

Abstract
Introduction
Immune checkpoints are regulatory pathways induced in activated T lymphocytes that regulate antigen responsiveness. These immune checkpoints are hijacked by tumors to promote dysfunction of anti-tumor effector cells and consequently of tumor escape from the host immune system.
Areas covered
PD1/PDL-1, a checkpoint pathway, has been extensively investigated in leukemia mouse models. Expression of PD-1 on the surface of activated immune cells and of its ligands, PD-L1 and PD-L2, on leukemic blasts has been documented. Clinical trials with PD-1 inhibitors in patients with hematological malignancies are ongoing with promising clinical responses.
Expert Opinion
Therapy of hematological cancers with antibodies blocking inhibitory receptors is expected to be highly clinically effective. Checkpoint inhibitory receptors and their ligands are co-expressed on hematopoietic cells found in the leukemic milieu. Several distinct immunological mechanisms are likely to be engaged by antibody-based checkpoint blockade. Co-expression of multiple inhibitory receptors on hematopoietic cells offers an opportunity for combining blocking antibodies to achieve more effective therapy. Up-regulation of receptor/ligand expression in the leukemic milieu may provide a blood marker predictive of response. Finally, chemotherapy-induced up-regulation of PD-1 on T cells after conventional leukemia therapy creates a solid rationale for application of checkpoint blockade as a follow-up therapy.
1. Introduction
Human tumors, including hematological malignancies, have developed multiple strategies for escape from the host immune system. Mechanisms used by tumors for escape have been extensively investigated in the last decade,1 and a better understanding of these mechanisms has facilitated the development of novel therapies aimed at arresting tumor immune evasion. One of the more recently discovered mechanisms of immune suppression operating in cancer involves immune cell intrinsic checkpoints that are induced on the surface of activated T cells.2 Several such checkpoint molecules serving as negative regulators of activated T cells are known, including cytotoxic T-cell antigen-4 (CTLA-4), programmed death-1 (PD-1), T cell immunoglobulin mucin-3 (TIM-3), lymphocyte activation gene-3 (LAG-3), B and T cell lymphocyte attenuator (BTLA) and others. Surface expression and inhibitory functions of these receptors are up-regulated in T cells present in the tumor microenvironment.3 While the presence of these inhibitory receptors on T cells is physiologically necessary to regulate cellular activation, their overexpression in disease leads to dysfunction of T cells and other immune effector cells.4-7 In the setting of cancer, chronic overexpression of checkpoint molecules results in T-cell dysfunction and impairs anti-tumor immunity.3
It has been observed in animal models of tumor growth that blocking of checkpoint receptors with antibodies (Abs) can restore anti-tumor immunity and prevent tumor progression.8, 9 One of the first checkpoint-blocking antibodies tested in preclinical studies and approved for therapy of patients with advanced melanoma in 2011 was ipilimumab, the anti-CTLA-4 Ab.8, 10-12 Its administration to patients with advanced melanoma and blockade of CTLA-4 provided first evidence that this immune therapy results in durable responses and improved survival in 10-15% of patients.12 The next anti-checkpoint Abs, pembrolizumab and nivolumab, approved for melanoma therapy, target PD-1. These antibodies are currently being actively investigated for the treatment of different cancers, including hematological malignancies. While more recent data for the blockade of the PD-1/PD-L1 pathway demonstrate durable responses in 30-35% of patients with advanced melanoma,13 the factors underlying molecular, cellular and functional aspects of checkpoint inhibition in cancer patients are not yet understood and are being intensively investigated. Our current insights into early studies combining anti-CTLA-4 with anti-PD-Abs suggest that this combination shows impressive response rates and a relatively low toxicity profile. The mechanisms responsible for these clinical successes are not entirely worked out, and the evidence indicating that only subsets of patients respond to this immune therapy suggests that more extensive studies are required for improving its anti-tumor activity.
While patients with advanced melanoma were the first cohort to be successfully treated with checkpoint inhibitors, efforts are underway to extend this therapy to other solid tumors and, more recently, to hematological malignancies. This is an exceedingly important effort that aims at providing potentially beneficial immunotherapy to the cancer patient population at large. The purpose of this review is to discuss the rationale for and consider the potential impact of checkpoint inhibition on disease control in acute myeloid leukemia (AML). Although in comparison to solid cancers, the data on checkpoint inhibition in leukemia are limited, preclinical data overwhelmingly indicate that hematological malignancies, including AML, which generally respond favorably to immune therapies, are also likely to benefit from checkpoint inhibition. As clinical trials with anti-PD-1 Ab checkpoint blockade in AML are being implemented, we anticipate that this immune therapy will rapidly move from the category of an experimental to an approved therapy for acute leukemias.
2. PD-1 Biology
Immune checkpoints are regulatory pathways that are induced in activated T cells and regulate the amplitude as well as the quality of T-cell antigen responses. These pathways are balanced by co-stimulatory and inhibitory signals and are critical in preventing autoimmunity and uncontrolled T-cell expansion that could facilitate oncogenic mutations. However, cancer has developed ways to exploit these immune cell-intrinsic checkpoints for escaping immune-mediated destruction.1 In cancer, expression and functions of checkpoint molecules on T cells are up-regulated, leading to reduction or elimination of anti-tumor immune activity.2 CTLA-4 and PD-1 are two of the most actively studied inhibitory receptors expressed by activated T-cells.
CTLA-4 receptor (CD 152) is present on T cells early in their activation stage, and it competes with CD28, a costimulatory receptor also expressed on T cells, for binding to CD80 (B7.1) and CD86 (B7.2) expressed on antigen-presenting cells (APC). Up-regulation of CTLA-4 activity interferes with co-stimulatory signals necessary for T-cell maturation and differentiation into effector cells. As discussed above, targeting of CTLA-4 with a blocking Ab, ipilimumab, in patients with advanced melanoma provided initial evidence that immune checkpoint blockade translates into clinical benefit in 10-15% of these patients by allowing restoration of more robust anti-cancer immune responses.12
The PD-1 receptor (CD279) is another inhibitory checkpoint also expressed by activated T-cells. (Figure 1) It is a type I transmembrane receptor member of the immunoglobulin superfamily and it binds two ligands: PD-L1 (B7-H1, CD274)7, 14 and PD-L2 (B7-DC, CD273),15, 16 both belonging to the B7 immunoglobulin superfamily. PD-1 differs from CTLA-4 in that its major role is to limit the inflammatory responses which occur in the periphery, when effector T cells recognize target antigens present on tissue cells. Uncontrolled T-cell response in this context leads to tissue damage. When the PD-1 receptor interacts with its ligands, PD-L1, which is expressed on most tissues and PD-L2, which is expressed on macrophages and dendritic cells, a signaling cascade is initiated that inhibits several of down-stream kinases involved in T-cell activation.17 Inflammatory signals, such as interferon-γ (IFN-γ) secreted by T-helper 1 cells (Th1), induce PD-L1 and PD-L2 expression on tissue cells. 18
Figure 1.
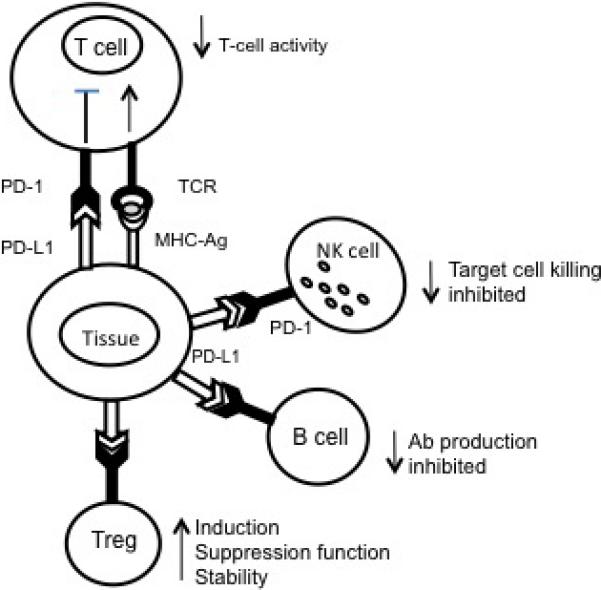
A diagram demonstrating interactions between PD-1 receptor expressed on immune cells and PD-L1. A PDL-1 expressing antigen-presenting cell delivers a negative signal to the PD-1 receptor expressed on immune cells, which blocks their effector functions. In T-cells, Ag-specific responses are blocked. In NK cells, the ability to mediate cytotoxicity is decreased. In B cells, the Ab production is inhibited. In contrast, the same negative signal enhances the development and suppressor functions of Treg. Checkpoint receptor engagement directly blocks functions of immune cells and also has indirect suppressive effects mediated by Treg.
NK: Natural killer; PD-1: Programmed death-1; PDL-1: Programmed death ligand-1; TCR: T-cell receptor.
In addition to activated effector T-cells, PD-1 is highly expressed on regulatory T cells (Treg), B-cells, and natural killer (NK) cells. When PD-1 expressed on Treg binds its ligand, induction of Treg and suppressor functions mediated by these cells are enhanced.19 Therefore, the PD-1 pathway not only suppresses functions of effector T cells, lytic capacity of NK cells and B-cell antibody production, but it also promotes stability and functions of Treg, thus contributing to maintenance of immune suppression in the microenvironment.
3. PD-1 Blockade
The critical role of PD-1 in immune suppression has been elucidated through a number of preclinical studies, many in the setting of chronic viral infections. CD8+ memory T cells seen in chronic viral infections such as human immunodeficiency virus (HIV), hepatitis C (HCV) or hepatitis B (HBV)20-22 have impaired proliferative and cytokine responses and are commonly referred to as “exhausted T cells.” The role of PD-1 in inducing dysfunction of CD8+ memory T cells was first demonstrated in mice chronically infected with lymphocytic choriomeningitis virus (LCMV). PD-1 was found to be selectively and significantly upregulated on exhausted T-cells in mice with chronic LCMV.23 Importantly, PD-1 blockade restored functions of these cells, resulting in a decreased viral load.23 Similar findings have subsequently been made for HIV-124 as well as chronic hepatitis.25
The availability of PD-1 knockout (KO) mice has further helped in clarifying the role PD-1 plays in immune regulation. The PD-1 KO mice consistently developed late-onset autoimmunity, with variable sites of autoimmune tissue damage dependent on the expression levels and persistence of tissue antigens in the background mouse strain. For example, non-obese (NOD) diabetic mice rapidly develop diabetes when PD-1 is knocked out;26 mice with the C57BL/6 background develop lupus-like autoimmunity,27 and mice with the BALB/c background acquire autoimmune-mediated dilated cardiomyopathy.28 The enhanced autoimmune phenomena in PD-1 KO mice appear to be primarily due to a higher frequency of tissue-invasive CD4+ and CD8+ T-cells polarized to the Th1 phenotype rather than increased autoantibody titers.26
The potential of the immune system for preventing cancer development has long been recognized.29 In part, immune surveillance and immune elimination of tumors is accomplished through the recognition of tumor associated antigens (TAA) by the adaptive immune system.1 Unfortunately, tumor cells frequently develop resistance to immune intervention and manage to escape from immune control. The responsible mechanisms may involve a loss or down-regulation in expression of TAA, alterations in the antigen-presenting machinery components in tumor cells or the development of resistance to cytotoxicity mediated by immune effector cells.30 More established tumors develop abilities to produce a variety of immunoinhibitory factors which alter the tumor microenvironment and induce suppression of anti-tumor functions in immune effector cells.31 The tumor microenvironment (TME) becomes enriched in Treg and myeloid-derived suppressor cells (MDSCs), which contribute to converting it into a highly immunosuppressive milieu. The PD-1/PD-L1 pathway participates in creating and maintaining tumor-associated immunosuppression.32 Tumors effectively convert this normally protective pathway, which is responsible for guarding against inflammation-induced tissue injury, to one that now protects the tumor from immune intervention. Tumors corrupt the PD-1/PD-L1 pathway by bombarding activated PD-1+ T cells with the ligand, thus inducing functional T-cell paralysis.33, 34
Several lines of evidence indicate that the PD-1/PD-L1 pathway is exploited by tumors. First, PD-L1 is found to be overexpressed on tumor cells in many different solid and hematological cancers. PD-L1 overexpression in tumors is driven in part by chronic exposure to the pro-inflammatory cytokine IFN-γ.7, 32 Expression of PD-L1 on tumor cells has been reported to be associated with poor prognosis in many tumor types, demonstrating that immune tolerance mediated by the PD-1/PD-L1 pathway has clinical significance.35-37 More recent studies indicate that levels of PD-L1 expression on tumor cells vary broadly, and it remains to be determined whether low expression or even absence of PD-L1 expression is associated with less effective therapy. Interestingly, clinical responses to anti-PD-1 Abs have been reported in patients whose tumors are negative for PD-L1.38 Second, numerous preclinical studies have reported the efficacy of the PD-1 signaling blockade in cancer. When anti-PD-L1 Abs were used to block this signaling pathway, increases in the frequency of tumor infiltrating CD8+ T cells and decreases in the frequency of Treg were seen that correlated with the arrest of tumor growth in mouse models of cancer.39, 40 Third, preclinical studies have also demonstrated that PD-1 blockade enhanced tumor responses to other forms of immunotherapy.41, 42 Finally, first-in-man clinical trials of Abs specific for PD-1 or PD-L1 performed in patients with different advanced malignancies confirmed clinical benefits of this immune therapy, serving as proof of principle. In a phase I study of the PD-L1 Ab, MDX-1105, in patients with different advanced malignancies, an objective response rate of 6 to 17% was seen, with stable disease in up to 41% of patients.43 In an additional dose-escalation trial of the anti-PD-1 Ab, MDX-1106, durable objective responses were seen in 18-28% of patients with non-small cell lung cancer, melanoma, and renal-cell cancer.34 These early studies have paved the way for many other clinical trials targeting the PD-1 pathway that are now being implemented in numerous institutions worldwide. Hematological malignancies, which have historically been sensitive to immunotherapy, promise to be yet another sensitive target for therapy with anti-PD-1 antibody.
4. PD-1 Pathway in Acute Myeloid Leukemia
4.1 Sensitivity of AML to immunotherapy
Beginning with allogeneic stem cell transplant, immune-based therapies have been often used for treatment of AML. Graft-versus-leukemia (GVL) effect of allogeneic hematopoietic stem cell transplantation (HSCT) is a well-established, successful form of immunotherapy. AML is the most common indication for HSCT in North America.44 Donor lymphocyte infusions (DLI) performed for relapsed AML, which rely solely on the GVL effect, lead to complete remission (CR) rates of 15-29%.45, 46 The CRs attained through DLI are frequently durable.47
Cytotoxic T lymphocytes (CTL) are well-established mediators of cancer-directed immunotherapy. Successful T-cell activation requires at least 3 signals. First, a specific antigenic peptide bound to a major histocompatibility complex on the APC must be recognized by an antigen-specific T-cell receptor (TCR) present on the surface of the cognate T cell. Subsequently, an antigen-independent co-stimulatory signal resulting from the interaction of CD28 on the T cell with CD80 or CD86 expressed on the antigen-presenting cell (APC) has to be generated to promote T-cell response. Next, cytokine-mediated stimulation of clonal T-cell expansion and functional maturation of T cells takes place. In the setting of HSCT for AML, the specific antigens recognized by TCRs to induce the GVL effect may include hematopoietic cell-restricted minor histocompatibility antigens on the recipient cells.48 However, TAA, i.e., immunogenic antigens expressed by leukemic blasts, play a critical role in successful T-cell based immunotherapy in the autologous setting.
Since the discovery of TAA in melanoma49, many AML blast-associated antigens have been identified.50 Some of these antigens can be considered leukemia-specific antigens (LSA), including fusion proteins DEK-CAN,51 and PML-RARα52 as well as antigens generated by gene mutations such as internal tandem duplications (ITD) of the FMS-like tyrosine kinase 3 (Flt3) gene and mutations in the nucleophosmin 1 (NPM1) gene.53, 54 Furthermore, many leukemia-associated antigens (LAA), i.e., antigens overexpressed by leukemia cells with limited expression by normal tissues, have also been characterized.50 Most LSA and LAA antigens are restricted in their expression to specific subgroups of AML, which limits their usefulness in antigen-targeted therapies, such as anti-leukemia vaccines. However, this is not a problem with checkpoint inhibition blockade, which is expected to be able to reverse immune tolerance to any endogenous antigen expressed by each patient's AML blasts.
4.2 Suppressive microenvironment in AML
Despite the sensitivity of AML to immune attack, the microenvironment in AML is immunosuppressive, facilitating immune tolerance of leukemia cells. In vitro studies have demonstrated that factors secreted by primary AML cells, particularly arginase II, can prevent T-cell activation and proliferation.55,56 HL-60 AML cells overexpress cyclooxygenase-2 (COX-2) and produce prostaglandin E2 (PGE2). They are also positive for indoleamine 2, 3-dioxygenase (IDO) upon induction with IFN-ɣ.57 Recently, Human Leukocyte Antigen-G (HLA-G), known to contribute to cancer cell immune escape, was found to be present on the surface of human AML blasts.58 Many other immunoinhibitiory soluble factors and cytokines produced by leukemic blasts or stromal cells in the leukemic bone marrow, including TGF-β1 and IL-10, may induce tolerance in hosts with AML.59
Treg are often increased in frequency in AML and contribute to creating an immunosuppressive microenvironment.60, 61 In mice, AML progression is associated with increased Treg infiltration at the site of disease. CTL adoptively transferred to leukemic mice have reduced proliferation and IFN-γ production at sites of Treg infiltration, with no effect on AML burden.62 However, Treg depletion with IL-2 diphtheria toxin (IL-2DT) prior to adoptive transfer of CTL significantly decreases the tumor burden and improves survival of mice with AML compared to control mice or those treated with either agent (IL-2DT or CTL) alone.62 Treg utilize a variety of mechanisms to mediate immunosuppression, including the production of inhibitory cytokines (IL-10, TGF-β) and suppressive factors such as adenosine or PGE2, competition for IL-2 or for co-stimulatory factors on APCs, which skews dendritic cell (DC) differentiation toward an immature and tolerogenic phenotype, and transfer of cyclic AMP to effector T-cells upon direct contact.31,61 Immature DC generated in the presence of Treg promote immunosuppression through expression of indoleamine 2,3-dioxygenase (IDO) which inhibits T cell proliferation by depleting tryptophan and promotes T-cell apoptosis by increasing levels of tryptophan metabolites.63 In mice, IDO was also shown to promote conversion of conventional T cells to Treg.64 Interestingly, AML cells also express IDO, as indicated above, and IDO activity is higher in patients with AML compared to normal controls.65, 66 In vitro, when IDO-expressing leukemic cells are co-incubated with T cells, the frequency of Treg and tryptophan catabolism increase, inhibiting naïve T-cell proliferation.65
Increases in the Treg frequency are observed in patients with AML at various stages of diagnosis and treatment. Compared to healthy controls, Treg percentages in the peripheral circulation are elevated at diagnosis in patients with AML,60, 67, 68 and higher frequencies of Treg at diagnosis are associated with poor prognosis.60, 67 In AML patients treated with cytotoxic chemotherapy or maintenance therapy, an increased frequency of strongly immunosuppressive Treg was observed, suggesting that therapy-resistant Treg contribute to leukemic relapse.69, 70
PD-1 is highly expressed on peripheral (inducible) Treg (pTreg),19 and in solid malignancies, its level of expression increases in pTreg accumulating in the tumor microenvironment.71 It has been reported that expression of PD-L1 in the tumor microenvironment controls the development of pTreg from human Th1 cells.19, 72 This participation of the PD-1/PD-L1 pathway in maintaining CD4+T-cell plasticity and in the generation of Treg demonstrates the interplay existing between immunosuppressive pathways. Amplification of the PD-1/PD-L1 pathway in the tumor microenvironment translates into expansion of Treg and suggests that blockade of this pathway will also relieve Treg-mediated immunosuppression.
4.3 The PD-1/PD-L1 pathway and immune suppression in AML
In AML, the PD-1/PD-L1 pathway is hijacked by malignant cells to facilitate immune escape. Many preclinical studies have demonstrated up-regulation of the PD-1/PD-L1 pathway in AML and the negative impact of this amplification on disease control. In mice injected with an AML cell line (C1498), the percentage of CD8+ T-cells expressing PD-1 dramatically increased in the liver, a major site of C1498 dissemination.73 Similarly, when C1498 cells were injected into mice and allowed to grow in vivo, PD-L1 expression on T cells increased compared to baseline.74
Functional consequences of the PD-1/PD-L1 pathway up-regulation in AML have been clearly demonstrated in PD-1 KO mice. When these mice are injected with C1498 AML cells, AML progression is slower than in wild-type (WT) mice, and the mice have significantly longer survival.73, 74 This appears to be due to augmented antigen-specific CD8+T-cell responses, as both the number of tumor specific CD8+ T-cells and their effector function were increased in PD-1 KO compared to WT mice.74 In addition to genetic PD-1 ablation, improved leukemic control has also been demonstrated with pharmacologic PD-1 inhibition. When a PD-1 blocking antibody was administered to WT mice with C1498 AML, the mice had lower AML burden, more CD8+ T cells infiltrating the liver and experienced longer survival than control mice.74
Expression of PD-1 and its ligands is also increased in hematopoietic cells of patients with AML. One study of 124 patients with myeloid malignancies, including 69 with myelodysplastic syndrome (MDS) and 9 with AML, sampled at various stages of treatment found that the PD-L1 mRNA expression level was upregulated by ≥2 fold in 36% and 25% of CD34+ cells in MDS and AML, respectively, compared to CD34+ normal control cells.75 PD-L2 was also upregulated in a smaller proportion of CD34+ cells, i.e., 12% in MDS and 33% in AML.75 In a smaller subset of patients, mRNA expression correlated perfectly with PD-L1 expression on CD34+ cells by immunohistochemistry. Expression levels of PD-L1, PD-L2, and PD-1 were also increased in peripheral blood mononuclear cells (PBMCs). In fact, expression levels of PD-L2 and PD-1 were higher in PBMCs than in CD34+ cells.75 Another cohort of 154 patients with AML demonstrated no significant increase in surface PD-L1 expression on leukemia cells at initial diagnosis compared to healthy controls. However, stimulation with IFN-γ significantly increased PD-L1 expression on AML blasts but not in normal controls.76 Interestingly, PD-L1 expression on myeloid precursor cells increased more dramatically with IFN-γ stimulation in samples of patients in complete remission or at relapse than in myeloid precursor cells of newly-diagnosed AML patients.76 These findings demonstrate that PD-L1 expression on myeloid precursor cells and leukemic blasts occurs in a substantial portion of patients with AML. At this time, it is not clear whether the frequency of positive cells or levels of expression can be related to disease progression or relapse. It would be important to establish how expression levels of PD-L1 on AML blasts and PD-1 on activated T cells or vice versa are regulated. Also, a better understanding of the timing required for up-regulation of expression levels requires studies in larger patient cohorts, where PD-1 and its ligands can be measured longitudinally. Nevertheless, the existing data suggest that activity of the PD-1/PD-L1 pathway, similar to Treg-mediated suppression,70 may be particularly increased upon recovery from cytotoxic chemotherapy.76 Consistent with these observations, PD-L1 expression in CD34+ cells prior to treatment did not appear to have prognostic significance in small cohorts of patients with AML. In 72 patients with MDS or AML tested prior to any treatment, PD-L1 expression in CD34+ cells was not associated with worse survival.75 However, in a smaller cohort of those 72 patients enrolled in a clinical trial and treated with hypomethylating agents and vorinostat, upregulation (≥ 2 fold) of PD-L1 or PD-L2 in peripheral blood mononuclear cells during therapy was associated with a significantly worse median survival: 6.6 months compared to 11.7 months in patients without demonstrable upregulation of PD-1 ligands. Similarly, Norde and colleagues found that in patients who relapsed late after allogeneic transplant, despite the presence of circulating alloreactive T-cells to hematopoietic cell-restricted minor histocompatibility antigens, PD-L1 was highly expressed on the leukemic cells at baseline or upon stimulation with IFN-γ.77 Furthermore, stimulation of allogeneic CD3+ T-cells with the PD-L1-expressing AML cells led to significantly enhanced T-cell proliferation and cytokine production when performed in presence of anti-PD-1 antibody compared to isotype controls.77 In aggregate, these findings suggest that the development of functionally- impaired T-cells during therapy through up-regulation of the PD-1 checkpoint leads to impaired control of leukemia and that PD-1 blockade restores anti-leukemia T-cell functions and thus is likely to offer therapeutic advantages.
4.5 Therapeutic potential of PD-1/PD-L1 Blockade in AML
In considering molecular mechanisms responsible for clinical success of the PD-1/PD-L1 pathway blockade with Abs, it is important to remember the broad cellular and tissue distribution of the receptor and its ligands as well as distinct effects these Abs are likely to exert upon interaction with normal vs malignant cells. Regardless of whether the blocking antibody targets the receptor or the ligand, the same two objectives are desired: (a) to eliminate or decrease immune suppression orchestrated by the tumor and (b) to simultaneously unleash the anti-tumor power of immune cells, converting them to fully competent anti-tumor effectors. In achieving either objective, the presence and expression levels of PD-1 and/or PD-L1 on target cells will determine antibody binding and interaction with its target. The PD-1 Ab will target various types of PD-1+ immune cells, especially activated immune cells, as well as PD-1 expressing leukemic cells. All these PD-1+ cells will be sensitive to elimination by antibody-dependent cell-mediated cytotoxicity (ADCC) mediated by FcγR+ immune cells, e.g., NK cells, monocytes/macrophages, provided that the antibody is of an IgG1 or IgG3 isotype. This includes elimination of regulatory cells (Treg, MDSC) as well as conventional immune effector cells overexpressing PD-1 (activated T cells, NK cells, B cells, macrophages) at levels sufficient to be recognized by the antibody. Thus, the PD-1 Abs have a profound dual impact on the entire immunoregulatory system, on the one hand blocking negative PD-L1 signaling, and on the other hand targeting activated PD-1+ immune cells for immune destruction (Figure 2).
Figure 2.
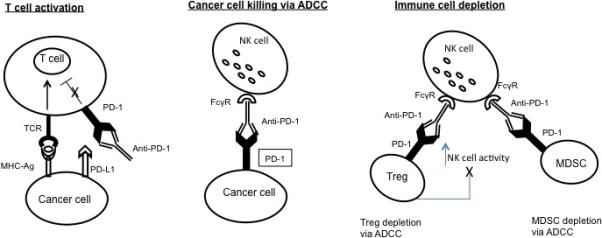
A diagram demonstrating blocking by anti-PD-1 Ab of PD-1 receptor-PD-L1 interactions. In the presence of a PD-L1+ tumor, checkpoint blockade with anti-PD-1 Ab not only unleashes immune cell effector functions from inhibition but provides a mechanism for tumor cell destruction by ADCC. In the presence of anti-PD-1 Ab, which targets all PD-1+ cells (i.e., immune cells, including Treg and MDSC, tumor cells), activated FcgR+ NK cells and monocytes effectively mediate ADCC. Tumorassociated antigens released by dying tumor cells are presented to activated, unleashed T effector cells and, in the absence of Treg, which are partially or completely depleted by ADCC, and which are no longer expanded or stabilized by PD-L1 signaling, swift anti-tumor immune responses are generated. The illustrated ADCC mechanisms will be effective only if mediated by the Ab with IgG1 or IgG3 isotypes.
ADCC: Antibody-dependent cytotoxicity; MDSC: Myeloid-derived suppressor cell; NK: Natural killer; PD-1: Programmed death-1; PDL-1: Programmed death ligand-1; TCR: T-cell receptor.
The end result of targeting PD-1 is likely to depend on the strength and persistence of environmental signals specifying the receptor expression and its functions. It is also possible that differences in levels of PD-1 expression between various immune cell subsets determine the cell sensitivity or resistance to checkpoint blockade. For example, Treg or MDSC, which overexpress PD-1 in the leukemic milieu could be especially sensitive to immune blockade as well as immune elimination, thus being very effectively removed or prevented from exerting immune suppression. Also, in the tumor microenvironment, overexpression of PDL-1 on tumor cells is used as a mechanism of tumor escape. Tumor infiltrating lymphocytes accumulating in the tumor microenvironment and overexpressing PD-1 lymphocytes are inhibited from mediating anti-tumor responses. PD-1 Ab blockade prevents tumor escape. In aggregate, the mechanisms through which a checkpoint antibody blockade exercises its immunorestorative effects appear to be complex and understanding of the molecular and cellular interactions involved in this process will require further examination.
The possibility that checkpoint blockade of tumor-induced immune suppression could make the delivery of other immunotherapies more effective has been also considered. In AML, combinations of PD-1 checkpoint inhibition with other immune-mediated therapies are under investigation. For example, the CD33/CD3-bispecific BITE antibody, AMG 330, is designed to redirect and activate T-cells to AML blasts, and it shows activity against AML cell lines and primary AML cells.78 In primary AML samples cultured ex-vivo, PD-1 was upregulated on activated T-cells upon addition of AMG 330, and PD-L1 expression was increased in 16/19 of the primary AML cell cultures.79 Antibody-dependent cytotoxicity (ADCC) of AML cells mediated by T cells in the presence of AMG 330 was significantly enhanced by PD-1/PD-L1 Ab blockade (75% vs 44% without PD-1 blockade).79 This study emphasizes the therapeutic potential of combining anti-leukemia Ab therapy with checkpoint blockade. It can be safely predicted that up-regulation of the PD-1/PD-L1 pathway in leukemia would impair responses to vaccines and that PD-1/PD-L1 blockade may enhance effective immune response to vaccination. Specifically, several approaches to dendritic cell-based vaccination of patients with hematologic malignancies have been recently evaluated in preclinical studies and early clinical trials 80 with promising results. It is likely that a combination of anti-leukemia vaccines with checkpoint inhibitors will improve and sustain immune responses generated by such vaccines. A clinical trial investigating a dendritic cell vaccine fused to autologous AML cells combined with PD-1 Ab blockade is currently underway in patients in complete remission. (NCT01096602)
5. Additional immune checkpoints in leukemia: LAG-3 and TIM-3
While PD-1 and CTLA-4 are the best understood checkpoint receptors with the most clinically advanced inhibitors, many additional immune checkpoints exist with functions that are non-redundant to PD-1 or CTLA-4. The notion that engagement of more than one checkpoint inhibitor or a series of checkpoint inhibitors might induce superior therapeutic responses has been introduced, based on the evidence for co-expression and synergy of inhibitory receptors on activated T cells81. This has already led to the introduction of combination therapies with, e.g., ipilimumab and nivolumab in patients with advanced melanoma82. In AML, co-expression of several inhibitory receptors on T cells has been evaluated in mouse models 41.
The lymphocyte activation gene-3 (LAG-3) is a homolog of CD483 that is expressed on subsets of T cells, NK cells, and B cells.84 Dual blockade of LAG-3 with PD-1 was first demonstrated to enhance anti-tumor responses in the setting of ovarian cancer.85 Since then, LAG-3 inhibition has been found to improve the effector function of adoptive immunotherapy in a murine model of leukemia.86 A particular challenge of cancer immunotherapy is posed by a need to break tolerance to TAA that represent self or modified self. This challenge is recapitulated in the Abl:Gag mice that express the Gag protein on normal hepatocytes; the Gag protein is also a tumor-associated antigen expressed by Friend virus-induced erythroleukemia (FBL). In Abl:Gag mice with murine FBL leukemia, dual blockade of PD-1 and CTLA-4 extended the life span of Gag-specific CD8+ CTLs.86 While additional blockade of LAG-3 did little to increase the persistence of the tumor-specific CD8+ T cells, it dramatically enhanced lytic activity of adoptively transferred CTLs and improved survival of mice compared to dual blockade of PD-1 and CTLA alone. Interestingly, triple-blockade of PD-1, CTLA, and LAG-3 in mice with FBL alone, even without adoptive transfer of Gag-specific CTLs, markedly increased survival compared to isotype controls.86 In sum, these results reveal an important role for LAG-3 in modulating T cell effector function against leukemia and suggest that blockade of multiple checkpoint pathways may offer enhanced therapeutic potential.
Dysfunctional T-cells present at tumor sites in human cancers are frequently characterized by sustained overexpression of PD-1. T-cell immunoglobulin and mucin domain-contained protein 3 (TIM-3), another cell-surface molecule present on dysfunctional tumor-associated T cells, is commonly co-expressed with PD-1.87, 88 In mice with solid tumors, TIM-3 and PD-1 co-expression was seen on the majority of tumor infiltrating lymphocytes, and these double-positive T cells were more dysfunctional than T cells expressing PD-1 alone.89 Co-expression of TIM-3 and PD-1 on T cells also characterizes dysfunctional T-cell phenotype in mice with AML.90 While treatment with a PD-L1 blocking Ab has led to a short period of improved leukemia control, TIM-3 inhibition did not reduce AML tumor burden. However, dual blockade of TIM-3 and PD-L1 significantly reduced tumor burden and prolonged survival of mice with advanced AML.90 Improved leukemia control seen with blockade of multiple immune checkpoints in preclinical models of leukemia implies cooperation and clearly offers greater potential for achieving remission. However, toxicities potentially associated with such dual or triple-checkpoint inhibition and its effectiveness in humans with leukemia remain to be determined.
Recent reports of cumulative expression of as many as five different inhibitory receptors (CTLA-4, PD-1, TIM-3, LAG-3 and BTLA) on CD8+ T cells infiltrating human solid tumors (A. Zippelius, personal communication) suggest that plans for further improvements in therapy with checkpoint inhibitors might require the use of carefully selected combinations of inhibitors. Sustained expression levels and cumulative co-expression of inhibitory receptors on effector T cells might vary in different cancers, might be time dependent and may or may not correlate with progressive T-cell dysfunction and disease stage. Should coordinate overexpression of inhibitory receptors prove to be a correlate of T-cell dysfunction and disease progression, their role as biomarkers of response to therapy could be considered. Thus, overexpression of multiple inhibitory receptors on T cells could be taken as an indication that immunosuppressed patients with advanced malignancies will be unlikely to respond to monotherapy and will require therapy with combinations of checkpoint inhibitors. Further efforts are now in progress to evaluate therapeutic efficacy and prognostic importance of multiple inhibitory receptor blockade. There is a good reason to expect, and some preclinical evidence to support the expectation, that the same considerations apply to hematological malignancies, and that checkpoint blockade of multiple inhibitory receptors may augment the anti-leukemic responses.
6. Potential Efficacy of PD-1 blockade in other hematologic malignancies
In AML, a considerable volume of pre-clinical data, including studies of PD-1 blockade in leukemic mice, sensitivity of AML to immunotherapy despite of the existing immunosuppressive microenvironment and expression of PD-1 and/or PD-L1 on leukemic blasts, suggest that clinical PD-1 blockade is a promising therapeutic option. Given the acceptable tolerability, strong pre-clinical rationale, and immunological activity of PD-1/PD-L1 blockade, several clinical trials of anti-PD1 mAbs have been either completed or are underway in patients with a variety of hematological diseases (see Table 1 for selected studies). These studies explore PD-1 blockade as a single agent at various time points of disease progression and in combination with other immunomodulatory agents. As these studies mature, much anticipated data on the most effective antibody dose, a full toxicity profile, effects on the immune system and the potential as biomarkers for response to therapy in hematological malignancies should become available and facilitate a more rapid progress in translating this therapeutic strategy to AML.
Table 1.
Ongoing Clinical Trials of PD-1 Checkpoint Blockade in Hematologic Malignancies.
| Hematologic Disease | Phase | Disease Stage | PD-1/PD-L1 Inhibitor | Combination Therapy | Reference |
|---|---|---|---|---|---|
| Blood Cancer | Phase 1B | Relapsed/Refractory | Pembrolizumab | Single-agent | NCT01953692 |
| Multiple Myeloma | Phase 1 | Relapsed/Refractory | Pembrolizumab | Lenalidomide and dexamethasone | NCT02036502 |
| CLL and Indolent NHL | Phase 2 | Relapsed/Refractory | Pembrolizumab | Single-agent | NCT02332980 |
| Multiple Myeloma | Phase 2 | After HDT/ASCT | Pembrolizumab | Revlimid | NCT02331368 |
| Mycosis Fungoides or Sezary Syndrome | Phase 2 | Relapsed/Refractory | Pembrolizumab | Single-agent | NCT02243579 |
| Multiple Myeloma | Phase 1/2 | Relapsed/Refractory | Pembrolizumab | Pomalidomide and dexamethasone | NCT02289222 |
| HD, NHL, or Multiple Myeloma | Phase 1 | Relapsed/Refractory | Nivolumab | Ipilimumab or Lirilumab | NCT01592370 |
| NHL | Phase 1/2 | Relapsed/Refractory | Nivolumab | Urelumab | NCT02253992 |
| AML | Randomized Phase 2 | CR after chemotherapy | Nivolumab | Single-agent | NCT02275533 |
| CML | Phase 1B | Progressing after ≥2 TKIs | Nivolumab | Dasatinib | NCT02011945 |
| CLL, FL, DLBCL | Phase 1/2A | Relapsed/Refractory | Nivolumab | Ibrutinib | NCT02329847 |
| HD | Phase 2 | Relapsed/Refractory | Nivolumab | Single-agent | NCT02181738 |
| FL | Phase 2 | Relapsed/Refractory | Nivolumab | Single-agent | NCT02038946 |
| DLBCL | Phase 2 | Relapsed/Refractory | Nivolumab | Single-agent | NCT02038933 |
| Multiple Myeloma | Phase 1/2 | Relapsed/Refractory | Pidilizumab | Lenalidomide | NCT02077959 |
| DLBCL, FL | Phase 1B | Relapsed/Refractory | MPDL-3280A | Obinuzutumab | NCT02220842 |
| DLBCL, MCL | Phase 1B/2 | Relapsed/Refractory | MEDI-0680 | MEDI-551 (Anti-CD19 Antibody) | NCT02271945 |
Abbreviations: CLL, Chronic lymphomic leukemia; NHL, Non-Hodgkin's Lymphoma, AML, Acute myeloid Leukemia; CR, Complete remission; CML, Chronic myelogenous leukemia; HD, Hodgkin's lymphoma; TKI, Tyrosine kinase inhibitor; FL, Follicular lymphoma; DLBCL, Diffuse large B-cell lymphoma; MCL, Mantle cell lymphoma
7. Conclusion
In cancer immunotherapy, finding a reliable and effective means for unleashing the immune system from tumor-induced suppression has been a difficult and elusive objective. Many strategies to mobilize dysfunctional tumor-associated T cells to fight cancer have been attempted, including activation of cellular pathways with various pharmacologic or biologic agents, TAA-specific vaccinations, adoptive immune cell transfers or elimination of regulatory cells responsible for immune suppression.91 Therefore, the realization that antibody blockade of immune checkpoints leads to at least a partial restoration of immune competence accompanied by anti-tumor effects created great expectations. Rapidly progressing in vivo studies in animal tumor models of cancer as well as in vitro studies with human immune cells showed that checkpoint blockade was effective in restoring anti-tumor immunity and arresting tumor growth. More recently, results of monotherapy clinical trials with ipilimumab or nivolumab confirmed durable clinical benefits of therapy for some patients with advanced solid cancers. At the same time, research has uncovered a complex interplay of immune regulatory molecules (co-inhibitory and co-stimulatory) that govern T-cell activation via multiple pairs of receptors/ligands broadly co-expressed on immune and tissue cells. Overexpression of inhibitory checkpoint receptors, such as CTLA-4 or PD-1, and their synergistic signaling in immune cells responding to cancer provided a rationale for antibody therapy targeting these receptors. Simultaneously, overexpression of the ligands on tumor cells explained dysfunction of the receptor-positive T cells in the TME. Blockade of either PD-1 or PDL1 is expected to effectively remove inhibitory signals hampering T cells and restore their anti-tumor activity.
However, the ever-present biological complexity may require additional mechanistic insights into the PD-1/PD-L1 pathway as it operates in AML before it can be therapeutically harnessed. In fact, checkpoint blockade in AML serves to illustrate multifactorial possibilities of this form of immunotherapy. In AML, sustained overexpression of PD-1, PD-L1, and PD-L2 on leukemic blasts and also on various activated immune cells in the bone marrow and in the periphery facilitates Ab checkpoint blockade. Such broadly-induced overexpression of the PD-1/PD-L1 pathway components together with co-expression of LAG-3, TIM-3 and potentially other inhibitory receptors presents an opportunity for more effective targeting and simultaneous engagement of several distinct mechanisms of disease control. These may include ADCC mediated by NK cells and/or monocytes, direct killing of blasts by re-activated LAA-specific CTL, simultaneous activation of Th1 effector T cells and dendritic cell for more effective antigen presentation and silencing of suppression mediated by Treg and/or MDSC. Synergy between all these mechanisms contributes a strong anti-leukemia environment. Also, chemotherapy-induced up-regulation of PD-1 on immune cells favors the implementation of checkpoint blockade following conventional leukemia therapies. While it is not yet clear that PD-1 is a dominant inhibitory receptor in AML, current research examining phenotypic and functional involvement of different inhibitory receptors is expected to provide the roadmap for a rational design of combinatorial check point blockade in patients with AML.
8. Expert opinion
Immunotherapy of AML emerges as a novel and potentially effective treatment option due to the rapid evolution of checkpoint blockade strategies. Based on pre-clinical data, it is expected but not yet proven that PD-1/PD-L1 blockade will eliminate immune suppression in AML. It is expected but not yet proven that PD-1/PD-L1 blockade in AML will show efficacy in inhibiting negative signaling in immune cells and, at the same time, induce death of leukemic blasts, providing immunogenic TAA that could serve to generate long-term anti-leukemia immunity. In addition, the ability to monitor PD-1 and PD-L1 expression levels or their co-expression with other checkpoint molecules on immune cell subsets in the tumor microenvironment and the circulation during therapy will likely provide a series of biomarkers predictive of response to checkpoint inhibition. Yet another potential benefit might emerge from future combinations of checkpoint inhibition with other form of anti-leukemia immunotherapy, all based on the proof of principle that release from tumor suppression allows for effective responses to immune interventions. These objectives of checkpoint blockade in AML appear to be achievable in the near future. Clinical trials with nivolumab or pembrolizumab in patients with various hematological diseases are currently ongoing, providing preliminary evidence of tolerable toxicity and promising efficacy. This is a good basis for up-coming clinical trials with checkpoint inhibitors in AML patients. There is much optimism associated with this therapy, because of emerging conviction that a relief from immune suppression is necessary for restoration of anti-leukemia functions in immune cells, for up-regulating anti-leukemia responses and ultimately for achieving complete remission. The future of therapy with checkpoint inhibitors for AML and other hematological malignancies is likely to depend on a skillful combination of two or more antibodies to inhibitory receptors or ligands. In addition, combinations of checkpoint inhibition with conventional therapies and pharmacologic inhibitors designed to optimize or increase immune stimulatory as well as anti-leukemia effects will be evaluated for improved efficacy with reduced toxicity. The future selection of combinatorial therapies for AML will be based upon further investigations of the inhibitory receptors co-expression, cooperation and predictive significance in longitudinally monitored AML patients treated with checkpoint inhibitors. Given the anticipated increased response rates to therapy with checkpoint inhibitors in patients with AML, it will be possible to establish reliable correlations between immune and clinical responses, facilitating the development of biomarkers of response and outcome. Such biomarkers will allow for the selection of patients most likely to respond to checkpoint immunotherapy. Overall, the availability and further development of this new immune therapy has the potential of changing the future clinical practice and improving treatment options available for patients with AML.
Article highlights.
PD-1, LAG-3, TIM-3 and possibly other inhibitory receptors are broadly co-expressed on various immune cells in the leukemic milieu
PD-L1 and potentially other ligands of inhibitory receptors are overexpressed in leukemic blasts
Mechanism responsible for recovery of anti-leukemia immune competence following checkpoint blockade involve ADCC, restored CTL and Th1 responses and reduced suppression by Treg or MDSC
Sustained co-expression of multiple inhibitory receptors on T cells in progressive disease offers an opportunity for simultaneous targeting of these receptors with antibody combinations
Chemotherapy up-regulates expression of checkpoint inhibitory receptors on T cells suggesting potential usefulness of checkpoint inhibitors as a follow-up therapy in leukemia
Overexpression of inhibitory receptors on T cells in advanced leukemia stages might serve as a biomarker for poor response to checkpoint inhibition and for outcome
Acknowledgments
TL Whiteside received National Institute of Health grant ROICA168628.
Footnotes
Financial and competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Contributor Information
Alison Sehgal, Division of Hematology/Oncology, University of Pittsburgh School of Medicine, University of Pittsburgh Cancer Institute, 5150 Centre Avenue, Pittsburgh, PA 15232.
Theresa L. Whiteside, University of Pittsburgh School of Medicine, University of Pittsburgh Cancer Institute, 5117 Centre Avenue, Pittsburgh, PA 15213.
Michael Boyiadzis, Division of Hematology-Oncology, University of Pittsburgh School of Medicine, University of Pittsburgh Cancer Institute, 5150 Centre Avenue, Pittsburgh, PA 15232.
References
- 1.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011 Mar 25;331(6024):1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 2.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012 Apr;24(2):207–12. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. doi: 10.1038/nrc3239. 04//print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 3(5):541–47. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 5.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative Disorders with Early Lethality in Mice Deficient in Ctla-4. Science. 1995 Nov 10;270(5238):985–88. doi: 10.1126/science.270.5238.985. 1995. [DOI] [PubMed] [Google Scholar]
- 6**.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Molecular and Cellular Biology. 2005 Nov 1;252(21):9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005. 2005. [This study describes distinct molecular mechanisms of T- cell activation medated by CTLA-4 and PD-1 receptors.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumorassociated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002 Aug;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 8*.Leach DR, Krummel MF, Allison JP. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science. 1996 Mar 22;271(5256):1734–36. doi: 10.1126/science.271.5256.1734. 1996. [This study provides first in vivo evidence for the role of CTLA-4 blockade in enhancing anti-tumor immunity in tumor-bearing mice.] [DOI] [PubMed] [Google Scholar]
- 9.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PDL1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002 Sep 17;99(19):12293–7. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proceedings of the National Academy of Sciences. 2003 2003 Apr 15;100(8):4712–17. doi: 10.1073/pnas.0830997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y-F, Zou J-P, Mu J, Wijesuriya R, Ono S, Walunas T, et al. Enhanced Induction of Antitumor T-Cell Responses by Cytotoxic T Lymphocyte-associated Molecule-4 Blockade: The Effect Is Manifested Only at the Restricted Tumor-bearing Stages. Cancer Research. 1997 1997 Sep 15;57(18):4036–41. [PubMed] [Google Scholar]
- 12*.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010 Aug 19;363(8):711–23. doi: 10.1056/NEJMoa1003466. [This is one of the first reports of the remarkable clinical effects of therapy with ipilimumab in patients with metaststic melanoma.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. New England Journal of Medicine. 2013;369(2):134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14*.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. The Journal of experimental medicine. 2000 Oct 2;192(7):1027–34. doi: 10.1084/jem.192.7.1027. [This study reports that the PD-L1 is a ligand of PD-1 on T cells, and their interaction inhibits lymphocyte activation.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15*.Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001 Apr 2;193(7):839–46. doi: 10.1084/jem.193.7.839. [This study identifies PD-L2 as a second ligand for PD-1 involved in blocking of T cell activation.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16*.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001 Mar;2(3):261–8. doi: 10.1038/85330. [This study identifies PD-L2 as a second ligand for PD-1 involved in blocking of T cell activation.] [DOI] [PubMed] [Google Scholar]
- 17.Keir ME, Francisco LM, Sharpe AH. PD-1 and its ligands in T-cell immunity. Curr Opin Immunol. 2007 Jun;19(3):309–14. doi: 10.1016/j.coi.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Wilke CM, Wei S, Wang L, Kryczek I, Kao J, Zou W. Dual biological effects of the cytokines interleukin-10 and interferon-gamma. Cancer Immunol Immunother. 2011 Nov;60(11):1529–41. doi: 10.1007/s00262-011-1104-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19**.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009 Dec 21;206(13):3015–29. doi: 10.1084/jem.20090847. [This study reports that in murine Treg, binding of PD-L1 to PD-1 up-regulates suppressor functions of these cells and prolongs their survival.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pantaleo G, Koup RA. Correlates of immune protection in HIV-1 infection: what we know, what we don't know, what we should know. Nat Med. 2004 Aug;10(8):806–10. doi: 10.1038/nm0804-806. [DOI] [PubMed] [Google Scholar]
- 21.Letvin NL, Walker BD. Immunopathogenesis and immunotherapy in AIDS virus infections. Nat Med. 2003 Jul;9(7):861–6. doi: 10.1038/nm0703-861. [DOI] [PubMed] [Google Scholar]
- 22.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005 Mar;5(3):215–29. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 23.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006 Feb 9;439(7077):682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 24.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006 Sep 21;443(7109):350–4. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 25.Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007 Mar;81(6):2545–53. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A. 2005 Aug 16;102(33):11823–8. doi: 10.1073/pnas.0505497102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motifcarrying immunoreceptor. Immunity. 1999 Aug;11(2):141–51. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 28.Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. 2003 Dec;9(12):1477–83. doi: 10.1038/nm955. [DOI] [PubMed] [Google Scholar]
- 29.Old LJ, Boyse EA. Immunology of Experimental Tumors. Annu Rev Med. 1964;15:167–86. doi: 10.1146/annurev.me.15.020164.001123. [DOI] [PubMed] [Google Scholar]
- 30.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annual review of immunology. 2011;29:235–71. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 31*.Whiteside TL. Induced regulatory T cells in inhibitory microenvironments created by cancer. Expert opinion on biological therapy. 2014 Oct;14(10):1411–25. doi: 10.1517/14712598.2014.927432. [This review emphasizes the presence in the tumor microenvironment of several distinct molecular pathways that inhibit anti-tumor immunity and favor tumor escape depending on the local context.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy--inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 2012 Dec 15;18(24):6580–7. doi: 10.1158/1078-0432.CCR-12-1362. [DOI] [PubMed] [Google Scholar]
- 33.Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci U S A. 2004 Dec 7;101(49):17174–9. doi: 10.1073/pnas.0406351101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012 Jun 28;366(26):2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009 Feb 1;15(3):971–9. doi: 10.1158/1078-0432.CCR-08-1608. [DOI] [PubMed] [Google Scholar]
- 36*.Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 2010 Apr 1;116(7):1757–66. doi: 10.1002/cncr.24899. [This study reports that the expression level of PD-L1 on the tumor serves as a biomarker of prognosis in malignant melanoma.] [DOI] [PubMed] [Google Scholar]
- 37.Mu CY, Huang JA, Chen Y, Chen C, Zhang XG. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol. 2011 Sep;28(3):682–8. doi: 10.1007/s12032-010-9515-2. [DOI] [PubMed] [Google Scholar]
- 38.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014 Oct 1;20(19):5064–74. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007 Apr 1;13(7):2151–7. doi: 10.1158/1078-0432.CCR-06-2746. [DOI] [PubMed] [Google Scholar]
- 40.Okudaira K, Hokari R, Tsuzuki Y, Okada Y, Komoto S, Watanabe C, et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int J Oncol. 2009 Oct;35(4):741–9. doi: 10.3892/ijo_00000387. [DOI] [PubMed] [Google Scholar]
- 41**.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proceedings of the National Academy of Sciences of the United States of America. 2010 Mar 2;107(9):4275–80. doi: 10.1073/pnas.0915174107. [This study reports on effects of combinatorial blockade of PD-1 and CTLA4 on Tcell migration to the tumor and reduction in suppressor cell subsets in mouse model of B16 melanoma.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Q, Xiao H, Liu Y, Peng Y, Hong Y, Yagita H, et al. Blockade of programmed death-1 pathway rescues the effector function of tumor-infiltrating T cells and enhances the antitumor efficacy of lentivector immunization. J Immunol. 2010 Nov 1;185(9):5082–92. doi: 10.4049/jimmunol.1001821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012 Jun 28;366(26):2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasquini MCZX. [March 1, 2015];Current use and outcome of hematopoietic stem cell transplantation: CIBMTR Summary Slides 2014. 2014 Available at: http://www.cibmtr.org.
- 45.Collins RH, Jr., Shpilberg O, Drobyski WR, Porter DL, Giralt S, Champlin R, et al. Donor leukocyte infusions in 140 patients with relapsed malignancy after allogeneic bone marrow transplantation. J Clin Oncol. 1997 Feb;15(2):433–44. doi: 10.1200/JCO.1997.15.2.433. [DOI] [PubMed] [Google Scholar]
- 46.Kolb HJ, Schattenberg A, Goldman JM, Hertenstein B, Jacobsen N, Arcese W, et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood. 1995 Sep 1;86(5):2041–50. [PubMed] [Google Scholar]
- 47.Schmid C, Labopin M, Nagler A, Bornhauser M, Finke J, Fassas A, et al. Donor lymphocyte infusion in the treatment of first hematological relapse after allogeneic stem-cell transplantation in adults with acute myeloid leukemia: a retrospective risk factors analysis and comparison with other strategies by the EBMT Acute Leukemia Working Party. J Clin Oncol. 2007 Nov 1;25(31):4938–45. doi: 10.1200/JCO.2007.11.6053. [DOI] [PubMed] [Google Scholar]
- 48.Goulmy E. Minor histocompatibility antigens: allo target molecules for tumor-specific immunotherapy. Cancer J. 2004 Jan-Feb;10(1):1–7. doi: 10.1097/00130404-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 49.Bodey B. Cancer-testis antigens: promising targets for antigen directed antineoplastic immunotherapy. Expert Opin Biol Ther. 2002 Aug;2(6):577–84. doi: 10.1517/14712598.2.6.577. [DOI] [PubMed] [Google Scholar]
- 50.Anguille S, Van Tendeloo VF, Berneman ZN. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia. 2012 Oct;26(10):2186–96. doi: 10.1038/leu.2012.145. [DOI] [PubMed] [Google Scholar]
- 51.Makita M, Azuma T, Hamaguchi H, Niiya H, Kojima K, Fujita S, et al. Leukemiaassociated fusion proteins, dek-can and bcr-abl, represent immunogenic HLA-DR-restricted epitopes recognized by fusion peptide-specific CD4+ T lymphocytes. Leukemia. 2002 Dec;16(12):2400–7. doi: 10.1038/sj.leu.2402742. [DOI] [PubMed] [Google Scholar]
- 52.Osman Y, Takahashi M, Zheng Z, Toba K, Liu A, Furukawa T, et al. Dendritic cells stimulate the expansion of PML-RAR alpha specific cytotoxic T-lymphocytes: its applicability for antileukemia immunotherapy. J Exp Clin Cancer Res. 1999 Dec;18(4):485–92. [PubMed] [Google Scholar]
- 53.Graf C, Heidel F, Tenzer S, Radsak MP, Solem FK, Britten CM, et al. A neoepitope generated by an FLT3 internal tandem duplication (FLT3-ITD) is recognized by leukemia-reactive autologous CD8+ T cells. Blood. 2007 Apr 1;109(7):2985–8. doi: 10.1182/blood-2006-07-032839. [DOI] [PubMed] [Google Scholar]
- 54.Greiner J, Ono Y, Hofmann S, Schmitt A, Mehring E, Gotz M, et al. Mutated regions of nucleophosmin 1 elicit both CD4(+) and CD8(+) T-cell responses in patients with acute myeloid leukemia. Blood. 2012 Aug 9;120(6):1282–9. doi: 10.1182/blood-2011-11-394395. [DOI] [PubMed] [Google Scholar]
- 55.Buggins AG, Milojkovic D, Arno MJ, Lea NC, Mufti GJ, Thomas NS, et al. Microenvironment produced by acute myeloid leukemia cells prevents T cell activation and proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways. J Immunol. 2001 Nov 15;167(10):6021–30. doi: 10.4049/jimmunol.167.10.6021. [DOI] [PubMed] [Google Scholar]
- 56*.Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. 2013 Aug 1;122(5):749–58. doi: 10.1182/blood-2013-01-480129. [This paper describes arginase production in AML which leads to suppression of anti-leukemia ativity.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iachininoto MG, Nuzzolo ER, Bonanno G, Mariotti A, Procoli A, Locatelli F, et al. Cyclooxygenase-2 (COX-2) inhibition constrains indoleamine 2,3-dioxygenase 1 (IDO1) activity in acute myeloid leukaemia cells. Molecules. 2013;18(9):10132–45. doi: 10.3390/molecules180910132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Locafaro G, Amodio G, Tomasoni D, Tresoldi C, Ciceri F, Gregori S. HLA-G expression on blasts and tolerogenic cells in patients affected by acute myeloid leukemia. J Immunol Res. 2014;2014:636292. doi: 10.1155/2014/636292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen X, Kline DE, Kline J. Peripheral T-cell tolerance in hosts with acute myeloid leukemia. Oncoimmunology. 2013 Aug 1;2(8):e25445. doi: 10.4161/onci.25445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.Szczepanski MJ, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009 May 15;15(10):3325–32. doi: 10.1158/1078-0432.CCR-08-3010. [This paper is the first to report on increased frequency and suppressor activity of Treg in patients with AML.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ustun C, Miller JS, Munn DH, Weisdorf DJ, Blazar BR. Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation? Blood. 2011 Nov 10;118(19):5084–95. doi: 10.1182/blood-2011-07-365817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou Q, Bucher C, Munger ME, Highfill SL, Tolar J, Munn DH, et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood. 2009 Oct 29;114(18):3793–802. doi: 10.1182/blood-2009-03-208181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004 Oct;4(10):762–74. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 64.Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites downregulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006 Jun 1;176(11):6752–61. doi: 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- 65.Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood. 2007 Apr 1;109(7):2871–7. doi: 10.1182/blood-2006-07-036863. [DOI] [PubMed] [Google Scholar]
- 66.Corm S, Berthon C, Imbenotte M, Biggio V, Lhermitte M, Dupont C, et al. Indoleamine 2,3-dioxygenase activity of acute myeloid leukemia cells can be measured from patients' sera by HPLC and is inducible by IFN-gamma. Leuk Res. 2009 Mar;33(3):490–4. doi: 10.1016/j.leukres.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 67.Shenghui Z, Yixiang H, Jianbo W, Kang Y, Laixi B, Yan Z, et al. Elevated frequencies of CD4(+) CD25(+) CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J Cancer. 2011 Sep 15;129(6):1373–81. doi: 10.1002/ijc.25791. [DOI] [PubMed] [Google Scholar]
- 68.Wang X, Zheng J, Liu J, Yao J, He Y, Li X, et al. Increased population of CD4(+)CD25(high), regulatory T cells with their higher apoptotic and proliferating status in peripheral blood of acute myeloid leukemia patients. Eur J Haematol. 2005 Dec;75(6):468–76. doi: 10.1111/j.1600-0609.2005.00537.x. [DOI] [PubMed] [Google Scholar]
- 69.Lichtenegger FS, Lorenz R, Gellhaus K, Hiddemann W, Beck B, Subklewe M. Impaired NK cells and increased T regulatory cell numbers during cytotoxic maintenance therapy in AML. Leukemia research. 2014 Aug;38(8):964–9. doi: 10.1016/j.leukres.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 70.Kanakry CG, Hess AD, Gocke CD, Thoburn C, Kos F, Meyer C, et al. Early lymphocyte recovery after intensive timed sequential chemotherapy for acute myelogenous leukemia: peripheral oligoclonal expansion of regulatory T cells. Blood. 2011 Jan 13;117(2):608–17. doi: 10.1182/blood-2010-04-277939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jie HB, Gildener-Leapman N, Li J, Srivastava RM, Gibson SP, Whiteside TL, et al. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. British journal of cancer. 2013 Nov 12;109(10):2629–35. doi: 10.1038/bjc.2013.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72**.Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med. 2011 Nov 30;3(111):111ra20. doi: 10.1126/scitranslmed.3003130. [This study shows that PD-1 and PD-L1 signaling is involved in conversion of Th1 effector cells into Treg.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73*.Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, Riddle M, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010 Oct 7;116(14):2484–93. doi: 10.1182/blood-2010-03-275446. [This study demonstrates that disruption of PD-1 signaling in improves cytotoxic lymphocyte proliferation and improves leukemia control in mice.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74*.Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009 Aug 20;114(8):1545–52. doi: 10.1182/blood-2009-03-206672. [This study demonstrates that PD-1 knockout mice have superior anti-tumor T-cell reponses, improved leukemia control, and better overall survival than wild-type mice.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014 Jun;28(6):1280–8. doi: 10.1038/leu.2013.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kronig H, Kremmler L, Haller B, Englert C, Peschel C, Andreesen R, et al. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur J Haematol. 2014 Mar;92(3):195–203. doi: 10.1111/ejh.12228. [DOI] [PubMed] [Google Scholar]
- 77.Norde WJ, Maas F, Hobo W, Korman A, Quigley M, Kester MG, et al. PD-1/PDL1 interactions contribute to functional T-cell impairment in patients who relapse with cancer after allogeneic stem cell transplantation. Cancer Res. 2011 Aug 1;71(15):5111–22. doi: 10.1158/0008-5472.CAN-11-0108. [DOI] [PubMed] [Google Scholar]
- 78.Laszlo GS, Gudgeon CJ, Harrington KH, Dell'Aringa J, Newhall KJ, Means GD, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific Tcell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014 Jan 23;123(4):554–61. doi: 10.1182/blood-2013-09-527044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krupka C KP, Kischel R, Zugmaier G, Kohnke T, Lichtenegger FS, Schnorfeil FM, Altmann T, Schneider S, Fiegl M, Spiekermann K, Sinclair AM, Newhall KJ, Frankel ST, Baeuerle P, Hiddemann W, Riethmuller G, Subklewe M. PD-1/PD-L1 Blocking Enhances CD33/CD3-Bispecific BiTE Antibody (AMG 330) Mediated Lysis of Primary AML Cells. Blood. 2014 Dec 6;124(21) 2014. [Google Scholar]
- 80.Pyzer AR, Avigan DE, Rosenblatt J. Clinical trials of dendritic cell-based cancer vaccines in hematologic malignancies. Hum Vaccin Immunother. 2014 Nov 2;10(11):3125–31. doi: 10.4161/21645515.2014.982993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81*.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012 Feb 15;72(4):917–27. doi: 10.1158/0008-5472.CAN-11-1620. [This study reports on synergistic functions of LAG3 and PD-1 in blocking Teffector cell antitumor responses and promoting tumor escape.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013 Jul 11;369(2):122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas- Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990 May 1;171(5):1393–405. doi: 10.1084/jem.171.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur J Immunol. 2005 Jul;35(7):2081–8. doi: 10.1002/eji.200526090. [DOI] [PubMed] [Google Scholar]
- 85.Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010 Apr 27;107(17):7875–80. doi: 10.1073/pnas.1003345107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Berrien-Elliott MM, Jackson SR, Meyer JM, Rouskey CJ, Nguyen TL, Yagita H, et al. Durable adoptive immunotherapy for leukemia produced by manipulation of multiple regulatory pathways of CD8+ T-cell tolerance. Cancer Res. 2013 Jan 15;73(2):605–16. doi: 10.1158/0008-5472.CAN-12-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010 Aug 17;107(33):14733–8. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takamura S, Tsuji-Kawahara S, Yagita H, Akiba H, Sakamoto M, Chikaishi T, et al. Premature terminal exhaustion of Friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. J Immunol. 2010 May 1;184(9):4696–707. doi: 10.4049/jimmunol.0903478. [DOI] [PubMed] [Google Scholar]
- 89.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore antitumor immunity. The Journal of experimental medicine. 2010 Sep 27;207(10):2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011 Apr 28;117(17):4501–10. doi: 10.1182/blood-2010-10-310425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Whiteside TL. Inhibiting the inhibitors: evaluating agents targeting cancer immunosuppression. Expert Opin Biol Ther. 2010 Jul;10(7):1019–35. doi: 10.1517/14712598.2010.482207. [DOI] [PMC free article] [PubMed] [Google Scholar]