

Abstract
PXR is a xenobiotic receptor that regulates drug metabolism by regulating the expression of drug-metabolizing enzymes including CYP3A4. It can be modulated by chemicals with different structures, functional groups and sizes. X-ray crystal structures of the ligand binding domain of human PXR (hPXR) alone or bound with agonists reveal a highly hydrophobic ligand binding pocket where the aromatic amino acid residue W299 appears to play a critical role in ligand binding. Here, we have investigated the role of W299 on the functional consequence of hPXR ligand binding. We first found that substitution of W299 with a hydrophobic residue retained its response to rifampicin, but substitution with a charged residue altered such agonist response in activating the transcription of CYP3A4. The activity of hPXR mutants on CYP3A4 expression correlates with the ability of hPXR mutants to interact with co-activator SRC-1. We further demonstrated that the effect of replacing W299 by residues with different side chains on hPXR’s function varied depending on the specific agonist used. Finally we interpreted the cellular activity of the hPXR mutants by analyzing reported crystallographic data and proposing a model. Our findings reveal the essential role of W299 in the transactivation of hPXR in response to agonist binding, and provide useful information for designing modulators of hPXR.
Keywords: PXR, CYP3A4, xenobiotic receptor, SAR, transcriptional regulation
Graphical abstract
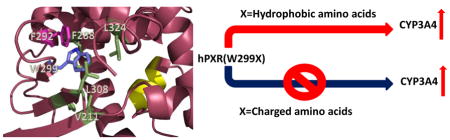
1. Introduction
Nuclear receptors are ligand activated transcription factors that control various biological functions including metabolism, development, reproduction, and aging [1, 2]. Pregnane X receptor (PXR) was first established as a xenobiotic nuclear receptor involved in drug metabolism by regulating the expression of cytochrome P450 [3], phase II enzymes, and drug transporters [4]. In addition to regulating drug metabolism, PXR has also been associated with many human diseases by regulating various signaling pathways. Binding of agonist to PXR causes dissociation of co-repressors, recruitment of co-activators, and subsequent activation of gene expression [5]. PXR consists of a highly conserved DNA binding domain (DBD), a C-terminal ligand binding domain (LBD), followed by an activation function 2 domain (AF-2) [3].
Numerous crystal structures of PXR LBD alone or in complex with agonists have been solved [6–13]. PXR has a hydrophobic, flexible, and promiscuous ligand binding pocket [6], and it can be activated by a diverse set of ligands including drug candidates, prescription drugs and herbal medicines such as rifampicin, paclitaxel, ritonavir, clotrimazole, TO901317, SR12813, and St. John’s Wort [14–16]. Activation of PXR may cause drug-drug interactions [17]. The transcriptional activity of PXR is controlled by ligand binding [18], and can also be modulated by posttranslational modifications at certain PXR residues [19–21]. Computational studies indicated that the most important hot spots of PXR for ligand binding are defined by W299, F288, and Y306, which are highly conserved hydrophobic aromatic acid residues [22]. However, the roles of these residues in ligand binding and the functional consequence have not yet been characterized in detail.
Here, we report the change of cellular activity of various W299 mutants of human PXR (hPXR) at both the basal and agonist-induced levels in human liver carcinoma cell line HepG2 and/or human intestinal epithelial cell line LS174T. We have found that depending on the nature of the side chain of the amino acid substituting W299, agonistic activation of CYP3A4 promoter can be substantially altered which could be interpreted by analyzing existing X-ray crystal structures of hPXR. This is the first report on detailed structural and functional analysis of the role of W299 on agonist-induced activation of PXR, which can aid in the development of modulators of PXR with potential implications in reducing PXR-mediated drug-drug interactions and human diseases.
2. Materials and Methods
2.1 Materials
HepG2 human liver carcinoma cells and LS174T human intestinal epithelial cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cell culture reagents were obtained from Invitrogen (Carlsbad, CA); anti-FLAG M2 and anti-β-actin antibodies, DMSO and compounds (rifampicin, SR12813, TO901317) were purchased from Sigma-Aldrich (St. Louis, MO); charcoal/dextran-treated fetal bovine serum (FBS) was purchased from Hyclone (Logan, UT); blocking buffer, anti-mouse, and anti-rabbit IR Dye secondary antibodies were from LI-COR Biosciences (Lincoln, NE).
2.2 Cell culture, plasmids, and transfection
HepG2 and LS174T cells were maintained in modified Eagle’s minimal essential medium (ATCC) with 10% FBS, 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. The pcDNA3-FLAG-hPXR expression plasmid (Flag-PXR) and the CYP3A4-luciferase reporter (CYP3A4-luc) were described previously [23]. The pcDNA3-FLAG-hPXR mutants containing various mutations as indicated were generated by Codex BioSolutions, Inc. (Gaithersburg, MD). Mutations were verified through nucleotide sequencing. Transfections were performed using FuGENE 6 (Roche Diagnostics, Indianapolis, IN) according to the manufacturer’s instructions.
2.3 Transient transfection and luciferase reporter gene assays
Detailed methods have been described previously [24]. Briefly, the cells were transfected with Flag-hPXR, CMV-Renilla (Promega, Madison, WI; as a transfection control and used for normalization), and CYP3A4-luc plasmids using FuGENE 6 (Roche Diagnostics). After 24 h, cells were seeded in 384-well plates (5000 cells/well) in phenol red-free medium containing 5% charcoal/dextran-treated FBS and incubated for another 24 h before compound treatment. Compounds were transferred using a pintool device. The cells were incubated with compounds for 24 h before processing using the Dual-Glo Luciferase Assay System (Promega). Renilla luciferase activity of CMV-Renilla was used to normalize the firefly luciferase activity of CYP3A4-luc, and the CYP3A4-luc/CMV-Renilla ratio (the relative CYP3A4 luciferase activity, in arbitrary unit, or a.u) was used to indicate the CYP3A4 promoter activity. DMSO was used as negative control. Curve-fitting software (GraphPad Prism 4.0; GraphPad Software, La Jolla, CA) was used to generate the curves.
2.4 Mammalian two-hybrid assay
The CheckMate mammalian two-hybrid system (Promega, Madison, WI) was performed as described previously [24]. It consists of VP16-hPXR, Gal4-SRC-1, and a luciferase reporter pG5-luc co-transfected into HepG2 cells. The Gal4 vector (pBIND) constitutively expresses Renilla luciferase, which was used as an internal transfection control. The Dual-Glo Luciferase Assay (Promega) was used to measure luciferase activity, which is an indicator of protein–protein interactions. The relative luciferase activity for pG5-luc was determined by normalizing firefly luciferase activity with Renilla luciferase activity, and expressed in arbitrary unit (a.u).
2.5 Western blot analysis
All cell extracts were harvested in 1× RIPA buffer from Cell Signaling Technology, Inc. (Danvers, MA), and samples were centrifuged at 12,000 × g at 4 °C for 25 min. The samples were then boiled in sample loading buffer (Invitrogen) containing SDS, and equal amounts of samples were resolved on 4–12% SDS-PAGE gradient gels and then transferred onto nitrocellulose membrane. The membrane was blocked and incubated with the indicated antibodies overnight at 4 °C. All Western blot analyses were performed on the Odyssey Infrared Imaging system (LI-COR Biosciences, Lincoln, NE). The intensity of each protein band was quantified using ImageJ 1.48 software [25]. The intensity of each protein band was normalized to that of β-actin to generate the relative intensity, with the relative intensity of the WT hPXR sample set as “1”.
2.6 RNA isolation and quantitative real-time polymerase chain reaction analysis (qRT-PCR)
The qRT-PCR experiment was performed as described previously [24]. Briefly, total RNA was isolated from HepG2 cells using Maxwell 16 LEV simplyRNA purification kits (Promega). qRT-PCR was performed using Taqman gene expression assays (Applied Biosystems, Carlsbad, CA) specific for CYP3A4, and β-actin (ACTB) was used as the reference gene according to the manufacturer’s protocol in an ABI 7900HT system (Applied Biosystems). The comparative Ct method was used for relative quantification of gene expression with the following formula: ΔCt = Ct (test gene) — Ct (ACTB); ΔΔCt (test gene) = ΔCt (test gene in treatment group) — ΔCt (test gene in vehicle control group); the fold changes of mRNA = 2−ΔΔCt, which indicated the relative mRNA level of the corresponding transcript relative to the control samples.
2.7 Structural analysis and docking
Crystal structures of hPXR-LBD-rifampicin (PDB: 1SKX), hPXR-LBD-TO901317 (PDB: 2O9I ), hPXR-LBD-SR12813 (PDB: 1NRL), and hPXR-LBD-compound 1 (PDB: 4XHD) were taken from the Protein Data Bank [9, 12, 13, 26] and analyzed using Pymol. Prediction of destabilizing effects of mutations on PXR were carried out using the program SIFT [27]. Simulated W299A mutations of hPXR were performed in Pymol and the volumes of the binding pocket for wild-type (WT) hPXR and mutant were measured using the software CASTp [28]. Docking studies were performed in AutoDock Vina version 1.1.1 [29] using PDBQT files generated in AutoDockTools (ADT) version 1.5.6 (http://mgltools.scripps.edu). The ligands were extracted from the pdb files and docked to their corresponding intact hPXR-LBD (containing wild-type W299) or hPXR-LBD containing simulated W299A mutation. Amino acid residues adjacent to W299 or A299 (V211, F288, Y306, L308, L324, H327) were made flexible for docking.
2.8 Statistical analysis
Results are expressed as the mean ± standard deviation from a representative experiment (n=3) and error bars indicate the standard deviation. Statistical analyses were performed using Student’s t-test. Differences were considered statistically significant for p ≤ 0.05 (*), and non-significant (NS) for p > 0.05.
3. Results
3.1 Functional analyses of W299 mutants
Several aromatic amino acid residues including F288, W299, Y306 in the hPXR ligand binding pocket have been reported to be critical for ligand binding [22], and W299 is conserved across all species [30]. In order to understand the contribution of the bulky residue W299 in ligand-induced CYP3A4 promoter activity, we have mutated W299 to hydrophobic (including other aromatic residues), uncharged polar and charged residues, and tested their activity by using CYP3A4-luc in luciferase reporter assays in either HepG2 or LS174T cells. In HepG2 cells, W299G and W299D mutants markedly reduced both basal and 5 μM rifampicin (RIF)-induced CYP3A4 promoter activity, when compared to WT hPXR (Figure 1). In contrast, mutations of W299 to residues with hydrophobic side chain, such as W299A, W299V, W299L, and W299F, retained the inducibility by RIF, similar to the WT hPXR. W299C, W299S, and W299C-F288C, while remained inducible by RIF, they were moderately less active than the WT hPXR. Interestingly, although W299A-F288A behaved similarly to WT hPXR, W299A-F288A-H327A and W299A-F288A-Y306A both markedly reduced their activity, similar to W299G and W299D.
Figure 1. The effect of different W299 mutants of hPXR on basal and rifampicin-induced CYP3A4 promoter activity in HepG2 cells.

HepG2 cells transiently transfected with WT hPXR or hPXR mutants as indicated, CYP3A4-luc, and CMV-Renilla were treated with DMSO or 5 μM RIF for 24 h prior to luciferase assay.
3.2 Differential regulation of W299 mutants by different hPXR agonists
To further investigate the effect of mutations at W299 and related residues on hPXR activity we used additional PXR agonists SR12813 (SR) and TO901317 (TO). As shown in Figure 2A, both W299D and W299A-F288A-Y306A were not substantially induced by any agonist tested. Interestingly, while RIF and SR activated W299A and WT hPXR similarly, TO robustly activated the WT hPXR but only weakly activated W299A. W299A-F288A retained its inducibility by RIF but markedly reduced its response to SR, and lost its inducibility by TO. W299G markedly reduced its response to both RIF and SR, and lost its inducibility by TO. Figure 2B showed the protein levels of all hPXR constructs, WT and mutated. We also evaluated the hPXR mutants in another cell line LS174T, and observed similar trend of responses to different agonists (Figure 3).
Figure 2. The activity and expression of WT hPXR and hPXR mutants in HepG2 cells.

(A) HepG2 cells transiently transfected with empty vector (EV), WT hPXR (hPXR) or hPXR mutant as indicated, CYP3A4-luc, and CMV-Renilla were treated with DMSO or different concentrations of agonists as indicated for 24 h prior to luciferase assay. (B) Western blot showing Flag-PXR (WT or mutants) protein levels in HepG2 cells upon (a) RIF [5 μM] and (b) DMSO treatment. The numbers below the protein bands indicate the relative intensity of the protein bands, with the wild-type Flag-PXR sample set as “1”. Anti-Flag antibody was used to detect Flag-PXR. Anti-β-actin was used to detect β-actin (as loading control).
Figure 3. The activity of WT hPXR and hPXR mutants in LS174T cells.

LS174T cells transiently transfected with empty vector (EV), WT hPXR (hPXR), or hPXR mutant as indicated, CYP3A4-luc, and CMV-Renilla were treated with DMSO or different concentrations of agonists as indicated for 24 h prior to luciferase assay.
3.3 Substituting W299 with charged residues compromises the function of hPXR
To further investigate the critical role of W299 in the activity of hPXR, we extended our studies by mutating W299 to several other amino acids with different side chains. Similar to W299D, substitution of W299 with other charged residues such as lysine and glutamate (W299E and W299K), causes a loss of function in HepG2 cells (Figure. 4). Similar to W299G (Figure 2A), W299N responded to RIF to a lesser extent when compared to the WT hPXR, but lost its inducibility by SR and TO (Figure 4). In contrast to both W299G and W299N, W299H responded to SR and TO to a lesser extent when compared to the WT hPXR, but lost it inducibility by RIF. Similar to W299A (Figure 2A), W299T was induced robustly by both RIF and SR, but not by TO (Figure 4). As expected, double mutant W299D-F288A exhibited a complete loss of function (Figure 4). The activities of WT hPXR and various W299 mutants with different side chains are summarized in Table 1 (based on data shown in Figures 1, 2A and 4).
Figure 4. The activity of WT and mutated hPXR in HepG2 cells.

HepG2 cells transiently transfected with empty vector (EV), WT hPXR (hPXR) or hPXR mutant as indicated, CYP3A4-luc, and CMV-Renilla were treated with DMSO or different concentrations of agonists as indicated for 24 h prior to luciferase assay.
Table 1.
Summary of activity of hPXR WT and mutants in HepG2 cells (CYP3A4 promoter activity; see Figures 1, 2A and 4 for details).
| Type of substituting residue(s) | WT hPXR or hPXR mutants | RIF activity | SR activity | TO activity |
|---|---|---|---|---|
| None | hPXR | +++ | +++ | +++ |
| Hydrophobic | W299A | +++ | +++ | + |
| W299V | +++ | not tested | not tested | |
| Charged | W299D | +/− | +/− | +/− |
| W299E | +/− | +/− | +/− | |
| W299K | +/− | +/− | +/− | |
| Double mutants | W299A-F288A | +++ | +/− | +/− |
| W299D-F288A | +/− | +/− | +/− | |
| Triple mutant | W299A-F288A-Y306A | − | − | − |
+++ strongly active;
+ weakly active;
+/− very weak or inactive;
− complete loss of function
3.4 Mutations at W299 affect the interactions of hPXR with SRC-1
To determine whether the activity of hPXR mutants as observed in the luciferase reporter assays correlates with their ability to interact with co-activator, we performed mammalian two-hybrid assay which measures the interaction between hPXR and the steroid receptor coactivator-1 (SRC-1) [31]; SRC-1 is known to mediate the ligand-induced activation of PXR [32, 33]. As expected, RIF and SR significantly increased the interaction between hPXR and SRC-1 (Figure 5). W299V behaves similarly to the WT hPXR in the luciferase reporter assay (Figure 1). The interaction between W299V and SRC-1 was also increased by both RIF and SR (Figure 5). On the other hand, at low concentration RIF and SR failed to enhance the interaction between SRC-1 and W299D or W299G, and the interactions were only marginally increased at higher concentrations (Figure 5), consistent with the luciferase reporter activity (Figure 1).
Figure 5. Mammalian two-hybrid assay to determine the interaction between WT or mutated hPXR and SRC-1.

Mammalian two-hybrid assays were performed in HepG2 cells transiently co-transfected with plasmids encoding Gal4-SRC-1 and the reporter gene pG5-luc, together with either empty vector pACT, or pACT fused to WT (pACT-hPXR) or mutated hPXR (W299V, W299G, W 299D) as indicated. The cells were treated with DMSO; RIF; or SR compound at the indicated concentrations. Luciferase assays were performed 24 h after the compound treatment. The relative luminescence for pG5-luc was determined by normalizing firefly luciferase activity to Renilla luciferase activity (Luciferase/Renilla).
To further verify the observations made in the luciferase reporter assays, we investigated the impact of W299 mutations on both basal and agonist-induced CYP3A4 mRNA expression levels at different RIF concentrations in HepG2 cells. HepG2 cells were transiently transfected with EV (control), WT hPXR, and the individual hPXR mutants. Overexpression of the WT hPXR or mutant in which W299 was substituted with a hydrophobic residue (W299A and W299V) markedly increased the RIF-induced CYP3A4 as compared to EV (Figure 6). RIF also induced CYP3A4 mRNA expression in EV-transfected cells through the endogenous hPXR. In contrast, the charged mutant W299D and triple mutant W299A-F288A-Y306A showed no substantial enhancement of RIF-induced CYP3A4 mRNA expression as compared to EV.
Figure 6. CYP3A4 mRNA in HepG2 cells transfected with empty vector (EV, to measure endogenous hPXR activity), WT hPXR (PXR) or hPXR mutant as indicated.

Real-time PCR analysis of CYP3A4 mRNA expression in HepG2 cells was performed after 48 h treatment with DMSO or RIF (5 or 10 μM).
3.5 Structural interpretation of the observed hPXR activities
Structural analysis provides a rational explanation for the observed functional consequences of these mutants at a molecular level. A large number of crystal structures of PXR-LBD in complex with various agonists have been reported, which show that W299 is an important amino acid residue that interacts with most of these ligands. In addition, W299 forms part of a hydrophobic pocket, interacting with several nonpolar residues such as F288, L324, L308, V211 and F292. Additional π-stacking contacts exist between W299 and F292 (Figure 7A). All these interactions are important in keeping the structural integrity of the LBD for ligand binding. Therefore, mutations from the hydrophobic W299 to a charged amino acid (such as E, H, D or N) would disrupt these hydrophobic effects, leading possibly to a partial unfolding of the LBD. However, mutation to a smaller and still hydrophobic residue such as alanine would not be able to impart such dramatic structural alteration. In addition, mutations to polar but uncharged residues such as in the case of W299S might not affect folding significantly as confirmed by the luciferase reporter assay data. Protein destabilizing effects of the mutations were predicted using the program SIFT, which anticipates that substitutions with V, M, F, I and L would be tolerated, while the rest of the amino acids would result in protein destabilization. These predictions are consistent with cell-based assays with the exception of alanine (discussed in the following paragraph). The residues serine and cysteine were also predicted to be destabilizing, but experimental data showed partial PXR activation. As the program cannot distinguish highly from mildly destabilizing mutations, it is possible that these mutants (W299S and W299C) are at the borderline in destabilizing hPXR.
Figure 7. Effects of W299 mutation on hPXR activity as explained by computational analysis of hPXR structures.

(A) W299 (blue stick) contacts several nonpolar residues (green and pink sticks) forming an important hydrophobic patch in the hPXR-LBD for ligand binding and structural integrity, which can be disrupted by replacement of W299 with charged residues (pdb code 4XHD). F292 (pink stick) is involved in π-stacking contact with W299. The AF-2 helix is represented in yellow. (B) TO docked to the W299A mutant (carbon atoms in pink) in relation to TO as observed in the WT crystal structure (carbon atoms in white). Hydrogen bonds to protein residues (lines with carbon atoms in white) are indicated with dashes. The sphere represents the region in the TO molecule to most likely clash against W299 (green stick) (C) SR docked to the W299A mutant (carbon atoms in pink) in relation to SR as observed in the WT crystal structure (carbon atoms in white). The t-butyl groups of the ligand (spheres) prevent the ligand from moving around due to steric hindrance even after additional space is created by replacement of W299 (green) with alanine. (D) RIF docked to the W299A mutant (carbon atoms in pink) in relation to RIF as observed in the WT crystal structure (carbon atoms in white). The large molecule RIF cannot move around even when additional space is created following W299 (green) replacement by alanine. The amino acid residues that were made flexible during docking are represented as blue surface (B–D).
Mutations of W299 to alanine, however, displayed some interesting and initially unexpected results depending on the agonist employed in our cell-based assays. Even though W299A was only weakly activated by TO (which robustly activated the WT hPXR), RIF and SR were able to induce W299A to almost a similar extent as the WT hPXR. This discrepancy among different agonists can be explained by comparing the crystal structures of hPXR-LBD in complex with any of the three agonists. Replacement of the bulkier tryptophan by the smaller alanine would open some space that allows for ligands to move around. It is estimated that the W299A mutation would increase the volume of the binding pocket by 173 Å3 for SR, 145 Å3 for RIF and 167 Å3 for TO. There are slight differences in volume between the three structures because the flexible ligand cavity conforms its shape depending on the ligand bound. In the case of TO, the additional space allows the ligand to wiggle, and therefore potentially compromise hydrogen bonds formed by E285, H327 and H407 (Figure 7B). However, the t-butyl moieties in SR (Figure 7C, represented as spheres) fixes the ligand in such a way that the added space that is formed would not be sufficient for the ligand to move around. RIF is such a large molecule that the additional space resulting from the mutation would also not be significant (Figure 7D). To test the hypothesis arising from the visual analysis of the crystal structures, we conducted docking studies with the simulated W299A mutant, where the side chains of neighboring residues (V211, F288, Y306, L308, L324, H327) were made flexible and allowed to refine during the docking process, because it is conceivable that the point mutation might affect the orientation of adjacent residues upon ligand binding. As expected, docking poses of SR (Figure 7C, carbon atoms in pink) and RIF (Figure 7D, carbon atoms in pink) in the W299A mutant are superimposable to the ligand structures observed in the crystal structures of the WT protein (Figures 7C and 7D, carbon atoms in white). However, docking results show that TO is noticeably displaced towards the void resulting from the W299A mutation (Figure 7B, carbon atoms in pink) when compared to the orientation of the ligand in the crystal structure (Figure 7B, carbon atoms in white). Hydrogen bonding is visibly compromised due to the reorientation of TO in the W299A mutant.
4. Discussion
PXR is an important xenobiotic nuclear receptor that controls the metabolism and excretion of harmful chemicals [34]. In addition, PXR is involved in various human diseases [35]. The activity of PXR can be regulated by small molecule modulators and post-translational modification of specific amino acid residues. Previous studies have identified such residues that are critical for PXR activity. Agonist binding causes PXR to recruit co-activator resulting in transcriptional up-regulation of its target genes [36]. Mutation of PXR residues that may not be posttranslationally modified but critical for ligand binding may affect ligand interaction and its functional consequences.
The natural variants of PXR protein have been identified and showed different basal and ligand-induced CYP3A4 promoter activity. These PXR protein variants may play a role in CYP3A4 expression and may be involved in altered sensitivities to carcinogens or atypical responses to drugs [37]. Interestingly, some of the previously identified naturally occurring variants (V140M, R148Q, D163G, R381W, A370T and I403V) are located within or close to the LBD of PXR [37–40]. However no naturally occurring mutation of W299 has been reported. PXR contains a promiscuous ligand binding pocket that consists of 28 amino acid residues [6]. Previous reports showed that mutation in the ligand binding pocket can influence the receptor activity upon ligand binding [6, 13]. W299 is present in the ligand binding pocket of PXR. It contains a large side chain and is involved in hydrophobic interactions with ligand [6, 13, 26]. It has been reported that mutation of W299 to alanine in CV-1 (monkey kidney cell line) cells has no effect on receptor activity; but the authors were unable to explain the role of this residue though this is an important residue for ligand binding [13]. However, the discrepancy can be explained by performing the experiment in more physiologically relevant human cellular systems such as HepG2 and LS174T cells. We have generated various W299 mutants of hPXR and analyzed their functional consequences on CYP3A4 promoter activity in HepG2 and/or LS174T cell lines. Table 1 summarizes the activities of the mutants based on the nature of the side chains of the substituting residues. Mutating W299 to an amino acid with a hydrophobic side chain (V, L, A, F) retained RIF-induced CYP3A4 promoter activity, comparable with WT hPXR. However, while RIF and SR activated W299A and WT hPXR similarly, TO robustly activated the WT hPXR but only weakly activated W299A. These observations suggest that mutation of W299 to even a hydrophobic residue might affect the response of hPXR to certain agonists. Mutation to amino acids with polar but uncharged side chain such as T or S retained the inducibility by RIF but may lose the inducibility by TO. W299N also retained moderate inducibility by RIF but not by SR and TO. However, mutating W299 to a residue with a charged side chain such as D substantially reduced its agonist-inducibility. Mutating W299 to E and K almost abolished the agonist-inducibility of hPXR. The behavior of W299H is interesting: while losing the inducibility by RIF, it retained moderate inducibility by SR and TO. A similar loss-of-function effect was also observed for the triple mutant W299A-F288A-Y306A. Our data from the mammalian two-hybrid assay suggest that substituting W299 with different residues affects the activity of hPXR, likely by affecting the interaction between hPXR and SRC-1. Together, our studies indicate that W299 is a critical residue; mutation of W299 to other residues might affect the response of hPXR to xenobiotics. These observations help explain why W299 is highly conserved.
Based on the analysis of the available crystal structures and docking studies, we propose that two major issues are at play by mutations at W299. Firstly, certain mutations can affect structural integrity of the protein leading to partial unfolding. As W299 forms part of a cluster of hydrophobic residues, substitution with a charged amino acid can be highly disruptive, as in the case with mutants such as W299D and W299E and W299K. This is consistent with the protein destabilizing effects of these mutants as predicted by the program SIFT. Secondly, mutating W299 to the smaller residue alanine increases the volume of the binding pocket, which can allow certain ligands to move around, and thus disrupting other important protein-ligand interactions. Such could be the case with the ligand TO, where hydrogen bonds in the vicinity could be susceptible to ligand movement. For ligands of larger size or less flexibility such as RIF and SR, the ligand is physically constrained that the extra space does not have a major impact on PXR activity. There is also the possibility that some of the mutants can mildly affect structural integrity, but the ligand acts as “glue” by providing additional interactions that hold the protein segments together. The strength of this “glue” would naturally depend on the type of the ligand: the large molecular size and considerable number of interactions of RIF with the ligand binding pocket would provide stronger protein structural integrity than TO, as observed in the reporter assays (Figure 2A).
In conclusion, we have found that the W299 residue of hPXR is important for not only ligand binding, but also transcriptional activation in response to the ligand binding. Our results showed that the W299 mutants behave differently according to the nature of the side chain of the substituted amino acid. Structural analysis provided explanations for the contrary effect of hydrophobic vs. charged residues at W299 of hPXR. hPXR consists of a promiscuous ligand binding pocket. However, the function of the promiscuous PXR can be altered by the mutation of a single residue. It will be interesting to examine the functional consequences of mutating other critical hydrophobic residues present in the LBD of hPXR, and determine if the functional consequence of mutation is residue specific or geometry/location specific. Because the residues present in the ligand binding pocket are critical for ligand binding, our data will be useful for structure-function studies of hPXR and hPXR-based drug design and development.
Acknowledgments
We thank other members of the Chen research laboratory for valuable discussions. This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital, and the National Institutes of Health [Grants R01GM086415, R01GM110034, and P30-CA21765]. The funding sources had no involvement in the writing or the decision to submit the manuscript for publication.
Footnotes
Conflict of interest
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lichti-Kaiser K, Brobst D, Xu CS, Staudinger JL. A Systematic Analysis of Predicted Phosphorylation Sites within the Human Pregnane X Receptor Protein. J Pharmacol Exp Ther. 2009;331:65–76. doi: 10.1124/jpet.109.157180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naar AM, Thakur JK. Nuclear receptor-like transcription factors in fungi. Genes Dev. 2009;23:419–32. doi: 10.1101/gad.1743009. [DOI] [PubMed] [Google Scholar]
- 3.Willson TM, Kliewer SA. PXR, CAR and drug metabolism. Nat Rev Drug Discov. 2002;1:259–66. doi: 10.1038/nrd753. [DOI] [PubMed] [Google Scholar]
- 4.Tolson AH, Wang H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv Drug Deliv Rev. 2010;62:1238–49. doi: 10.1016/j.addr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee M, Chen T. Differential regulation of CYP3A4 promoter activity by a new class of natural product derivatives binding to pregnane X receptor. Biochem Pharmacol. 2013;86:824–35. doi: 10.1016/j.bcp.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, et al. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–33. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 7.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, et al. 2.1 A crystal structure of human PXR in complex with the St. John’s wort compound hyperforin. Biochemistry. 2003;42:1430–8. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 8.Xue Y, Moore LB, Orans J, Peng L, Bencharit S, Kliewer SA, et al. Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol Endocrinol. 2007;21:1028–38. doi: 10.1210/me.2006-0323. [DOI] [PubMed] [Google Scholar]
- 9.Khan JA, Camac DM, Low S, Tebben AJ, Wensel DL, Wright MC, et al. Developing Adnectins That Target SRC Co-Activator Binding to PXR: A Structural Approach toward Understanding Promiscuity of PXR. J Mol Biol. 2015;427:924–42. doi: 10.1016/j.jmb.2014.12.022. [DOI] [PubMed] [Google Scholar]
- 10.Cheng Y, Redinbo MR. Activation of the human nuclear xenobiotic receptor PXR by the reverse transcriptase-targeted anti-HIV drug PNU-142721. Protein Sci. 2011;20:1713–9. doi: 10.1002/pro.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teotico DG, Bischof JJ, Peng L, Kliewer SA, Redinbo MR. Structural basis of human pregnane X receptor activation by the hops constituent colupulone. Mol Pharmacol. 2008;74:1512–20. doi: 10.1124/mol.108.050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue Y, Chao E, Zuercher WJ, Willson TM, Collins JL, Redinbo MR. Crystal structure of the PXR-T1317 complex provides a scaffold to examine the potential for receptor antagonism. Bioorg Med Chem. 2007;15:2156–66. doi: 10.1016/j.bmc.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, et al. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol Endocrinol. 2005;19:1125–34. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 14.Shukla SJ, Sakamuru S, Huang R, Moeller TA, Shinn P, Vanleer D, et al. Identification of clinically used drugs that activate pregnane X receptors. Drug Metab Dispos. 2011;39:151–9. doi: 10.1124/dmd.110.035105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mani S, Dou W, Redinbo MR. PXR antagonists and implication in drug metabolism. Drug Metab Rev. 2013;45:60–72. doi: 10.3109/03602532.2012.746363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang TK. Activation of pregnane X receptor (PXR) and constitutive androstane receptor (CAR) by herbal medicines. AAPS J. 2009;11:590–601. doi: 10.1208/s12248-009-9135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harmsen S, Meijerman I, Beijnen JH, Schellens JH. The role of nuclear receptors in pharmacokinetic drug-drug interactions in oncology. Cancer Treat Rev. 2007;33:369–80. doi: 10.1016/j.ctrv.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Saradhi M, Sengupta A, Mukhopadhyay G, Tyagi RK. Pregnane and Xenobiotic Receptor (PXR/SXR) resides predominantly in the nuclear compartment of the interphase cell and associates with the condensed chromosomes during mitosis. Biochim Biophys Acta. 2005;1746:85–94. doi: 10.1016/j.bbamcr.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Smutny T, Mani S, Pavek P. Post-translational and Post-transcriptional Modifications of Pregnane X Receptor (PXR) in Regulation of the Cytochrome P450 Superfamily. Curr Drug Metab. 2013;14:1059–69. doi: 10.2174/1389200214666131211153307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elias A, High AA, Mishra A, Ong SS, Wu J, Peng JM, et al. Identification and characterization of phosphorylation sites within the pregnane X receptor protein. Biochem Pharmacol. 2014;87:360–70. doi: 10.1016/j.bcp.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang YM, Chai SC, Lin WW, Chai XJ, Elias A, Wu J, et al. Serine 350 of human pregnane X receptor is crucial for its hetrodimerization with retinoid X receptor alpha and transactivation of target genes in vitro and in vivo. Biochem Pharmacol. 2015;96:357–68. doi: 10.1016/j.bcp.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ngan CH, Beglov D, Rudnitskaya AN, Kozakov D, Waxman DJ, Vajda S. The structural basis of pregnane X receptor binding promiscuity. Biochemistry. 2009;48:11572–81. doi: 10.1021/bi901578n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin W, Wu J, Dong H, Bouck D, Zeng FY, Chen T. Cyclin-dependent kinase 2 negatively regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells. J Biol Chem. 2008;283:30650–7. doi: 10.1074/jbc.M806132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee M, Chen T. Thiazide-like diuretic drug metolazone activates human pregnane X receptor to induce cytochrome 3A4 and multidrug-resistance protein 1. Biochem Pharmacol. 2014;92:389–402. doi: 10.1016/j.bcp.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–28. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 27.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–W7. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Binkowski TA, Naghibzadeh S, Liang J. CASTp: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2003;31:3352–5. doi: 10.1093/nar/gkg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trott O, Olson AJ. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J Comput Chem. 2010;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reschly EJ, Krasowski MD. Evolution and function of the NR1I nuclear hormone receptor subfamily (VDR, PXR, and CAR) with respect to metabolism of xenobiotics and endogenous compounds. Curr Drug Metab. 2006;7:349–65. doi: 10.2174/138920006776873526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pondugula SR, Brimer-Cline C, Wu J, Schuetz EG, Tyagi RK, Chen T. A phosphomimetic mutation at threonine-57 abolishes transactivation activity and alters nuclear localization pattern of human pregnane x receptor. Drug Metab Dispos. 2009;37:719–30. doi: 10.1124/dmd.108.024695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 33.Sui YP, Ai N, Park SH, Rios-Pilier J, Perkins JT, Welsh WJ, et al. Bisphenol A and Its Analogues Activate Human Pregnane X Receptor. Environ Health Perspect. 2012;120:399–405. doi: 10.1289/ehp.1104426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kliewer SA, Willson TM. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J Lipid Res. 2002;43:359–64. [PubMed] [Google Scholar]
- 35.Banerjee M, Robbins D, Chen T. Targeting xenobiotic receptors PXR and CAR in human diseases. Drug Discov Today. 2015;20:618–28. doi: 10.1016/j.drudis.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banerjee M, Robbins D, Chen T. Modulation of Xenobiotic Receptors by Steroids. Molecules. 2013;18:7389–406. doi: 10.3390/molecules18077389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hustert E, Zibat A, Presecan-Siedel E, Eiselt R, Mueller R, Fuss C, et al. Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug Metab Dispos. 2001;29:1454–9. [PubMed] [Google Scholar]
- 38.Koyano S, Kurose K, Ozawa S, Saeki M, Nakajima Y, Hasegawa R, et al. Eleven novel single nucleotide polymorphisms in the NR1I2 (PXR) gene, four of which induce non-synonymous amino acid alterations. Drug Metab Pharmacokinet. 2002;17:561–5. doi: 10.2133/dmpk.17.561. [DOI] [PubMed] [Google Scholar]
- 39.Koyano S, Kurose K, Saito Y, Ozawa S, Hasegawa R, Komamura K, et al. Functional characterization of four naturally occurring variants of human pregnane X receptor (PXR): One variant causes dramatic loss of both DNA binding activity and the transactivation of the CYP3A4 promoter/enhancer region. Drug Metab Dispos. 2004;32:149–54. doi: 10.1124/dmd.32.1.149. [DOI] [PubMed] [Google Scholar]
- 40.Lamba J, Lamba V, Schuetz E. Genetic variants of PXR (NR1I2) and CAR (NR1I3) and their implications in drug metabolism and pharmacogenetics. Curr Drug Metab. 2005;6:369–83. doi: 10.2174/1389200054633880. [DOI] [PubMed] [Google Scholar]