

Abstract
Epileptic activity may be more prevalent in early stage Alzheimer’s disease (AD) than previously believed. Several studies report spontaneous seizures and interictal discharges in mouse models of AD undergoing age-related Aβ accumulation. The mechanism by which Aβ-induced neuronal excitability can trigger epileptiform activity remains unknown. Here, we systematically examined field excitatory postsynaptic potentials in stratum radiatum and population spikes in the adjacent stratum pyramidale of CA1 in wild-type mouse hippocampal slices. Soluble Aβ oligomers (oAβ) blocked hippocampal LTP and EPSP-spike (E-S) potentiation, and these effects were occluded by prior treatment with the glutamate uptake inhibitor TBOA. In accord, oAβ elevated glutamate levels in the hippocampal slice medium. Recording population spikes (PS) revealed that oAβ increased PS frequency and reduced LTP, and the latter effect was occluded by pretreatment with the GABAA antagonist picrotoxin. Whole-cell recordings showed that oAβ significantly increased spontaneous EPSC frequency. Decreasing neuronal activity by increasing GABA tone or partially blocking NMDAR activity prevented oAβ impairment of hippocampal LTP. Finally, treating slices with two antiepileptic drugs rescued the LTP inhibition induced by oAβ. We conclude that soluble Aβ oligomers at the low nanomolar levels present in AD brain increase neuronal excitability by disrupting glutamatergic/GABAergic balance, thereby impairing synaptic plasticity.
Introduction
The risk of unprovoked epileptic seizures is rather high in early-onset (often familial) Alzheimer’s disease, perhaps as much as ~90-fold higher than in an age-matched reference population (Amatniek et al., 2006). Importantly, epileptic activity may also be more prevalent in the early stages of ‘sporadic’ late-onset AD than was previously thought (Vossel et al., 2013), but the underlying mechanisms are poorly understood. In line with the increasing awareness of the occurrence of subclinical and clinical seizures in AD patients, several laboratories have reported spontaneous seizures and interictal discharges in mouse models of AD undergoing age-related Aβ accumulation (Palop et al., 2007; Westmark et al., 2008; Minkeviciene et al., 2009; Vogt et al., 2011; Bezzina et al., 2015). Such studies suggest that the hyperexcitability induced by oligomeric Aβ assemblies (oAβ) may involve depolarization of the resting membrane potential (Minkeviciene et al., 2009), depression of the slow after-hyperexcitability (Driver et al., 2007), depletion of calbindin and ectopic expression of neuropeptide Y (Palop et al., 2007). Although epileptiform activity and its relevant effects are common features of AD mouse models that likely contribute to Aβ-induced cognitive impairment, the neurophysiological basis by which Aβ-induced neuronal hyperexcitability can trigger epileptiform activity remains to be elucidated.
Neuronal activity in the brain depends on a balanced regulation between excitatory and inhibitory neurotransmission. A loss of excitation/inhibition balance in favor of excitation can result in epileptiform activity. The excitatory glutamatergic system plays a key role in generating and spreading epileptic discharges. Extracellular glutamate concentrations are tightly controlled by a family of membrane transporters predominantly expressed by perisynaptic astrocytes (Danbolt, 2001). Glutamate transporter (GT) expression has been found to be decreased in postmortem AD brain and in APP tg mouse models of AD (Masliah et al., 1996, 2000; Jacob et al., 2007). As a potentially primary or early pathogenetic factor, oAβ was shown by us to inhibit glutamate uptake in mouse hippocampal synaptosomes, similar to the effect of a known glutamate uptake blocker, TBOA (Li et al., 2009). Several mouse models of seizure states have been generated by GT impairments (Danbolt 2001; Maragakis & Rothstein 2004; Milh et al, 2007), and increases in extracellular glutamate concentrations have been observed in the tissues of epileptic mice and humans (Dabolt 2001; Eid et al. 2008). On the other hand, reducing GABAergic tone favors increased excitability, as some reports have shown that neuronal hyperactivity is associated with decreased GABAergic inhibition, and GABA administration significantly improved cognitive function in APP Tg mice (Busche et al., 2008; Sun et al., 2012). Recent findings also suggest that Aβ-mediated synaptic suppression occurs in part through inhibition of the GABAA receptor (Orr et al., 2014). These results suggest that oAβ can interrupt glutamate uptake, increase extracellular glutamate levels and thereby activate extrasynaptic GluN2B receptors, and can also reduce GABAA receptor-mediated inhibition, thus promoting neuronal hyperexcitability. Here, we demonstrate directly that soluble Aβ oligomers increase neuronal excitability and impair hippocampal LTP and E-S coupling by inducing an imbalance between glutamatergic and GABAergic transmission. Further, we show that the application of antiepileptic drugs to the hippocampus can prevent oAβ-mediated impairment of LTP.
Materials and Methods
Aβ preparations
Secreted human Aβ peptides were collected and prepared from the conditioned medium (CM) of a CHO cell line (7PA2) that stably expresses human APP751 containing the V717F AD mutation (Podlisny et al, 1995; Welzel et al, 2014). Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, 1% penicillin/streptomycin, 2 mM L-glutamine, and 200 mg/ml G418 for selection. Upon reaching ~95% confluency, the cells were washed and cultured overnight (~15 h) in serum-free medium. CM was collected, spun at 1500 × g to remove dead cells and debris, and stored at 4°C. The CM was concentrated 10-fold with a YM-3 Centricon filter (Walsh et al., 2005). Aliquots of concentrated 7PA2 CM were stored at −80°C. Once the 7PA2 CM is added to the ACSF perfusate, the total Aβ concentration is around 5.5 ± 0.9 ng/ml as measured by Aβ ELISA (Walsh et al., 2002).
Synthetic Aβ(1–40)S26C peptide was synthesized and purified using reverse phase HPLC by Dr. James I. Elliott at Yale University, (New Haven, CT). Peptide mass and purity (>99%) were confirmed by electrospray/ion trap mass spectrometry and reverse phase HPLC. Oxidatively cross-linked Aβ(1–40)S26C dimer ([Aβ40S26C]2) was prepared as described previously (O’Nuallain et al., 2010; O’Malley et al., 2014). Following oxidation the [Aβ40S26C]2 was treated with 7 M guanidinium HCl to remove aggregates and dimer isolated using a Superdex 75 10/30 HR column (GE Healthcare Biosciences, Pittsburgh, PA) eluted in 20 mM sodium phosphate, pH 7.4. The peak fraction of [Aβ40S26C]2 was collected, the concentration determined (ɛ275 = 2272 M−1 cm−1) and the sample diluted to 20 µM in elution buffer. [Aβ40S26C]2 readily assembles to form kinetically trapped protofibrillar assemblies under quiescent conditions (O’Nuallain et al., 2010; O’Malley et al., 2014); thus aliquots (120 μl) of the diluted sample were incubated in the wells of a black polystyrene, 96-well plate (Fisher Scientific) at 37°C for 5 days until a maximal ThT fluorescence was attained. Thereafter, material from 40 wells was pooled, gently mixed and aliquoted into 50 μl lots and stored at −80°C until required.
Hippocampal slice preparation
Mice (C57BL/6×129, wild-type, both genders), were sacrificed by isoflurane anesthesia at age 3~4 wk for patch-clamping or at 6~8 wk for field recordings. Brains were quickly removed and submerged in ice-cold oxygenated sucrose-replaced artificial cerebrospinal fluid (ACSF) cutting solution containing (in mM) 206 sucrose, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 1 CaCl2, 1 MgCl2, 26 NaHCO3, 10 D-glucose, pH 7.4, 315 mOsm. Transverse slices (350 μm thick) were cut with a vibroslicer from the middle portion of each hippocampus. After dissection, slices were incubated in ACSF containing (in mM): 124 NaCl, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 2.5 CaCl2, 26 NaHCO3, 10 D-glucose, pH 7.4, 310 mOsm, in which they were allowed to recover for at least 90 min before recording. A single slice was then transferred to the recording chamber and submerged beneath continuously perfused ACSF saturated with 95% O2 and 5% CO2. Slices were incubated in this chamber for 20 min before stimulation at RT (~24°C).
Electrophysiology
Standard field excitatory postsynaptic potentials (fEPSP) and population spikes (PS) were recorded in the CA1 region of hippocampus. A bipolar stimulating electrode (FHC Inc., Bowdoin, ME) was placed in the Schaffer collaterals to deliver test and conditioning stimuli. Two borosilicate glass recording electrodes filled with ACSF were positioned in stratum radiatum and stratum pyramidale of CA1, 200–300 μm from the stimulating electrode. fEPSP (from stratum radiatum) and population spikes (from stratum pyramidale) in CA1 were induced by test stimuli at 0.05 Hz with an intensity that elicited a fEPSP amplitude of 40~50% of maximum. Test responses were recorded for 30–60 min prior to beginning the experiment, to ensure stability of the response. To induce LTP, two consecutive trains (1 s) of stimuli at 100 Hz separated by 20 s, a protocol that induces LTP lasting ~1.5 hr in wild-type mice of this genetic background were applied to the slices. The field potentials were amplified 100× using Axon Instruments 200B amplifier and digitized with Digidata 1322A. The data were sampled at 10 kHz and filtered at 2 kHz. Traces were obtained by pClamp 9.2 and analyzed using the Clampfit 9.2.
Whole-cell recordings were made from the somata of visually identified CA1 pyramidal neurons. Patch pipettes (5~7 MΩ) were filled with an internal solution containing (in mM): 120 CsGluconate, 5 MgCl2, 0.6 EGTA, 30 HEPES, 4 MgATP, 0.4 Na2GTP, 10 phosphocreatine-Tris, 5 QX-314; 290 mOsm; pH was adjusted to 7.2 with CSOH. Spontaneous excitatory postsynaptic currents (sEPSCs) were collected at a membrane holding potential of −70 mV, which is close to the calculated reverse potential of GABA. In order to measure simultaneously the excitatory and inhibitory inputs on the same neuron, spontaneous inhibitory postsynaptic currents (sIPSCs) were also measured on the same neuron by raising the holding potential to 0–10 mV, a potential close to the reverse potential of excitatory input, without visualizing a negative rebound deflection. Recorded neuronal activities were detected by custom software (DClamp: available at www.ieeg.org/?q=node/34). Evoked excitatory postsynaptic currents (eEPSC) was induced by tungsten wire electrodes placed in stratum radiatum ~300 µm away from the recording pyramidal neuron in CA1. Electrical stimulation was delivered every 0.05 Hz. NMDA-mediated EPSCs were recorded at +45 mV in ACSF containing 10 μM bicuculline and 10 μM NBQX, or else at −70 mV in 0.1 mM Mg2+ ACSF containing bicuculline and NBQX. In some experiments, we blocked active synaptic NMDAR by applying MK-801 (20 μM) in the above buffer and changed the stimulation protocol to 0.125 Hz using paired-pulse stimulation (100 ms interpulse interval), because this effects presynaptic release of glutamate, which in turn facilitates the activation of synaptic NMDAR currents. After washout of the MK801, the remaining EPSC can be considered as extrasynaptic NMDA-currents. Series resistance was kept at 15~30 MΩ and monitored throughout each recording. Neurons with negative resting membrane potentials less than −60 mV were discarded. Neuron input resistance and patching access resistance were repetitively monitoring during the experiment. Cells with changes in those parameters of more than 15–20% were excluded from the analysis. All patch clamp experiments were performed at 24°C.
Measuring glutamate concentrations
Fresh brain slices (350 μm thickness) were prepared as described above. The slices were incubated at room temperature in ACSF with continuous oxygenization for 1 hr prior to treatment in order to recover. The slices were then treated with either 10 nM pure synthetic [Aβ40S26C]2 solution or blank vehicle for 2 hr by incubating at room temperature with continuous oxygenation. The solution from each incubation chamber was sampled every hour. Fresh solution was added to corresponding chambers after sampling to maintain the same volume. The glutamate level in each sample was determined by Amplex® Red Glutamic Acid/Glutamate Oxidase Assay Kit (Invitrogen™, Grand Island, New York).
Drug treatments
Paired control and experimental slices from a single mouse were maintained together in a single chamber, except during drug treatments. DL-2-Amino-5-phosphonopentanoic acid (AP5), MK-801, GABA, Ro 25-6981, picrotoxin, topiramate and levetiracetan were from Tocris. DL-threo-β-benzyloxyaspartic acid (TBOA) was from Sigma. Among these, picrotoxin, Ro 25-6981 and TBOA were each dissolved in dimethylsulfoxide (DMSO). The DMSO concentration used in experiments was <0.01%. All compounds were added to the perfusion ACSF for at least a 20 min baseline recording before HFS was applied. When combined with oAβ (7PA2 CM or [S26C]2), these compounds were added 10 min before the oAβ and then the baseline was recorded for at least 30 min prior to HFS. The compounds and oAβ were then perfused for the entire duration of the recording.
Statistical analysis
LTP values expressed here are those at 60 min after the conditioning stimulus, unless stated otherwise. Results are expressed as means ± SEM from at least 4 independent recordings. Statistical comparisons were made using the t test or one-way or two-way ANOVA (Student-Newmann–Keuls test was used for post hoc comparison), One-way or two-way repeated-measures ANOVA (Holm–Sidak method) was used for multiple comparisons. In all cases, p < 0.05 is considered statistically significant.
Results
A glutamate uptake blocker occludes Aβ-mediated inhibition of E-S potentiation
It has been widely demonstrated that soluble Aβ oligomers (oAβ) of various sources can inhibit hippocampal LTP recorded from the stratum radiatum of CA1 region (e.g., Hu et al., 2009; Li et al., 2011; 2013). To investigate whether oAβ-impaired hippocampal LTP also involves the excitatory postsynaptic potential (EPSP)-spike (E–S) component, two recording electrodes were placed in the stratum radiatum and the adjacent stratum pyramidale simultaneously to record the fEPSP and population spikes (PS), respectively. We first quantified the degree of LTP inhibition by cell-derived Aβ oligomers in the conditioned medium (CM) of 7PA2 cells (hAPP V717F-expresing stable CHO cells) (Li et al., 2009; 2011 Li et al., 2013), a well-characterized cellular source of various low MW human Aβ species that have potent bioactivity in hippocampus at low nM concentrations (Welzel et al. 2014). The PS amplitude recorded from the pyramidale layer was reduced significantly from 556 ± 39% (in oAβ-free CHO- CM, n=10) to 292 ± 93% (in oAβ-rich 7PA2 CM, n=9) (Fig. 1A), while the simultaneously recorded fEPSP decreased in a manner consistent with previous reports (158 ± 6% vs. 119 ± 5%, respectively) (Fig. 1B). When we calculated the ratio of PS amplitude to fEPSP slope, referred to as E-S coupling or E-S potentiation, this ratio was decreased by oAβ treatment (394 ± 56% vs. 152 ± 16%) (Fig. 1C). We previously showed that soluble oAβ can enhance LTD in part by decreasing glutamate uptake (Li, et al., 2009). Application of the non-selective inhibitor of glutamate transporters, DL-TBOA, also inhibited LTP in a similar manner to oAβ: the PS amplitude and fEPSP slope were each decreased by TBOA (10 µM) (547 ± 86% vs. 209 ± 10%, and 151 ± 10% vs. 120 ± 5%; n=8 vs. n=9) (Fig. 1D). Accordingly, TBOA reduced the E-S coupling (vehicle 366 ± 66% vs. TBOA 162 ± 10%) (Fig. 1E).
Figure 1. Soluble Aβ inhibited EPSP-spike coupling in the CA1 region of hippocampus is occluded by a glutamate uptake inhibitor.
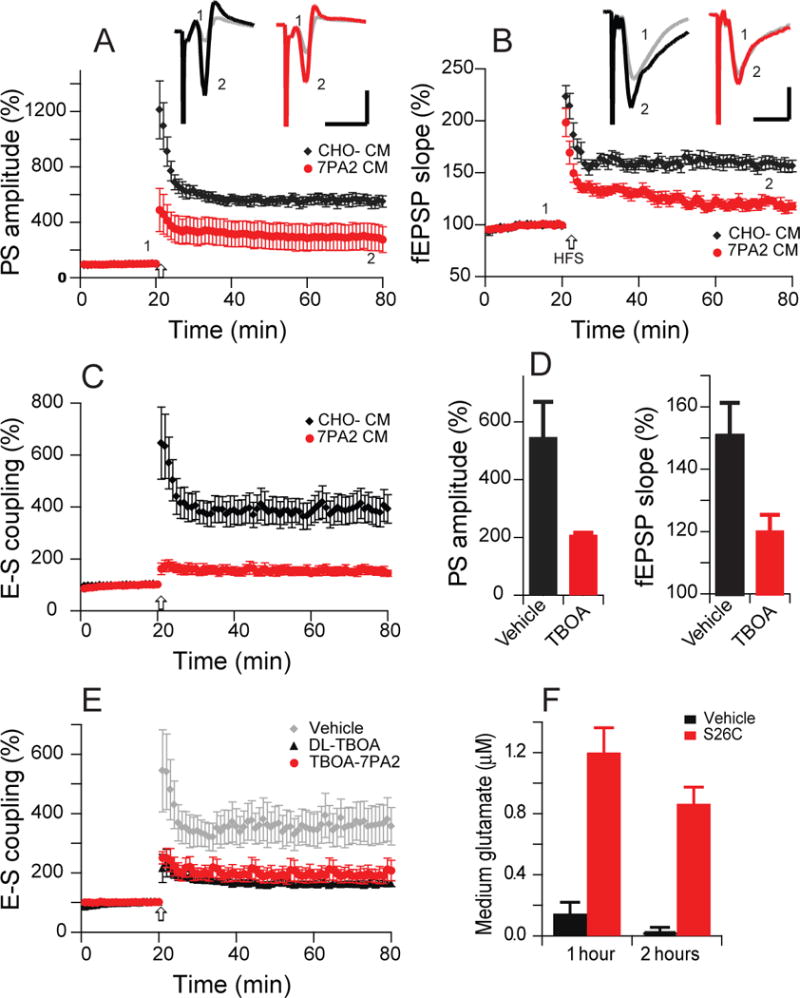
(A) 7PA2 CM rich in soluble Aβ inhibited population spike-LTP (red, n=9) induced by high-frequency stimulation (HFS, arrow). CHO- CM had no effect on LTP (black, n=10). (B) 7PA2 CM also inhibited simultaneously recorded fEPSP-LTP (red), and CHO- CM had no effect on this regular LTP (black). (C) Potentiation of EPSP-spike coupling (E-S potentiation) is reduced by the soluble oAβ-rich 7PA2 CM (red) but is normal in CHO- CM treated slices (black). E-S coupling is expressed as the ratio of population spike amplitude (A) to fEPSP rising slope (B). (D) Hippocampal population spike-LTP (left) and fEPSP-LTP (right) were also blocked by the glutamate uptake inhibitor, TBOA (10 μM) (red, n=8) compared to vehicle (black, n=9). (E) Potentiation of E-S coupling is inhibited by DL-TBOA (black, n=8) compared to vehicle (grey, n=6), while pre-treatment with TBOA (10 μM) does not further change the 7PA2 CM effect on E-S coupling (red, n=7). (F) Glutamate concentrations in brain slices incubated in medium (as during LTP recordings) for 1 and 2 hr after adding pure, soluble [Aβ40S26C]2 (10 nM). Inset traces are typical population spikes (PSs) (A) or field excitatory postsynaptic potentials (fEPSPs) (B) recorded before (gray) and after (black or red) HFS for each condition. Horizontal calibration bars: 10 ms; vertical bars: 1 mV (A) or 0.5 mV (B).
If soluble oAβ inhibits LTP by interfering with glutamate uptake, then fully inhibiting glutamate transporters first should occlude the oAβ from further inhibiting LTP. To test this, we treated slices with DL-TBOA at a dose of 10 µM for 10 min, then added the 7PA2 CM. The PS amplitude and fEPSP slope showed no further decrease from the oAβ-rich 7PA2 CM (PS 315 ± 52% and fEPSP 124 ± 3%, n=6). E-S coupling was also similar to TBOA alone or 7PA2 CM alone (184 ± 27%, p>0.05. Fig. 1E), indicating that blocking glutamate uptake occludes oAβ-mediated impairment of E-S coupling.
To verify that this blocking of glutamate uptake actually increases extracellular glutamate levels in our preparation, we treated hippocampal slices with either vehicle or pure, synthetic Aβ oligomers [Aβ40S26C]2 at 10 nM concentration in a bubbling container and collected the slice medium at 1 and 2 hr post-treatment, the same times at which we had done the above LTP recordings. We found the glutamate concentrations in the slice medium to be markedly and significantly increased by oligomeric [Aβ40S26C]2 treatment after both 1 and 2 hr (Fig. 1F), suggesting that soluble Aβ oligomers per se interrupt glutamate uptake.
Impairment of E-S potentiation by Aβ oligomers involves changes in GABAergic tone
The LTP typically measured by changes in the field EPSP slope provides a measurement of the excitatory drive to the dendritic fields of pyramidal neurons, while the extracellularly recorded PS reflect the numbers of synchronously firing neurons in the stratum pyramidale. To confirm that oAβ per se inhibited the LTP of PS as seen by treatment with the 7PA2 CM (Fig. 1A), we treated the hippocampal slices instead with pure, synthetic [Aβ40S26C]2 oAβ for 30 min before an HFS. Again, the LTPs of both PS and fEPSP were found to be significantly reduced (PS: Veh: 537 ± 34%, n=7, vs. S26C: 259 ± 27%, n=6, p<0.001) (Fig. 2A), (fEPSP: Veh: 537 ± 34%, n=7, vs. S26C: 259 ± 27%, n=6, p<0.001) (Fig. 2B), suggesting that oAβ can induce a suppression of PS potentiation that could depress the E-S potentiation. A previous report demonstrated that Aβ-impaired LTP of fEPSP in hippocampus does not involve the activation of GABAergic receptors (Raymond et al., 2003). This undetectable change may due to the slope of fEPSP being insensitive to GABAA receptor antagonism, in contrast to the amplitude of the population spike (Wigstrom and Gustafsson, 1985). We therefore asked whether the GABAA receptor antagonist, picrotoxin (PTX, 50 μM), has any effect on the LTPs of PS and fEPSP. Reducing inhibitory tone with PTX increased neuronal excitability, thus significantly depressing the LTP of PS (207 ± 20% n=7, vs 586 ± 41%n=8, p<0.001) (Fig. 2C), while the LTP of fEPSP slope recorded simultaneously showed a slight and insignificant decrease (147 ± 2% vs. 155 ± 7%, p>0.05) (Fig. 2D), suggesting that the PS may be a better parameter for the synaptic plasticity changes that involve a GABAergic mechanism. Interestingly, pre-treating with PTX occluded the ability of subsequent S26C oligomer application to impair LTP of PS (195 ± 24%, n=7) (Fig. 2E, red). We also applied a lower dose of PTX (10 μM) to the hippocampal slices and found that the field EPSP LTP slope was unchanged (data not shown), while the LTP of PS was significantly reduced (270 ± 32%, n=15 vs. 199 ± 18%, n=13, p<0.05) (Fig 2F), suggesting that the LTP of PS amplitude involves in part a GABAergic mechanism. More importantly, PTX (10 μM) pretreatment occluded 7PA2 CM from further reducing the PS-LTP significantly (Fig. 2F, red). Together, these results suggest that Aβ oligomers impair hippocampal plasticity in part via a mechanism involving GABAergic function.
Figure 2. Reduced activity of GABAA receptors is involve in soluble Aβ-mediated impairment of LTP in the CA1 region of hippocampus.
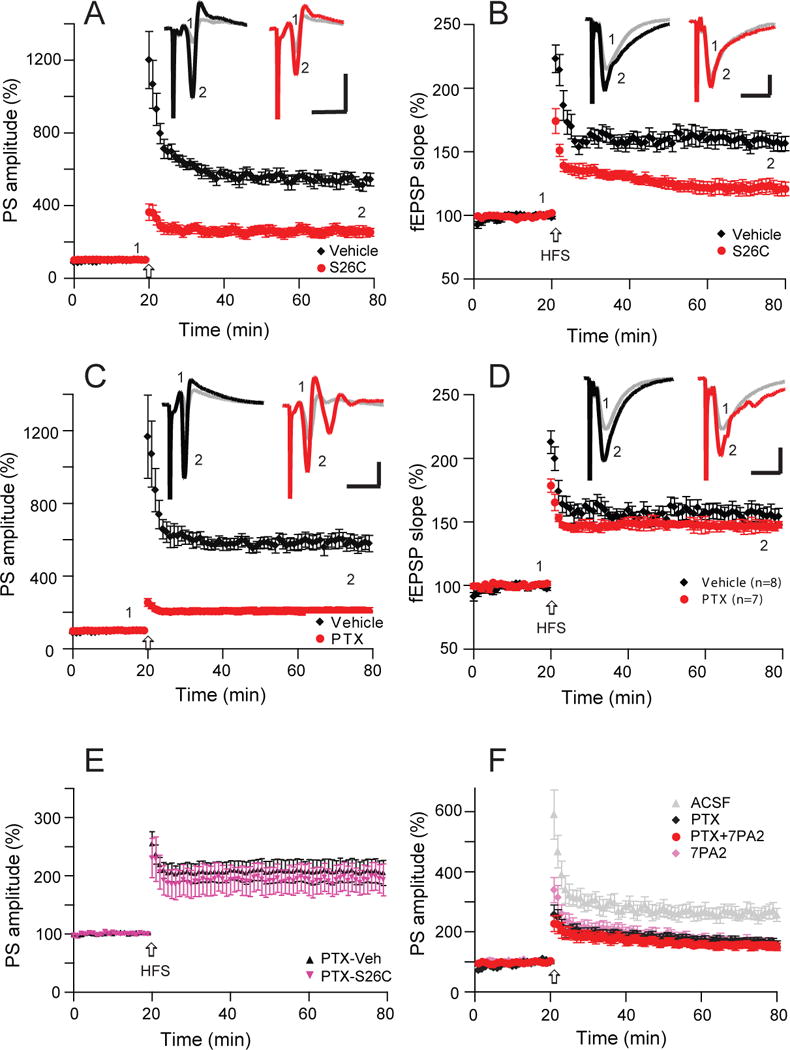
(A) Synthetic Aβ40S26C dimer significantly reduces population spike LTP (red, n=6) induced by high-frequency stimulation (HFS, arrow) in comparison with the vehicle effect on LTP (black, n=7). (B) Synthetic Aβ40S26C dimer also inhibited simultaneously recorded fEPSP-LTP (red), while the vehicle had no effect on this regular LTP (black). (C) A GABAA antagonist (picrotoxin, 50 μM) significantly reduce population spike LTP (red, n=8). (D) The same dose of GABAA antagonist (picrotoxin, 50 μM) has no effect on the simultaneously recorded usual field EPSP (blue, n=8). (E) Hippocampal population spike-LTP was also reduced by the GABAA receptor antagonist, picrotoxin (50 μM) (black, n=7), while pretreatment with picrotoxin prevented the S26C-dimer induced further decrease of PS-LTP (red, n=6). (F) A low dose of a GABAA antagonist (picrotoxin, 10 μM) occluded 7PA2 CM-mediated impairment of population spike LTP (red, n=8). Inset traces are typical population spikes (A, C) and field excitatory postsynaptic potentials (fEPSPs) (B, D) recorded before (gray) and after (black or red) HFS for each condition. Horizontal calibration bars: 10 ms; vertical bars: 2 mV (A, C) or 1 mV (B, D).
Aβ oligomers increase population spike frequency
To further explore the role of neuronal excitability in the adverse effects of soluble Aβ oligomers, we recorded from the pyramidal cell layer of CA1 region, where any hyperexcitability can be more readily characterized as an increased propensity for populations of neurons to fire in synchrony, generating population spikes (‘pop-spikes’ or PS). In contrast to the field EPSP waveforms, which showed modest or no changes when neurons increased their excitatory properties in either Mg2+-free buffer, a common method to induce epileptiform activity in rodent models of epilepsy (e.g., Yuen and Sander, 2012) (Fig. 3: A1,3) or after blocking inhibitory interneuron activity with PTX (50 μM) (Fig. 3: A2,3), the PS profile showed more marked changes (induction of additional PSs) under Mg-free (Fig. 3: B1,3) or PTX-containing conditions (Fig. 3: B2,3).
Figure 3. Soluble Aβ increase the number of population spikes recorded from stratum pyramidale.
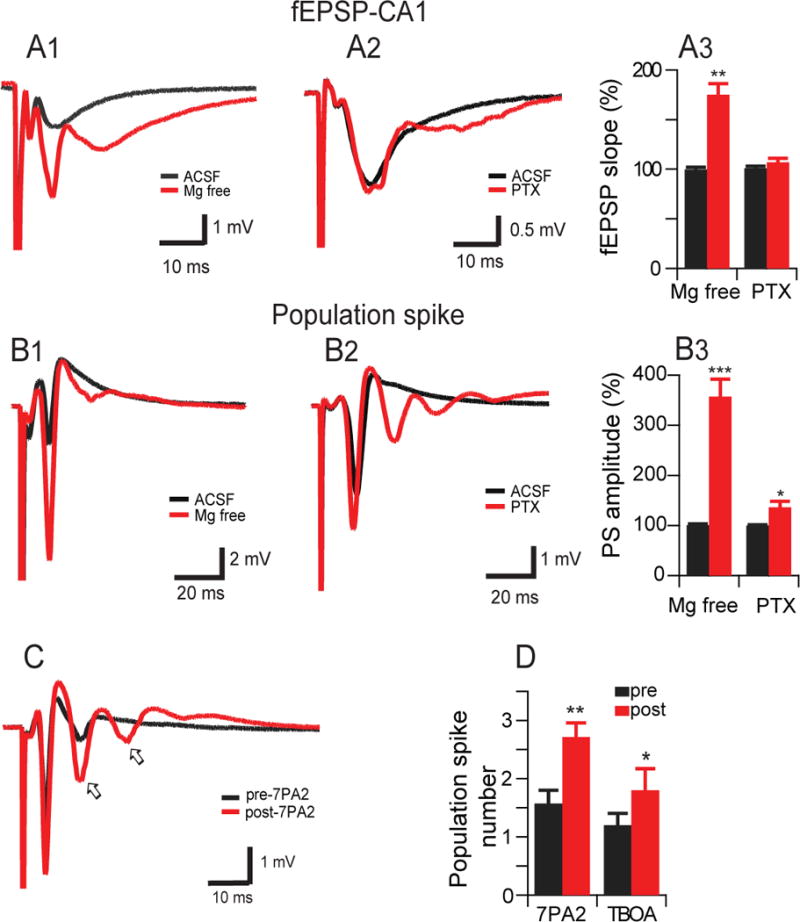
(A) Representative field potentials recorded in the stratum radiatum of CA1 hippocampal slices in response to a single electrical stimulus applied to the Schaffer collateral/commissural fibers in normal ASCF (black traces) and in 0 Mg2+ ACSF (A1) or picrotoxin (50 μM) (A2) (red traces) and summary data (A3). (B) Representative field potentials recorded in the stratum pyramidale of CA1 hippocampal slices in response to a single electrical stimulus applied to the stratum radiatum in normal ASCF (black traces) and 0 Mg2+ ACSF (B1) or picrotoxin (50 μM) (B2) (red traces) and summary data (B3). (C) Representative traces from soluble oAβ shows increased number of population spikes recorded from stratum pyramidale. (D) summary data of population spike numbers recorded before (pre, black) and after (post, red) addition of 0 Mg2+-ACSF treatment with 7PA2 CM (8 slices) and low dose of TBOA (1 μM) (6 slices).
Low-magnesium medium can decrease seizure thresholds in acute brain slices as a model of epilepsy (Tancredi et al., 1990; Yuen and Sander, 2012). To further explore how soluble Aβ oligomers induce neuronal hyperexcitability, the oligomer-rich 7PA2 CM was added to the 0-Mg2+ ACSF buffer, and the PS number (Fig. 3C, arrows) was significantly increased (before adding 7PA2 CM: 1.6 ± 0.6; after: 2.6 ± 0.8; n=13; p<0.01) (Fig. 3D). Similarly, adding DL-TBOA (1 μM) alone also increased the PS number (Fig. 3D). The fact that 7PA2 CM and TBOA both induced epileptic activity in Mg2+-free buffer that was similar to that in PTX-containing buffers suggests that oAβ-induced neuronal hyperexcitability involves the loss of glutamatergic and GABAergic functional balance.
Aβ oligomers enhance coupling between fEPSPs and population spikes
To further investigate the mechanism of the hyperexcitability induced by oAβ, we analyzed the coupling of excitatory inputs and firing efficiency, i.e., E-S coupling. We had already shown that the potentiation of E-S coupling is significantly reduced in the presence of oAβ (Fig. 1C) or TBOA (Fig. 1E). Here, a range of stimulation intensities were plotted for the E-S relationship: PTX (50 μM) induced a large leftward shift in the E-S coupling curve, as shown by a decrease from 0.71 to 0.44 in the E50 (the value of the normalized fEPSP slope at which the PS amplitude was 50% of its maximal response). (Fig. 4A). Similarly, application of oAβ-rich 7PA2 CM shifted the E-S curve to the left (red) compared to its control (pre-7PA2 CM) curve (blue), so that the E50 decreased from 0.68 to 0.57 (Fig. 4B). Therefore, oAβ-induced hyperexcitability is associated with a relative decrease in GABAergic inhibition.
Figure 4. Aβ oligomers enhances extracellular field EPSP (fEPSP)-to-spike (E-S) coupling in the CA1 region of hippocampus.
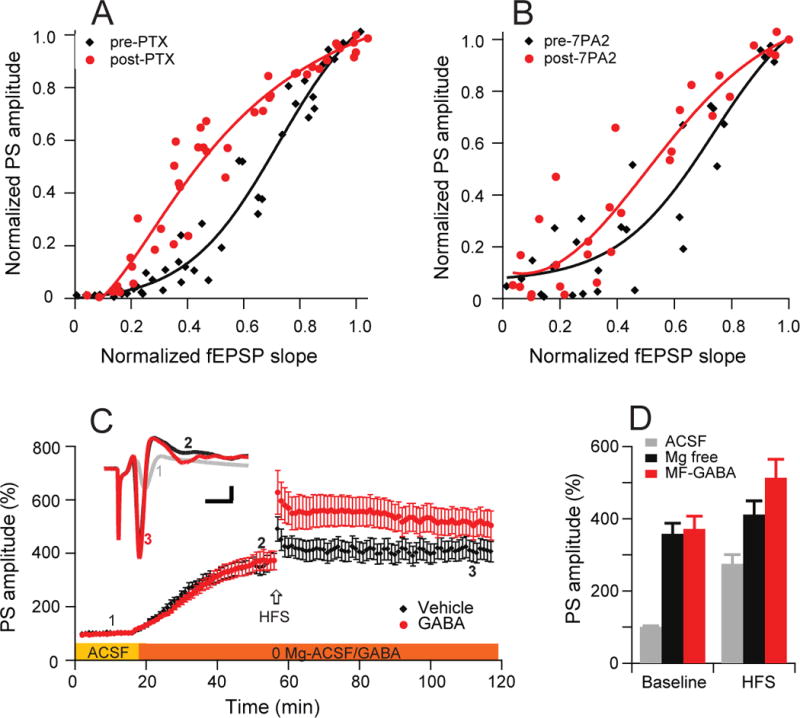
(A) GABAA receptor antagonist, picrotoxin (50 μM) induce a leftward shift of the E-S coupling curves, indicating that a larger PS was produced by a given fEPSP slope. The amplitudes of PS normalized to maximal amplitude are plotted against the fEPSP slope normalized to maximal slope in the before (black) and after PTX (red) administration. (B) The Aβ oligomers-rich 7PA2 CM applications produce similar leftward shift of E-S coupling curves as PTX did. (C) Removal of Mg2+ from ACSF induces significant enhancement of amplitudes of population spikes and reduces further HFS (arrow) -induced LTP in vehicle (black, n=10) or GABA (2.5 μM) (red, n=8) treated slices. (D) Summary data of PS amplitude changes in either Mg2+ free buffer (at 60 min time point) or upon HFS-induced LTP (120 min time point). Inset traces are typical population spikes recorded in normal ACSF (gray) and before (black) and after (red) HFS in 0- Mg2+ACSF. Horizontal calibration bars: 10 ms; vertical bars: 1 mV.
To further investigate the effect of neuronal excitability on the LTP of PS, we increased neuronal activity by fully removing Mg2+ from the ACSF perfusate and found that the PS amplitude was gradually and significantly enhanced up to 358 ± 30% after 40 min (n=10) (Fig. 4C, black). Interestingly, a subsequent HFS induced a small LTP (411 ± 38%, p<0.05) (Fig. 4C, black - after 60 min). In order to reduce neuronal excitability, we added a low dose (2.5 μM) of GABA itself to the Mg2+-free ACSF. Although the GABA alone did not significantly alter the PS amplitude vs. no GABA (372 ± 35%, n=8), it did induce significant LTP after an HFS (513 ± 51% at 110 min, n=8; p<0.01 vs. vehicle) (Fig. 4C). Figure 4D summarizes these baseline and HFS effects of Mg2+-free +/− GABA conditions on PS amplitude. These results further suggest that neuronal hyperexcitability perturbs hippocampal LTP and that oAβ-impaired LTP may be mediated by an abnormal balance of glutamatergic (excitatory) and GABAergic (inhibitory) activity.
Soluble Aβ oligomers increase neuronal activity by increasing spontaneous EPSC frequency
To further investigate whether oAβ-induced neuronal excitability occurs through glutamate-mediated EPSCs and/or GABAergic IPSCs, we performed whole-cell patch clamping of the CA1 pyramidal neurons. Spontaneous EPSC (sEPSC) was recorded at a holding potential of −70 mV, while sIPSC was recorded at a holding potential of 0mV. These spontaneous currents were compared in the same neuron before vs. 30 min after treatment with oAβ-rich 7PA2 CM. The sEPSC frequency was significantly enhanced after 7PA2 CM administration (0.82 ± 0.18 Hz before and 4.72 ± 0.78 Hz after, n=6, p<0.01) (Fig. 5A, top 3 rows; Fig. 5B top), while the frequency of sIPSCs was not significantly altered (P>0.05), although a weak trend of decrease was observed (Fig. 5A, lower 3 rows; Fig. 5B top). The mean amplitude of the sEPSC decreased (20.63 ± 0.97 vs. 13.59 ± 1.05 pA, n=6, p<0.01) while that of the sIPSC did not change (Fig. 5B bottom). We conclude that the enhanced sEPSC frequency provides further evidence that soluble oAβ can increase neuronal firing probability.
Figure 5. Application of oAβ increases the frequency of spontaneous excitatory postsynaptic currents (sEPSC).
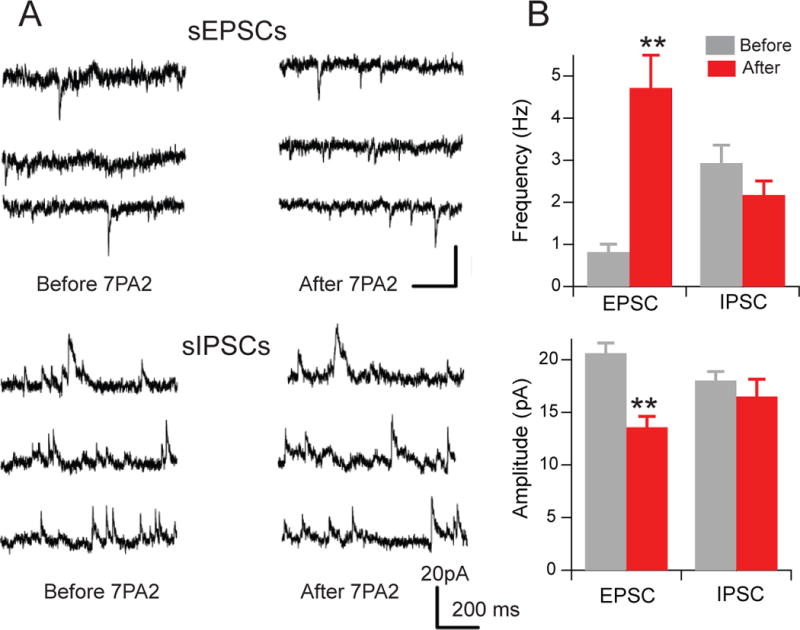
(A) Representative traces of voltage-clamp recordings from CA1 pyramidal cells held at −70 mV (upper 3 rows) for sEPSCs and at 0 mV (lower 3 rows) for sIPSCs before (left column) and after (right column) oAβ-rich 7PA2 CM treatment. (B) Summary data of the sEPSC and sIPSC change in frequency (upper) and amplitude (lower) after 7PA2 CM treatment.
Decreasing the neuronal activity prevents oAβ-impaired hippocampal LTP
If neuronal hyperexcitability and/or frank seizure activity inhibit hippocampal LTP, normalizing this hyperexcitability should restore the LTP. To investigate whether reducing neuronal activity can prevent the oAβ-mediated inhibition of synaptic plasticity (i.e., HFS-induced LTP), we pretreated hippocampal slices with 2.5 μM GABA (Fig. 6A). Then, we added sources of soluble oAβ. We found that oAβ-impaired LTP was partially restored when oAβ treatment was coupled with GABA (GABA + pure synthetic [Aβ40S26C]2: 132 ± 3%, n=6, vs. GABA alone: 134 ± 4%, n=6, and [Aβ40S26C]2 alone: 116 ± 4%, n=8) (Fig. 6A, B). More specifically, we found that the hyperactivity induced by oAβ is mainly mediated by NMDAR, because partial blockade of NMDAR excitatory activity by a low dose of the antagonist AP5 (2 µM) prevented the oAβ-mediated impairment of LTP (AP5: 136 ± 2%, n=5 vs. AP5 + 7PA2: 135 ± 8%, n=5, p>0.05; 7PA2 CM alone: 108 ± 7%, n=6,) (Fig. 6C,D). To further investigate how the low (2 µM) dose of AP5 influenced the responses, the PS amplitude and fEPSP slope in low (0.25 mM) Mg2+ buffer were compared before and after treatment with 2 μM AP5. Here, the fEPSP was significantly reduced (92 ± 3%, n=8, p<0.05) (Fig.6E). Interestingly, whole-cell recordings confirmed that 2 μM AP5 mainly acted upon the extrasynaptic NMDAR EPSC component (Fig. 6F), because it further decreased the remaining NMDA-mediated response after MK-801 had blocked the active synaptic NMDA component. The latter finding further supports our previous report that antagonists of GluN2B-containing NMDAR can specifically rescue LTP inhibition induced by oAβ (Li et al., 2011).
Figure 6. Decreased neuronal activity prevents LTP impairment induced by Aβ oligomers.
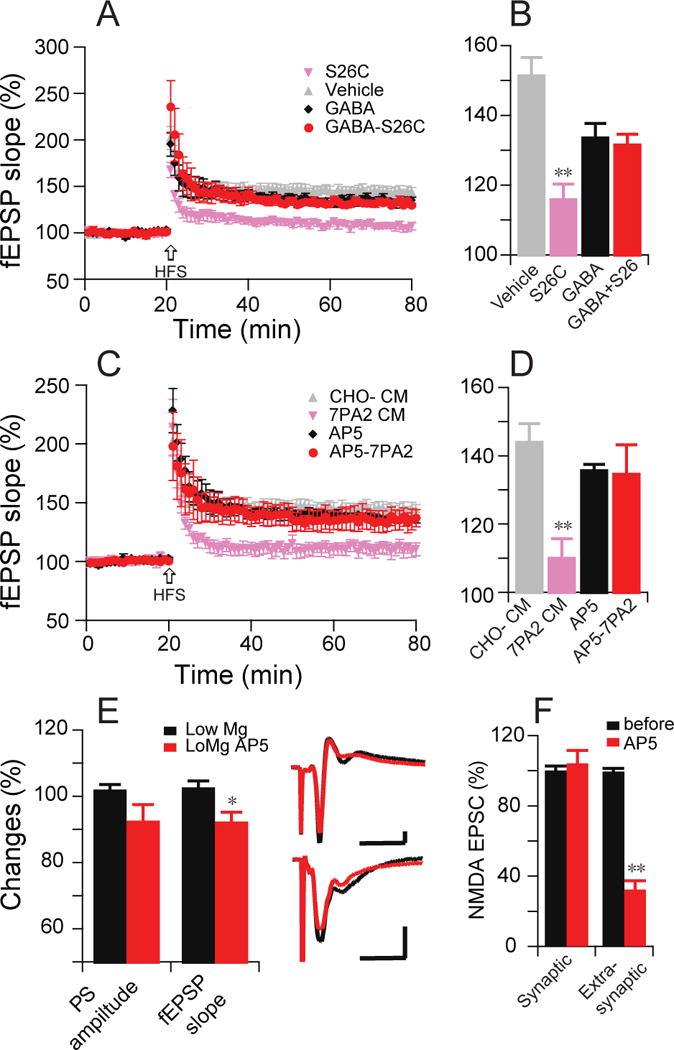
(A) Pure, synthetic [Aβ40S26C]2 oligomers (10 nM) prevent HFS-induced hippocampal LTP (pink, n=7), while the vehicle does not (grey, n=6). Effects on LTP of a low dose of GABA (2.5 µM) alone (black, n=6) or combined with [Aβ40S26C]2 oligomers (red, n=6). (B) Summary data at 60 min post-HFS from (A). (C) A low dose of NMDAR antagonist AP5 (2 μM) alone (black, n=5) or combined with 7PA2 CM (red, n=5): effect on HFS-induced fEPSP LTP in CA1 region. (D) Summary data at 60 min post-HFS from (C). (E) Effect of a small dose of AP5 (2 μM) in the low Mg2+-ACSF buffer increased population spike amplitudes and conventional fEPSP slopes. Inset traces are typical PS (upper) or fEPSPs (lower) recorded before (black) and after (red) AP5 treatment. Horizontal calibration bars: 10 ms; vertical bars: 0.5 mV. (F) Same low-dose AP5 effect on the whole-cell patch isolated NMDAR-mediated currents.
Anticonvulsants restore oAβ-impaired hippocampal LTP
We sought further support for our central hypothesis that soluble oAβ interferes with glutamate transporter function, resulting in rises of extracellular glutamate and thereby triggering epileptic activation. To confirm that 0Aβ impairs LTP through hyper-excitatory or epileptiform activity, we assessed the effects of two well-studied antiepileptic drugs in our LTP experiments. First, the anticonvulsant, topiramate (20 μM) applied to hippocampal slices had no significant effect on LTP by itself but prevented the 7PA2 CM-mediated inhibition of both PS-LTP (430 ± 28%, n=5 vs. 423 ± 50%, n=6, p>0.05) (Fig.7A) and fEPSP-LTP (151 ± 10%, n=5 vs. 142 ± 6%, n=6, p>0.05) (Fig.7B) that were recorded simultaneously from stratum pyramidale and stratum radiatum, respectively. Topiramate likewise prevented synthetic [Aβ40S26C]2 impairment of fEPSP-LTP (139 ± 4%, n=5 vs. 134 ± 3%, n=5, p>0.05) (Fig.7C). Another anticonvulsant, levetiracetam (100 µM), also restored oAβ-inhibition of LTP (147 ± 7%, n=5 vs. 143 ± 9%, n=6, p>0.05) (Fig. 7D). The effects of these two distinct anti-epileptic compounds further support our conclusion that the hippocampal LTP impairment induced by oAβ involves neuronal hyperexcitability.
Figure 7. Antiepileptic drugs prevent LTP impairment induced by Aβ oligomers.
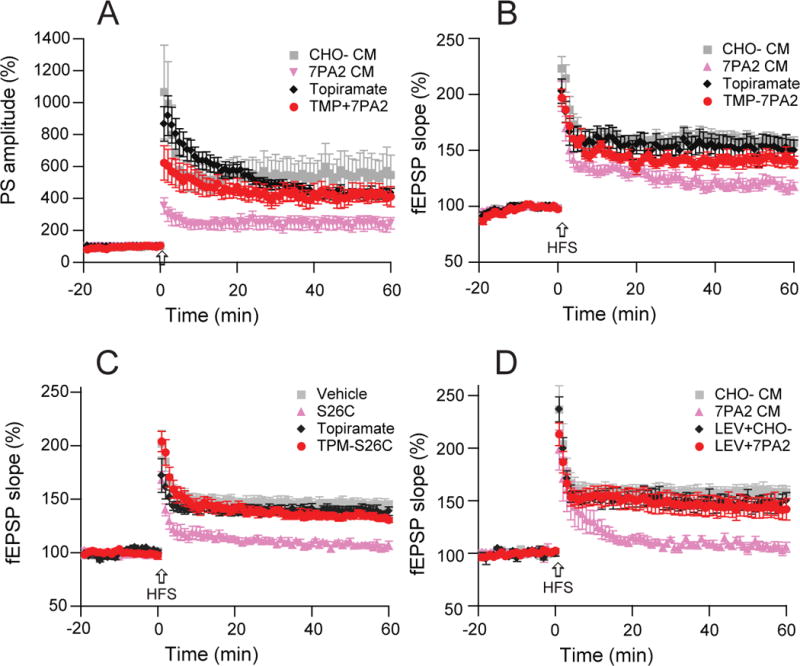
(A) Topiramate (20 μM) 20 min pretreatment of hippocampal slices prevents 7PA2 CM-reduced population spike-LTP as recorded from stratum pyramidale of CA1 (red, n=7) compared to 7PA2 CM alone (pink, n=7). (B) Topiramate, also prevented 7PA2 CM-impaired conventional fEPSP-LTP recorded simultaneously from stratum radiatum of CA1. (C) Topiramate (20 µM) has no significant effect on the hippocampal LTP (black, n=5), while topiramate added 20 min before pure synthetic [Aβ40S26C]2 (10 nM) prevents the impaired hippocampal LTP (red, n=5). Grey triangles are the [Aβ40S26C]2 oligomers effect on LTP. (D) Levetiracetam (100 µM) has no significant effect on the hippocampal LTP (blue, n=5), while levetiracetam added 20 min before 7PA2 CM prevents oAβ impairment of hippocampal LTP (red, n=6).
Discussion
Here we demonstrate that soluble Aβ oligomers – both naturally cell-secreted and pure synthetic – impair hippocampal LTP in a manner similar to the glutamate uptake inhibitor, TBOA, and these oligomer effects can be occluded by TBOA. Mechanistically, the oAβ disruption of synaptic function is mediated in part by an increase in NMDAR-dependent neuronal excitability and a simultaneous dysfunction of GABAergic interneuron activity. Reducing neuronal activity or treating with antiepileptic drugs can prevent soluble oAβ from impairing LTP.
Our findings that TBOA occludes oAβ-inhibited LTP and that oAβ increases glutamate concentrations in brain slice medium extend multiple reports that Aβ can inhibit glutamate uptake by rat cortical synaptosomes (Keller et al., 1997, Lauderback et al., 1999), cultured neurons (Fernández-Tomé et al., 2004), astrocytes (Harris et al., 1996; Harkany et al., 2000), oocytes (Gu et al., 2004) and human fibroblasts (Zoia et al., 2011). Collectively, these extensive findings are also consistent with our earlier report that icv injection of oAβ into rat brain causes a rapid increase in interstitial fluid (ISF) levels of glutamate without altering ISF GABA or aspartate levels (O’Shea et al. 2008).
The hydrophobic Aβ oligomers would be expected to bind principally to hydrophobic membrane lipid surfaces and thereby could secondarily interfere with the structure and function of synaptic transmembrane transporters (glutamate transporters), leading to increases of extracellular glutamate concentration. Elevations in extracellular glutamate have been reported in animal models of epilepsy (Zhang et al., 1991; Janjua et al., 1992,) and during spontaneous seizures in humans (During and Spencer, 1993). Moreover, raised glutamate levels have specifically been observed secondary to alterations in glutamate transporter function (Rothstein et al., 1996, Masliah et al., 2000). It has been shown that inhibition of glutamate transporters produces NMDAR-mediated seizures in the immature neocortex (Demarque et al., 2004) and increases excitability in local neocortical networks (Campbell and Hablitz 2004; 2008). NMDAR play an important role in glutamate-mediated excitotoxicity, which is also believed to be a contributing mechanism in AD. In line with this theory, oAβ has been shown to rapidly increase NMDAR-mediated currents in rat dentate gyrus (Wu et al., 1995), to increase NMDA-evoked hippocampal neuronal firing in vivo (Molnár et al., 2004), to enhance extrasynaptic NMDA EPSC in hippocampal slices (Li et al., 2009, 2011), and to stimulate active NMDAR in oocytes (Texidó et al., 2011). These enhanced NMDAR responses all suggest a possible mechanism for oAβ-induced neuronal hyperexcitability. Specifically, the GluN2B subtype of NMDA receptors directly modulates neuronal excitability and contributes to seizures (Moddel et al.,2005; Ying et al.,2004), and blockade of this receptor can increase the threshold for epileptiform discharges in lesioned cortex (DeFazio and Hablitz, 2000). In accord, reducing NMDAR activity by a low dose of AP5 (2 μM) (Fig. 6C) or by pharmacologically blocking GluN2B receptors with ifenprodil (Li et al., 2011) prevents oAβ-mediated synaptotoxicity.
The neurotransmitter glutamate serves a double role as an excitatory transmitter and as precursor for GABA. Thus, that soluble Aβ oligomers interfere with glutamate uptake could exert complex effects on the excitation-inhibition balance in epileptic networks. For example, a decrease in glutamate uptake into cells will also reduce GABA production. A previous report suggested that synthetic oAβ impairment of hippocampal LTP does not involve GABAergic inhibition (Raymond et al., 2003). This report could be due to the fEPSP slope measurement being insensitive to GABAergic inhibition (Wigstrom and Gustafsson, 1985), as we showed in Figs. 2D and 3A2. In contrast to the fEPSP slope, the quantification of population spikes is more sensitive to GABAergic inhibition (Fig. 2C; Fig. 3B); accordingly, we were able to observe GABAergic involvement in the LTP of PS (Fig. 2C). Our findings are consistent with a recent report that synthetic Aβ42 impairs LTP of PS (Orr et al., 2014), but our naturally secreted human oAβ species (7PA2 CM) and our pure, synthetic Aβ40-S26C dimers/oligomers each also blocked fEPSP LTP. There is evidence that hyperactive neurons in cortical circuits of hAPP/PS1 transgenic mice are associated with decreased GABAergic inhibition (Busche et al., 2008). Similarly, GABA administration during early life (before age 2 mos) significantly improved cognitive function in APP/PS1 mice (Sun et al., 2012). Other studies showed significantly elevated GABA levels in APP/PS1 mice compared with their wild-type littermates by HPLC analysis, and this included an increased GABA release from astrocytes (Jo et al., 2014). Such studies suggest that oAβ-induced increases in GABAergic neurotransmission and a chronic imbalance between glutamatergic and GABAergic transmission are likely to contribute to synaptic dysfunction in AD.
In recent years, some reports have shown that oAβ-induced synaptic dysfunction occurs, in part, through alteration of neuronal intrinsic excitability or disruption of the modulation of intrinsic excitability in AD brain (Zhang and Linden, 2003). Administrations of soluble oAβ to cultured neurons (Cuevas et al., 2011), brain slices (Minkeviciene et al.,, 2009; Varga et al.,2014; Ren et al., 2014) or living animals (Orbán et al.,2011; Busche et al., 2012) all induced significant hyperexcitability in hippocampal neurons. These findings are consistent with several AD mouse models in which elevated levels of Aβ are associated with altered neuronal activity, spontaneous seizures, and epileptiform discharges (Palop et al.,2007; Brown et al., 2011; Kerrigan et al., 2014). Our findings here that biochemically well-defined soluble forms of Aβ oligomers increase population spike frequency and impair LTP were mimicked by low-dose TBOA and by the GABA antagonist picrotoxin. oAβ induced a leftward shift of the E-S coupling curves indicating that a given fEPSP slope could produce a larger PS. Further, oAβ also increased sEPSC frequency, suggesting that oAβ increases overall neuronal excitability. The decrease of sEPSC amplitudes may be due to the increased extracellular glutamate level causing AMPAR desensitization, as shown (Li et al., 2009). Importantly, we found that reducing neuronal activity by enhancing inhibitory tone or by a low dose of an NMDAR antagonist or by treatment with certain antiepileptic drugs during oAβ exposure could each prevent oAβ-impaired synaptic dysfunction, consistent with the conclusion that oAβ-induced hyperexcitability impairs hippocampal LTP.
LTP is widely viewed as a correlate of the cellular mechanisms of learning and memory. Thus, this study of hippocampal LTP can help characterize the role of epilepsy in oAβ-induced cognitive impairment. As a model of epileptogenesis, kindling is associated with long-lasting facilitation of synaptic transmission and shares several features with LTP (Geinisman et al., 1988). Experimentally, kindling or seizure-like activity can severely attenuate or totally negate the ability to generate LTP in vitro (Schubert et al., 2005; Salmani et al., 2011). The maintenance phase of LTP can also be disrupted by seizures or become altered in seizure-prone animals (Hesse and Teyler, 1976; Schubert et al., 2005). Mechanistically, LTP and seizure activity share similar signaling pathways; thus, preventing neuronal seizure activity can restore tetanus-induced LTP impairment (Albensi et al., 2006)Consistent with these concepts, we show here that two antiepileptic drugs, topiramate and levetiracetam, that had no significant effect on LTP alone could both prevent oAβ-impaired hippocampal LTP, supporting the idea that oAβ-induced hyperexcitability can prevent electrical tetanus induced LTP. Also consistent are reports that levetiracetam suppressed epileptiform activity in hAPP mice and improved learning and memory in this oAβ-driven AD model (Sanchez et al., 2012) and even in patients with amnestic MCI, who usually have substantial brain levels of oAβ (Bakker et al., 2012). Taken together, our new data suggest that brain over-activation or epileptiform activity induced by Aβ oligomers may play an important role in the pathogenesis of early synaptic functional impairment in AD.
Acknowledgments
We thank Nina Shepardson and Sumin Kim for preparing the 7PA2 CM and CHO- CM. Supported by a grant (to SL) from Alzheimer’s Association (NIRG-12-242825), NIH grants AG 027443 and AG 036694 (to DJS) and grants (to PX) from 973 State Key Development Program for Basic Research of China (2011CB510000) and the National Natural Science Foundation of China (81071032 and 81271428).
Abbreviations
- AD
Alzheimer’s disease
- oAβ
soluble Aβ oligomers
- AP5
DL-2-Amino-5-phosphonopentanoic acid
- fEPSP
field excitatory postsynaptic potentials
- E-S
EPSP-spike
- GT
glutamate transporter
- IPSC
inhibitory postsynaptic currents
- sEPSC
spontaneous excitatory postsynaptic currents
- PS
population spike
- PTX
picrotoxin
- TBOA
DL-threo-β-benzyloxyaspartic acid
Footnotes
Any Conflict of Interest: No
References
- Albensi BC, Oliver DR, Toupin J, Odero G. Electrical stimulation protocols for hippocampal synaptic plasticity and neuronal hyper-excitability: are they effective or relevant? Exp Neurol. 2007;204(1):1–13. doi: 10.1016/j.expneurol.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia. 2006;47:867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch SB, He S, Dong Y. Inverse relationship between seizure expression and extrasynaptic NMDAR function following chronic NMDAR inhibition. Epilepsia. 2010;51(Suppl 3):102–105. doi: 10.1111/j.1528-1167.2010.02621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzina C, Verret L, Juan C, Remaud J, Halley H, Rampon C, Dahan L. Early onset of hypersynchronous network activity and expression of a marker of chronic seizures in the tg2576 mouse model of Alzheimer’s disease. PLoS One. 2015;10(3):e0119910. doi: 10.1371/journal.pone.0119910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JT, Chin J, Leiser SC, Pangalos MN, Randall AD. Altered intrinsic neuronal excitability and reduced Na+ currents in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2011;32:2109.e1–14. doi: 10.1016/j.neurobiolaging.2011.05.025. [DOI] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2012;109:8740–8745. doi: 10.1073/pnas.1206171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SL, Hablitz JJ. Glutamate transporters regulate excitability in local networks in rat neocortex. Neuroscience. 2004;127:625–635. doi: 10.1016/j.neuroscience.2004.05.030. [DOI] [PubMed] [Google Scholar]
- Campbell SL, Hablitz JJ. Decreased glutamate transport enhances excitability in a rat model of cortical dysplasia. Neurobiol Dis. 2008;32:254–261. doi: 10.1016/j.nbd.2008.07.003. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas ME, Haensgen H, Sepúlveda FJ, Zegers G, Roa J, Opazo C, Aguayo LG. Soluble Aβ(1–40) peptide increases excitatory neurotransmission and induces epileptiform activity in hippocampal neurons. J Alzheimers Dis. 2011;23:673–687. doi: 10.3233/JAD-2011-091717. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Hablitz JJ. Alterations in NMDA receptors in a rat model of cortical dysplasia. J Neurophysiol. 2000;83:315–321. doi: 10.1152/jn.2000.83.1.315. [DOI] [PubMed] [Google Scholar]
- Demarque M, Villeneuve N, Manent JB, Becq H, Represa A, Ben-Ari Y, Aniksztejn L. Glutamate transporters prevent the generation of seizures in the developing rat neocortex. J Neurosci. 2004;24:3289–3294. doi: 10.1523/JNEUROSCI.5338-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driver JE, Racca C, Cunningham MO, Towers SK, Davies CH, Whittington MA, LeBeau FE. Impairment of hippocampal gamma-frequency oscillations in vitro in mice overexpressing human amyloid precursor protein (APP) Eur J Neurosci. 2007;26:1280–1288. doi: 10.1111/j.1460-9568.2007.05705.x. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341:1607–1610. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC. Glutamate and astrocytes–key players in human mesial temporal lobe epilepsy? Epilepsia. 2008;49(Suppl 2):42–52. doi: 10.1111/j.1528-1167.2008.01492.x. [DOI] [PubMed] [Google Scholar]
- Fernández-Tomé P, Brera B, Arévalo MA, de Ceballos ML. Beta-amyloid25–35 inhibits glutamate uptake in cultured neurons and astrocytes: modulation of uptake as a survival mechanism. Neurobiol Dis. 2004;15:580–589. doi: 10.1016/j.nbd.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Frasca A, Aalbers M, Frigerio F, Fiordaliso F, Salio M, Gobbi M, Cagnotto A, Gardoni F, Battaglia GS, Hoogland G, Di Luca M, Vezzani A. Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol Dis. 2011;43:507–515. doi: 10.1016/j.nbd.2011.04.024. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Morrell F, deToledo-Morrell L. Remodeling of synaptic architecture during hippocampal “kindling”. Proc Natl Acad Sci U S A. 1988;85:3260–3264. doi: 10.1073/pnas.85.9.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu QB, Zhao JX, Fei J, Schwarz W. Modulation of Na(+),K(+) pumping and neurotransmitter uptake by beta-amyloid. Neuroscience. 2004;126:61–67. doi: 10.1016/j.neuroscience.2004.03.022. [DOI] [PubMed] [Google Scholar]
- Harkany T, Abrahám I, Timmerman W, Laskay G, Tóth B, Sasvári M, Kónya C, Sebens JB, Korf J, Nyakas C, Zarándi M, Soós K, Penke B, Luiten PG. beta-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur J Neurosci. 2000;12:2735–2745. doi: 10.1046/j.1460-9568.2000.00164.x. [DOI] [PubMed] [Google Scholar]
- Harris ME, Wang Y, Pedigo NW, Jr, Hensley K, Butterfield DA, Carney JM. Amyloid beta peptide (25–35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem. 1996;67:277–286. doi: 10.1046/j.1471-4159.1996.67010277.x. [DOI] [PubMed] [Google Scholar]
- Hesse GW, Teyler TJ. Reversible loss of hippocampal long term potentiation following electronconvulsive seizures. Nature. 1976;264:562–564. doi: 10.1038/264562a0. [DOI] [PubMed] [Google Scholar]
- Hsu KS, Ho WC, Huang CC, Tsai JJ. Transient removal of extracellular Mg(2+) elicits persistent suppression of LTP at hippocampal CA1 synapses via PKC activation. J Neurophysiol. 2000;84:1279–1288. doi: 10.1152/jn.2000.84.3.1279. [DOI] [PubMed] [Google Scholar]
- Hu NW, Klyubin I, Anwyl R, Rowan MJ. GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc Natl Acad Sci U S A. 2009;106:20504–20509. doi: 10.1073/pnas.0908083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauderback CM, Harris-White ME, Wang Y, Pedigo NW, Jr, Carney JM, Butterfield DA. Amyloid beta-peptide inhibits Na+-dependent glutamate uptake. Life Sci. 1999;65:1977–1981. doi: 10.1016/s0024-3205(99)00459-2. [DOI] [PubMed] [Google Scholar]
- Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, Ravid R, Roggendorf W, Riederer P, Grünblatt E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis. 2007;11:97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- Janjua NA, Kabuto H, Mori A. Increased plasma glutamic acid in a genetic model of epilepsy. Neurochem Res. 1992;17:293–296. doi: 10.1007/BF00966673. [DOI] [PubMed] [Google Scholar]
- Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee da Y, Hong J, Kim HY, Oh SJ, Park SJ, Lee H, Yoon BE, Kim Y, Jeong Y, Shim I, Bae YC, Cho J, Kowall NW, Ryu H, Hwang E, Kim D, Lee CJ. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med. 2014;20:886–896. doi: 10.1038/nm.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, Mattson MP. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 1997;69:273–284. doi: 10.1046/j.1471-4159.1997.69010273.x. [DOI] [PubMed] [Google Scholar]
- Kerrigan TL, Brown JT, Randall AD. Characterization of altered intrinsic excitability in hippocampal CA1 pyramidal cells of the Aβ-overproducing PDAPP mouse. Neuropharmacology. 2014;79:515–524. doi: 10.1016/j.neuropharm.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Glutamate transporters: animal models to neurologic disease. Neurobiol Dis. 2004;15:461–473. doi: 10.1016/j.nbd.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol. 1996;40:759–766. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- Masliah E, Alford M, Mallory M, Rockenstein E, Moechars D, Van Leuven F. Abnormal glutamate transport function in mutant amyloid precursor protein transgenic mice. Exp Neurol. 2000;163:381–387. doi: 10.1006/exnr.2000.7386. [DOI] [PubMed] [Google Scholar]
- Milh M, Becq H, Villeneuve N, Ben-Ari Y, Aniksztejn L. Inhibition of glutamate transporters results in a “suppression-burst” pattern and partial seizures in the newborn rat. Epilepsia. 2007;48:169–174. doi: 10.1111/j.1528-1167.2006.00839.x. [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Möddel G, Jacobson B, Ying Z, Janigro D, Bingaman W, González-Martínez J, Kellinghaus C, Prayson RA, Najm IM. The NMDA receptor NR2B subunit contributes to epileptogenesis in human cortical dysplasia. Brain Res. 2005;1046:10–23. doi: 10.1016/j.brainres.2005.03.042. [DOI] [PubMed] [Google Scholar]
- Molnár Z, Soós K, Lengyel I, Penke B, Szegedi V, Budai D. Enhancement of NMDA responses by beta-amyloid peptides in the hippocampus in vivo. Neuroreport. 2004;15:1649–1652. doi: 10.1097/01.wnr.0000134471.06244.d2. [DOI] [PubMed] [Google Scholar]
- O’Malley TT, Oktaviani NA, Zhang D, Lomakin A, O’Nuallain B, Linse S, Benedek GB, Rowan MJ, Mulder FA, Walsh DM. Aβ dimers differ from monomers in structural propensity, aggregation paths and population of synaptotoxic assemblies. Biochem J. 2014;461:413–426. doi: 10.1042/BJ20140219. [DOI] [PubMed] [Google Scholar]
- O’Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, Collinge J, Walsh DM. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–14419. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orbán G, Völgyi K, Juhász G, Penke B, Kékesi KA, Kardos J, Czurkó A. Different electrophysiological actions of 24- and 72-hour aggregated amyloid-beta oligomers on hippocampal field population spike in both anesthetized and awake rats. Brain Res. 2010;1354:227–235. doi: 10.1016/j.brainres.2010.07.061. [DOI] [PubMed] [Google Scholar]
- Orr AL, Hanson JE, Li D, Klotz A, Wright S, Schenk D, Seubert P, Madison DV. β-Amyloid inhibits E-S potentiation through suppression of cannabinoid receptor 1-dependent synaptic disinhibition. Neuron. 2014;82:1334–1345. doi: 10.1016/j.neuron.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea SD, Smith IM, McCabe OM, Cronin MM, Walsh DM, O’Connor WT. Intracerebroventricular administration of amyloid b-protein oligomers selectively increases dorsal hippocampal dialysate glutamate levels in the awake rat. Sensors. 2008;8:7428–7437. doi: 10.3390/s8117428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Ireland DR, Abraham WC. NMDA receptor regulation by amyloid-beta does not account for its inhibition of LTP in rat hippocampus. Brain Res. 2003;968:263–272. doi: 10.1016/s0006-8993(03)02269-8. [DOI] [PubMed] [Google Scholar]
- Ren SC, Chen PZ, Jiang HH, Mi Z, Xu F, Hu B, Zhang J, Zhu ZR. Persistent sodium currents contribute to Aβ1–42-induced hyperexcitation of hippocampal CA1 pyramidal neurons. Neurosci Lett. 2014;580:62–67. doi: 10.1016/j.neulet.2014.07.050. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Salmani ME, Fathollahi Y, Mirnajafizadeh J, Semnanian S. Epileptogenic insult alters endogenous adenosine control on long-term changes in synaptic strength by theta pattern stimulation in hippocampus area CA1. Synapse. 2011;65:189–197. doi: 10.1002/syn.20834. [DOI] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, Mucke L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M, Siegmund H, Pape HC, Albrecht D. Kindling-induced changes in plasticity of the rat amygdala and hippocampus. Learn Mem. 2005;12:520–526. doi: 10.1101/lm.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Meng X, Zhang J, Li Y, Wang L, Qin X, Sui N, Zhang Y. GABA attenuates amyloid toxicity by downregulating its endocytosis and improves cognitive impairment. J Alzheimers Dis. 2012;31:635–649. doi: 10.3233/JAD-2012-120535. [DOI] [PubMed] [Google Scholar]
- Texidó L, Martín-Satué M, Alberdi E, Solsona C, Matute C. Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium. 2011;49:184–190. doi: 10.1016/j.ceca.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Tancredi V, Hwa GG, Zona C, Brancati A, Avoli M. Low magnesium epileptogenesis in the rat hippocampal slice: electrophysiological and pharmacological features. Brain Res. 1990;511:280–290. doi: 10.1016/0006-8993(90)90173-9. [DOI] [PubMed] [Google Scholar]
- Varga E, Juhász G, Bozsó Z, Penke B, Fülöp L, Szegedi V. Abeta(1–42) enhances neuronal excitability in the CA1 via NR2B subunit-containing NMDA receptors. Neural Plast. 2014;2014:584314. doi: 10.1155/2014/584314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, Pimplikar SW. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol Aging. 2011;32:1725–1729. doi: 10.1016/j.neurobiolaging.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, Seeley WW, Geschwind MD, Gorno-Tempini ML, Shih T, Kirsch HE, Garcia PA, Miller BL, Mucke L. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark CJ, Westmark PR, Beard AM, Hildebrandt SM, Malter JS. Seizure susceptibility and mortality in mice that over-express amyloid precursor protein. Int J Clin Exp Pathol. 2008;1:157–168. [PMC free article] [PubMed] [Google Scholar]
- Welzel AT, Maggio JE, Shankar GM, Walker DE, Ostaszewski BL, Li S, Klyubin I, Rowan MJ, Seubert P, Walsh DM, Selkoe DJ. Secreted amyloid β-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity. Biochemistry. 2014;53:3908–3921. doi: 10.1021/bi5003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigström H, Gustafsson B. Facilitation of hippocampal long-lasting potentiation by GABA antagonists. Acta Physiol Scand. 1985;125:159–172. doi: 10.1111/j.1748-1716.1985.tb07703.x. [DOI] [PubMed] [Google Scholar]
- Wu J, Anwyl R, Rowan MJ. beta-Amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. Neuroreport. 1995;6:2409–2413. doi: 10.1097/00001756-199511270-00031. [DOI] [PubMed] [Google Scholar]
- Ying Z, Bingaman W, Najm IM. ncreased numbers of coassembled PSD-95 to NMDA-receptor subunits NR2B and NR1 in human epileptic cortical dysplasia. Epilepsia. 2004;45:314–321. doi: 10.1111/j.0013-9580.2004.37703.x. [DOI] [PubMed] [Google Scholar]
- Yuen AW, Sander JW. Can magnesium supplementation reduce seizures in people with epilepsy? A hypothesis. Epilepsy Res. 2012;100:152–156. doi: 10.1016/j.eplepsyres.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
- Zhang WQ, Hudson PM, Sobotka TJ, Hong JS, Tilson HA. Extracellular concentrations of amino acid transmitters in ventral hippocampus during and after the development of kindling. Brain Res. 1991;540:315–318. doi: 10.1016/0006-8993(91)90527-3. [DOI] [PubMed] [Google Scholar]
- Zoia CP, Riva C, Isella V, Proserpio P, Terruzzi A, Arban S, Salerno D, Cassina V, Mantegazza F, Tremolizzo L, Ferrarese C. Nonfibrillar Abeta 1–42 inhibits glutamate uptake and phosphorylates p38 in human fibroblasts. Alzheimer Dis Assoc Disord. 2011;25:164–172. doi: 10.1097/WAD.0b013e3181f9860f. [DOI] [PubMed] [Google Scholar]