

Summary
High‐mobility group box 1 (HMGB1) has been implicated in angiogenesis and rheumatoid arthritis (RA). The aim of this study was to define more clearly the role of HMGB1 in the synovial angiogenesis and pathogenesis of an immune model of arthritis. BALB/c mice were injected with monoclonal anti‐collagen antibody cocktail followed by lipopolysaccharide to induce arthritis. HMGB1 and vascular endothelial growth factor (VEGF) were over‐expressed in the areas of the synovium where more inflammation and neoangiogenesis were present. The selective blockade of HMGB1 or VEGF resulted alternatively in a lower severity of arthritis evaluated by the arthritis index. Furthermore, exogenous HMGB1 administration caused a worsening of arthritis, associated with VEGF up‐regulation and increased synovial angiogenesis. The selective inhibition of VEGF also resulted in no induction of arthritis in mice receiving exogenous HMGB1. Cytokine enzyme‐linked immunosorbent assay (ELISA) analyses performed on peripheral blood and synovial fluid demonstrated a significant reduction of interleukin (IL)−1β, IL‐6 and tumour necrosis factor (TNF)‐α in mice where HMGB1 and VEGF pathways were blocked. Interestingly, the selective blockade of HMGB1 and VEGF resulted in an increase of the peripheral IL‐17A concentration. The development of arthritis mediated by HMGB1 and the synovial angiogenesis can be blocked by inhibiting the VEGF activity. The proinflammatory and proangiogenic cytokine IL‐17A was increased when HMGB1 is inhibited, but the synovial angiogenesis was nevertheless reduced in this model of arthritis. Taken together, these findings shed new light on the role of this nuclear protein in the pathogenesis of arthritis in an RA‐like model.
Keywords: angiogenesis, high‐mobility group box 1, rheumatoid arthritis, vascular endothelial growth factor
Introduction
High‐mobility group box‐1 (HMGB1) is a highly conserved nuclear protein that acts as a cytokine when released into the extracellular space by necrotic and inflammatory cells, and is involved in inflammatory response and tissue regeneration 1, 2. HMGB1 is released passively during cellular necrosis by almost all cells and secreted actively by immune cells such as monocytes and macrophages 3. Several studies established the ‘alarmin’ activity of HMGB1 and its extracellular translocation in the settings of immune cell function. Among endogenous danger molecules, HMGB1 interacts with a variety of receptor systems, including the receptor for advanced glycation end‐products (RAGE) and the Toll‐like receptors (TLRs) 4. In addition to its effects on inflammation, HMGB1 plays a relevant role in angiogenesis, myogenesis and skeletal muscle activity 2, 4, 5. Of immunologically mediated diseases, rheumatoid arthritis (RA) shows the definitive evidence for involvement of HMGB1 in pathogenesis and development. In patients with RA, extracellular HMGB1 release could perpetuate synovitis by up‐regulating the expression of tumour necrosis factor (TNF)‐α, interleukin (IL)‐1β and IL‐6 and other proinflammatory factors 6. Finally, very interesting data demonstrate that HMGB1 blockade therapy with antibodies ameliorates collagen‐induced arthritis in both mice and rats 7.
Synovium from patients affected by RA is characterized by an abnormal angiogenesis, the formation of new blood vessels 8. Neovascularization is a complex process involving several factors including TNF‐α, fibroblast growth factor (FGF), IL‐8, transforming growth factor (TGF)‐β and vascular endothelial growth factor (VEGF) 9. VEGF is implicated in the stimulation of normal postnatal angiogenesis involved in wound healing and pathological conditions, such as diabetic retinopathy and tumour growth 10. VEGF stimulates endothelial cell (ECs) proliferation and chemotaxis, increases vascular permeability and induces the secretion of matrix metalloproteases (MMPs) 11. Changes in vascular permeability enhance tissue swelling, whereas stimulation of MMPs function is important in synovitis and cartilage destruction observed in RA 12. Interestingly, HMGB1 plays a key role in angiogenesis through multiple mechanisms, including up‐regulation of VEGF, promoting the homing of endothelial progenitor cells (EPCs) to ischaemic tissues and inducing ECs migration and sprouting 13. Furthermore, it is demonstrated that RAGE blockade inhibits HMGB1‐induced neovascularization and EC proliferation in vitro 14 and that exogenous HMGB1 administration enhances angiogenesis in vivo 2, 15. Interestingly, recent data suggest that this nuclear protein induces angiogenesis in rheumatoid arthritis via HIF‐1α activation 16.
Although HMGB1 has often been implicated in the pathogenesis of RA, the mechanisms of action by which this nuclear protein may determine the development of the disease are not understood completely. Teleologically, HMGB1 is considered an ‘alarmin’ because it is released within a defence response; thereafter, it would lead to the onset of an inflammatory damage and of pathological angiogenesis. The aim of this study was to explore the role of HMGB1 in the pathogenesis of an immune mediated RA‐like model, with particular attention to the neoangiogenesis of the synovial pannus in the experimental model of collagen antibody‐induced arthritis (CAIA).
Material and methods
Mouse anti‐collagen antibody‐induced arthritis model (CAIA)
All investigations were approved by the Catholic University School of Medicine Institutional Animal Care and Use Committee. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by Directive 2010/63/EU of the European Parliament. Male BALB/c mice (The Jackson Laboratory, Bar Harbor, ME, USA) aged 8–12 weeks were used for experiments. All the animals were allowed free access to food and water throughout the study. A cocktail of five monoclonal antibodies, clone A2‐10 [immunoglobulin (Ig)G2a], F10‐21 (IgG2a), D8‐6 (IgG2a), D1‐2G (IgG2b) and D2‐112 (IgG2b), recognizing the conserved epitopes on various species of type II collagen (Chondrex Inc., Redmond, WA, USA), was used according to the manufacturer's instructions and as described previously 17. Mice were injected with the cocktail of antibodies intravenously (i.v.). Three days later the mice were injected with lipopolysaccharide (LPS) from Escherichia coli 0111:B4 (Chondrex Inc.) intraperitoneally (i.p.) to trigger arthritis development. Animals were evaluated every 3 days after the infusion of the antibody cocktail for arthritis incidence and each paw was evaluated and scored individually on a scale of 0–4, with 4 indicating the most severe inflammation 18. An arthritis index (AI) that expressed a cumulative score for all paws (maximum possible value = 16) was calculated for each animal 19. Two independent observers blinded to the identity of the mice performed all arthritis evaluations.
Experimental design and groups
To investigate the role of HMGB1 in pathological synovial angiogenesis in a model of arthritis (CAIA) in mice, three groups of mice (n = 5 per group) were studied: CAIA mice treated with HMGB1, CAIA mice treated with HMGB1 selective inhibitor BoxA and untreated control CAIA mice. To define and clarify further the HMGB1–VEGF interaction, 15 additional CAIA mice were used. To meet the European ethical directives and in order to avoid interexperimental variability, the experiments were carried out simultaneously and controls were examined only once.
Exogenous HMGB1 protein administration
In 10 CAIA mice, HMGB1 protein (HMGBiotech, Milan, Italy) was administered in a single dose by i.p. injection, 1 h before the induction of the arthritis, at a concentration of 800 ng per mouse in 0·2 ml of phosphate‐buffered saline (PBS).
In‐vivo inhibition of HMGB1 function
The activity of HMGB1 was systemically inhibited in vivo in 10 CAIA mice by an i.p. injection of the HMGB1 inhibitor BoxA (HMGBiotech), 1 h before the induction of the arthritis, at a concentration of 800 ng per mouse in 0·2 ml of PBS.
In‐vivo inhibition of VEGF activity
To examine the effects of VEGF in pathogenesis of CAIA, we blocked VEGF activity by using a selective and specific inhibitor sFlt‐1, a soluble form of the Flt‐1 VEGF receptor (VEGFR) 20. This isoform inhibits VEGF activity by directly sequestering VEGF and functioning as a dominant negative inhibitor against VEGFRs. The plasmid was kindly provided by Professor Kensuke Egashira.
sFlt‐1 plasmid (100 μg/30 μl PBS) was injected into the right femoral muscle of five CAIA mice treated with HMGB1 protein and five CAIA mice treated with BoxA, using a 27‐gauge needle 1 day before the induction of arthritis. To ensure VEGF inhibition, changes in VEGFR‐1 (Flt‐1) and VEGFR‐2 (Flk‐1) phosphorylation were evaluated (see Supporting information) 2. A separate group of five CAIA animals received an equal amount of empty plasmid via i.m. injection within the same time schedule.
Laser Doppler analysis
A laser Doppler perfusion imager (LDPI) system (PeriScan PIM II; Perimed, Järfälla, Sweden) was used to measure hindlimb blood perfusion before and after the arthritis induction and then followed at 7‐day intervals, until the end of the study, for a total follow‐up of 21 days after antibody injection 21. Before evaluation, excess hairs were removed from the limbs using depilatory cream and animals were placed on a heating plate at 40°C 20. The imager was positioned 40 cm above the surface of the limbs for all mice. Subsequent image analysis was performed with the manufacturer's dedicated software, which displayed a colour‐coded image of tissue perfusion on a monitor. The results were expressed as the ratio between the perfusion of the sum of the four limbs to that measured before induction of arthritis.
Histological assays
Thirty animals were included in this longitudinal trial. All the animals were killed 21 days after immunization. For cartilage staining, safranin O‐fast green was used on the joints. Immunohistochemical analysis was realized using a labelled streptavidin–biotin–peroxidase method (LSABPx). Sections were cut at a thickness of 3 mm and mounted onto slides coated with the adhesive 3‐aminopropyltriethoxysilane and dried in a 60°C oven for 4 h to ensure maximum adhesion. After dewaxing and rehydration, slides were placed in antigen retrieval solution and treated for 30 min in the microwave oven at 250 W followed by cooling for 20 min at room temperature. Endogenous peroxidase was blocked with 3% hydrogen peroxide for 5 min. After several washing steps with phosphate‐buffered saline, sections were incubated with the following antibodies: IL‐6 (rabbit polyclonal antibody, dilution 1 : 100, retrieval with citrate buffer; TCM Tecnochimica Moderna, Rome, Italy); HMGB1 [rabbit polyclonal antibody, dilution 1 : 300 retrieval with Tris/ethylenediamine tetraacetic acid (EDTA)/citrate solution (TEC) buffer; ThermoFisher Scientific, Carlsbad, CA, USA]; VEGF (A‐20 sc‐152 rabbit polyclonal antibody, dilution 1 : 100 without retrieval; Santa Cruz Biotechnology, Santa Cruz, CA, USA), CD31 (rat monoclonal antibody, clone SZ31, dilution 1 : 20 EDTA buffer; Optistain, ThermoFisher Scientific) for 30 min in the humid chamber at room temperature. Visualization of the reaction was performed with the Dako LSAB 2 Kit peroxidase (Dako, Glostrup, Denmark), which contains labelled streptavin–biotin for primary rabbit/mouse antibody and diaminobenzidine. Sections were counterstained briefly with haematoxylin, dehydrated, cleared and coverslipped. Negative controls were obtained by substitution of the primary antibody with rabbit non‐immune serum or omitting the primary antibody. The area and the positive cells were measured using an NIH image analysis system (ImageJ version 1·41; National Institutes of Health, Bethesda, MD, USA). Capillary density and was measured by counting six random high‐power (magnification ×200) fields from each preparation of synovial tissue on an inverted light microscope, and were expressed by the number of CD31+ cells per square field. Two operators extracted the results independently.
ELISA
Sera and joint fluid were harvested from all animals 10 days after the induction of arthritis and kept frozen until biochemical analysis. Multi‐Analyte ELISArray kits (Qiagen, Valencia, CA, USA) designed for mice were used for the quantitative measurement of 12 different cytokines and chemokines (e.g. IL‐1β, IL‐6, TNF‐α, VEGF and IL‐17A). For the serum fluid, a total of five measurements per treatment were made and the graphs show the mean with standard deviation. The joint fluid was collected by cutting open capsules of ankle joints and by washing the joint cavity with 5 μl PBS, and 1 μl of withdrawn joint fluid was diluted 10‐fold to assess the concentrations of cytokines/chemokines. The synovial fluid measurements were made by combining the samples of a pool of five mice per group. We read the absorbance at 450 nm within 30 min of stopping the reaction. Standards were used, standard curves were plotted and the experimental protein values were calculated. Results were expressed as cytokine/chemokine fold increases, calculated as the ratio between protein levels in HMGB1‐, BoxA‐, sFlt‐1‐treated mice and control CAIA mice. The measurement was performed only once, and therefore a standard deviation was not determined.
Statistical analysis
Statistical analysis was performed using stata software version 10·0; StataCorp, College Station, TX, USA). The data are expressed as the mean ± standard error of the mean (s.e.m.). Comparison among groups was carried out using analysis of variance (anova) followed by Fisher's post‐hoc test. Repeated anova measures were used to assess the improvement in perfusion over time within groups. Significance was set at a probability value (P) of < 0·05.
Results
HMGB1 and VEGF are over‐expressed in the areas of the synovial inflammation
Severe arthritis was observed in control CAIA mice (Fig. 1). Histological and immunohistochemical analyses confirmed the presence of an inflammatory infiltrate at the level of the synovium of arthritic mice associated with the local over‐expression of IL‐6 (Fig. 2). Immunohistochemical analysis also confirmed that VEGF and HMGB1 were expressed more locally in mice with more severe arthritis (see Supporting information). ELISA analysis performed on the synovial fluid showed that the levels of IL‐1β, IL‐6, TNF‐α, VEGF and IL‐17A were elevated in control CAIA mice (Fig. 3). The immunostaining for CD31, a specific marker of endothelial cells, revealed an increased amount of blood vessels in the synovium of mice with arthritis, compared to control CAIA mice (Fig. 2). The LDPI analysis confirmed an improved blood flow at the level of the arthritic joint 7, 14 and 21 days after the induction of the arthritis (Fig. 4).
Figure 1.
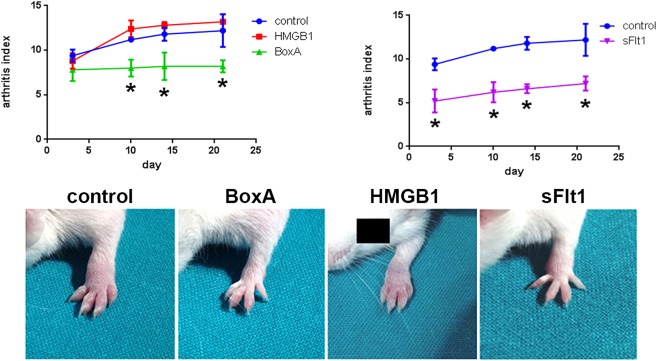
Arthritis index (AI) evaluation. An AI that expressed a cumulative score for all paws (maximum possible value = 16) was calculated for each animal. Animals were evaluated every 3 days after the infusion of the antibody cocktail for arthritis incidence and each paw was evaluated and scored individually on a scale of 0–4, with 4 indicating the most severe inflammation. Less severe arthritis was observed in mice in which high‐mobility group box 1 (HMGB1) was blocked by BoxA administration. The arthritis induced in HMGB1‐treated mice was similar to that observed in control collagen antibody induced arthritis (CAIA) mice. Arthritis induction was reduced in mice treated with the vascular endothelial growth factor (VEGF) inhibitor sFlt1; n = 5 mice per group. *P < 0.05 versus control CAIA mice.
Figure 2.
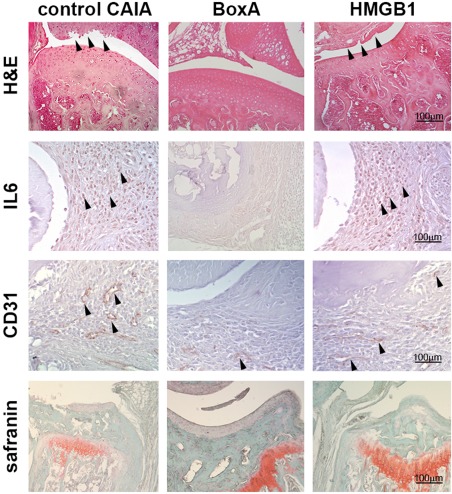
Representative micrographs illustrating the haematoxylin and eosin staining, the immunohistochemical staining of interleukin (IL)‐6, CD31, a specific marker of endothelial cells and the safranin staining in control arthritic, BoxA‐treated and high‐mobility group box 1 (HMGB1)‐treated mice obtained on day 21 of arthritis. For immunohistochemical staining, positive staining appears in brown. Magnification ×200; n = 10 sections per joints; n = 5 mice per group.
Figure 3.
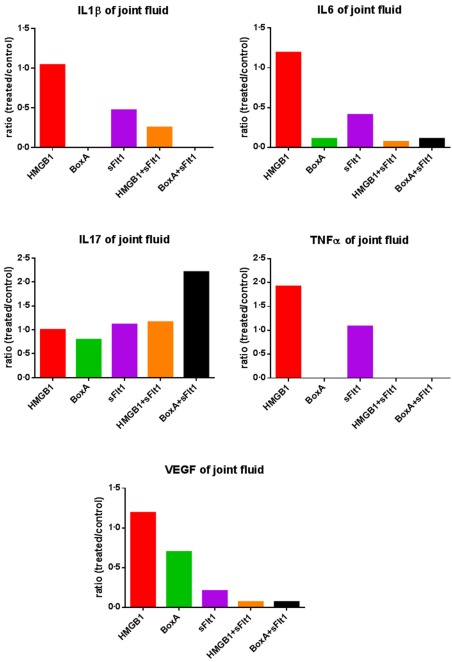
Enzyme‐linked immunosorbent assay (ELISA) analyses performed on synovial fluid demonstrated a reduction of interleukin (IL)‐1β, IL‐6 and tumour necrosis factor (TNF)‐α in mice where high‐mobility group box 1 (HMGB1) and vascular endothelial growth factor (VEGF) pathways were blocked. Interestingly, the selective blockade of HMGB1 and VEGF resulted in an increase of the synovial IL‐17A concentration. The synovial fluid measurements were made by combining the samples of a pool of five mice per group. Results were expressed as cytokine/chemokine fold increases, calculated as the ratio between protein levels in HMGB1‐, BoxA‐, sFlt‐1‐treated mice and control arthritic mice. The measurement was performed only once, and a standard deviation has not been determined; n = 5 mice per group.
Figure 4.
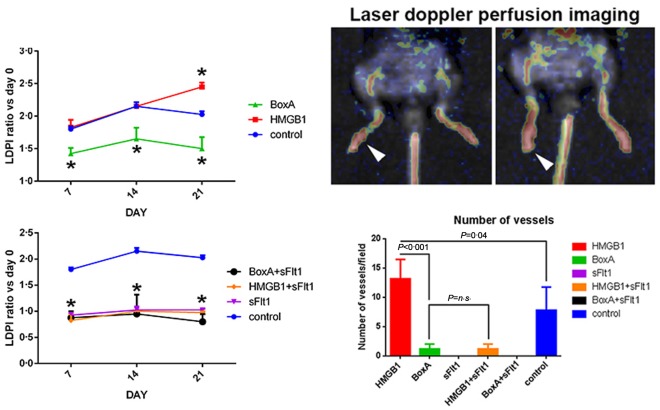
Foot blood flow monitored in vivo by laser Doppler perfusion imaging (LDPI) in untreated control arthritic, BoxA‐treated and high‐mobility group box 1 (HMGB1)‐treated mice. Representative evaluation of an HMGB1‐treated mice 7 (left) and 21 days (right) after arthritis induction. In colour‐coded images, red indicates normal perfusion while yellow indicates reduced perfusion and blue indicates a marked reduction in blood flow in the hindlimb. The results are expressed as the ratio between the perfusion of the sum of the four limbs with that measured before the induction of arthritis. The blood flow of the arthritic joint is expressed as the ratio between perfusion of day 0 versus days 7, 14 and 21. *P < 0·05 versus control collagen antibody‐induced arthritis (CAIA) mice. The number of vessels per field, obtained by evaluation of the CD31‐positive cells, was increased significantly in HMGB1‐treated mice with respect to control CAIA mice; n = 5 mice per group.
Selective blockade of HMGB1 results in a lower severity of arthritis
To confirm the importance of the HMGB1 role in the CAIA model we selectively blocked the HMGB1 pathway by administration of exogenous BoxA. Clinical assessment by the AI showed a reduction of arthritis induced in BoxA‐treated mice (P = 0·003) (Fig. 1). Histological assay confirmed that in these mice there was a reduced inflammatory infiltrate and joint erosions were not present (Fig. 2). The immunostaining for the inflammatory markers showed a decreased expression of IL‐6 in mice in which the HMGB1 activity was blocked systemically (Fig. 2). ELISA analyses performed on serum revealed a reduction of inflammatory cytokines in mice treated with the selective HMGB1 inhibitor BoxA (Fig. 5). According to the systemic findings, ELISA evaluation of synovial fluid revealed reduced levels of IL‐1β, IL‐6, TNF‐α, VEGF and IL‐17A (Fig. 3). Interestingly, immunohistochemical analysis and LDPI assessment demonstrated a significant reduction of angiogenesis and blood flow in mice in which the HMGB1 activity was inhibited (P < 0·001) (Fig. 4).
Figure 5.
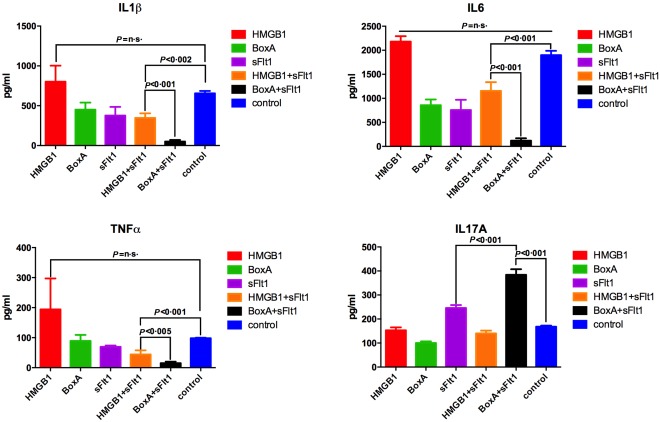
Enzyme‐linked immunosorbent assay (ELISA) analyses performed on serum demonstrated a significant reduction of interleukin (IL)‐1β, IL‐6 and tumour necrosis factor (TNF)‐α in mice where high‐mobility group box 1 (HMGB1) and vascular endothelial growth factor (VEGF) pathways were blocked. Interestingly, the selective blockade of HMGB1 and VEGF resulted in an increase of the peripheral IL‐17A concentration. For the serum fluid, a total of five measurements per treatment were made and the graphs show the mean with the standard deviation; n = 5 mice per group.
Systemic inhibition of VEGF induces lower severity of arthritis
By analysing the joints we noted that the VEGF concentration was higher in mice in which a more severe arthritis was observed. In fact, ELISA performed on the synovial fluid and immunohistochemical analyses of synovium revealed that VEGF expression was greater in mice treated with HMGB1 and in control CAIA mice, compared with BoxA‐treated mice (Fig. 3). Starting from the hypothesis that VEGF could have a causal role in the development of arthritis in this model, we selectively blocked its function using the sFlt1 vector. Interestingly, arthritis induction was reduced in mice treated with the VEGF inhibitor sFlt1, compared with control CAIA mice (P < 0·001) (Fig. 1). Moreover, arthritis was almost absent in mice in which the activity of VEGF and HMGB1 had been blocked at the same time (P < 0·001) (Fig. 6). Systemic levels of proinflammatory cytokines were reduced in mice in which the VEGF activity was abolished compared to controls and to HMGB1‐treated mice (Fig. 5). Interestingly, serum levels of IL‐1β, IL‐6 and TNF‐α were reduced in HMGB1‐treated mice in which the VEGF activity was inhibited compared to control CAIA mice (Fig. 5).
Figure 6.
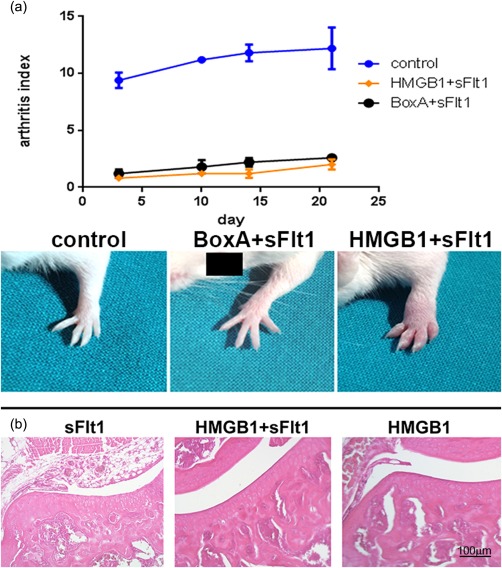
(a) Arthritis index (AI) evaluation. Arthritis was almost absent in mice in which the activity of vascular endothelial growth factor (VEGF) and of high‐mobility group box 1 (HMGB1) had been blocked at the same time. *P < 0·01 versus control collagen antibody‐induced arthritis (CAIA) mice. (b) Haematoxylin and eosin sections of joints from control arthritic, sFlt1‐ and HMGB1+sFlt1‐treated arthritic mice obtained on day 21 of arthritis. Magnification ×200; n = 10 sections per joints; n = 5 mice per group.
Exogenous HMGB1 administration causes worsening of arthritis associated with VEGF up‐regulation and increased synovial angiogenesis
More severe arthritis was observed in mice treated with exogenous HMGB1 and was similar to that observed in control CAIA mice [P = not significant (n.s.)] (Fig. 1). LDPI revealed that blood perfusion of arthritic limbs was represented more in mice treated with HMGB1 compared with the other group of mice. Notably, the perfusion evaluation demonstrated that angiogenesis was increased significantly even compared to control CAIA mice, considering day 21. Histological and immunohistochemical analyses confirmed that in HMGB1‐treated mice severe inflammation and erosion were present (Fig. 2). Interestingly, ELISA analysis of synovial fluid revealed an increased VEGF concentration in these mice, compared with control CAIA mice (Fig. 3). The immunostaining for the vascular endothelial specific marker CD31 showed an increased number of vessels in HMGB1‐treated mice (P = 0·046) (Fig. 4), suggesting an enhanced synovial angiogenesis.
Selective inhibition of VEGF also results in the lack of induction of arthritis in mice receiving exogenous HMGB1
Given the relationship between HMGB1 and VEGF, we tried to evaluate the effect of exogenous administration of HMGB1 in mice in which the VEGF activity was blocked systematically. Surprisingly, HMGB1 administration did not induce arthritis in the CAIA model when VEGF pathway was inhibited (P < 0·001) (Fig. 6a). Histological and immunohistochemical evaluations confirmed the absence of inflammation and erosion in these mice (Fig. 6b). ELISA analysis performed on the synovial fluid showed that the levels of IL‐1β, IL‐6, TNF‐α and VEGF were reduced in HMGB1/sFlt1‐treated mice, compared to control CAIA mice (Fig. 3).
Discussion
The traditional approach for understanding human joint chronic inflammation such as RA has implicated several mechanisms that contribute to the onset and perpetuation of synovial injury, including production of proinflammatory molecules 22. In this scenario HMGB1 represents an interesting and very attractive player. When HMGB1 is released in cases of cellular damage into the extracellular space, it is able to activate several inflammatory pathways and release of cytokines, such as TNF‐α, IFN‐γ, IL‐1, IL‐6 and IL‐8 23. Interestingly, many studies have been performed and are ongoing to determine whether this protein is crucial in damage responses and whether it is useful to modulate its pathway, or whether it is only an epiphenomenon in activated inflammatory cascades. Furthermore, it is interesting to note that cells undergoing programmed death do not release this protein 3. This phenomenon seems to indicate that this nuclear protein represents an ancestral defence mechanism that has been maintained over time and that is activated during cell injury.
An alternative point of view on synovial biology in RA has recently proposed a new approach derived from oncology. According to this view, the rheumatoid synovium could be seen as a tumour‐like mass invading articular cartilage. Indeed, as a cancer, the synovium of a patient with RA also requires nutrients and oxygen to grow; in this context, angiogenesis plays a major role. The formation of new tissue needs a blood supply, and the formation of new blood vessels serves to ensure an adequate blood flow. In rheumatoid synovium there is a relevant increase in vascular density. In partial contrast with these data, however, the pathological synovial tissue presents marked hypoxia and acidosis. Hypoxia, through the secretion of hypoxia‐inducible factors (HIFs), determines an increase of the angiogenic stimulus. Nevertheless, it is unclear whether this new vessel formation is functionally effective or whether they are primarily disorganized vascular structures. Probably, as in many other conditions, an initially protective response to the pathogen stimulus becomes uncontrolled and harmful.
The role of HMGB1 in RA has been studied from many viewpoints, and most findings confirm that this nuclear protein may play an important part in the pathogenesis of the disease. This alarmin exerts proinflammatory effects by induction of cytokine production, recruitment and activation of antigen‐presenting cells 24. There are different theories concerning the molecular mechanisms by which these effects are induced. First, HMGB1 promotes inflammation both by direct interaction with Toll‐like receptor (TLR)−2 and TLR‐4, resulting in protein kinase B (Akt), nuclear factor‐kappa B (NF‐κB) and TNF‐α pathways activation 6, 25. Moreover, HMGB1 signalling through RAGE induces chemotaxis, maturation and migration of immune cells, as well as the up‐regulation of cell surface receptors 25. Accordingly, in a murine model of arthritis, HMGB1 local administration causes articular injury by stimulating monocyte/macrophages to secrete a subset of proinflammatory cytokines and chemokines 26. In addition, the alarmin induces synovial cell proliferation via the activation of the signal transducer and activator of transcription‐1 (STAT‐1)/ suppressor of cytokine signaling‐1 (SOCS‐1) signal pathway in an experimental model of arthritis 27. Furthermore, HMGB1 promotes the development of inflammation by up‐regulating TLR‐2 and IL‐23 from CD14+ monocytes from patients with rheumatoid arthritis 24. Regarding the formation of new vessels, several reports have suggested that HMGB1 induces up‐regulation of proangiogenic factors, promotes the homing of EPCs to ischaemic tissues and induces ECs migration and sprouting in vitro and in vivo 13, 14, 15. Interestingly, recent data suggest that HMGB1 is involved in angiogenesis in arthritis via the HIF‐1α pathway.
The first evidence of the present study is that HMGB1 is involved in the pathogenesis of CAIA and that its administration is able to facilitate, but not to increase, the development of arthritis in this animal model. This finding is consistent with previous data in literature but adds a new element: alarmin is important in the pathogenesis of arthritis but already exerts its highest biological effects at physiological concentrations. Another relevant discovery of this study is that HMGB1 systemic administration induces VEGF up‐regulation in the synovial tissue. VEGF is one of the most proangiogenic factors and is involved in neovessel formation. Accordingly, in mice treated with HMGB1 we noted an enhanced angiogenesis of the arthritic synovium. Furthermore, the formation of new vascular structures is dependent upon VEGF. Indeed, in mice where the activity of this proangiogenic cytokine was blocked, angiogenesis induced by HMGB1 administration has not been demonstrated.
Because we have found that HMGB1 and VEGF are linked closely in this animal model of experimental arthritis, we sought to investigate the relationships between them through the selective inhibition of the two proteins. Initially, we blocked the systemic activity of HMGB1 and confirmed that VEGF local expression and synovial neoangiogenesis is reduced in this condition. Interestingly, in CAIA mice treated with BoxA, the HMGB1 inhibitor, the severity and progression of arthritis are decreased, compared with control CAIA mice. This is consistent with previous evidence 6, 26 whereby this nuclear protein is implicated in the pathogenesis of arthritis. To investigate further the relationship with VEGF, we systemically blocked the activity of this proangiogenic factor by the administration of the sFlt‐1 plasmid. As expected, in CAIA mice treated with sFlt‐1 we also observed a reduced degree of synovial neoangiogenesis in animals subject to exogenous administration of HMGB1. This is further confirmation that angiogenesis induced by HMGB1 is dependent upon VEGF. The beneficial effects of the VEGF blockade alone has been demonstrated previously 28, 29. To our knowledge, this is the first demonstration that HMGB1 administration is able to induce synovial angiogenesis in an experimental model of rheumatoid arthritis and that the formation of new vessels is dependent upon VEGF and can be prevented by systemic inhibition of the VEGF activity. Surprisingly, even the severity and pattern of arthritis were limited in CAIA mice when the VEGF activity was blocked, compared with control arthritic mice. A possible explanation could be due to the differences we noted when we measured the levels of proinflammatory cytokines at systemic and intra‐articular levels. While systemic levels of IL‐1, TNF‐α and IL‐6 are elevated significantly in HMGB1‐treated animals in which VEGF activity was blocked, the levels of these inflammatory cytokines are reduced in the synovial fluid of the same mice compared to control CAIA mice. Thus, it is not difficult to suppose that the impaired synovial neovascularization of these animals minimizes the spread of inflammatory systemic factors. An additional finding of this study is that the severity and progression of arthritis are reduced further when both the HMGB1 and VEGF activity are inhibited in CAIA mice. Furthermore, a reduction of IL‐1, TNF‐α and IL‐6 was detected in the synovial fluid of animals treated concomitantly with BoxA and sFlt‐1 compared to arthritic control mice.
A controversial role is played by IL‐17A in our model. This proinflammatory and proangiogenic cytokine is extremely important in several pathological conditions, including carcinogenesis and arthritis 30, 31, 32, 33. Our data show that IL‐17A levels are increased when VEGF is blocked systemically, suggesting a possible compensatory response given by this cytokine. The IL‐17A up‐regulation, however, did not result in an increased synovial angiogenesis, probably because its proangiogenic role is closely dependent upon VEGF activity.
There are several limitations in this study, and we list the most relevant. First, we performed an experimental animal model of antibody to collagen‐induced arthritis. Clearly, the human scenario is much more complex than the one that can be recreated in the CAIA model. Collagen‐induced arthritis, for instance, is highly related to IL‐1 and TNF‐α, but it is much less dependent upon IL‐6 activity 34. It would not be possible, however, to block selectively the pathways of HMGB1 and VEGF in conditions more similar to the real patient. Moreover, our data do not explain fully why the additional blockade of HMGB1 and VEGF result in a reduction of arthritis more significant than that obtained with the isolated block of the two pathways. Another important concern is that the CAIA model we performed requires LPS administration. It should be noted that HMGB1 functions and activities could be stimulated directly by LPS. This is why we also used animals that received exogenous HMGB1. As we found that the arthritic control mice and HMGB1‐treated animals showed no substantial differences in the development of arthritis, we believe that interactions with LPS were not relevant to the experiment. Furthermore, the synovial fluid evaluation was performed without a control protein detected in the sample: we analysed the ratio between the treated and control groups. This specific bias could be responsible for potential mistakes.
In conclusion, we have demonstrated for the first time that the proinflammatory activity of HMGB1 in the model of collagen‐induced arthritis is dependent upon VEGF, that HMGB1 directly induces the formation of new synovial blood vessels and that synovial HMGB1‐dependent angiogenesis can be prevented by the selective inhibition of VEGF. Further experiments and evaluations are needed to define the role and relationships of these two key proteins in the pathophysiology of human inflammation, but data obtained in this study shed new light on the possible therapeutic implications of the HMGB1/VEGF pathway modulation.
Disclosure
The authors declare that they have no disclosures.
Author contributions
F. B., A. F. and G. F. conceived the study, participated in its design, performed the mouse anti‐collagen antibody‐induced arthritis model, performed data analysis and reviewed the manuscript. V. A., E. S. and F. A. performed the immunohistochemical analysis. B. T. and F. A. carried out the immunoassays. E. G. and G. F. participated in the design of the study and helped draft the manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Supporting Information
Acknowledgements
The authors gratefully acknowledge the contribution of Dr Maria Emiliana Caristo, Director of the Animal House, Catholic University School of Medicine, Rome, Italy. The authors also gratefully acknowledge the contribution of Professor Kensuke Egashira, from the Department of Cardiovascular Medicine, Kyushu University, Fukuoka, Japan. This work was supported by the Catholic University School of Medicine.
References
- 1. Palumbo R, Sampaolesi M, De Marchis F et al Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol 2004; 164:441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Biscetti F, Straface G, De Cristofaro R et al High‐mobility group box‐1 protein promotes angiogenesis after peripheral ischemia in diabetic mice through a VEGF‐dependent mechanism. Diabetes 2010; 59:1496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418:191–5. [DOI] [PubMed] [Google Scholar]
- 4. Pisetsky DS, Erlandsson‐Harris H, Andersson U. High‐mobility group box protein 1 (HMGB1): an alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res Ther 2008; 10:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biscetti F, Ghirlanda G, Flex A. Therapeutic potential of high mobility group box‐1 in ischemic injury and tissue regeneration. Curr Vasc Pharmacol 2011; 9:677–81. [DOI] [PubMed] [Google Scholar]
- 6. Kokkola R, Sundberg E, Ulfgren AK et al High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum 2002; 46:2598–603. [DOI] [PubMed] [Google Scholar]
- 7. Kokkola R, Li J, Sundberg E et al Successful treatment of collagen‐induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum 2003; 48:2052–8. [DOI] [PubMed] [Google Scholar]
- 8. Szekanecz Z, Koch AE. Mechanisms of disease: angiogenesis in inflammatory diseases. Nat Clin Pract Rheumatol 2007; 3:635–43. [DOI] [PubMed] [Google Scholar]
- 9. Brenchley PE. Angiogenesis in inflammatory joint disease: a target for therapeutic intervention. Clin Exp Immunol 2000; 121:426–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Biscetti F, Straface G, Pitocco D, Zaccardi F, Ghirlanda G, Flex A. Peroxisome proliferator‐activated receptors and angiogenesis. Nutr Metab Cardiovasc Dis 2009; 19:751–9. [DOI] [PubMed] [Google Scholar]
- 11. Wary KK, Thakker GD, Humtsoe JO, Yang J. Analysis of VEGF‐responsive genes involved in the activation of endothelial cells. Mol Cancer 2003; 2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dvorak HF, Detmar M, Claffey KP, Nagy JA, van de Water L, Senger DR. Vascular permeability factor/vascular endothelial growth factor: an important mediator of angiogenesis in malignancy and inflammation. Int Arch Allergy Immunol 1995; 107:233–5. [DOI] [PubMed] [Google Scholar]
- 13. Schlueter C, Weber H, Meyer B et al Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol 2005; 166:1259–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mitola S, Belleri M, Urbinati C et al Cutting edge: extracellular high mobility group box‐1 protein is a proangiogenic cytokine. J Immunol 2006; 176:12–5. [DOI] [PubMed] [Google Scholar]
- 15. Limana F, Germani A, Zacheo A et al Exogenous high‐mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C‐kit+ cell proliferation and differentiation. Circ Res 2005; 97:e73–83. [DOI] [PubMed] [Google Scholar]
- 16. Park SY, Lee SW, Kim HY, Lee WS, Hong KW, Kim CD. HMGB1 induces angiogenesis in rheumatoid arthritis via HIF‐1α activation. Eur J Immunol 2015; 45:1216–27. [DOI] [PubMed] [Google Scholar]
- 17. Bender AT, Spyvee M, Satoh T et al Evaluation of a candidate anti‐arthritic drug using the mouse collagen antibody induced arthritis model and clinically relevant biomarkers. Am J Transl Res 2013; 5:92–102. [PMC free article] [PubMed] [Google Scholar]
- 18. Brand DD, Latham KA, Rosloniec EF. Collagen‐induced arthritis. Nat Protoc 2007; 2:1269–75. [DOI] [PubMed] [Google Scholar]
- 19. Palmblad K, Sundberg E, Diez M et al Morphological characterization of intra‐articular HMGB1 expression during the course of collagen‐induced arthritis. Arthritis Res Ther 2007; 9:R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Biscetti F, Straface G, Arena V et al Pioglitazone enhances collateral blood flow in ischemic hindlimb of diabetic mice through an Akt‐dependent VEGF‐mediated mechanism, regardless of PPARgamma stimulation. Cardiovasc Diabetol 2009; 8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferrell WR, Balint PV, Egan CG, Lockhart JC, Sturrock RD. Metacarpophalangeal joints in rheumatoid arthritis: laser Doppler imaging – initial experience. Radiology 2001; 220:257–62. [DOI] [PubMed] [Google Scholar]
- 22. Firestein GS. Starving the synovium: angiogenesis and inflammation in rheumatoid arthritis. J Clin Invest 1999; 103:3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li W, Sama AE, Wang H. Role of HMGB1 in cardiovascular diseases. Curr Opin Pharmacol 2006; 6:130–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He Z, Shotorbani SS, Jiao Z et al HMGB1 promotes the differentiation of Th17 via up‐regulating TLR2 and IL‐23 of CD14+ monocytes from patients with rheumatoid arthritis. Scand J Immunol 2012; 76:483–90. [DOI] [PubMed] [Google Scholar]
- 25. Suda K, Takeuchi H, Ishizaka A, Kitagawa Y. High‐mobility‐group box chromosomal protein 1 as a new target for modulating stress response. Surg Today 2010; 40:592–601. [DOI] [PubMed] [Google Scholar]
- 26. Pullerits R, Jonsson IM, Verdrengh M et al High mobility group box chromosomal protein 1, a DNA binding cytokine, induces arthritis. Arthritis Rheum 2003; 48:1693–700. [DOI] [PubMed] [Google Scholar]
- 27. Guo HF, Liu SX, Zhang YJ, Liu QJ, Hao J, Gao LX. High mobility group box 1 induces synoviocyte proliferation in rheumatoid arthritis by activating the signal transducer and activator transcription signal pathway. Clin Exp Med 2011; 11:65–74. [DOI] [PubMed] [Google Scholar]
- 28. Miotla J, Maciewicz R, Kendrew J, Feldmann M, Paleolog E. Treatment with soluble VEGF receptor reduces disease severity in murine collagen‐induced arthritis. Lab Invest 2000; 80:1195–205. [DOI] [PubMed] [Google Scholar]
- 29. Sone H, Kawakami Y, Sakauchi M et al Neutralization of vascular endothelial growth factor prevents collagen‐induced arthritis and ameliorates established disease in mice. Biochem Biophys Res Commun 2001; 281:562–8. [DOI] [PubMed] [Google Scholar]
- 30. Benedetti G, Miossec P. Interleukin 17 contributes to the chronicity of inflammatory diseases such as rheumatoid arthritis. Eur J Immunol 2014; 44:339–47. [DOI] [PubMed] [Google Scholar]
- 31. Lee SY, Yoon BY, Kim JI et al Interleukin‐17 increases the expression of Toll‐like receptor 3 via the STAT3 pathway in rheumatoid arthritis fibroblast‐like synoviocytes. Immunology 2014; 141:353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fazaa A, Ben Abdelghani K, Abdeladhim M, Laatar A, Ben Ahmed M, Zakraoui L. The level of interleukin‐17 in serum is linked to synovial hypervascularisation in rheumatoid arthritis. Joint Bone Spine 2014; 81:550–1. [DOI] [PubMed] [Google Scholar]
- 33. Yang B, Kang H, Fung A, Zhao H, Wang T, Ma D. The role of interleukin 17 in tumour proliferation, angiogenesis, and metastasis. Mediators Inflamm 2014; 2014:623759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hata H, Sakaguchi N, Yoshitomi H et al Distinct contribution of IL‐6, TNF‐alpha, IL‐1, and IL‐10 to T cell‐mediated spontaneous autoimmune arthritis in mice. J Clin Invest 2004; 114:582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Supporting Information