

Abstract
AIM: To investigate the role of Gadd45a in hepatic fibrosis and the transforming growth factor (TGF)-β/Smad signaling pathway.
METHODS: Wild-type male BALB/c mice were treated with CCl4 to induce a model of chronic liver injury. Hepatic stellate cells (HSCs) were isolated from the liver of BALB/c mice and were treated with small interfering RNAs (siRNAs) targeting Gadd45a or the pcDNA3.1-Gadd45a recombinant plasmid. Cellular α-smooth muscle actin (α-SMA), β-actin, type I collagen, phospho-Smad2, phospho-Smad3, Smad2, Smad3, and Smad4 were detected by Western blots. The mRNA levels of α-SMA, β-actin, and type I collagen were determined by quantitative real-time (qRT)-PCR analyses. Reactive oxygen species production was monitored by flow cytometry using 2,7-dichlorodihydrofluorescein diacetate. Gadd45a, Gadd45b, anti-Gadd45g, type I collagen, and SMA local expression in liver tissue were measured by histologic and immunohistochemical analyses.
RESULTS: Significant downregulation of Gadd45a, but not Gadd45b or Gadd45g, accompanied by activation of the TGF-β/Smad signaling pathways was detected in fibrotic liver tissues of mice and isolated HSCs with chronic liver injury induced by CCl4 treatment. Overexpression of Gadd45a reduced the expression of extracellular matrix proteins and α-SMA in HSCs, whereas transient knockdown of Gadd45a with siRNA reversed this process. Gadd45a inhibited the activity of a plasminogen activator inhibitor-1 promoter construct and (CAGA)9 MLP-Luc, an artificial Smad3/4-specific reporter, as well as reduced the phosphorylation and nuclear translocation of Smad3. Gadd45a showed protective effects by scavenging reactive oxygen species and upregulating antioxidant enzymes.
CONCLUSION: Gadd45a may counteract hepatic fibrosis by regulating the activation of HSCs via the inhibition of TGF-β/Smad signaling.
Keywords: Antioxidant, Gadd45a, Hepatic fibrosis, Hepatic stellate cells, Transforming growth factor-β/Smad signaling
Core tip: The data in this paper provide evidence that Gadd45a may exert a protective effect against hepatic fibrosis induced by CCl4, via the inhibition of canonical transforming growth factor-β/Smad signaling and fibrogenic gene expression. We also propose a molecular basis for the antioxidant potential of Gadd45a in the hepatic fibrosis process. Although clinical methods for the direct targeting of hepatic stellate cells remain under development, our study provides an important advance in the understanding of the biologic functions of Gadd45a and a potential target for the treatment of hepatic fibrosis.
INTRODUCTION
Hepatic fibrosis is a wound-healing response characterized by the accumulation of extracellular matrix (ECM) following liver injury, and may lead to cirrhosis and hepatocellular carcinoma. Therefore, hepatic fibrosis is responsible for significant morbidity and mortality[1,2]. All types of liver fibrosis, regardless of etiology, cause excessive deposition of ECM, such as collagens, proteoglycans, fibronectin, and a variety of glycoproteins[3]. Activated hepatic stellate cells (HSCs) play a vital role in the fibrogenic and inflammatory response by increasing cell proliferation, excessive deposition of fibrous collagen-rich ECM, and expression of α-smooth muscle actin (α-SMA)[4,5]. Moreover, activated HSCs also contribute to the immunoregulatory response to injury through the production of chemokines and cytokines[6]. Although the role of HSCs in hepatic fibrosis has been well established, the detailed underlying mechanisms remain incompletely understood.
Gadd45 proteins, a family of p53-regulated and DNA damage-inducible proteins, have been found to negatively regulate cell growth and apoptosis[7-9]. The three proteins, known as Gadd45a, Gadd45b, and Gadd45g, share 55-58% amino acid homology and function as stress sensors in response to various physiologic or environmental stresses, including methyl methanesulfonate, hypoxia, ionizing radiation, UV radiation, cisplatin, growth factor withdrawal, and medium depletion, finally resulting in cell cycle arrest, DNA repair, genomic stability, and apoptosis[8-12]. Among the three proteins, Gadd45a can interact with key cell regulators such as p21[13,14], cdc2/cyclinB1[15], proliferating cell nuclear antigen[14,16], p38[17], and MAP kinase kinase kinase (MEKK4)[18]. Recent reports have suggested that downregulation of Gadd45a and Gadd45g proteins contributes to nuclear factor (NF)-κB-mediated cell survival, as inhibition of NF-κB in cancer cells results in the Gadd45a- and Gadd45g-dependent induction of apoptosis via the activation of c-Jun N-terminal kinase (JNK)[19]. Furthermore, all three Gadd45 proteins can bind to and activate MEKK4[18], thus leading to the activation of both JNK and p38; in addition, it has been proposed that the induction of Gadd45 is required for normal activation of JNK and p38 in response to cellular stress induced by UV radiation[20]. Therefore, Gadd45 proteins have been shown to play similar but not identical roles, depending on the particular stress response pathway that is activated[21,22].
In addition, Gadd45 proteins play crucial roles in transforming growth factor (TGF)-β-mediated apoptosis of hepatocytes and myeloid cells[23]. TGF-β induces the phosphorylation of Smad2 and Smad3 proteins through a heteromeric complex of type I and type I serine/threonine kinase receptors[24,25]. In fibrotic liver, enhanced TGF-β signaling stimulates transdifferentiation of HSCs to α-SMA-positive myofibroblasts that are the main producers of excessive ECM proteins[26,27]. Therefore, the aim of this study was to clarify the role of both Gadd45- and Smad-independent TGF-β signaling pathways so that a promising therapeutic strategy for the treatment of hepatic fibrosis can be developed[28].
MATERIALS AND METHODS
Model of chronic liver injury
Wild-type male BALB/c mice (6-8 wk) were obtained from the Shanghai Laboratory Animal Company (Shanghai, China), maintained on a 12-h light/dark cycle at 25 °C, and were provided free access to commercial rodent food and tap water before the experiments. To induce chronic liver injury, mice were administered CCl4 by gavage, biweekly for up to 4 wk. CCl4 was diluted in mineral oil as described previously[29]. The first dose of CCl4 was 100 μL/kg body weight, and subsequent doses were adjusted, based on weight changes 48 h after the most recent dose. Control animals received mineral oil only. These experiments were conducted in accordance with the Shanghai Jiao Tong University Guidelines for the Care and Use of Laboratory Animals.
HSC isolation and transient transfection
HSCs were isolated by perfusion with collagenase and cultured as described previously[30]. In brief, HSCs were isolated from the liver of BALB/c mice by perfusion with Hanks’ balanced salt solution (HBSS), then 0.4% protease (Sigma-Aldrich, St. Louis, MO, United States), followed by 0.01% collagenase (Sigma-Aldrich) solutions in Dulbecco’s modified Eagle medium (DMEM)/F12 medium. After gentle mechanical disruption, the resulting suspension was incubated at 37 °C in a 0.4% collagenase solution in DMEM/F12 with 0.5 mg/mL DNAase for 20 min. The supernatant was then centrifuged and resuspended in 20% Optiprep (Sigma-Aldrich) diluted in HBSS and overlaid with 11.2% Optiprep, then overlaid with HBSS and centrifuged at 1400 × g for 17 min. HSCs were harvested from the interface of the Optiprep and HBSS layers, and were washed twice with complete RPMI 1640 containing penicillin, streptomycin, and amphotericin B (Lonza, Basel, Switzerland). For overexpression or silencing of Gadd45 proteins, Lipofectamine Plus (Invitrogen of Thermo Fisher Scientific, Waltham, MA, United States) was used to transfect small interfering RNAs (siRNAs) targeting Gadd45a or pcDNA3.1-Gadd45a plasmid into HSCs according to the manufacturer’s protocol. Cells were collected for quantitative real-time (qRT)-PCR and Western blot detection after 48 h.
Western blot analysis
Cellular proteins were extracted with lysis buffer and loaded on a 10%-12% sodium dodecyl sulfate-polyacrylamide gel. After 1 h of separation by electrophoresis, the proteins were transferred electrophoretically to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, United States). The membranes were blocked with 5% nonfat dried milk in tris-buffered saline with tween for 1 h and then incubated with primary antibodies against α-SMA, β-actin (Abcam, Cambridge, United Kingdom), type I collagen, phospho-Smad2, phospho-Smad3, Smad2, Smad3, and Smad4 (Cell Signaling Technology, Inc., Danvers, MA, United States) at the appropriate concentrations (1:1000). Membranes were probed with anti-mouse or anti-rabbit immunoglobulin (Ig)G secondary antibodies conjugated to horseradish peroxidase and then visualized using an enhanced chemiluminescence detection system (EMD Millipore).
qRT-PCR analyses
Total RNA from microglia was extracted using TRIzol reagent (Omega Bio-Tek, Inc., Norcross, GA, United States), and 2 μL of purified RNA was transcribed to cDNA by using the PrimeScript RT reagent kit according to the manufacturer’s instructions. The cDNA was used as the template, and qPCR was performed with the Roche LightCycler 480 system (Roche Holding AG, Basel, Switzerland) by using SYBR Premix Ex Taq. The threshold cycle (Ct) values were used for calculating the relative expression ratios of SK channel genes. The relative gene expression was calculated with the ∆∆Ct equation: ∆Ct = Cttarget - CtGAPDH, ∆∆Ct = ∆Cttarget - ∆Ctcontrol, where 2-∆∆Ct = relative expression, and was directly proportional to gene expression. The gene-specific primers used in this study are shown in Table 1.
Table 1.
Primers used in this study
| Gene | Forward primer | Reverse primer |
| Collagen I | GGAATCCGGGGTTTACCTGG | AGGCGACCCTCTGATACCTT |
| Fibronectin | GAGCCGGACAACTTCTGGTC | ATTTGCTGAGCCTGCCTCTT |
| α-SMA | TCTTCCAGCCTTCCTTTA | ATGTCAATGTCACACTTCA |
| Gadd45g | CAATGTGACCTTCTGTGT | ATCAGCGTAAAATGGATCT |
| Gadd45a | GACGAATCCACATTCATCTC | TTGATCCATGTAGCGACTT |
| Gadd45b | ATTGACGAGGAGGAGGAG | GTTGTCACAGCAGAAGGA |
| GADPH | CTCTGGTAAAGTGGATATTGT | GGTGGAATCATATTGGAACA |
SMA: Smooth muscle actin.
Measurement of reactive oxygen species production
Measurement of reactive oxygen species (ROS) production was monitored by flow cytometry using 2,7-dichlorodihydrofluorescein diacetate. This dye is a stable nonpolar compound that readily diffuses into cells and is hydrolyzed by intracellular esterases to yield 2,7-dichlorodihydrofluorescein, which is then trapped within the cells. Hydrogen peroxide and low-molecular-weight peroxides produced by the cells oxidize 2,7-dichlorodihydrofluorescein to the highly fluorescent compound 2,7-dichlorofluorescein. Thus, the fluorescence intensity is proportional to the amount of peroxide produced by the cells. After treatment, HSCs were incubated in 20 μmol/L 2,7-dichlorodihydrofluorescein diacetate for 30 min. The cells were harvested, washed once, and resuspended in phosphate-buffered saline. Fluorescence was monitored using a flow cytometer.
Histologic and immunohistochemical analysis
Liver tissue was collected by perfusion, fixed with HBSS containing 4% paraformaldehyde, and embedded in paraffin. Serial 4 μm sections were subjected to hematoxylin and picrosirius red staining using standard procedures. For immunohistochemical analysis, deparaffinized sections were incubated with anti-Gadd45a, anti-Gadd45b, anti-Gadd45g, anti-type I collagen, or anti-α-SMA primary antibodies followed by horseradish peroxidase-conjugated anti-rabbit IgG secondary antibodies, according to the manufacturer’s instructions. α-SMA-positive areas, indicated by the brown color, within the fibrotic region were then observed.
Statistical analysis
Data are expressed as mean ± SD. Statistical significance was determined by the Student’s t test for comparison between means or one-way analysis of variance with the post hoc Dunnett’s test. Differences were considered significant at P < 0.05.
RESULTS
Chronic CCl4 injury increases fibrogenic protein expression and ECM deposition
As shown in Figure 1A, the relative mRNA expression of hepatic collagen I, fibronectin, and α-SMA was enhanced in mice treated with CCl4. Collagen I and α-SMA expression reached a peak after 4 wk, and fibronectin expression reached a peak after 3 wk. Moreover, Western blot analysis showed similar results, with a gradual increase of collagen I, fibronectin, and α-SMA protein expression levels in a time-dependent manner (Figure 1B). Furthermore, picrosirius red staining showed that the deposition of collagen fibers was increased in the liver of mouse models (Figure 1C). These results demonstrate that the chronic liver injury model was successfully established.
Figure 1.
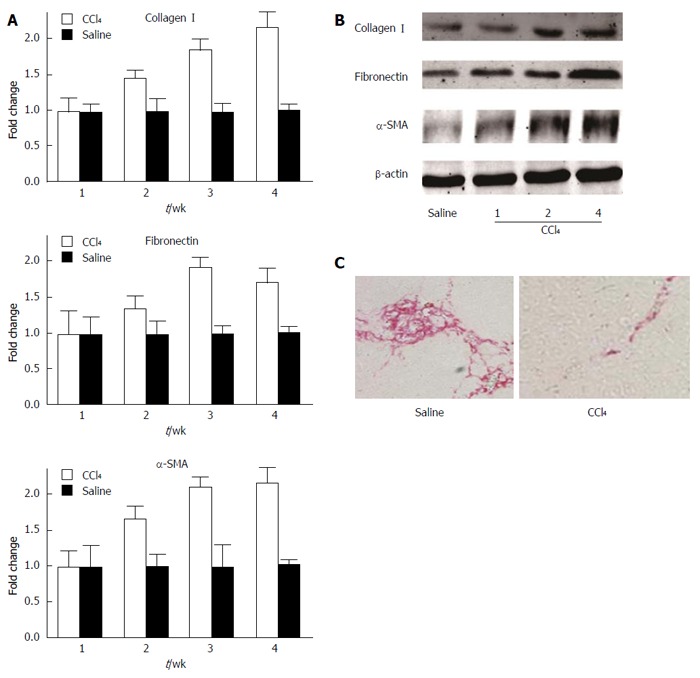
Expression of fibrogenic proteins and extracellular matrix deposition in the liver of mice exposed to CCl4. A: Relative mRNA expression of collagen I, fibronectin, and α-SMA after 1, 2, 3, and 4 wk of CCl4 treatment, as determined by quantitative RT-PCR; B: Protein expression of collagen I, fibronectin, and α-SMA after 1, 2, and 4 wk of CCl4 treatment, as determined by Western blot; C: Picrosirius red staining shows hepatic collagen deposition in mice treated with CCl4 for 4 wk; saline treatment served as the control group. SMA: Smooth muscle actin.
Chronic CCl4 injury suppresses Gadd45a expression and activates TGF-β/Smad signaling
To investigate the potential role of Gadd45 in chronic liver injury, the relative mRNA and protein expression levels of Gadd45 protein in the liver of CCl4- and saline-treated mice were examined. As shown in Figure 2A, the relative mRNA expression of Gadd45a, but not Gadd45b or Gadd45g, was significantly reduced in CCl4-treated mice in a time-dependent manner when compared with the saline-treated group. The protein expression results, assessed by Western blot analysis, were consistent with the mRNA expression data (Figure 2B). The immunofluorescence results also showed that CCl4 was able to inhibit the expression of Gadd45a, but not of Gadd45b or Gadd45g (Figure 2C). The possible underlying mechanism was also explored. As shown in Figure 2D, CCl4 remarkably enhanced TGF-β expression and TGF-β-stimulated phosphorylation of Smad2, Smad3, and Smad4 (Figure 2D).
Figure 2.
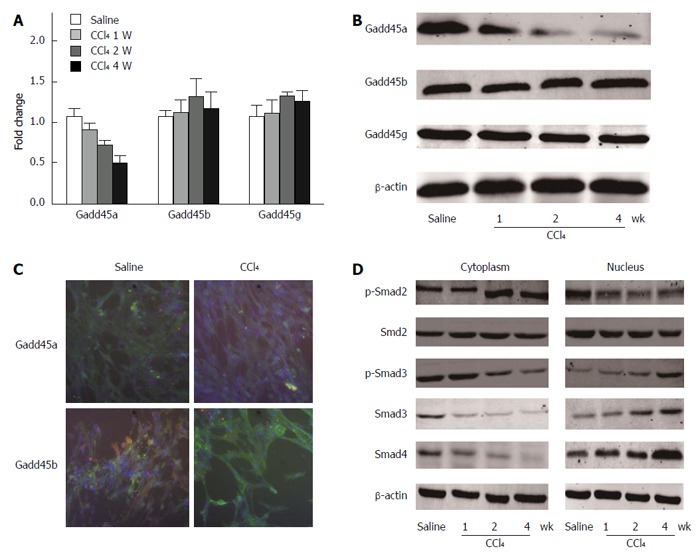
Chronic CCl4 injury suppresses Gadd45a expression and activates TGF-β/Smad signaling. A: Relative mRNA expression levels of Gadd45a, Gadd45b, and Gadd45g in the livers of mice treated with CCl4 for 1, 2, and 4 wk; the saline-treated group served as the control group; B: Relative protein expression levels of Gadd45a, Gadd45b, and Gadd45g in the livers of mice treated with CCl4 for 1, 2, and 4 wk; C: Immunofluorescence staining of Gadd45a, Gadd45b, and Gadd45g (in green) in HSCs treated with CCl4 for 4 wk (nuclei are stained blue with DAPI; scale bars = 10 μm); D: Expression levels of p-Smad2, p-Smad3, Smad2, Smad3, and Smad4 were analyzed in HSCs treated with CCl4 for 1, 2, and 4 wk. HSC: Hepatic stellate cells; TGF: Transforming growth factor.
Gadd45a inhibits the expression of ECM proteins and α-SMA in HSCs treated with CCl4
As shown in Figure 3A, the expression levels of collagen I, fibronectin, and α-SMA in vitro were increased in the CCl4-treated group when compared to the saline-treated group. Moreover, Gadd45a expression was strongly decreased in the CCl4-treated group, whereas no differences in the expression of Gadd45b or Gadd45g were observed between the CCl4-treated and the saline-treated groups (Figure 3B). The aforementioned results were consistent with the in vivo results. Furthermore, as shown in Figure 3C, the expression of collagen I, fibronectin, and α-SMA was suppressed after overexpression of Gadd45a in HSCs treated with CCl4. However, the expression of collagen I, fibronectin, and α-SMA was increased after Gadd45a silencing in HSCs.
Figure 3.
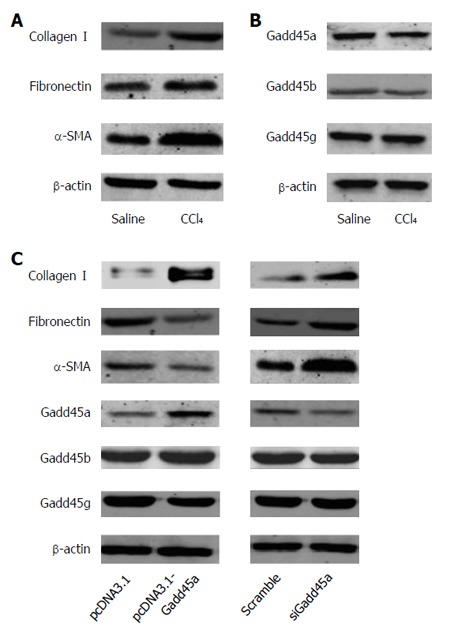
Gadd45a inhibits the expression of extracellular matrix proteins and α-smooth muscle actin in CCl4-treated hepatic stellate cells. A: Expression of type I collagen, fibronectin, and α-SMA in HSCs treated with CCl4 vs saline for 24 h; B: Expression of Gadd45a, Gadd45b, and Gadd45g in HSCs treated with CCl4 vs saline for 24 h; C: Expression of type I collagen, fibronectin, α-SMA, Gadd45a, Gadd45b, and Gadd45g in HSCs transfected with pcDNA3.1-Gadd45a or siRNA-Gadd45a. HSC: Hepatic stellate cell; SMA: Smooth muscle actin.
Gadd45a inhibits hepatic fibrosis through the suppression of TGF-β/Smad signaling
To determine whether Gadd45a suppresses the expression of profibrogenic proteins via the inhibition of TGF-β signaling, the role of Gadd45a on the expression of plasminogen activator inhibitor-1 (PAI-1), whose promoter contains three binding sites for Smad3/Smad4, was first assessed. PAI-1 expression was remarkably decreased in HSCs treated with pcDNA3.1-Gadd45a in both the CCl4 and saline groups. In contrast, PAI-1 expression was significantly increased in HSCs treated with Gadd45a siRNA in both the CCl4 and saline groups (Figure 4A). Moreover, (CAGA)9 MLP-Luc, an artificial Smad3/4-specific reporter, was used to further confirm the role of Gadd45a on the canonical TGF-β/Smad signaling pathways[31]. Gadd45a remarkably inhibited the CCl4-induced activity of (CAGA)9 MLP-Luc (Figure 4B). However, knockdown of Gadd45a increased the activity of (CAGA)9 MLP-Luc to some extent.
Figure 4.
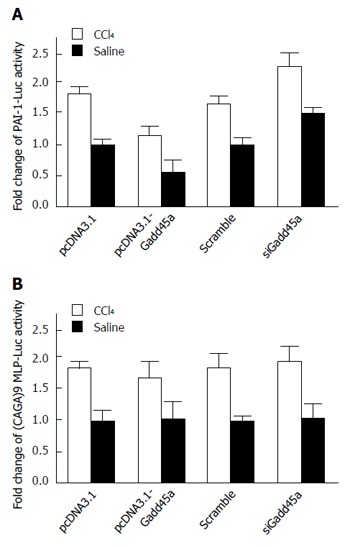
Gadd45a inhibits hepatic fibrosis through the suppression of transforming growth factor-β/Smad signaling. A: Effects of Gadd45 on PAI-1-Luc activity in HSCs treated with pcDNA3.1, Gadd45a, and Gadd45a siRNA in both the CCl4 and saline groups; B: Effects of Gadd45 on (CAGA)9 MLP-Luc activity in HSCs treated with pcDNA3.1, Gadd45a, and Gadd45a siRNA in both the CCl4 and saline groups. HSC: Hepatic stellate cell; TGF: Transforming growth factor.
Gadd45a exerts ROS scavenging effects via upregulating the expression of antioxidant enzymes
CCl4 is known to induce liver fibrosis partly due to increased ROS production[32], and Gadd45 exerts its antioxidant properties in chronic liver injury[33]. However, whether Gadd45a is able to inhibit CCl4-mediated ROS generation in HSCs is still unclear. As shown in Figure 5A, overexpression of Gadd45a significantly reduced the levels of intracellular ROS in HSCs treated with CCl4 for 24 h, whereas Gadd45a knockdown enhanced ROS production. Next, we further explored the expression of antioxidant enzymes, such as HO-1 and NQO1, which are required for scavenging ROS. As expected, the relative mRNA expression of HO-1 and NQO1 was reduced in HSCs treated with CCl4. Interestingly, the expression of HO-1 and NQO1 was increased when HSCs overexpressed Gadd45a in both the CCl4 and saline groups. HO-1 and NQO1 expression was reduced when the HSCs were treated with Gadd45a siRNA (Figure 5B). The protein expression, as detected by Western blot, showed similar results (Figure 5C). The expression of HO-1 and NQO1 was upregulated in Gadd45a-overexpressed HSCs treated with CCl4 but downregulated by Gadd45a knockdown.
Figure 5.
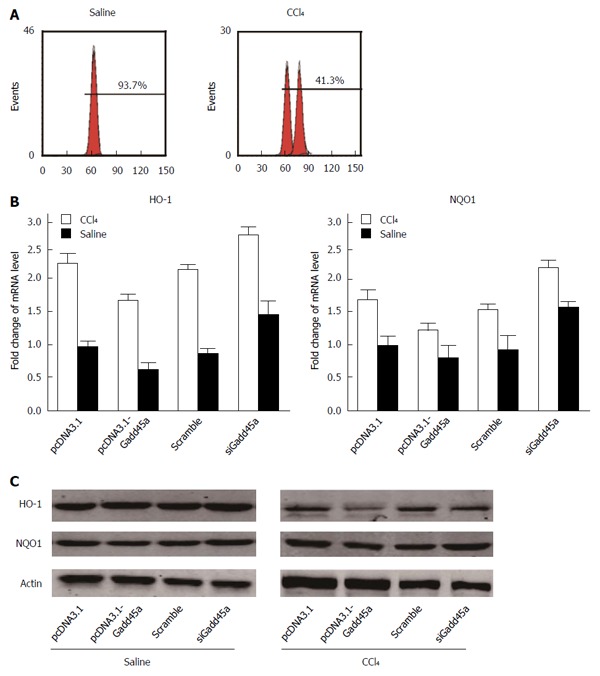
Gadd45a exerts reactive oxygen species scavenging effects by upregulating the expression of antioxidant enzymes. A: The effects of Gadd45a on ROS generation in CCl4-pretreated HSCs treated with pcDNA3.1, PCDNA3.1-Gadd45a, scrambled siRNA, and Gadd45a siRNA were detected by flow cytometry; B: Effects of Gadd45a on HO-1 and NQO1 RNA expression in CCl4-pretreated HSCs treated with pcDNA3.1, PCDNA3.1-Gadd45a, scrambled siRNA, and Gadd45a siRNA; C: Effects of Gadd45a on HO-1 and NQO1 protein expression in CCl4-pretreated HSCs treated with pcDNA3.1, PCDNA3.1-Gadd45a, scrambled siRNA, and Gadd45a siRNA. HSC: Hepatic stellate cells; ROS: Reactive oxygen species.
Gadd45a inhibits the TGF-β/Smad signaling pathways partly due to its antioxidant properties
To clarify the role of ROS in Gadd45a-mediated inhibition of the TGF-β/Smad signaling pathways, the effect of glutathione, a potent ROS scavenger, was explored in HSCs. As shown in Figure 6A, glutathione was able to suppress the relative mRNA expression of type I collagen and α-SMA to some extent when compared to DMSO. Similar results of protein expression were obtained by Western blot (Figure 6B), suggesting that antioxidants play an important role in the inhibitory effect on hepatic fibrosis induced by Gadd45a. Thus, the requirement of antioxidant function of Gadd45a for the inhibition of TGF-β/Smad was further investigated. As shown in Figure 6C, the expression of TGF-β, as well as the phosphorylation of Smad2, Smad3, and Smad4, were inhibited in the group exposed to the combination of pcDNA3.1-Gadd45a and glutathione.
Figure 6.
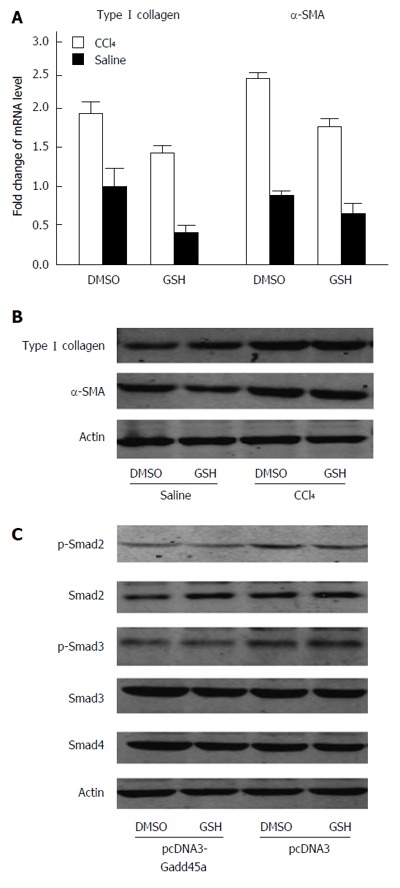
Gadd45a inhibits the transforming growth factor-β/Smad signaling pathways partly due to its antioxidant properties. A: Relative mRNA expression of type I collagen and α-SMA as determined by quantitative RT-PCR. Hepatic stellate cells were stimulated with CCl4 in the presence or absence of glutathione for 24 h; B: Western blot analyses of type I collagen and α-SMA expression; C: Activation of the TGF-β/Smad signaling pathways was measured by Western blot analyses of p-Smad2, p-Smad3, Smd2, Smad3, and Smad4. GSH: Glutathione; SMA: Smooth muscle actin; TGF: Transforming growth factor.
DISCUSSION
To date, there are no antifibrotic drugs approved for clinical use to prevent and reverse hepatic fibrosis and cirrhosis[34,35], partly because the basic mechanisms underlying cirrhosis are still elusive despite decades of study. Therefore, there is an urgent need to identify effective antifibrotic targets to prevent or treat hepatic fibrosis. Recently, the regulation of Gadd45a expression in response to mechanical stress and the association of Gadd45a genetic variants with acute lung injury/ventilator-induced lung injury susceptibility has been demonstrated[36]. In this study, liver fibrosis induced by CCl4 administration in BALB/c mice was accompanied by the downregulation of Gadd45a and upregulation of TGF-β/Smad, suggesting that these proteins play an important role in the fibrosis process that develops during chronic liver injury.
Next, using HSCs, the antifibrotic effect of Gadd45 was found to be associated with suppression of HSC activation and reduced expression of ECM components and antifibrolytic proteins. In addition, Gadd45 suppressed CCl4-induced fibrogenic gene expression through inhibition of TGF-β/Smad signaling. Moreover, TGF-β has been identified as a potent profibrogenic factor that contributes to liver fibrosis in patients[37,38]. Growing evidence has suggested that TGF-β exhibits its profibrotic activity through multiple biologic mechanisms, such as activation of HSCs to ECM-producing myofibroblasts and induction of epithelial-mesenchymal transition of hepatocytes and cholangiocytes[38-40]. Furthermore, the canonical Smad-dependent pathway is involved in TGF-β stimulation of excessive ECM deposition by regulating both ECM proteins and inhibitors of ECM degradation. Altogether, inhibition of the TGF-β/Smad signaling pathways is an attractive therapeutic target for the prevention of liver fibrosis[28]. In this study, overexpression of Gadd45a repressed CCl4-stimulated expression of both ECM proteins and α-SMA in HSCs. Moreover, Gadd45a inhibited HSC activation as shown by the reduction of CCl4-enhanced α-SMA expression.
During hepatic injury, increased ROS in liver tissue derived from Kupffer cells, infiltrated neutrophils, or hepatocytes and can damage hepatocytes and activate HSCs, playing a central role in liver fibrosis[2,41]. TGF-β can mediate ROS production, which further enhances profibrogenic signals of TGF-β through Smad-independent pathways[28]. Thus, Gadd45a exhibits a protective effect against oxidative stress by scavenging ROS and upregulating antioxidant enzymes. In this regard, we hypothesized that Gadd45 may suppress TGF-β/Smad signaling through induction of antioxidant genes. Our findings demonstrated that glutathione contributes to the suppression of the Smad-dependent pathway of TGF-β signaling by Gadd45a. Although we showed that Gadd45a inhibits TGF-β/Smad3-mediated HSC activation and ROS generation, the precise mechanisms by which Gadd45a prevents liver fibrosis induced by chronic CCl4 administration require further investigation.
Overall, these findings provide evidence that Gadd45a exerts a protective effect against hepatic fibrosis induced by CCl4 via the inhibition of canonical TGF-β/Smad signaling and fibrogenic gene expression. We also propose a molecular basis for the antioxidant potential of Gadd45a in the hepatic fibrosis process. Although clinical methods for the direct targeting of HSCs remain under development, our study provides an important advance in the understanding of the biologic functions of Gadd45a and a potential target for the treatment of hepatic fibrosis.
ACKNOWLEDGMENTS
We thank Medjaden Bioscience Limited for assisting in the preparation of this manuscript.
COMMENTS
Background
Hepatic fibrosis is a wound-healing response characterized by the accumulation of extracellular matrix from activated hepatic stellate cells (HSCs). Gadd45 proteins have been shown to play similar but not identical roles, depending on the particular stress response pathway that is activated. But the role of Gadd45a in hepatic fibrosis and transforming growth factor (TGF)-β/Smad signaling pathway is still unclarified.
Research frontiers
The data in this paper provide evidence that Gadd45a may exert a protective effect against hepatic fibrosis induced by CCl4, via the inhibition of canonical TGF-β/Smad signaling and fibrogenic gene expression. The authors also propose a molecular basis for the antioxidant potential of Gadd45a in the hepatic fibrosis process.
Innovations and breakthroughs
Gadd45a inhibits hepatic fibrosis through blockade of TGF-β/Smad signaling. Gadd45a inhibits canonical TGF-β/Smad signaling partly due to its antioxidant properties. Gadd45a may counteract hepatic fibrosis by regulating the activation of HSCs via the inhibition of TGF-β/Smad signaling.
Applications
This study provides an important advance in the understanding of the biologic functions of Gadd45a and a potential target for the treatment of hepatic fibrosis.
Peer-review
The cell fate determination protein (Gadd45a) was connected with hepatic fibrosis and may be considered as a potential target for the treatment of hepatic fibrosis.
Footnotes
Supported by Medicine and Health Research Programs of Zhejiang Province, No. 2013KYB252; and the Science Foundation of the Science and Technology Commission of Ruian City, No. 201302012.
Institutional animal care and use committee statement: Animals (Shanghai Center of Experimental Animal, Chinese Academy of Sciences) were housed and fed with free access to food and water. All procedures were performed following approval of the Institutional Animal Care and Use Committee of the Chinese Academy of Sciences.
Conflict-of-interest statement: The authors declare no conflicts of interest.
Data sharing statement: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 15, 2015
First decision: June 19, 2015
Article in press: November 9, 2015
P- Reviewer: Zhang YP S- Editor: Qi Y L- Editor: Filipodia E- Editor: Wang CH
References
- 1.Friedman SL. Liver fibrosis -- from bench to bedside. J Hepatol. 2003;38 Suppl 1:S38–S53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 2.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 4.Gäbele E, Brenner DA, Rippe RA. Liver fibrosis: signals leading to the amplification of the fibrogenic hepatic stellate cell. Front Biosci. 2003;8:d69–d77. doi: 10.2741/887. [DOI] [PubMed] [Google Scholar]
- 5.Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. 2007;131:1728–1734. doi: 10.5858/2007-131-1728-HSCALF. [DOI] [PubMed] [Google Scholar]
- 6.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 7.Jin S, Antinore MJ, Lung FD, Dong X, Zhao H, Fan F, Colchagie AB, Blanck P, Roller PP, Fornace AJ, et al. The GADD45 inhibition of Cdc2 kinase correlates with GADD45-mediated growth suppression. J Biol Chem. 2000;275:16602–16608. doi: 10.1074/jbc.M000284200. [DOI] [PubMed] [Google Scholar]
- 8.Salvador JM, Brown-Clay JD, Fornace AJ. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv Exp Med Biol. 2013;793:1–19. doi: 10.1007/978-1-4614-8289-5_1. [DOI] [PubMed] [Google Scholar]
- 9.Schäfer A. Gadd45 proteins: key players of repair-mediated DNA demethylation. Adv Exp Med Biol. 2013;793:35–50. doi: 10.1007/978-1-4614-8289-5_3. [DOI] [PubMed] [Google Scholar]
- 10.Fornace AJ, Alamo I, Hollander MC. DNA damage-inducible transcripts in mammalian cells. Proc Natl Acad Sci USA. 1988;85:8800–8804. doi: 10.1073/pnas.85.23.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geifman-Holtzman O, Xiong Y, Holtzman EJ. Gadd45 stress sensors in preeclampsia. Adv Exp Med Biol. 2013;793:121–129. doi: 10.1007/978-1-4614-8289-5_7. [DOI] [PubMed] [Google Scholar]
- 12.Hoffman B, Liebermann DA. Gadd45 in modulation of solid tumors and leukemia. Adv Exp Med Biol. 2013;793:21–33. doi: 10.1007/978-1-4614-8289-5_2. [DOI] [PubMed] [Google Scholar]
- 13.Kearsey JM, Coates PJ, Prescott AR, Warbrick E, Hall PA. Gadd45 is a nuclear cell cycle regulated protein which interacts with p21Cip1. Oncogene. 1995;11:1675–1683. [PubMed] [Google Scholar]
- 14.Vairapandi M, Balliet AG, Fornace AJ, Hoffman B, Liebermann DA. The differentiation primary response gene MyD118, related to GADD45, encodes for a nuclear protein which interacts with PCNA and p21WAF1/CIP1. Oncogene. 1996;12:2579–2594. [PubMed] [Google Scholar]
- 15.Vairapandi M, Balliet AG, Hoffman B, Liebermann DA. GADD45b and GADD45g are cdc2/cyclinB1 kinase inhibitors with a role in S and G2/M cell cycle checkpoints induced by genotoxic stress. J Cell Physiol. 2002;192:327–338. doi: 10.1002/jcp.10140. [DOI] [PubMed] [Google Scholar]
- 16.Vairapandi M, Azam N, Balliet AG, Hoffman B, Liebermann DA. Characterization of MyD118, Gadd45, and proliferating cell nuclear antigen (PCNA) interacting domains. PCNA impedes MyD118 AND Gadd45-mediated negative growth control. J Biol Chem. 2000;275:16810–16819. doi: 10.1074/jbc.275.22.16810. [DOI] [PubMed] [Google Scholar]
- 17.Bulavin DV, Kovalsky O, Hollander MC, Fornace AJ. Loss of oncogenic H-ras-induced cell cycle arrest and p38 mitogen-activated protein kinase activation by disruption of Gadd45a. Mol Cell Biol. 2003;23:3859–3871. doi: 10.1128/MCB.23.11.3859-3871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95:521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 19.Zerbini LF, Wang Y, Czibere A, Correa RG, Cho JY, Ijiri K, Wei W, Joseph M, Gu X, Grall F, et al. NF-kappa B-mediated repression of growth arrest- and DNA-damage-inducible proteins 45alpha and gamma is essential for cancer cell survival. Proc Natl Acad Sci USA. 2004;101:13618–13623. doi: 10.1073/pnas.0402069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta SK, Gupta M, Hoffman B, Liebermann DA. Hematopoietic cells from gadd45a-deficient and gadd45b-deficient mice exhibit impaired stress responses to acute stimulation with cytokines, myeloablation and inflammation. Oncogene. 2006;25:5537–5546. doi: 10.1038/sj.onc.1209555. [DOI] [PubMed] [Google Scholar]
- 21.Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ, Liebermann DA, Bottinger EP, Roberts AB. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J Biol Chem. 2003;278:43001–43007. doi: 10.1074/jbc.M307869200. [DOI] [PubMed] [Google Scholar]
- 22.Yang Z, Song L, Huang C. Gadd45 proteins as critical signal transducers linking NF-kappaB to MAPK cascades. Curr Cancer Drug Targets. 2009;9:915–930. doi: 10.2174/156800909790192383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith ML, Ford JM, Hollander MC, Bortnick RA, Amundson SA, Seo YR, Deng CX, Hanawalt PC, Fornace AJ. p53-mediated DNA repair responses to UV radiation: studies of mouse cells lacking p53, p21, and/or gadd45 genes. Mol Cell Biol. 2000;20:3705–3714. doi: 10.1128/mcb.20.10.3705-3714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Feng X, We R, Derynck R. Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature. 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 26.Hemmann S, Graf J, Roderfeld M, Roeb E. Expression of MMPs and TIMPs in liver fibrosis - a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol. 2007;46:955–975. doi: 10.1016/j.jhep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Inagaki Y, Okazaki I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56:284–292. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic Biol Med. 2010;48:1–15. doi: 10.1016/j.freeradbiomed.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang G, Yang HR, Wang L, Wildey GM, Fung J, Qian S, Lu L. Hepatic stellate cells preferentially expand allogeneic CD4+ CD25+ FoxP3+ regulatory T cells in an IL-2-dependent manner. Transplantation. 2008;86:1492–1502. doi: 10.1097/TP.0b013e31818bfd13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moles A, Tarrats N, Fernández-Checa JC, Marí M. Cathepsins B and D drive hepatic stellate cell proliferation and promote their fibrogenic potential. Hepatology. 2009;49:1297–1307. doi: 10.1002/hep.22753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adachi M, Brenner DA. High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology. 2008;47:677–685. doi: 10.1002/hep.21991. [DOI] [PubMed] [Google Scholar]
- 33.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 34.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 35.Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50:1294–1306. doi: 10.1002/hep.23123. [DOI] [PubMed] [Google Scholar]
- 36.Mitra S, Wade MS, Sun X, Moldobaeva N, Flores C, Ma SF, Zhang W, Garcia JG, Jacobson JR. GADD45a promoter regulation by a functional genetic variant associated with acute lung injury. PLoS One. 2014;9:e100169. doi: 10.1371/journal.pone.0100169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, Hu H, Yin JQ. Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver Int. 2006;26:8–22. doi: 10.1111/j.1478-3231.2005.01192.x. [DOI] [PubMed] [Google Scholar]
- 38.Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med. 2006;10:76–99. doi: 10.1111/j.1582-4934.2006.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007;282:22089–22101. doi: 10.1074/jbc.M700998200. [DOI] [PubMed] [Google Scholar]
- 40.Chen YL, Lv J, Ye XL, Sun MY, Xu Q, Liu CH, Min LH, Li HP, Liu P, Ding X. Sorafenib inhibits transforming growth factor β1-mediated epithelial-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology. 2011;53:1708–1718. doi: 10.1002/hep.24254. [DOI] [PubMed] [Google Scholar]
- 41.Lotersztajn S, Julien B, Teixeira-Clerc F, Grenard P, Mallat A. Hepatic fibrosis: molecular mechanisms and drug targets. Annu Rev Pharmacol Toxicol. 2005;45:605–628. doi: 10.1146/annurev.pharmtox.45.120403.095906. [DOI] [PubMed] [Google Scholar]