

Abstract
The insulin-like growth factor (IGF) signaling pathway is an important pathway in the process of hepatocarcinogenesis, and the IGF network is clearly dysregulated in many cancers and developmental abnormalities. In hepatocellular carcinoma (HCC), only a minority of patients are eligible for curative treatments, such as tumor resection or liver transplant. Unfortunately, there is a high recurrence of HCC after surgical tumor removal. Recent research efforts have focused on targeting IGF axis members in an attempt to find therapeutic options for many health problems. In this review, we shed lights on the regulation of members of the IGF axis, mainly by microRNAs in HCC. MicroRNAs in HCC attempt to halt the aberrant expression of the IGF network, and a single microRNA can have multiple downstream targets in one or more signaling pathways. Targeting microRNAs is a relatively new approach for identifying an efficient radical cure for HCC.
Keywords: Hepatocellular carcinoma, Insulin-like growth factors, Insulin-like growth factor receptors, Epigenetics, microRNAs
Core tip: Recent research efforts have focused on targeting the insulin-like growth factor (IGF) axis in an attempt to identify therapeutic options for many health problems. Here, we review the regulation of IGF axis members in hepatocellular carcinoma (HCC), mainly by microRNAs. MicroRNAs work by halting the aberrant expression of the IGF network, as demonstrated by the fact that a single microRNA can have multiple downstream targets in one or more signaling pathways. Use of this approach in an attempt to find an efficient radical cure for HCC.
INTRODUCTION
The insulin-like growth factor (IGF) axis is a highly conserved signaling pathway, with a crucial role in cellular and tissue regeneration through its proliferative and anti-apoptotic activities. This pathway has been shown to be dysregulated in many diseases [1]. Key players of the IGF network include IGF-I and IGF-II ligands that bind to their membrane bound receptor, IGF-type I receptor (IGF-1R), and IGF-type 2 receptor/mannose-6-phosphate receptor (IGF-2R, IGF-II/M6PR). The bioavailability of IGF ligands is controlled through complex formation with IGF binding proteins 1-6 (IGFBP 1-6). In the circulation under physiological conditions, 70% of IGF-II is bound to the most abundant IGFBP-3[2].
Upon binding of IGF ligands to IGF-1R,which is expressed on most cells, its intrinsic tyrosine kinase activity is activated with successive phosphorylation and activation of the downstream intracellular substrates: insulin receptor-substrates 1, 2, and 4 (IRS1, IRS2, and IRS4)[3-5] as well as src homology 2 domain containing (SHC) protein, which is responsible for the stimulation of growth factor receptor-bound protein 2 (Grb2)[6] binding. The phosphatidylinositol 3-kinase (PI3K)[7] pathway is another downstream pathway that is stimulated by IGF ligands. Via the PI3K pathway, PKB/Akt suppresses many apoptosis-stimulating proteins, such as Bad[8] and caspases 9[9], or mitosis-promoting signals through GSK-3β reduction mediated β-catenin degredation[10]. Signaling of Sos/Ras/Raf/MAPK-ERK kinase 1 (MEK1)/extracellular-signal kinase (ERK)[11,12] and translocation of signal transducer and activator of transcription 3 (STAT-3) into the nucleus[13] transmit additional anti-apoptotic and mitogenic stimuli. This triggered regulatory network ends up over stimulating the expression of multiple target genes, such as p27Kip1, c-fos, c-myc, cyclin B, and vascular endothelial growth factor (VEGF). Thus, critical cellular processes are managed via IGF/IGF-1R signaling network, including apoptosis and proliferation.
IGF signaling is important during development, where some mutations in IGF-I and IGF-II have been shown to impair normal birth weight in igfI-null[14,15] and igfII-null mice[15], respectively. On the contrary, overexpression of IGF axis members in some tissues led to drastic outcomes. For example; overexpression of IGF-I protein led to non-small-cell-lung cancer[16], while IGF-II overexpression led to breast cancer[17] and hepatocellular carcinoma (HCC)[18].
EPIGENETIC REGULATION OF OUR GENOME
It is clear that regulation of the IGF axis is quite important, although the precise mechanisms of this regulation remain unclear. Many attempts, however, are being made to target components of the IGF pathway to alleviate various types of pathology.
Epigenetic regulation refers to any changes in genetic phenotype or expression due to mechanisms other than DNA sequence mutations, for example, microRNAs and gene methylation. IGF-2R was imprinted in humans due to fetal overgrowth, teratogenesis, and carcinogenesis [19]. IGF-II is also highly affected by epigenetic mechanism and is found to be highly upregulated in liver tissues of HCC patients due to hypomethylation of its promoter (P4)[20].
DNA methylation
Of the types of epigenetic modification in mammals, DNA methylation is considered to be the most extensively analyzed. DNA methylation maintains the stability of gene silencing, a key process in the regulation of gene expression. DNA methylation in mammals takes place almost exclusively by covalent modification of cytosine residues. This modification is done by adding a methyl group to a cytosine ring at the fifth position in CpG dinucleotides and is mediated by a family of enzymes called DNA methyltransferases (DNMTs)[21]. The majority of CpG dinucleotides are localized in so-called “CpG islands” rather than being evenly distributed across the human genome[22,23]. CpG islands are short DNA stretches that are preferentially found at the 5’ end of genes and comprise about 60% of the sequence of the promoters of human gene[24]. During developmental stages as well as tissue differentiation, most of the CpG islands remain unmethylated. Under normal conditions, these CpG sites are mostly in the methylated state[25]. Interestingly, some CpG sites in some promoters were found to be methylated during development, which resulted in long-term transcriptional silencing.
MicroRNAs
MicroRNAs are small noncoding RNAs that also contribute to the epigenetic regulation of the IGF axis. These non-coding RNAs consist of non-coding DNA sequences that are in full control of gene expression and function, as they collectively target up to 90% of human genes[26]. In addition, some studies revealed that one cluster of two to five microRNAs was able to regulate as much as 14% of all genes[27]. MiRNA transcription depends on genomic localization. MiRNAs can be located in introns of coding or non-coding genes or in exons. Transcription is dependent on the host gene[28,29]. miRNAs that are independently expressed have their own promoter; however, some miRNAs are naturally organized in clusters that share the same transcriptional regulation [30]. The biogenesis of miRNAs involves several processing and trimming steps by RNA endonucleases, with Drosha and dicers giving rise to mature microRNAs (approximately 22 nucleotides), after being transcribed by RNA polymerase II to form its genomic sequence. Mature miRNAs are then incorporated into RNA-induced silencing complex (RISC) through which miRNAs mainly exert their regulatory function by base pairing to a complementary target region in the 3’ untranslated region (3’UTR) of their target gene transcript. Transcripts are regulated either through their degradation or translational repression[31]. More or less, perfect complementarities lead to complete target mRNA degradation. However, imperfect complementarities result in decreased translational expression of the target gene without affecting mRNA level[32,33].
In this review, we shed light on the epigenetic regulation of the IGF axis in HCC and provid a summary of most of the microRNAs involved in the process (Table 1). This information may provide insight on means to restore the expression of those aberrantly expressed microRNAs and the normal expression of IGF axis members.
Table 1.
microRNAs that regulate key players of the insulin-like growth factor axis in hepatocellular carcinoma
| Downstream IGF member | Upstream microRNAs | Net effect on IGF member expression |
| IGF-I | miR-190b | Downregulated |
| miR-486-5p | Upregulated | |
| IGF-II | miR-615-5p | |
| Let-7a | Downregulated | |
| miR-96 | ||
| miR-182 | Upregulated | |
| miR-155 | ||
| IGFBPs: | miR-17-5p | Downregulated |
| IGFBP3 | ||
| IGF-1R | miR-145 | |
| miR-122 | ||
| miR-99a | ||
| miR-223 | Downregulated | |
| miR-486-5p | ||
| miR-182 | ||
| miR-1275 | ||
| miR-181a | ||
| miR-96 | ||
| miR-155 | Upregulated | |
| IGF-2R | miR-211 | Downregulated |
IGF: Insulin-like growth factor.
IGF-I
IGF-I is a potent and crucial growth factor in the IGF signaling network. IGF-I is released by different tissues, including the liver[34]. IGF-I is the key mediator of growth hormone (GH) function in different developmental stages[35]. GH is secreted by the pituitary gland, and it binds to GH receptors in the liver to activate several signaling pathways leading to transcription of several genes, including IGF-I. Hepatocytes are the fundamental source of IGF-I in the liver, with a minimal contribution from non-parenchymal cells[36]. Maximum levels of circulating IGF-I are reached starting from birth up to puberty and then they significantly decline thereafter with age[37]. IGF-I is a crucial ligand for the IGF pathway, and it is highly expressed and known to highly promote the growth of several tumors. Interestingly, this is not the case in liver cancer, where it has been strongly suggested that IGF-I is an anti-tumorgenic factor in HCC. These suggestions were based on several findings: (1) in cases with liver cirrhosis as the type of chronic liver damage, IGF-I expression was minimal or even totally blocked in the most severe cases[38]. Despite minimal IGF-I, cirrhosis progressed into HCC, which suggests that decreased expression of IGF-I is a protumorigenic signal for hepatocarcinogenesis[39]; (2) Low IGF-1 expression levels were also seen in HCC compared to healthy volunteers[40], regardless of the extent of liver function impairment[39]; and (3) decreased IGF-I expression was strongly associated with high tumor invasiveness and poor prognosis[41].
Despite its importance in growth and differentiation, epigenetic regulation of IGF-I has not been yet extensively researched.
microRNAs and IGF-I in HCC
To date, miR-190b is the only microRNA described to regulate IGF-I directly in HCC cells. Ectopic expression of miR-190b resulted in decreased IGF-I expression in hepatoma cell lines. Insulin resistance may be a part of the physiopathologic significance of decreased IGF-I expression in HCC development and progression[42]. Interestingly, one study demonstrated a reciprocal relationship between IGF-I and anti-miR-122. HepG2 cells secreted IGF-I that was able to suppress miR-122 expression in Huh-7 cells. In this experiment, IGF-I hindered intercellular exosomal transfer of miR-122, thereby ensuring its own proliferative signals by repressing growth by retarding miR-122 in neighboring cells[43].
In a recent study by our group, overexpression of the tumor suppressor miR-486-5p in natural killer cells of HCC patients resulted in elevated expression of IGF-I mRNA, and this was directly correlated with induced perforin and NKG2D activating receptor expression(a representatives of natural killer cell cytotoxicity)[44].
IGF-II
IGF-II is a well described growth factor that is involved in neonatal development and progression of many cancers[45]. In contrast to IGF-I, GH does not regulate IGF-II release[2]. Both parenchymal and non-parenchymal liver cells can produce IGF-II[36], and IGF-II expression reaches its maximal level during the fetal stage, where it functions in a pivotal role in fetal development [46]. Nevertheless, after birth, IGF-II levels decline gradually and reach a constant steady state for life[47]. IGF-II has high affinity towards IGF-1R through which it regulates cell growth[2]. More interestingly, IGF-II binds to IGF-2R to induce IGF-II internalization and breakdown[48]. Extensive research has found that the IGF-II ligand is the most epigenetically regulated member among the IGF family.
IGF-II overexpression has been evidenced in 16%-40% of human HCC[49]. Experimental induction of IGF-II expression was positively correlated with enhanced cell growth; moreover, its inhibition promoted apoptosis [50,51].
DNA methylation and IGF-II in HCC
Genomic imprinting is a genetic phenomenon where genes are expressed in a parent-of-origin-specific manner. The IGF-II gene is maternally imprinted in humans[52-53], which emphasizes the importance of gene dosage; and normal development requires controlled accurate expression of IGF2. Moreover, Loss Of Imprinting (i.e., bi-allelic expression) of the IGF-II gene in different tumors explains dysregulation of IGF-II imprinting, which plays a major role in tumorigenesis. IGF-II LOI has been demonstrated in colorectal carcinomas[54], Wilms tumor[55], juvenile nasopharyngeal angiofibromas[56], and childhood acute lymphoblastic leukemia[57]. In the case of bi-allelic expression of IGF-II from both parental IGF-II alleles, the incline in IGF-II production was believed to be the major carcinogenic mechanism[58]. In HCC, IGF-II LOI has been frequently reported to be involved in tumor development. In a study where 71 HCC tissues were analyzed, LOI of the IGF-II locus was reported in 89% of the tissues[59]. This observation suggests that loss of parental-specific imprinting at the IGF-II locus is associated with HCC.
microRNAs and IGF-II in HCC
Emerging studies on the regulation of IGF-II by microRNA are a step towards a new therapeutic approach for the control of many cancers and developmental abnormalities. A link between IGF-II and its intronic-derived miR-483 has been revealed. The miR-483 sequence is located in intron 2 of the IGF-II gene, and the relationship between the intronic miR-483 and its host gene is quite uncommon. miR-483-3p was found to be co-expressed with its host gene IGF-II, although some tumors exhibited a high expression of miR-483-3p without a concomitant increase in IGF-II. Therefore, it was suggested that miR-483-3p might work in cooperation with IGF-II or independently of IGF-II, where it acts as an autonomous oncogene. In addition, inhibition of miR-483-3p by oligonucleotides did not affect IGF-II expression and led to miR-483-3p inhibition only in HepG2 cell lines[60]. While miR-483-3p expression defects had no impact on post-transcriptional IGF-II expression regulation in intrauterine growth restriction [61], it was reported that miR-483-5p was downregulated after Hepa1-6 cells treatment with chromeceptin (IGF-II transcripts inhibitor), since the expression of the entire transcript of IGF-II was silenced.
We reported on several microRNAs that highly controlled IGF-II; beginning with miR-615-5p, which is exclusively expressed in HCC tissues as a compensatory anti-tumor mechanism and totally absent in healthy liver tissues. miR-615-5p intended overexpression was able to markedly suppress IGF-II expression in HuH-7 and HepG2 cells, resulting in inhibited cellular viability, migration, proliferation, and colony-forming ability[62]. MicroRNAs are known to knock down the expression of their target gene. However, in HuH-7 and HepG2 cells, our results showed that overexpression of the oncomiR-155 remarkably induced the expression of IGF-II, which was a highly predicted downstream target by several computational algorithmic tools. The concomitant overexpression of miR-155 and its predicted downstream target IGF-II was highly correlated with enhanced cell proliferation, migration, viability, and clonogenicity[63-65] in addition to the single-clustered hepatic metastamiRs, miR-96 and miR-182, which induced IGF-II mRNA expression following overexpression in HuH-7 cells[66].
The expression of IGF-II is regulated by mechanisms other than gene imprinting and miRNAs. A recently discovered family of mRNA binding proteins called IGF-II mRNA binding proteins (IGF2BPs) was shown to greatly influence IGF-II expression. Of the three IGF2BPs, IGF2BP-1 was reported to repress the IGF-II mRNA translation in growing chicken by binding to its 5’-UTR at multiple sites[67]. Both IGF2BP-2 and 3 are upregulated in HCC, and their expression is strongly correlated with tumor grade, poor prognosis, and metastasis[68]. The regulatory effects of IGF2BP-2 and 3 on IGF-II expression have never been investigated in HCC.
Our research group performed bioinformatics analysis, and it was predicted that let-7a can target both IGF2BP-2 and 3 with a very promising score. This was confirmed via forced expression of let-7a in Huh7 cells, which resulted in a significant downregulation of both IGF2BP-2 and 3 mRNAs. Furthermore, transfecting Huh7 cells with inhibitors of let-7a resulted in restored expression of both IGF2BP-2 and 3 mRNAs. These finding show that let-7a can also downregulate IGF-II indirectly through targeting its regulators, the IGF2BPs[69-70].
microRNA-DNA methylation interplay in IGF-II regulation
Previously, we showed that the let-7a gene was more hypermethylated in HCC tissues than in healthy liver biopsies. The relationship between hypermethylated let-7a3 and IGF-II was further investigated, and it was shown that their expression in HCC tissues demonstrated an inverse relationship. This relationship was further confirmed by forced expression of let-7a in HuH-7 cells, which resulted in IGF-II expression repression[71].
The impact of reactivation of the let-7a-3 gene was determined by utilizing the demethylating drug 5-aza-2’-deoxycytidine (Decitabine). Decitabine passively removes methylation patterns by inhibiting DNMT-1[71]. DNMT-1 is called the maintenance DNMT as it is responsible for the inheritance of methylation patterns in cells over cell divisions[72]. Thus, inhibition of DNMT-1 by Decitabine caused gradual loss of methylation and relief of expression of epigenetically silenced genes[71]. In our work, treating Huh7 cells with high doses of Decitabine for 5 d resulted in significant upregulation of let-7a. Interestingly, the reactivation of let-7a expression resulted in a significant downregulation of IGF-II[69].
IGFBPs
IGFBPs are six high affinity binding proteins (IGFBP1-6) that share 36% homology[73,74]. IGFBPs genes are transcribed in a cell specific manner under tight hormonal and growth factor control[75]. Although IGFBPs are expressed widely among tissues, one or two classes are preferentially produced in each tissue[76]. Liver is the principle source of IGFBPs[36], and circulating IGFBPs bind efficiently to IGF-I and IGF-II[76-77]. Since IGBPs are abundantly expressed, 99% of circulating IGF-I is bound to IGFBPs[75,78]. IGFBPs function by decreasing the bioavailability of IGF ligands, weakening IGF-1R axis. Therefore, some IGFBPs have anti-proliferative effects in human HCC. For instance, ectopic treatment of HepG2 cells by IGFBP3 was able to reverse the carcinogenic effect of exogenously-supplied IGF-I[79]. Consequently, as a cancer hallmark of HCC, IGFBPs were poorly expressed in human HCC[80].
microRNAs and IGFBPs
Recently, research on the biological roles of IGFBPs has expanded. Continuous accumulation of data indicates that IGFBPs are not just responsible for modulating IGF bioactivity but are also responsible for important biological actions independent of their abilities to bind IGFs[81].
Our previous work described miR-17-5p as an oncomiR in HCC that contributed to increase in the proliferation and migration of HuH-7 cells upon miR-17-5p overexpression[82]. In further studies, our group demonstrated a direct relationship between miR-17-5p and IGFBP-3 mRNA expression in HCC patients. However, bioinformatics as well as gain and loss of function experiments of miR-17-5p in HuH-7 cells showed downregulation of IGFBP-3 mRNA following overexpression of miR-17-5p in HuH-7 cells[83]. In addition, miR-96 and miR-182 forced overexpression in HuH-7 cells resulted in IGFBP-3 mRNA expression[65].
IGF-1R
IGF-1R is an important transmembrane gate for IGFs that opens the way for IGF mitogenic pathway activation through its intrinsic tyrosine kinase activity[84]. Binding of IGF-I and IGF-II potently activates the IGF-1R [85]. Consequently, IGF-1R undergoes conformational changes, autophosphorylation of specific tyrosines, and activation of certain docking proteins, insulin-receptor substrate proteins (IRS-1 to -4)[1].
Healthy mature hepatocytes do not express IGF-1R, whereas HCC cells exhibit overexpression and overactivation IGF-1R[86]. Overexpression of IGF-1R in HCC has been extended to its downstream components, such as IRS-1, which correlates with tumor growth [87].
microRNAs and IGF-1R
IGF1R is widely regulated by microRNAs in HCC. miR-145 was shown to be downregulated in HCC, and restoration of its expression in an in vitro model, HKCl-C2, was associated with inhibition of cellular viability and proliferation. The underlying tumor suppressing mechanism of miR-145 was reported to be via targeting IGF-1R together with its downstream docking proteins IRS1 and IRS2 (24690171)[88] and an important circuit involving GSK-3p and CCAAT enhancer binding protein (C/EBP)α. IGF-1R and miR-122 relationship was unveiled, where IGF-1R results in activation of the downstream Akt that led to GSK-3p kinases activation (via de-phosphorylation), which then phosphorylated the transactivator C/EBPα that bound to miR-122 promoter, inducing miR-122 expression. Authors have experimentally validated IGF-1R as a direct target of miR-122 in HepG2 cells, where miR-122 overexpression resulted in IGF-1R downregulation. More interestingly, miR-122 was able to abolish growth signaling through the IGF-1R/Akt/GSK-3p/cyclin D1 pathway, which subsequently resulted in DNA replication and cell cycle progression [89]. miR-99a was also suggested to target IGF-1R in HCC cells. Forced overexpression of miR-99a downregulated IGF-1R, which led to cell cycle arrest, inhibited colonigenicity, and growth of HCC cells[90]. Moreover, overexpression of miR-223 in hepatoma, leukemia, and HeLa cells revealed potent IGF-1R downregulation that resulted in suppressed proliferation, growth rate, and colony formation in vitro and in vivo tumorigenicity in nude mice[91].
In our research group, several microRNAs were described that regulate IGF-1R in HCC. The single-clustered hepatic metastamiRs miR-96 and miR-182 were able to regulate the expression of IGF-IR in a paradoxical manner in Huh-7 cell lines, where forced expression of miR-96 induced IGF-IR mRNA expression in contrast to miR-182, which inhibited IGF-IR mRNA expression in HuH-7 cells[66], in addition to oncomiR-155 that induced IGF-1R mRNA expression upon miR-155 intended overexpression in HuH-7 cells. This finding was closely correlated to increased HuH-7 cell viability, proliferation, migration, and clonogenicity[64,66]. Additionally, we demonstrated that forced overexpression of miR-181a negatively regulated IGF-1R and an IGF-1R regulatory protein, Decorin, in HCC cell lines[92]. Using an immunomodulatory approach, our group showed for the first time that forced expression of miR-486-5p in natural killer cells isolated from HCC patients was able to downregulate IGF-1R together with its downstream signaling proteins STAT3 and mTOR. This downregulation was inversely correlated with the cytotoxicity of HCC-NK cells, which was represented by overexpression of perforin and NKG2D activating receptor upon miR-486-5p overexpression[44].
IGF-2R
IGF-2R is expressed from the IGF-IIR gene, and only 10% of IGF-2Rs are expressed on the cell surface[93]. Its extracellular domain has 15 homologous tandem repeats that enable the receptor to bind to M6P containing proteins as well as M6P free factors[94]. IGF-II is among the M6P free proteins[95] in addition to IGF-I that binds the receptor with lower affinity[95]. The IGF-II-IGF2R complex travels to the endosomal compartment; where the IGF-II is degraded, and the receptor is re-expressed on the cell membrane to capture more ligands[96]. Therefore, in the IGF axis, IGF-2R is the scavenger receptor that regulates IGF-II bioavailability. Accordingly, IGF-2R was demonstrated as a tumor suppressor factor among the mitogenic IGF family [97-99].
IGF-2R expression profile in HCC meets the features of a tumor suppressor, as it is downregulated and closely related with increased IGF-II expression and cellular proliferation in vitro and in vivo[100,101].
DNA methylation of IGF2R
IGF2R gene imprinting profile varies among species[99,102]. Polymorphic expression of IGF2R was evidenced in humans, where the majority showed a biallelic IGF2R expression while some showed imprinted expression[19,103]. These individuals with imprinted expression were liable to develop HCC due to decreased expression and function of IGF2R protein[104].
IGF-2R regulation by microRNAs
IGF-2R regulation by microRNAs exhibits the lowest records to date. This finding might be due to the lack of an intrinsic tyrosine kinase activity, making it not involved in many processes of cellular division or development.
Based on an algorithmic analysis, it was found that miR-453 needs less energy for binding to the 3’UTR of IGF-2R than miR-657, which shows a stronger hybridization of the former to IGF-2R[105]. In malignant melanomas, miR-211 was found to be silenced; and IGF2R, as shown by its expression induction in cell lines, was suppressed. Therefore, silencing of miR-211 may lead to melanoma invasiveness and further progression through loss of control on IGF-2R[106].
CONCLUSION
In the new and rapidly emerging field of microRNAs, implementation of the short non-coding RNAs and RNA interference based treatment in clinical practice is becoming a possibility. In conclusion, the UTRs of IGFs, IGFBP, and IGF receptors have been shown to be targeted by numerous microRNAs that ultimately regulate their targets at the mRNA and/or protein level. This regulation by a big group of upstream microRNAs could be a potentially new area of research for harnessing the undesirable effects of IGF network deregulation, or on the other hand, inducing their developmental functions. It was remarkable to find several microRNAs that can regulate multiple targets solely within one pathway, such as miR-155, miR-96, miR-182, and let-7a (Figure 1). Regulation of such crucial IGF signaling pathway components by microRNAs is still considered a new area for research that needs further investigation. Since most of the studies that investigated IGF axis regulation by microRNAs were done in cancers associated with upregulated mitogenic IGF members, correction of this pathway could potentially tame the wild effects of the IGF proteins.
Figure 1.
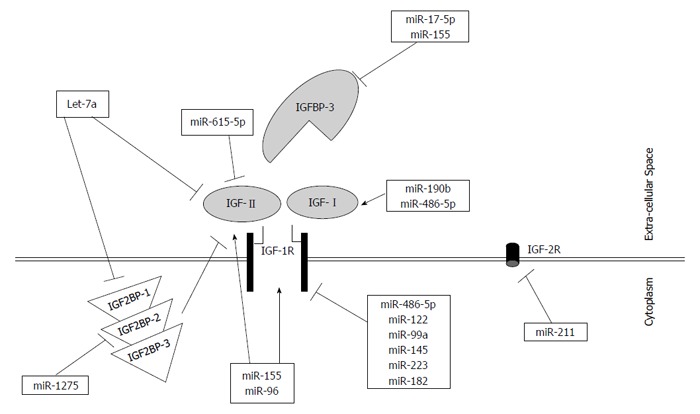
Regulation of insulin-like growth factor signaling pathway by microRNAs in hepatocellular carcinoma. A schematic representation indicating the insulin-like growth factor (IGF) signaling pathway with its different members. MicroRNAs were shown to impact differentially IGF members. Some microRNAs induce the expression of their downstream IGF target (showed as an arrow), while others inhibit the expression of their downstream targets (showed as dash-ended line). Interestingly, some microRNAs showed an inhibitory effect on their downstream target directly as well as indirectly, such as miR-1275 and Let-7a.
ACKNOWLEDGMENTS
The authors acknowledge the English instructor, Ms Sara Magdy, and the English department at the German University in Cairo (GUC) for revising the manuscript.
Footnotes
Conflict-of-interest statement: The authors have no conflict of interest to report.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 7, 2015
First decision: August 31, 2015
Article in press: December 8, 2015
P- Reviewer: Wang K S- Editor: Qi Y L- Editor: Filipodia E- Editor: Wang CH
References
- 1.Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–518. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- 2.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 3.Myers MG, Sun XJ, White MF. The IRS-1 signaling system. Trends Biochem Sci. 1994;19:289–293. doi: 10.1016/0968-0004(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 4.Fantin VR, Sparling JD, Slot JW, Keller SR, Lienhard GE, Lavan BE. Characterization of insulin receptor substrate 4 in human embryonic kidney 293 cells. J Biol Chem. 1998;273:10726–10732. doi: 10.1074/jbc.273.17.10726. [DOI] [PubMed] [Google Scholar]
- 5.He W, Craparo A, Zhu Y, O’Neill TJ, Wang LM, Pierce JH, Gustafson TA. Interaction of insulin receptor substrate-2 (IRS-2) with the insulin and insulin-like growth factor I receptors. Evidence for two distinct phosphotyrosine-dependent interaction domains within IRS-2. J Biol Chem. 1996;271:11641–11645. doi: 10.1074/jbc.271.20.11641. [DOI] [PubMed] [Google Scholar]
- 6.Skolnik EY, Lee CH, Batzer A, Vicentini LM, Zhou M, Daly R, Myers MJ, Backer JM, Ullrich A, White MF. The SH2/SH3 domain-containing protein GRB2 interacts with tyrosine-phosphorylated IRS1 and Shc: implications for insulin control of ras signalling. EMBO J. 1993;12:1929–1936. doi: 10.1002/j.1460-2075.1993.tb05842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamothe B, Bucchini D, Jami J, Joshi RL. Interaction of p85 subunit of PI 3-kinase with insulin and IGF-1 receptors analysed by using the two-hybrid system. FEBS Lett. 1995;373:51–55. doi: 10.1016/0014-5793(95)01011-3. [DOI] [PubMed] [Google Scholar]
- 8.del Peso L, González-García M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 9.Fujita E, Jinbo A, Matuzaki H, Konishi H, Kikkawa U, Momoi T. Akt phosphorylation site found in human caspase-9 is absent in mouse caspase-9. Biochem Biophys Res Commun. 1999;264:550–555. doi: 10.1006/bbrc.1999.1387. [DOI] [PubMed] [Google Scholar]
- 10.Sharma M, Chuang WW, Sun Z. Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta-catenin accumulation. J Biol Chem. 2002;277:30935–30941. doi: 10.1074/jbc.M201919200. [DOI] [PubMed] [Google Scholar]
- 11.Cristofanelli B, Valentinis B, Soddu S, Rizzo MG, Marchetti A, Bossi G, Morena AR, Dews M, Baserga R, Sacchi A. Cooperative transformation of 32D cells by the combined expression of IRS-1 and V-Ha-Ras. Oncogene. 2000;19:3245–3255. doi: 10.1038/sj.onc.1203664. [DOI] [PubMed] [Google Scholar]
- 12.Peruzzi F, Prisco M, Dews M, Salomoni P, Grassilli E, Romano G, Calabretta B, Baserga R. Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol Cell Biol. 1999;19:7203–7215. doi: 10.1128/mcb.19.10.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zong CS, Zeng L, Jiang Y, Sadowski HB, Wang LH. Stat3 plays an important role in oncogenic Ros- and insulin-like growth factor I receptor-induced anchorage-independent growth. J Biol Chem. 1998;273:28065–28072. doi: 10.1074/jbc.273.43.28065. [DOI] [PubMed] [Google Scholar]
- 14.Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N, Stewart TA. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993;7:2609–2617. doi: 10.1101/gad.7.12b.2609. [DOI] [PubMed] [Google Scholar]
- 15.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 16.Kim WY, Kim MJ, Moon H, Yuan P, Kim JS, Woo JK, Zhang G, Suh YA, Feng L, Behrens C, et al. Differential impacts of insulin-like growth factor-binding protein-3 (IGFBP-3) in epithelial IGF-induced lung cancer development. Endocrinology. 2011;152:2164–2173. doi: 10.1210/en.2010-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richardson AE, Hamilton N, Davis W, Brito C, De León D. Insulin-like growth factor-2 (IGF-2) activates estrogen receptor-α and -β via the IGF-1 and the insulin receptors in breast cancer cells. Growth Factors. 2011;29:82–93. doi: 10.3109/08977194.2011.565003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Tayebi HM, Salah W, El Sayed IH, Salam EM, Zekri AR, Zayed N, Salem ES, Esmat G, Abdelaziz AI. Expression of insulin-like growth factor-II, matrix metalloproteinases, and their tissue inhibitors as predictive markers in the peripheral blood of HCC patients. Biomarkers. 2011;16:346–354. doi: 10.3109/1354750X.2011.573095. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Goodyer CG, Deal C, Polychronakos C. Functional polymorphism in the parental imprinting of the human IGF2R gene. Biochem Biophys Res Commun. 1993;197:747–754. doi: 10.1006/bbrc.1993.2542. [DOI] [PubMed] [Google Scholar]
- 20.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787–3800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 21.Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. 2007;61:24R–29R. doi: 10.1203/pdr.0b013e3180457684. [DOI] [PubMed] [Google Scholar]
- 22.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 23.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Leung FC. An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics. 2004;20:1170–1177. doi: 10.1093/bioinformatics/bth059. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 26.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 27.Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, Taccioli C, Zanesi N, Garzon R, Aqeilan RI, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–5171. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin SL, Miller JD, Ying SY. Intronic microRNA (miRNA) J Biomed Biotechnol. 2006;2006:26818. doi: 10.1155/JBB/2006/26818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 32.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 33.Cummins JM, Velculescu VE. Implications of micro-RNA profiling for cancer diagnosis. Oncogene. 2006;25:6220–6227. doi: 10.1038/sj.onc.1209914. [DOI] [PubMed] [Google Scholar]
- 34.Cohick WS, Clemmons DR. The insulin-like growth factors. Annu Rev Physiol. 1993;55:131–153. doi: 10.1146/annurev.ph.55.030193.001023. [DOI] [PubMed] [Google Scholar]
- 35.Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA. 1999;96:7324–7329. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scharf JG, Dombrowski F, Ramadori G. The IGF axis and hepatocarcinogenesis. Mol Pathol. 2001;54:138–144. doi: 10.1136/mp.54.3.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lelbach A, Muzes G, Feher J. The insulin-like growth factor system: IGFs, IGF-binding proteins and IGFBP-proteases. Acta Physiol Hung. 2005;92:97–107. doi: 10.1556/APhysiol.92.2005.2.1. [DOI] [PubMed] [Google Scholar]
- 38.Fernández-Rodriguez CM, Prada I, Andrade A, Moreiras M, Guitián R, Aller R, Lledó JL, Cacho G, Quiroga J, Prieto J. Disturbed synthesis of insulinlike growth factor I and its binding proteins may influence renal function changes in liver cirrhosis. Dig Dis Sci. 2001;46:1313–1320. doi: 10.1023/a:1010631800505. [DOI] [PubMed] [Google Scholar]
- 39.Mazziotti G, Sorvillo F, Morisco F, Carbone A, Rotondi M, Stornaiuolo G, Precone DF, Cioffi M, Gaeta GB, Caporaso N, et al. Serum insulin-like growth factor I evaluation as a useful tool for predicting the risk of developing hepatocellular carcinoma in patients with hepatitis C virus-related cirrhosis: a prospective study. Cancer. 2002;95:2539–2545. doi: 10.1002/cncr.11002. [DOI] [PubMed] [Google Scholar]
- 40.Su WW, Lee KT, Yeh YT, Soon MS, Wang CL, Yu ML, Wang SN. Association of circulating insulin-like growth factor 1 with hepatocellular carcinoma: one cross-sectional correlation study. J Clin Lab Anal. 2010;24:195–200. doi: 10.1002/jcla.20320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaseb AO, Morris JS, Hassan MM, Siddiqui AM, Lin E, Xiao L, Abdalla EK, Vauthey JN, Aloia TA, Krishnan S, et al. Clinical and prognostic implications of plasma insulin-like growth factor-1 and vascular endothelial growth factor in patients with hepatocellular carcinoma. J Clin Oncol. 2011;29:3892–3899. doi: 10.1200/JCO.2011.36.0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hung TM, Ho CM, Liu YC, Lee JL, Liao YR, Wu YM, Ho MC, Chen CH, Lai HS, Lee PH. Up-regulation of microRNA-190b plays a role for decreased IGF-1 that induces insulin resistance in human hepatocellular carcinoma. PLoS One. 2014;9:e89446. doi: 10.1371/journal.pone.0089446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basu S, Bhattacharyya SN. Insulin-like growth factor-1 prevents miR-122 production in neighbouring cells to curtail its intercellular transfer to ensure proliferation of human hepatoma cells. Nucleic Acids Res. 2014;42:7170–7185. doi: 10.1093/nar/gku346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bassiouni AA, Rahmoon MA, Youness RA, Gomaa AI, Waked I, Esmat G, El Tayebi HM, Ahmed I. Abdelaziz Improving Natural Killer Cells Cytotoxicty And Harnessing Hepatocyte Cancer Progression By Mir-486-5p. In: European Associaton for the study of Liver. Hepatolopy. 2015;62:S495. [Google Scholar]
- 45.Schirmacher P, Held WA, Yang D, Chisari FV, Rustum Y, Rogler CE. Reactivation of insulin-like growth factor II during hepatocarcinogenesis in transgenic mice suggests a role in malignant growth. Cancer Res. 1992;52:2549–2556. [PubMed] [Google Scholar]
- 46.Yaseen MA, Wrenzycki C, Herrmann D, Carnwath JW, Niemann H. Changes in the relative abundance of mRNA transcripts for insulin-like growth factor (IGF-I and IGF-II) ligands and their receptors (IGF-IR/IGF-IIR) in preimplantation bovine embryos derived from different in vitro systems. Reproduction. 2001;122:601–610. [PubMed] [Google Scholar]
- 47.Daughaday WH, Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989;10:68–91. doi: 10.1210/edrv-10-1-68. [DOI] [PubMed] [Google Scholar]
- 48.Oka Y, Rozek LM, Czech MP. Direct demonstration of rapid insulin-like growth factor II Receptor internalization and recycling in rat adipocytes. Insulin stimulates 125I-insulin-like growth factor II degradation by modulating the IGF-II receptor recycling process. J Biol Chem. 1985;260:9435–9442. [PubMed] [Google Scholar]
- 49.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989–5005. doi: 10.1038/onc.2010.236. [DOI] [PubMed] [Google Scholar]
- 50.Lund P, Schubert D, Niketeghad F, Schirmacher P. Autocrine inhibition of chemotherapy response in human liver tumor cells by insulin-like growth factor-II. Cancer Lett. 2004;206:85–96. doi: 10.1016/j.canlet.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 51.Yao X, Hu JF, Daniels M, Shiran H, Zhou X, Yan H, Lu H, Zeng Z, Wang Q, Li T, et al. A methylated oligonucleotide inhibits IGF2 expression and enhances survival in a model of hepatocellular carcinoma. J Clin Invest. 2003;111:265–273. doi: 10.1172/JCI15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wollmann H, Berger F. Epigenetic reprogramming during plant reproduction and seed development. Curr Opin Plant Biol. 2012;15:63–69. doi: 10.1016/j.pbi.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 53.Krassas GE, Pontikides N, Kaltsas T, Dumas A, Frystyk J, Chen JW, Flyvbjerg A. Free and total insulin-like growth factor (IGF)-I, -II, and IGF binding protein-1, -2, and -3 serum levels in patients with active thyroid eye disease. J Clin Endocrinol Metab. 2003;88:132–135. doi: 10.1210/jc.2002-021349. [DOI] [PubMed] [Google Scholar]
- 54.Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenburg S, Wu Y, He X, Powe NR, Feinberg AP. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299:1753–1755. doi: 10.1126/science.1080902. [DOI] [PubMed] [Google Scholar]
- 55.Ogawa O, Becroft DM, Morison IM, Eccles MR, Skeen JE, Mauger DC, Reeve AE. Constitutional relaxation of insulin-like growth factor II gene imprinting associated with Wilms’ tumour and gigantism. Nat Genet. 1993;5:408–412. doi: 10.1038/ng1293-408. [DOI] [PubMed] [Google Scholar]
- 56.Coutinho-Camillo CM, Brentani MM, Butugan O, Torloni H, Nagai MA. Relaxation of imprinting of IGFII gene in juvenile nasopharyngeal angiofibromas. Diagn Mol Pathol. 2003;12:57–62. doi: 10.1097/00019606-200303000-00008. [DOI] [PubMed] [Google Scholar]
- 57.Vorwerk P, Wex H, Bessert C, Hohmann B, Schmidt U, Mittler U. Loss of imprinting of IGF-II gene in children with acute lymphoblastic leukemia. Leuk Res. 2003;27:807–812. doi: 10.1016/s0145-2126(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 58.Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- 59.Poirier K, Chalas C, Tissier F, Couvert P, Mallet V, Carrié A, Marchio A, Sarli D, Gicquel C, Chaussade S, et al. Loss of parental-specific methylation at the IGF2 locus in human hepatocellular carcinoma. J Pathol. 2003;201:473–479. doi: 10.1002/path.1477. [DOI] [PubMed] [Google Scholar]
- 60.Veronese A, Lupini L, Consiglio J, Visone R, Ferracin M, Fornari F, Zanesi N, Alder H, D’Elia G, Gramantieri L, et al. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 2010;70:3140–3149. doi: 10.1158/0008-5472.CAN-09-4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tabano S, Colapietro P, Cetin I, Grati FR, Zanutto S, Mandò C, Antonazzo P, Pileri P, Rossella F, Larizza L, et al. Epigenetic modulation of the IGF2/H19 imprinted domain in human embryonic and extra-embryonic compartments and its possible role in fetal growth restriction. Epigenetics. 2010;5:313–324. doi: 10.4161/epi.5.4.11637. [DOI] [PubMed] [Google Scholar]
- 62.El Tayebi HM, Hosny KA, Esmat G, Breuhahn K, Abdelaziz AI. miR-615-5p is restrictedly expressed in cirrhotic and cancerous liver tissues and its overexpression alleviates the tumorigenic effects in hepatocellular carcinoma. FEBS Lett. 2012;586:3309–3316. doi: 10.1016/j.febslet.2012.06.054. [DOI] [PubMed] [Google Scholar]
- 63.El Tayebi HM, Hosny KA, Esmat G, Breuhahn K, Abdelaziz AI. 268 Mir-155 is an inducer for the insulin-like growth factor ii mitogenic signaling pathway in HCV-progressed hepatocellular carcinoma. J Hepatol. 2012;56:S112–S112. [Google Scholar]
- 64.El Tayebi HM, Assal RA, Hosny KA, Esmat G, Abdelaziz AI. Transcriptional activation of the IGF-II/IGF-1R axis and inhibition of IGFBP-3 by miR-155 in hepatocellular carcinoma. Oncol Lett. 2015;10:3206–3212. doi: 10.3892/ol.2015.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.El Tayebi HM, Waly AA. miR-155 induces mTOR expression via activation of IGF-II/IGF-1R axis and inhibitionof IGFBP-3 in Hepatocellular Carcinoma. In: American Association for Cancer Research (AACR), editor. Philadelphia: AACR; 2014. [Google Scholar]
- 66.Assal RA, El Tayebi HM, Hosny KA, Esmat G, Abdelaziz AI. A pleiotropic effect of the single clustered hepatic metastamiRs miR-96-5p and miR-182-5p on insulin-like growth factor II, insulin-like growth factor-1 receptor and insulin-like growth factor-binding protein-3 in hepatocellular carcinoma. Mol Med Rep. 2015;12:645–650. doi: 10.3892/mmr.2015.3382. [DOI] [PubMed] [Google Scholar]
- 67.Nielsen J, Christiansen J, Lykke-Andersen J, Johnsen AH, Wewer UM, Nielsen FC. A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol Cell Biol. 1999;19:1262–1270. doi: 10.1128/mcb.19.2.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jeng YM, Chang CC, Hu FC, Chou HY, Kao HL, Wang TH, Hsu HC. RNA-binding protein insulin-like growth factor II mRNA-binding protein 3 expression promotes tumor invasion and predicts early recurrence and poor prognosis in hepatocellular carcinoma. Hepatology. 2008;48:1118–1127. doi: 10.1002/hep.22459. [DOI] [PubMed] [Google Scholar]
- 69.Waly AA, El Tayebi HM, Hosny KA, Esmat G, Abdelaziz AI. Unshielding Igf-Ii Mrna By Targeting Igf2bps Through Demethylating The Let-7a-3 Gene In Hcc Cell Lines. In: EASL HCC Summit. Hepatology; 2014. [Google Scholar]
- 70.Waly AA, El Tayebi HM, Hosny KA, Esmat G, Abdelaziz AI. 1097 Hypermethylation of the microRNA let-7a-3 gene represses the primary and mature let-7a-3 with an inverse correlation to IGF-II mRNA in HCV-induced Hepatocellular Carcinoma. Hepatology. 2013;58 Suppl 1:S448–S449. [Google Scholar]
- 71.Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 72.Hermann A, Schmitt S, Jeltsch A. The human Dnmt2 has residual DNA-(cytosine-C5) methyltransferase activity. J Biol Chem. 2003;278:31717–31721. doi: 10.1074/jbc.M305448200. [DOI] [PubMed] [Google Scholar]
- 73.Rosenfeld RG, Lamson G, Pham H, Oh Y, Conover C, De Leon DD, Donovan SM, Ocrant I, Giudice L. Insulinlike growth factor-binding proteins. Recent Prog Horm Res. 1990;46:99–159; discussion 159-63. doi: 10.1016/b978-0-12-571146-3.50009-2. [DOI] [PubMed] [Google Scholar]
- 74.Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr Rev. 1999;20:761–787. doi: 10.1210/edrv.20.6.0382. [DOI] [PubMed] [Google Scholar]
- 75.Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst. 2000;92:1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 76.Mohan S, Baylink DJ. IGF-binding proteins are multifunctional and act via IGF-dependent and -independent mechanisms. J Endocrinol. 2002;175:19–31. doi: 10.1677/joe.0.1750019. [DOI] [PubMed] [Google Scholar]
- 77.Wu J, Zhu AX. Targeting insulin-like growth factor axis in hepatocellular carcinoma. J Hematol Oncol. 2011;4:30. doi: 10.1186/1756-8722-4-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rajaram S, Baylink DJ, Mohan S. Insulin-like growth factor-binding proteins in serum and other biological fluids: regulation and functions. Endocr Rev. 1997;18:801–831. doi: 10.1210/edrv.18.6.0321. [DOI] [PubMed] [Google Scholar]
- 79.Huynh H, Chow PK, Ooi LL, Soo KC. A possible role for insulin-like growth factor-binding protein-3 autocrine/paracrine loops in controlling hepatocellular carcinoma cell proliferation. Cell Growth Differ. 2002;13:115–122. [PubMed] [Google Scholar]
- 80.Alexia C, Fallot G, Lasfer M, Schweizer-Groyer G, Groyer A. An evaluation of the role of insulin-like growth factors (IGF) and of type-I IGF receptor signalling in hepatocarcinogenesis and in the resistance of hepatocarcinoma cells against drug-induced apoptosis. Biochem Pharmacol. 2004;68:1003–1015. doi: 10.1016/j.bcp.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 81.Oh Y, Müller HL, Lamson G, Rosenfeld RG. Insulin-like growth factor (IGF)-independent action of IGF-binding protein-3 in Hs578T human breast cancer cells. Cell surface binding and growth inhibition. J Biol Chem. 1993;268:14964–14971. [PubMed] [Google Scholar]
- 82.El Tayebi HM, Omar K, Hegy S, El Maghrabi M, El Brolosy M, Hosny KA, Esmat G, Abdelaziz AI. Repression of miR-17-5p with elevated expression of E2F-1 and c-MYC in non-metastatic hepatocellular carcinoma and enhancement of cell growth upon reversing this expression pattern. Biochem Biophys Res Commun. 2013;434:421–427. doi: 10.1016/j.bbrc.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 83.Habashy DA, Hosny KA, Esmat G, Abdelaziz AI. MicroRNA-17-5p promotes tumorgenesis through the repression of IGFBP-3 and enhancement of Insulin-like growth factor signaling. In: European Society of Pathology., editor. Lisbon: Virchow Archives; 2013. [Google Scholar]
- 84.Riedemann J, Macaulay VM. IGF1R signalling and its inhibition. Endocr Relat Cancer. 2006;13 Suppl 1:S33–S43. doi: 10.1677/erc.1.01280. [DOI] [PubMed] [Google Scholar]
- 85.Roth RA, Kiess W. Insulin-like growth factor receptors: recent developments and new methodologies. Growth Regul. 1994;4 Suppl 1:31–38. [PubMed] [Google Scholar]
- 86.Aleem E, Nehrbass D, Klimek F, Mayer D, Bannasch P. Upregulation of the insulin receptor and type I insulin-like growth factor receptor are early events in hepatocarcinogenesis. Toxicol Pathol. 2011;39:524–543. doi: 10.1177/0192623310396905. [DOI] [PubMed] [Google Scholar]
- 87.Tanaka S, Wands JR. Insulin receptor substrate 1 overexpression in human hepatocellular carcinoma cells prevents transforming growth factor beta1-induced apoptosis. Cancer Res. 1996;56:3391–3394. [PubMed] [Google Scholar]
- 88.Law PT, Ching AK, Chan AW, Wong QW, Wong CK, To KF, Wong N. MiR-145 modulates multiple components of the insulin-like growth factor pathway in hepatocellular carcinoma. Carcinogenesis. 2012;33:1134–1141. doi: 10.1093/carcin/bgs130. [DOI] [PubMed] [Google Scholar]
- 89.Zeng C, Wang R, Li D, Lin XJ, Wei QK, Yuan Y, Wang Q, Chen W, Zhuang SM. A novel GSK-3 beta-C/EBP alpha-miR-122-insulin-like growth factor 1 receptor regulatory circuitry in human hepatocellular carcinoma. Hepatology. 2010;52:1702–1712. doi: 10.1002/hep.23875. [DOI] [PubMed] [Google Scholar]
- 90.Li D, Liu X, Lin L, Hou J, Li N, Wang C, Wang P, Zhang Q, Zhang P, Zhou W, et al. MicroRNA-99a inhibits hepatocellular carcinoma growth and correlates with prognosis of patients with hepatocellular carcinoma. J Biol Chem. 2011;286:36677–36685. doi: 10.1074/jbc.M111.270561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jia CY, Li HH, Zhu XC, Dong YW, Fu D, Zhao QL, Wu W, Wu XZ. MiR-223 suppresses cell proliferation by targeting IGF-1R. PLoS One. 2011;6:e27008. doi: 10.1371/journal.pone.0027008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Assal RA, El Tayebi HM, Hosny KA, Esmat G, Abdelaziz AI. Despite repressing IGF-1R, miR-181a halts the tumor supressor activity of Decorin by enhancing tumor progression in Hepatocellular Carcinoma. In: European Association for the Study of Liver (EASL), editor. Vienna: Austria, Hepatology; 2015. [Google Scholar]
- 93.Killian JK, Jirtle RL. Genomic structure of the human M6P/IGF2 receptor. Mamm Genome. 1999;10:74–77. doi: 10.1007/s003359900947. [DOI] [PubMed] [Google Scholar]
- 94.Morgan DO, Edman JC, Standring DN, Fried VA, Smith MC, Roth RA, Rutter WJ. Insulin-like growth factor II receptor as a multifunctional binding protein. Nature. 1987;329:301–307. doi: 10.1038/329301a0. [DOI] [PubMed] [Google Scholar]
- 95.Gary-Bobo M, Nirdé P, Jeanjean A, Morère A, Garcia M. Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr Med Chem. 2007;14:2945–2953. doi: 10.2174/092986707782794005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Espelund U, Bruun JM, Richelsen B, Flyvbjerg A, Frystyk J. Pro- and mature IGF-II during diet-induced weight loss in obese subjects. Eur J Endocrinol. 2005;153:861–869. doi: 10.1530/eje.1.02028. [DOI] [PubMed] [Google Scholar]
- 97.De Souza AT, Hankins GR, Washington MK, Orton TC, Jirtle RL. M6P/IGF2R gene is mutated in human hepatocellular carcinomas with loss of heterozygosity. Nat Genet. 1995;11:447–449. doi: 10.1038/ng1295-447. [DOI] [PubMed] [Google Scholar]
- 98.De Souza AT, Hankins GR, Washington MK, Fine RL, Orton TC, Jirtle RL. Frequent loss of heterozygosity on 6q at the mannose 6-phosphate/insulin-like growth factor II receptor locus in human hepatocellular tumors. Oncogene. 1995;10:1725–1729. [PubMed] [Google Scholar]
- 99.Mills JJ, Falls JG, De Souza AT, Jirtle RL. Imprinted M6p/Igf2 receptor is mutated in rat liver tumors. Oncogene. 1998;16:2797–2802. doi: 10.1038/sj.onc.1201801. [DOI] [PubMed] [Google Scholar]
- 100.Chen Z, Ge Y, Landman N, Kang JX. Decreased expression of the mannose 6-phosphate/insulin-like growth factor-II receptor promotes growth of human breast cancer cells. BMC Cancer. 2002;2:18. doi: 10.1186/1471-2407-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.O’Gorman DB, Costello M, Weiss J, Firth SM, Scott CD. Decreased insulin-like growth factor-II/mannose 6-phosphate receptor expression enhances tumorigenicity in JEG-3 cells. Cancer Res. 1999;59:5692–5694. [PubMed] [Google Scholar]
- 102.Barlow DP, Stöger R, Herrmann BG, Saito K, Schweifer N. The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature. 1991;349:84–87. doi: 10.1038/349084a0. [DOI] [PubMed] [Google Scholar]
- 103.Kalscheuer VM, Mariman EC, Schepens MT, Rehder H, Ropers HH. The insulin-like growth factor type-2 receptor gene is imprinted in the mouse but not in humans. Nat Genet. 1993;5:74–78. doi: 10.1038/ng0993-74. [DOI] [PubMed] [Google Scholar]
- 104.De Souza AT, Yamada T, Mills JJ, Jirtle RL. Imprinted genes in liver carcinogenesis. FASEB J. 1997;11:60–67. doi: 10.1096/fasebj.11.1.9034167. [DOI] [PubMed] [Google Scholar]
- 105.Lv K, Guo Y, Zhang Y, Wang K, Jia Y, Sun S. Allele-specific targeting of hsa-miR-657 to human IGF2R creates a potential mechanism underlying the association of ACAA-insertion/deletion polymorphism with type 2 diabetes. Biochem Biophys Res Commun. 2008;374:101–105. doi: 10.1016/j.bbrc.2008.06.102. [DOI] [PubMed] [Google Scholar]
- 106.Levy C, Khaled M, Iliopoulos D, Janas MM, Schubert S, Pinner S, Chen PH, Li S, Fletcher AL, Yokoyama S, et al. Intronic miR-211 assumes the tumor suppressive function of its host gene in melanoma. Mol Cell. 2010;40:841–849. doi: 10.1016/j.molcel.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]