

Abstract
Heterogeneity is a hallmark of glioblastoma with intratumoral heterogeneity contributing to variability in responses and resistance to standard treatments. Promoter methylation status of the DNA repair enzyme O6-methylguanine DNA methyltransferase (MGMT) is the most important clinical biomarker in glioblastoma, predicting for therapeutic response. However, it does not always correlate with response. This may be due to intratumoral heterogeneity, with a single biopsy unlikely to represent the entire lesion. Aberrations in other DNA repair mechanisms may also contribute. This study investigated intratumoral heterogeneity in multiple glioblastoma tumors with a particular focus on the DNA repair pathways. Transcriptional intratumoral heterogeneity was identified in 40% of cases with variability in MGMT methylation status found in 14% of cases. As well as identifying intratumoral heterogeneity at the transcriptional and epigenetic levels, targeted next generation sequencing identified between 1 and 37 unique sequence variants per specimen. In-silico tools were then able to identify deleterious variants in both the base excision repair and the mismatch repair pathways that may contribute to therapeutic response. As these pathways have roles in temozolomide response, these findings may confound patient management and highlight the importance of assessing multiple tumor biopsies.
Glioblastoma, a grade IV tumor of glial cell origin, is the most common and lethal primary brain tumor in adults. Tumors are genetically unstable and highly heterogeneous, displaying variable responses to therapy from patient to patient. Despite aggressive treatment involving maximal surgical resection followed by radiotherapy (RT) and concomitant and adjuvant chemotherapy (temozolomide, TMZ) median survival is just 15 months with a 5-year survival rate of less than 10%1. Ultimately, most patients recur as treatment resistance develops and there is no established salvage treatment that has clearly shown an improved survival benefit2.
Individual tumors contain a complex hierarchy of genetically distinct subclones with diverse morphologies and biological behaviors. Recent data suggest that this diversity or ‘intratumoral heterogeneity’ is one of the major reasons behind treatment failure. These diverse subclones have been shown to co-exist within the same tumor specimen3,4,5,6,7,8,9,10,11,12. It can be hypothesized that selecting therapy based on the analysis from a single biopsy specimen may not be representative of the entire lesion and could result in treatment failure. RT and alkylating chemotherapy can further exacerbate this heterogeneity by introducing additional alterations into the tumor12,13.
Such intratumoral heterogeneity undermines the usefulness of clinical biomarkers in glioblastoma and has significant implications for patient prognosis and management. Promoter methylation of the DNA repair enzyme O6-methylguanine DNA methyltransferase (MGMT) is the major clinically used predictive and prognostic biomarker of survival following RT and TMZ chemotherapy. Glioblastoma patients with methylated MGMT promoters have a more favorable outcome when treated with combination RT and TMZ versus RT alone14. However, a subset of patients with MGMT promoter methylation have poor outcome and some patients with non-methylated promoters have long survival. Intratumoral heterogeneity may be a contributing factor to this variability in response. Other cellular DNA repair mechanisms including the mismatch repair (MMR) and base excision repair (BER) pathways may also contribute and intratumoral heterogeneity in these pathways would further confound results.
The molecular subtype of the tumor can also be used as a predictive biomarker of outcome and response to therapy. Large scale molecular analysis performed by the TCGA and others has shown that glioblastoma encompasses multiple molecular subclasses of disease, each having slightly different outcomes and responses to standard of care therapy. The proneural subtype has been associated with increased survival yet no benefit was seen with aggressive therapy15 whereas the mesenchymal subtype has been associated with a worse outcome but a significant benefit from aggressive therapy16. Future stratification of patients based on molecular subtyping may improve therapy selection and outcome.
In the current study, multiple spatially distinct biopsies were taken from the same glioblastoma tumor in order to investigate intratumoral heterogeneity in key glioma biomarkers including MGMT promoter methylation and transcriptional subtype. We also examined whether tumors displayed intratumoral heterogeneity in the MMR and BER DNA repair pathways which may promote intrinsic or acquired treatment resistance.
Results
Transcriptional profiling of tumor biopsies
Two different limited gene panels classified tumor specimens into transcriptional subtypes with strong concordance
Gene expression profiling was performed using 2 different gene panels to accurately classify the tumor biopsies into transcriptional subgroups. Genes were chosen based on previous studies as classifiers for the proneural, mesenchymal and classical transcriptional subtypes. Hierarchical clustering was performed and specimens were divided into 1 of 3 different cluster groups using the 30-gene panel (Fig. 1, A1 and SI Fig. 1). The blue group featured significantly higher mean expression of EGFR than either of the other groups (p < 0.0001, one-way ANOVA, Kruskal-Wallis test) suggestive of a classical subtype15. This was supported by increased mean expression of Notch (NOTCH3, JAG1) and Sonic Hedgehog (GLI2) signaling pathway genes for this group. The green group featured highest mean expression of CHI3L1, CD44, SERPINE1 and CTGF characteristic of the mesenchymal subgroup15,17,18. The red group was characterized by highest mean expression for the proneural markers DLL3, OLIG2, ASCL1 and PDGFR15,16,17,18,19.
Figure 1. Transcriptional profiling and cluster analysis reveals intratumoral heterogeneity in 43% (6 of 14) of tumors.
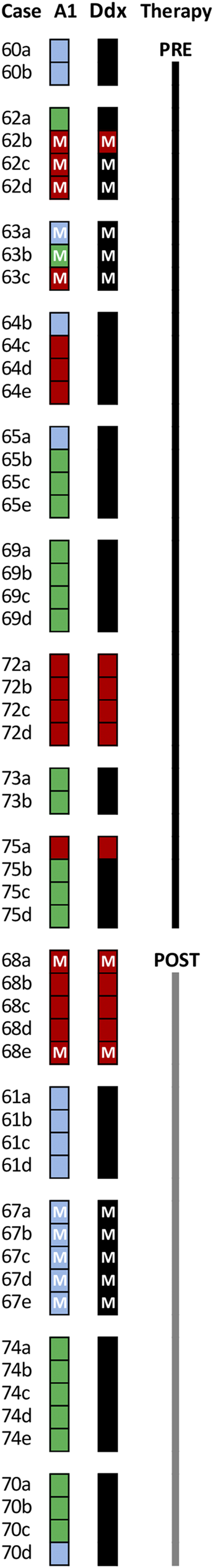
Intratumoral heterogeneity is defined as specimens from the same patient clustering into different groups by at least one analysis method (A1 or Ddx). Specimens were assigned to 1 of 3 groups by hierarchal clustering based on Euclidean distance of RT-qPCR results for A1 using 30 genes (SI Fig. 1, SI Table 3) and into 2 groups for Ddx by the 9 genes used for the Decision Dx panel (SI Fig. 2). “M” signifies a MGMT promoter methylation positive specimen. Specimens are grouped by patient and ordered by therapy. The 3 cluster groups for A1 are colored and concur with gene expression levels expected for the classical (blue), mesenchymal (green) and proneural (red) subgroups. The specimens in the red cluster group for Ddx have the highest individual scores using the Ddx algorithm suggestive of a better outcome and proneural subtype. The specimens in the black cluster group are the remaining specimens (with lower Ddx scores) and thus comprise specimens of the classical and mesenchymal subtypes.
We also performed gene expression profiling using the 9-gene Decision Dx panel (Ddx;16). These 9 genes were used to interrogate the cohort by both hierarchical cluster analysis and the Ddx score using ΔCt values in a weighted model for prediction of overall survival (Fig. 1, Ddx, SI Fig. 2)16. The top 11 ranked specimens according to the Ddx score comprised a distinct group by cluster analysis. A high Ddx score which characterized this cluster group (red group, Fig. 1, SI Fig. 2) has previously been shown to be predictive of a proneural subtype16.
While only 3 genes (CHI3L1, OLIG2 and RTN1) were common to both the 30-gene panel and the 9-gene Ddx panel (SI Figs 1 and 2), there was strong concordance between the 2 different gene-panels for the prediction of the proneural (red) subtype with only 11% (6 of 54) of specimens discordant between A1 and Ddx. These 6 samples were ranked in the next 10 by the Ddx algorithm (SI Fig. 2) indicating different cut-off points for the different clustering algorithms with the Ddx being the most stringent for the proneural subtype.
Figure 2. Gene expression analysis of MMR and BER pathways genes identifies both aberrant expression and intratumoral heterogeneity.

The gene expression of Base Excision Repair (BER) genes (APEX1 and PARP1) and Mismatch Repair (MMR) genes (MSH6, MSH2, MLH1 and PMS2) was examined in tumor biopsies pre- and post-adjuvant therapy (indicated by circle and diamond symbols, respectively) relative to normal brain tissue (indicated by the broken line). Results were generated using Taqman assays and TBP as the reference for normalization. A 2-fold change in gene expression relative to normal brain is indicated by the solid grey line. Closed symbols indicate MGMT promoter unmethylated samples, open symbols indicate MGMT promoter methylated samples.
Transcriptional subtyping identified intratumoral molecular heterogeneity in 40% of cases
Having classified the tumor specimens into the 3 different transcriptional sub-groups we next investigated whether the molecular heterogeneity between tumors was also evident within patients with the premise that intratumoral heterogeneity may confound the clinical utility of molecular sub-classification. Ddx identified 2 patients with intratumoral heterogeneity. In both cases, intratumoral heterogeneity was also identified by the 30-gene A1 analysis (Fig. 1). The A1 analysis further revealed heterogeneity in 40% (6 of 14) of patients. There was no significant difference in the occurrence of intratumoral heterogeneity in tumors collected pre-therapy or following recurrence (2-tailed Fisher’s Exact test). Interestingly, we did not find a significant dilution effect with differing amounts of tumour tissue. For example, specimens from the same patient which differed in tumour content by 40% were still classified as the same tumour subtype e.g. 72a (60% tumour content) and 72d (100% tumour content) and conversely, specimens taken from the same patient in which tumour content was the same (100%) were classified into different subtypes e.g. 64b and 64c. These results suggest that intratumoral heterogeneity may lead to a non-consensus transcriptional subtype in up to 40% of cases where the results are based on a single biopsy specimen.
Intratumoral heterogeneity in MGMT promoter methylation status was identified in 14% of tumors and was independent of transcriptional heterogeneity
MGMT promoter methylation status was determined using a 4 CpG island assay that detects methylation at 4 sites in exon 1 of the human MGMT gene (Chr 10: 131,265,519-131,265,537), a region strongly correlated with protein expression and patient outcome20. MGMT methylation was identified in 30% (4 of 14) of cases (Table 1). Two cases were uniformly methylated across all biopsies. However, 14% (2 of 14) of cases demonstrated intratumoral heterogeneity in methylation levels with the percentage of methylation varying up to 4-fold within each case (Table 1, SI Table 4). Of note, methylation status appeared to be independent of transcriptional classification by either A1 or Ddx (Fig. 1). Similar to our results for transcriptional subtyping, we did not find a significant dilution effect with differing amounts of tumour tissue. Specimens from the same patient which differed in tumour content by 40% were still classified into the same methylation group e.g. 63a (100% tumour content) and 63c (60% tumour content) were classified as being methylated and conversely, specimens taken from the same patient in which tumour content was the same (100%) were classified into different methylation groups e.g. 68a (methylated) and 68b (unmethylated).
Table 1. Cohort details including MGMT promoter methylation status that demonstrates intratumoral heterogeneity (M: Methylated promoter, U: Unmethylated promoter, RT: radiotherapy, TMZ: temozolomide chemotherapy).
| Case # | Surgery | Treatment |
MGMT promoter methylation status (M: > 13%, U: < 13%) |
|||||
|---|---|---|---|---|---|---|---|---|
| Biopsy A | Biopsy B | Biopsy C | Biopsy D | Biopsy E | Biopsy F | |||
| 60 | 1 | Naïve | U | U | ||||
| 62 | 1 | Naïve | U (4.3) | M (20.3) | M (32.8) | M (16.3) | ||
| 63 | 1 | Naïve | M | M | M | |||
| 64 | 1 | Naïve | U | U | U | U | U | |
| 65 | 1 | Naïve | U | U | U | U | U | |
| 69 | 1 | Naïve | U | U | U | U | ||
| 72 | 1 | Naïve | U | U | U | U | ||
| 73 | 1 | Naïve | U | U | ||||
| 75 | 1 | Naïve | U | U | U | U | ||
| 68* | 2 | RT | M (33.0) | U (9.8) | U (8.8) | U (11.8) | M (16.5) | |
| 61 | 2 | RT-TMZ | U | U | U | U | ||
| 67 | 2 | RT-TMZ | M | M | M | M | M | |
| 74 | 2 | RT-TMZ | U | U | U | U | U | U |
| 70 | 3 | RT-TMZ | U | U | U | U | ||
For cases where variability was detected, the percentage methylation is shown in brackets.
*IDH1 mutation identified; secondary glioblastoma.
These results support previous reports of intratumoral heterogeneity for MGMT methylation levels and the possibility that reliance on a single biopsy specimen may not provide an accurate consensus of MGMT methylation for the tumor, reducing the clinical value of this biomarker.
Intratumoral heterogeneity was detected for the expression of MMR and BER pathway genes APEX1, PARP1, MSH2 and PMS2
Alterations to the MMR and BER pathways may confer resistance to TMZ independent of MGMT methylation. To determine whether alterations in these pathways exist in our cohort, we first assessed the gene expression levels of components of the pathways: MMR (MSH2, MSH6, MLH1 and PMS2) and BER (APEX1 and PARP1). Results were compared with those from normal brain (broken line, Fig. 2) and a 2-fold increase in expression for BER genes or 2-fold decrease for MMR genes set as arbitrary cut-offs for aberrant expression (solid line; 1(log2); Fig. 2). Intratumoral heterogeneity was defined as a greater than 2-fold range in gene expression with values spanning the cut-offs for aberrant expression.
APEX1 was notable for being aberrantly elevated in 86% of cases in our cohort with heterogeneity of expression noted in 29% (4 of 14) of cases (Fig. 2a). PARP1 was aberrantly elevated in at least one specimen in 71% of our cohort with intratumoral expression heterogeneity noted in 50% (7 of 14) of cases (Fig. 2b). Intratumoral heterogeneity with markedly reduced levels of the MSH2 and PMS2 was observed in 14% (2 of 14) and 21% (3 of 14) of cases respectively (Fig. 2c,f). No cases demonstrated intratumoral heterogeneity or markedly reduced levels of the MMR genes MLH1 or MSH6 (Fig. 2d,e).
There was no correlation between MGMT promoter methylation level and the expression of any of these genes. However, specimens from 3 of the 4 cases with methylated MGMT (open circles, Fig. 2) displayed aberrant levels of at least one of these genes. The intratumoral heterogeneity observed suggests the presence of clonal populations, some of which may confer decreased sensitivity to TMZ, confounding the predictive value of MGMT promoter methylation.
Mutational analysis
Next generation sequencing of MMR and BER genes confirms intratumoral heterogeneity
Having identified the presence of inappropriate expression of members of the MMR and BER pathways we next assessed these genes at the DNA level. We performed next generation sequencing for the 6 genes of interest (APEX1, PARP1, MSH2, MSH6, MLH1 and PMS2) on multiple specimens from 12 cases with up to 3000x depth of coverage (average = 847x) to definitively determine whether intratumoral heterogeneity exists at this level and thus would confirm the presence of clonal or sub-clonal populations within a tumor.
After filtering for possible sequencing artifacts we identified a total of 523 sequence variants across the 6 genes in the 48 specimens sequenced from 12 cases. We found that each specimen had between 40 and 80 sequence variants in total (Fig. 3). Of these, 35 to 56 were common across each case; up to 20 were shared between at least 2 specimens of each case, and 1 to 37 variants were unique to each specimen. All specimens had sequence variants not present in the other specimens from that case, clearly demonstrating the presence of intratumoral heterogeneity.
Figure 3. Next generation sequencing of MMR and BER genes identifies intratumoral heterogeneity at the DNA level.
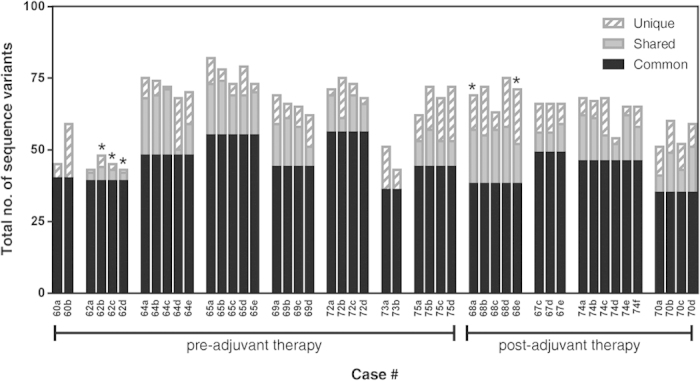
Targeted next generation exome sequencing results for 6 genes (APEX1, PARP1, MSH2, MSH6, MLH2, PMS2) performed on the Illumina Miseq platform and analyzed with Avadis NGS software. The total number of sequence variants identified for all the genes in each specimen are graphed, and specimens are grouped by case. ‘Common’ variants (those present in all specimens from a case) are illustrated in solid black bars; ‘Shared’ variants (those present in more than one but not all specimens/case) are illustrated in solid grey bars and ‘unique’ variants (those present only in one specimen/case) are illustrated in diagonal grey stripped bars. Specimens with MGMT promoter methylation are indicated with an asterisk (*).
Damaging DNA variants identified in DNA repair pathways
Having identified both the presence of sequence variants and intratumoral heterogeneity of these variants, we next sought to determine the functional significance of the variants identified. All variants were assessed by 2 software tools (PROVEAN and SIFT) to identify those predicted to have functional consequences. Eleven sequence variants were predicted by these software programs to be damaging and/or deleterious (SI Table 5). Four of these variants were present in sufficient abundance to be confirmed by Sanger sequencing. PARP1 S383Y (ENST00000366794, rs3219062), PMS2 S46I (ENST00000265849, rs121434629) and APEX1 Q51H (ENST00000216714, rs1048945) were found in all specimens from a case (with supporting reads of approximately 50% with a coverage of up to 3000x), suggestive of germline mutations. The last DNA variant confirmed by Sanger sequencing is novel and only present in an isolated specimen from a single case (specimen 68e; PMS2 P573fs, ENST00000265849 with supporting reads of 50%) confirming the presence of intratumoral heterogeneity.
The level of sequence heterogeneity varies between clinical groupings
We next determined the level of sequence heterogeneity for each gene across clinically relevant groupings of our cohort: MGMT promoter methylated versus unmethylated and pre and post-therapy. MGMT promoter methylation hinders DNA repair implying that such specimens may have increased sequence heterogeneity relative to unmethylated specimens. Our results support this with sequence heterogeneity across all genes found to be 2-fold higher in MGMT promoter methylated versus unmethylated cases (Fig. 4a).
Figure 4. Sequence heterogeneity of BER and MMR genes.
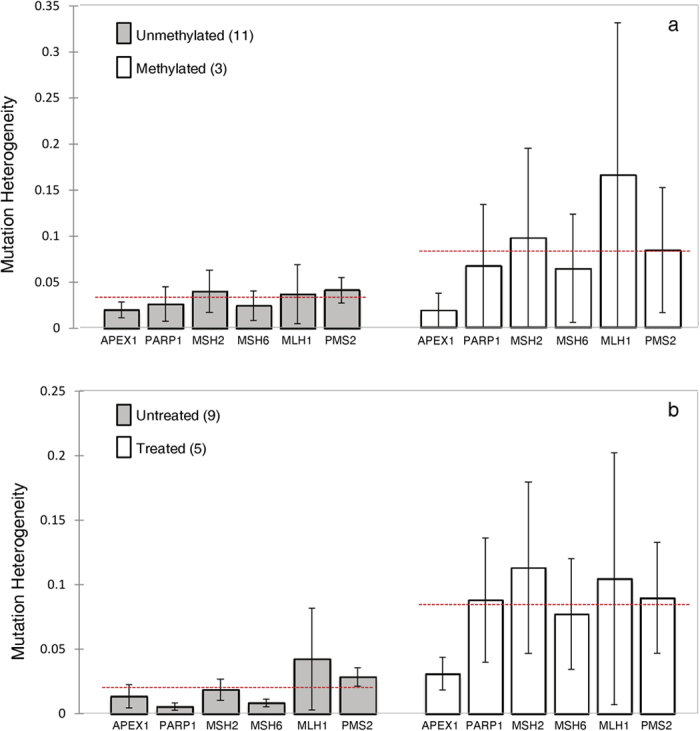
Sequence heterogeneity was calculated for each BER (APEX1 and PARP1) and MMR (MSH6, MSH2, MLH1 and PMS2) gene for each biopsy as described in the methods. Samples were then stratified by MGMT promoter methylation (a) or treatment status (b). The red dotted line indicates the mean sequence heterogeneity of the samples. For cases in which MGMT promoter methylation heterogeneity was identified, case 62 was included in the methylated group with biopsy 62a being excluded from the analysis (as this single biopsy was determined to be unmethylated), and case 68 was included in the unmethylated group with biopsies 68a and 68e also being excluded (as they were determined to be methylated).
Standard of care treatment acts by damaging DNA, thereby triggering apoptosis of cells with damaged DNA. However, in the presence of resistance mechanisms apoptosis may not be triggered leading to accumulation of mutations or alternatively, the repair of the DNA damage or both in combination resulting in increased sequence heterogeneity. Comparison of post-therapy versus pre-therapy cases found that as anticipated there was 4-fold higher heterogeneity in post-therapy cases relative to pre-therapy cases (Fig. 4b).
Discussion
Intratumoral heterogeneity appears to be responsible, at least in part, for resistance to standard treatments and the variable responses seen to treatment8,9,21. In this study we identified intratumoral heterogeneity at the transcriptional, methylation and mutational levels in multiple spatially distinct biopsies taken from up to 14 different glioblastoma cases.
Molecular classification of tumors has identified different subtypes which may be important in treatment stratification response15,18. However, a single biopsy specimen is unlikely to represent the molecular landscape of the entire tumor. Using gene clustering techniques, we observed that biopsies taken from different locations within the same tumor were classified into different molecular subtypes, in agreement with previous research10. Such results not only highlight the intratumoral heterogeneity that exists but also the importance of taking multiple tumor biopsies and genomically analyzing them in order to correctly classify tumors.
The most important clinical biomarker used for glioblastoma is MGMT promoter methylation status. Similar to our results for molecularly classifying the tumors, intratumoral heterogeneity in MGMT status was identified in our cohort. These findings reiterate that basing clinical management on the findings of a single biopsy specimen and a single clinical biomarker such as MGMT may have devastating consequences for the patients. Intratumoral heterogeneity in MGMT promoter methylation status has been previously demonstrated in some22 but not in all studies23. In addition, area-specific intratumoral heterogeneity for MGMT protein expression has also been identified24,25. Similar patterns of non-uniform expression have been shown with other markers26,27,28,29,30,31 demonstrating that this is not a unique feature of MGMT expression. Spatial heterogeneity has also been demonstrated by comparative genome hybridization analysis which revealed different chromosomal aberrations in different areas of the same individual tumor10,31. Thus the classification of tumors based on a single biopsy specimen can be misleading for patient stratification and patient management. However, it must be remembered that certain changes such as mutations of IDH1 are considered early events in tumor development12 and are consequently found ubiquitously in tumor tissue. Distinguishing additional early driver mutations from late changes may better assist in patient stratification for treatment selection.
MGMT promoter methylation status is not always predictive of response to therapy. We have demonstrated intratumoral heterogeneity for MGMT and thus reliance on a single biopsy specimen for analysis may be misleading. However, previous studies have also demonstrated that other DNA repair mechanisms are involved. Aberrant expression of MMR and BER may confer decreased sensitivity to TMZ thus confounding the predictive value of MGMT methylation status. Up-regulation of BER components32,33 or loss of MMR components34 has been shown to promote resistance to treatment. In addition, in MGMT methylated patients, treatment with the alkylating agent TMZ has been shown to induce a hypermutated phenotype, resulting in DNA repair deficiency and enhanced chemo-resistance12,35,36,37,38. More recently, MSH6 mutations independent of MGMT methylation status, have been used as biomarkers for identifying patients intrinsically deficient in DNA repair capacity making them more resistant to chemotherapy39. Similarly, in our cohort, predicted deleterious DNA variants were identified in both the BER and MMR pathways independent of MGMT methylation status. The consequences of such variants may lead to inactivation of these repair pathways and failure of treatment.
While personalized medicine for glioblastoma patients may still be in its infancy, the layers of complexity that are added due to intratumoral heterogeneity are staggering. Modulation of the DNA repair pathways may be of benefit in patients32,40,41,42,43 however, correctly stratifying patients for these molecular targeted treatments needs to be carefully considered. Methods which can identify phylogenetic clonality and common driver mutations would greatly enhance the ability to select appropriate treatments for these individual patients. Current practice is to take a single biopsy specimen for both diagnosis and molecular testing for MGMT promoter methylation or other parameters. However, our results suggest that taking multiple biopsy specimens would be more representative of the entire tumor landscape and may uncover particular aberrations which may be targetable by newly developed treatments.
Taken together our results demonstrate intratumoral heterogeneity at the epigenetic level through MGMT status, the gene expression level, and the DNA mutational level through sequence variation. The identified intratumoral heterogeneity highlights the importance of taking multiple biopsies from the same tumor in order to correctly classify patients. This intratumoral heterogeneity may be the reason for variations in treatment response and thus should be considered thoroughly when analyzing tumors to stratify patients. Caution must be taken when interpreting the results of a genomic biomarker from a single biopsy specimen.
Methods
Clinical Characteristics
Glioblastoma samples were from patients diagnosed and treated at Royal North Shore (RNS) and North Shore Private Hospitals between August 2012 and July 2013. All cases were consented and approved by Northern Sydney Local Health District Human Research Ethics Committee, under protocols 0211-171M and 1306-212M. All procedures were performed in accordance with the approved guidelines and regulations. Informed consent was obtained from all subjects. Tumor histology was reviewed by neuropathologist (J.C.) and a diagnosis of glioblastoma confirmed. Clinical data collected included age at diagnosis, gender, treatment and survival, with a minimum of 15 months follow-up on all cases (Supplementary Information [SI] Table 1). Adjuvant therapy included TMZ and RT or RT alone.
Fresh Frozen Tumor Tissue
A cohort of 14 glioblastoma cases was available. For each case, up to six tumor tissue biopsies (5–10 mm3) were harvested at surgery, from regions at least 1cm apart. Tissue was immediately snap frozen and stored at −80 °C until further analysis.
Tumor Tissue Quality Control
In order to assess tumor tissue quality a small section of each biopsy (3–5 mm3) was cut, fixed in formalin, and stained with haematoxylin and eosin (H&E) for scoring by neuropathologist (J.C.). Slides were scored for percentage volume of tumor, necrosis and non-tumor tissue.
Extraction of DNA and RNA
DNA and RNA was extracted from all remaining tissue from each biopsy using the Allprep DNA/RNA/Protein kit (Qiagen, Valencia, CA) as per manufacturer’s instructions. For all samples, purity was assessed using a Nanodrop ND-1000 (Thermo Scientific, Wilmington, DE) and for deep sequencing, DNA samples were further quantified using the Qubit dsDNA HS Assay Kit (Q32851, Life Technologies, Mulgrave, Victoria), performed on the Qubit Fluorometer 1.0 (Life Technologies).
Analysis of IDH1 mutation status
IDH1 mutation status was determined by Sanger sequencing of exon 4. DNA was amplified by polymerase chain reaction (PCR), using primers spanning exon 4 (5′-CATTTGTCTGAAAAACTTTGCTT-3′ (forward) and 5′-TCACATTATTGCCAACATGAC-3′ (reverse); amplicon size: 359 bp). PCR products were purified using the DNA Clean and Concentrator Kit (Zymo Research, Irvine, CA), and commercially sequenced (Australian Genome Research Facility, Westmead, Australia). Immunohistochemical screening for IDH1 mutation-positive cases was also performed by Pathology North, RNS Hospital, using the monoclonal antibody against IDH1 p.R132H (Clone H09; Dianova, Germany).
Determination of MGMT Promoter Methylation Status
DNA extracted from frozen tumor tissue was tested for MGMT promoter methylation by commercial pyrosequencing (University of Sydney). The assay threshold was determined by averaging the percent methylation at all 4 CpG sites in 4 non-neoplastic brain tissue samples (previously confirmed as being unmethylated by both pyrosequencing and methylation-specific PCR), and applying 2 standard deviations as previously reported by Dunn et al.44. Control samples were analyzed in 3 to 5 independent pyrosequencing runs, giving a mean of 5.49% (SD 3.85) and a positive methylation assay threshold of 13%.
Gene Expression Profiling
Following quality analysis and quantification, 1 μg RNA was treated with DNase 1, amplification grade (Life Technologies) and reverse transcribed using the SuperScript III First Strand Synthesis SuperMix Kit (Life Technologies) according to the manufacturer’s instructions. Gene expression was then analyzed commercially using the Fluidigm 96.96 BioMark HD System (Ramaciotti Gene Analysis Centre, Randwick, NSW, Australia) as per manufacturer’s instructions. The NormFinder algorithm45 was used to compare 5 endogenous control genes included on the array (TBP, ACTB, GAPDH, IPO8 and SDHA), identifying TBP as being the most stable control gene. Relative expression of target genes was determined using the 2−delta-delta Ct method (Fluidigm Real-time PCR analysis software), normalizing expression to TBP and a commercial pooled normal brain control sample (calibrator; Ambion, Life Technologies).
Genes of interest were then validated in triplicate using the Applied Biosystems 7900HT real time PCR system and individual TaqMan assays following the manufacturer’s instructions. Results were analyzed using Data Assist Software v3.0 (Life Technologies) and fold changes in each individual gene expression were calculated independently using the 2−delta-delta Ct method after normalizing to TBP.
Classification of Transcriptional Subtypes
Ct values from the Fluidigm and AB7900HT system (LGALS3/TBP only) were imported into the HTqPCR package in Bioconductor46 and unreliable data filtered out by applying a Ct cut off value of 40. Genes with errors detected in less than 1% of samples were retained in the analysis by imputing median values for those samples. Samples with Ct values greater than 40 for CCND2 were given a Ct = 40 to retain these in the analysis. TBP was used for delta Ct normalization. The Euclidean distance metric was used for hierarchical clustering of samples into transcriptional subclasses using a panel of genes previously found to be differentially expressed between subtypes47 or the DecisionDx-GBM®16 gene expression panel.
Targeted Next Generation Sequencing
An amplicon library was created using 250 ng of genomic DNA from tumor specimens as per the manufacturer’s instructions (TruSeq Custom Amplicon Library Kit, Illumina, Scoresby, Victoria). A custom oligo panel was designed using Illumina Design Studio software and the UCSC genome browser to improve coverage, avoiding regions of low complexity sequence and CpG rich sequence. Primers were designed against all exonic regions of the six MMR/BER genes of interest (APEX1, PARP1, MSH2, MSH6, MLH1 and PMS2) using a total of 183 amplicons, representing 99% of the coding sequence. The exome library was then pooled and sequenced on the Illumina MiSeq platform as 250-bp paired end reads (MiSeq Reagent Kit v2, 300 cycles) according to the Illumina protocol. Primary analysis of deep sequencing data was performed by the MiSeq Illumina reporter software (using default settings), including alignment of sequence reads to the human genome reference GRCh37 (hg19).
DNA Variant Identification
Sequenced reads were mapped to the hg19 reference human genome (Ensembl Gene annotation 2014.01.02) using Avadis NGS v1.6 software (Strand Life Sciences, India). After filtering out low-quality and duplicate reads, single nucleotide variants (SNVs) and short insertions/deletions were identified using pipeline analysis in the Avadis software and default criteria (≥20X coverage, an average base quality ≥20 with a confidence score cutoff greater than 50, ≥10 reads coverage of the specific variant location and ≥2 reads of the variant).
Identification of Predicted Significant variants
PROVEAN (Protein Variant Effect Analyzer) online software tool v1.1.348 was used to predict which variants may have damaging or deleterious effects according to PROVEAN and SIFT (Scale-Invariant Feature Transform) predictions.
Sanger sequencing
To validate the DNA variants predicted to be damaging or deleterious, Sanger sequencing was performed. Primers were designed using Primer 3 (v.0.4.0) to amplify ~200 bp amplicon containing the region of interest (SI Table 5). PCR reactions were carried out in a total of 30 μL which included ~100 ng genomic DNA, 6 μL HOT FIREPOL blend mix (Solis BioDyne, Tartu, Estonia) and 0.5 uM primers with cycling conditions (95 °C for 14 mins; 28 cycles of 95 °C for 15 secs, 58 °C for 30 secs, 72 °C for 1 min; 72 °C for 7 mins). Amplicons were purified and commercially sequenced as described above.
Sequence heterogeneity calculation
The sequence variants were used to calculate the sequence heterogeneity between the different biopsy specimens from each patient using MATLAB® R2013a (Mathworks, MA). Each gene was analysed individually. The total number of DNA variants in all biopsies for each gene was first identified (i.e. the total number of all DNA variants identified from all biopsies combined together). The number of DNA variants for each individual biopsy was then compared to this number to generate a ‘sequence variant percentage’ for each biopsy or, the number of DNA variants in an individual biopsy divided by the total number of DNA variants identified overall. This sequence variant percentage was then used to compare individual biopsy from the same patient together with similar percentages implying low heterogeneity. Samples were then stratified according to MGMT promoter methylation or treatment status.
Additional Information
How to cite this article: Parker, N. R. et al. Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci. Rep. 6, 22477; doi: 10.1038/srep22477 (2016).
Supplementary Material
Acknowledgments
This work was funded by The Sydney Neuro-Oncology Group.
Footnotes
Author Contributions N.R.P., P.K., J.F.P., V.M.H. and H.R.W. conceived the project and experiments. N.R.P., A.L.H., P.K., T.D. and R.J.I. designed and performed the experiments. N.R.P., A.L.H., P.K., T.D., R.J.I., Y.Z., Z.J.C., F.V., J.C. and V.M.H. analysed the data. N.R.P., T.D., H.R.W. and V.M.H. advised on the research. N.R.P., A.L.H., P.K., H.R.W. and V.M.H. wrote the manuscript. All authors reviewed and revised the manuscript.
References
- Stupp R. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10, 459–466 (2009). [DOI] [PubMed] [Google Scholar]
- Omuro A. & DeAngelis L. M. Glioblastoma and other malignant gliomas: a clinical review. JAMA 310, 1842–1850 (2013). [DOI] [PubMed] [Google Scholar]
- Patel A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabilsi N. H. et al. Multiplex mapping of chromatin accessibility and DNA methylation within targeted single molecules identifies epigenetic heterogeneity in neural stem cells and glioblastoma. Genome Res 24, 329–339 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szerlip N. J. et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci USA 109, 3041–3046 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snuderl M. et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20, 810–817 (2011). [DOI] [PubMed] [Google Scholar]
- Nickel G. C. et al. Characterizing mutational heterogeneity in a glioblastoma patient with double recurrence. PLoS One 7, e35262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonavia R., Inda M. M., Cavenee W. K. & Furnari F. B. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res 71, 4055–4060 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro J. R., Yung W. K. & Shapiro W. R. Isolation, karyotype, and clonal growth of heterogeneous subpopulations of human malignant gliomas. Cancer Res 41, 2349–2359 (1981). [PubMed] [Google Scholar]
- Sottoriva A. et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA 110, 4009–4014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little S. E. et al. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res 72, 1614–1620 (2012). [DOI] [PubMed] [Google Scholar]
- Johnson B. E. et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson D. A. et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 343, 72–76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegi M. E. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352, 997–1003 (2005). [DOI] [PubMed] [Google Scholar]
- Verhaak R. G. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman H. et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol 12, 49–57 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat K. P. et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 24, 331–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips H. S. et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173 (2006). [DOI] [PubMed] [Google Scholar]
- Brennan C. et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One 4, e7752 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bady P. et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol 124, 547–560 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung W. K., Shapiro J. R. & Shapiro W. R. Heterogeneous chemosensitivities of subpopulations of human glioma cells in culture. Cancer Res 42, 992–998 (1982). [PubMed] [Google Scholar]
- Parkinson J. F. et al. Variation of O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation in serial samples in glioblastoma. J Neurooncol 87, 71–78 (2008). [DOI] [PubMed] [Google Scholar]
- Grasbon-Frodl E. M. et al. Intratumoral homogeneity of MGMT promoter hypermethylation as demonstrated in serial stereotactic specimens from anaplastic astrocytomas and glioblastomas. Int J Cancer 121, 2458–2464 (2007). [DOI] [PubMed] [Google Scholar]
- Mollemann M., Wolter M., Felsberg J., Collins V. P. & Reifenberger G. Frequent promoter hypermethylation and low expression of the MGMT gene in oligodendroglial tumors. Int J Cancer 113, 379–385 (2005). [DOI] [PubMed] [Google Scholar]
- Hegi M. E. et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol 26, 4189–4199 (2008). [DOI] [PubMed] [Google Scholar]
- Hermanson M. et al. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res 52, 3213–3219 (1992). [PubMed] [Google Scholar]
- Nabeshima K. et al. Expression of c-Met correlates with grade of malignancy in human astrocytic tumours: an immunohistochemical study. Histopathology 31, 436–443 (1997). [DOI] [PubMed] [Google Scholar]
- Koga K. et al. Expression of angiopoietin-2 in human glioma cells and its role for angiogenesis. Cancer Res 61, 6248–6254 (2001). [PubMed] [Google Scholar]
- Bello L. et al. Alpha(v)beta3 and alpha(v)beta5 integrin expression in glioma periphery. Neurosurgery 49, 380–389; discussion 390 (2001). [DOI] [PubMed] [Google Scholar]
- Nishikawa R. et al. Immunohistochemical analysis of the mutant epidermal growth factor, deltaEGFR, in glioblastoma. Brain Tumor Pathol 21, 53–56 (2004). [DOI] [PubMed] [Google Scholar]
- Jung V. et al. Evidence of focal genetic microheterogeneity in glioblastoma multiforme by area-specific CGH on microdissected tumor cells. J Neuropathol Exp Neurol 58, 993–999 (1999). [DOI] [PubMed] [Google Scholar]
- Silber J. R. et al. The apurinic/apyrimidinic endonuclease activity of Ape1/Ref-1 contributes to human glioma cell resistance to alkylating agents and is elevated by oxidative stress. Clin Cancer Res 8, 3008–3018 (2002). [PubMed] [Google Scholar]
- Bobola M. S., Kolstoe D. D., Blank A., Chamberlain M. C. & Silber J. R. Repair of 3-methyladenine and abasic sites by base excision repair mediates glioblastoma resistance to temozolomide. Front Oncol 2, 176 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 7, 335–346 (2006). [DOI] [PubMed] [Google Scholar]
- Yip S. et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 15, 4622–4629 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill D. P. et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 13, 2038–2045 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsberg J. et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer 129, 659–670 (2011). [DOI] [PubMed] [Google Scholar]
- Shinsato Y. et al. Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen S. A. et al. Novel MSH6 Mutations in Treatment-Naive Glioblastoma and Anaplastic Oligodendroglioma Contribute to Temozolomide Resistance Independently of MGMT Promoter Methylation. Clin Cancer Res 20, 4894–4903 (2014). [DOI] [PubMed] [Google Scholar]
- Venere M. et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ 21, 258–269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J. B. et al. N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro Oncol 13, 471–486 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barazzuol L. et al. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat Oncol 8, 65 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed M. Z. et al. Development and evaluation of human AP endonuclease inhibitors in melanoma and glioma cell lines. Br J Cancer 104, 653–663 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn J. et al. Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br J Cancer 101, 124–131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen C. L., Jensen J. L. & Orntoft T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64, 5245–5250 (2004). [DOI] [PubMed] [Google Scholar]
- Dvinge H. & Bertone P. HTqPCR: high-throughput analysis and visualization of quantitative real-time PCR data in R. Bioinformatics 25, 3325–3326 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak R. G. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y. A Fast Computation of Pairwise Sequence Alignment Scores Between a Protein and a Set of Single-Locus Variants of Another Protein. In: In Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine (BCB’ 12) Edited by ACM, vol. 12. New York, NY, USA, 414–417 (2012).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.