protein kinase V from rat cerebrum. J Biol Chem 1993;268:9143–9147. [PubMed] [Google Scholar]
- 217.Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR. Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation. J Biol Chem 1994;269:15520–15527. [PubMed] [Google Scholar]
- 218.Gao Y, Davies SP, Augustin M, Woodward A, Patel UA, Kovelman R, Harvey KJ. A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem J 2013;451:313–328. [DOI] [PubMed] [Google Scholar]
- 219.Li G, Hidaka H, Wollheim CB. Inhibition of voltage-gated Ca2+ channels and insulin secretion in HIT cells by the Ca2+/calmodulin-dependent protein kinase II inhibitor KN-62: comparison with antagonists of calmodulin and L-type Ca2+ channels. Mol Pharmacol 1992;42:489-8. [PubMed] [Google Scholar]
- 220.Rezazadeh S, Claydon TW, Fedida D. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J Pharmacol Exp Ther 2006;317:292–299. [DOI] [PubMed] [Google Scholar]
- 221.Braun AP, Schulman H. A non-selective cation current activated via the multifunctional Ca(2+)-calmodulin-dependent protein kinase in human epithelial cells. J Physiol 1995;488:37–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun 1995;212:806–812. [DOI] [PubMed] [Google Scholar]
- 223.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA 2009;106:2342–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Witczak CA, Jessen N, Warro DM, Toyoda T, Fujii N, Anderson ME, Hirshman MF, Goodyear LJ. CaMKII regulates contraction- but not insulin-induced glucose uptake in mouse skeletal muscle. Am J Physiol Endocrinol Metab 2010;298:E1150–E1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Backs J, Stein P, Backs T, Duncan FE, Grueter CE, McAnally J, Qi X, Schultz RM, Olson EN. The gamma isoform of CaM kinase II controls mouse egg activation by regulating cell cycle resumption. Proc Natl Acad Sci USA 2010;107:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW. CaMKII binding to GluN2B is critical during memory consolidation. EMBO J 2012;31:1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Schulman H, Anderson ME. Ca/calmodulin-dependent protein kinase II in heart failure. Drug Discov Today Dis Mech 2010;7:e117–e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kumarswamy R, Thum T. Non-coding RNAs in cardiac remodeling and heart failure. Circ Res 2013;113:676–689. [DOI] [PubMed] [Google Scholar]
- 229.Cha MJ, Jang JK, Ham O, Song BW, Lee SY, Lee CY, Park JH, Lee J, Seo HH, Choi E, Jeon WM, Hwang HJ, Shin HT, Choi E, Hwang KC. MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIdelta. Biochem Biophys Res Commun 2013;435:720–726. [DOI] [PubMed] [Google Scholar]
- 230.He J, Jiang S, Li FL, Zhao XJ, Chu EF, Sun MN, Chen MZ, Li H. MicroRNA-30b-5p is involved in the regulation of cardiac hypertrophy by targeting CaMKIIdelta. J Investig Med 2013;61:604–612. [DOI] [PubMed] [Google Scholar]
- 231.Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N, Lin H, Xiao L, Maguy A, Qi XY, Li Y, Gao X, Dong D, Zhang Y, Bai Y, Ai J, Sun L, Lu H, Luo XY, Wang Z, Lu Y, Yang B, Nattel S. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest 2013;123:1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]