

Abstract
Plants respond differently to environmental conditions. Among various abiotic stresses, salt stress is a condition where excess salt in soil causes inhibition of plant growth. To understand the response of plants to the stress conditions, identification of the responsible genes is required. Clustering is a data mining technique used to group the genes with similar expression. The genes of a cluster show similar expression and function. We applied clustering algorithms on gene expression data of Solanum tuberosum showing differential expression in Capsicum annuum under salt stress. The clusters, which were common in multiple algorithms were taken further for analysis. Principal component analysis (PCA) further validated the findings of other cluster algorithms by visualizing their clusters in three-dimensional space. Functional annotation results revealed that most of the genes were involved in stress related responses. Our findings suggest that these algorithms may be helpful in the prediction of the function of co-expressed genes.
Keywords: Clustering algorithms, Gene expression, Co-expression, Functional annotation, Abiotic stress
1. Introduction
Heat, cold, drought and salinity are common abiotic stress conditions for the plants. These stresses affect both plant growth and production [1]. Salt stress presents an increasing threat to agriculture due to its effect on plant growth [2]. Reduced availability of water, increased respiration rate, altered mineral distribution, membrane instability, failure in the maintenance of turgor pressure are some of the events that prevails during salt stress [3]. The response of plants to salinity consists of numerous processes that function in coordination to reduce the cellular hyper-osmolarity and ion disequilibrium [4]. It is important to analyze the function of stress-inducible genes not only to understand the molecular mechanisms of stress tolerance but also to improve the stress tolerance of crops by gene manipulation. Gene expression analysis or expression profiling refers to the study of response of an organism against environmental changes [5]. The expression profiles of thousands of genes results from microarray technology, a preferred method to identify genes involved in abiotic stress responses [6], [7]. In addition, analysis of relationship between genes, their functions and classification is also required. Because of the large number of genes and the complexity of biological networks, clustering algorithms are found to be useful exploratory technique for the analysis of gene expression data [8]. To make some meaningful biological inference from large number of genes or samples, they are required to be clustered together to obtain co-expressed genes. The co-expressed genes may have similarity in their expression levels. Cluster analysis is one of the primary statistical tool for data analysis [9], [10]. The clustering algorithms have been proven useful for identifying biologically relevant groups of genes or samples and can be applied for gene function discovery process. Clustering algorithms can be classified mainly as unsupervised like Hierarchical clustering (HCL) [11], K-means clustering (KMC) [12], Principal Component Analysis (PCA) [13] and Self Organizing Map (SOM) [14] or supervised like Support Vector Machine (SVM) [15]. These clustering techniques are based on the hypothesis that the genes in a cluster might share common function and regulatory elements [16]. The cluster analysis routinely run as a first step of data analysis and separates a set of objects into several subsets based on their similarity [17], [18]. None of the clustering algorithms available provides evidence to support that clustering of genes having similar expression patterns is more likely to have similar biological function [19]. The genes with similar expression profiles are said to be co-expressed genes [20]. Co-expressed genes may help in revealing useful biological information as they are functionally related [11], [21]. Hence, grouping of genes with similar expression levels can reveal the function of those genes, which were previously uncharacterized. To find out the utility of clustering techniques, the gene expression profiles of the genes of Solanum tuberosum showing differential expression under salt stress at six different time points in Capsicum annuum were retrieved and analyzed. The clusters, obtained from different algorithms showed common patterns of genes having similarity in their expression and function.
2. Materials and methods
2.1. Retrieval of gene expression data
The normalized gene expression data of S. tuberosum showing differential expression in Capsicum annuum were retrieved. As per experimented details provided in GEO database, the ESTs of S. tuberosum were used as spotted PCR amplified cDNA array. After the application of 150 mM NaCl at different time periods in C. annuum, the leaf tissue samples were collected including control plants. The RNA from these tissues were isolated and hybridized with cDNA arrays. The genes, showing response under salt stress at six different time points were considered as six samples and were downloaded from Gene Expression Omnibus (GEO) database at NCBI (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE8158) [22]. The sample ids were GSM201905, GSM201906, GSM201907, GSM2019058, GSM201909 and GSM201910 (Supplementary Table 1). The filtering of dataset was done in order to obtain highly expressed and significant genes.
2.2. Clustering of genes
Clustering of the genes having similar expression profile was done using the clustering tool, Genesis 1.7.6 [20]. The genes expression data was imported in the form of a text file. Different algorithms like, hierarchical clustering, k-means clustering, principal component analysis and self-organized map algorithms were applied for clustering of the gene expression data. The number of runs for each clustering algorithms were minimum 1000 cycles.
2.3. Functional annotation
The functional annotation of the genes was done using already annotated genes of Arabidopsis thaliana, using Gene Ontology (GO), a feature available in the program Genesis. For the GO annotation, a mapping file, having the GO annotations for A. thaliana, was created using Batch SOURCE (http://smd.stanford.edu/cgi-bin/source//sourceBatchSearch) available in Stanford SOURCE database (http://source.stanford.edu/). This file was imported to Genesis for creating GO tree. From this tree, the genes of C. annuum, induced in salt stress were assigned their functions.
3. Results
3.1. Retrieval and filtering of gene expression data
The differential expression data were retrieved from GEO database. The data were in form of 17,453 genes having corresponding expression values at different time periods of salt stress responses. These genes were of S. tuberosum showing differential expression in C. annuum. The mean of intensity values of six samples were calculated for each gene. The genes having the mean of intensity value greater than + 0.585 were considered as highly up regulated and less than − 0.585 as highly down regulated. Walia et al. has reported 0.585 as cut off log2 value of expression ratio to get the significant genes [23]. After filtering, sixty genes were obtained and taken for further analyses.
3.2. Clustering results to find co-expressed genes
3.2.1. Principal component analysis (PCA)
PCA algorithm was performed prior to clustering to obtain different patterns or dimensions formed from the expression data. PCA calculates a new system of coordinates. The directions of the coordinate system calculated by PCA are known as eigenvectors. Six different time points (principal components) were observed in the data, based on eigenvectors. The eigenvalues for all the principal components (PCs) have been plotted (Fig. 1). First PC revealed largest eigenvalues which mean that it accounts for high variation in expression value of the genes. The variation in expression values of the genes decreased successively in other PCs.
Fig. 1.
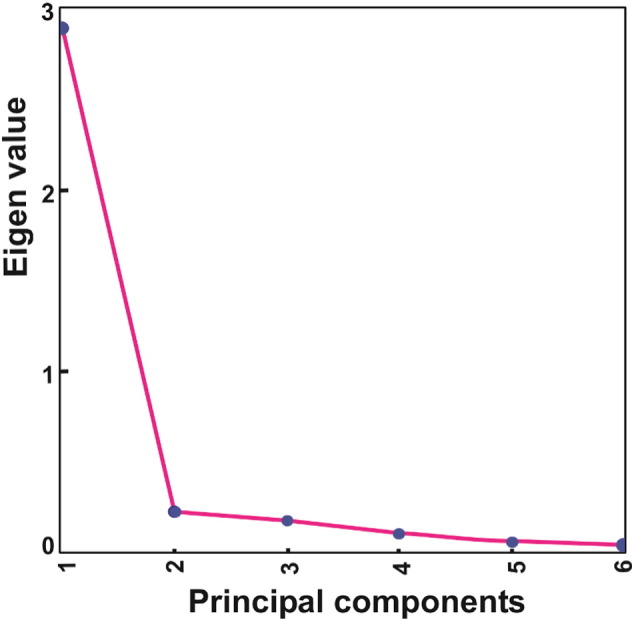
Eigenvalue plot of the principal components.
The six principal components (PC) represent six samples of the gene expression data. The PC containing large eigenvalue represents high variability among its genes.
3.2.2. Hierarchical clustering (HCL)
HCL algorithm was performed to obtain the number of patterns hidden in the dataset using average linkage method. Based on these patterns the number of clusters was identified. These patterns are useful to set the number of clusters in other algorithms. Five clusters were identified among 60 genes (Fig. 2). Cluster 5 showed highest number of genes, which were down regulated. Cluster 2 showed most of the up-regulated genes. In the heatmap, for each gene red, green and black colors indicates up, down and no expression respectively. These colors are representative of a relative scale (− 3.0 to + 3.0) derived from the expression values. The white dots in the heatmap represent the gene showing maximum expression at a particular time point. The expression images of the clusters having similar genes belonging to a particular cluster have been plotted (Fig. 3). Clusters 1 and 2 consisted of upregulated genes whereas clusters 3, 4 and 5 had downregulated genes. The magenta curve represents the mean expression values at different time points. In each expression view, the x-axis represents time points whereas the y-axis represents expression values.
Fig. 2.

Heatmap of clustered genes showing five clusters.
The hierarchical clustering of the genes is represented in heatmap. The red and green color shows the up and down expression respectively. The range of expression values is − 0.3 to 0.3. The genes are represented in form of their accession number followed by their function.
Fig. 3.

Expression views of hierarchical clustering.
The clusters resulted from hierarchical clustering are shown. The number of genes lying in each cluster is mentioned. Multiple black lines represent each gene showing its expression values. In each cluster, the pink line represents the mean of expression values of all the genes.
3.2.3. K-Means clustering (KMC)
KMC algorithm is based on given number of clusters. The number of clusters can be chosen randomly or estimated by performing a hierarchical clustering of the dataset [24]. In this study, we took the number of clusters as five based on result of HCL algorithm. Out of 5 obtained clusters, clusters 1 and 3 had downregulated and clusters 2, 4 and 5 were with upregulated genes. The mean of all the genes belonging to a particular cluster have been represented in magenta color. The x-axis represents time points whereas the y-axis represents expression values (Fig. 4).
Fig. 4.

Expression views of K-means clustering.
The clusters resulted from K-means clustering are shown. The number of genes lying in each cluster is mentioned. Multiple black lines represent each gene showing its expression values. In each cluster, the pink line represents the mean of expression values of all the genes.
3.2.4. Self-organizing map (SOM)
SOM clustering algorithm has also been applied to the dataset for clustering. The number of clusters was set as 6 (2 × 3), on the basis of clusters obtained from HCL algorithm. However, SOM analysis resulted in six clusters. Five clusters showed the presence of genes and cluster 3 did not had any gene (Fig. 5).
Fig. 5.

Expression images of SOM algorithm.
The clusters resulted from SOM clustering shown. The number of genes lying in each cluster is mentioned. Multiple black lines represent each gene showing its expression values. In each cluster, the pink line represents the mean of expression values of all the genes.
3.2.5. Cluster comparison analysis
The comparison of clusters obtained from common clustering techniques might provide additional information as compared to single method approach [20]. In comparison, three clusters of HCL and KMC algorithms were found to be similar to each other showing same pattern of expression. One cluster from SOM and HCL algorithms and one cluster from all three algorithm revealed similarity in their expression.
3.2.6. Visualization of clustering algorithms through PCA
PCA algorithm was used to visualize the clusters obtained from HCL, KMC and SOM algorithms. The five groups of genes obtained from each algorithm have been shown in the 3D space of PCA analysis (Fig. 6). PCA analysis demonstrates measure of variability among genes [25]. The genes of cluster 2 of HCL showed highest variability. The value of average distance from the mean was 0.77, which is highest among all the clusters made from HCL algorithm. Cluster 4 of KMC revealed 0.73 average distance from the mean indicating highest variability among its genes. In SOM algorithm, highest variability of 0.94 was observed among genes of cluster 4. The analysis suggests that the variability among genes within a cluster depends upon the values of average distance from the mean.
Fig. 6.

Clusters of HCL (a), KMC (b) and SOM (c) in 3-D space.
The clustered genes are represented in balls of different colors in three-dimensional space. The balls of different colors represent genes. The clusters, which are similar in their expression values, lie close to each other.
3.2.7. Functional annotation of genes
The ontological classification of selected differentially expressed genes was done. Biological process, molecular function and cellular location classification were taken into consideration for functional assignment of the genes. Some of the genes could not be assigned any function. The genes, for which function could not be assigned, were found to be present in cluster 5 of HCL and cluster 1 of KMC. Both the clusters were similar as could be seen in the centroid views (Fig. 7a). Some other genes having no function belonged to cluster 1 of HCL and cluster 5 of KMC, which were also similar to each other (Fig. 7b). Thus, these eight genes were assigned of their functions according to their co-expression present in the same clusters (Table 1). Most of these genes belonged to function related to stress responses.
Fig. 7.

Centroid views of comparison of clusters made from HCL and KMC.
Three clusters of HCL and KMC are represented in a comparative manner. The pink line in each cluster represents the pattern of mean expression values in each sample.
Table 1.
Functional assignment to the genes, having no any prior information.
| Genes | Functions |
|---|---|
| BQ516961 | Biological process | response to cold and salt stress |
| BQ515216 | Response to light stimulus | response to stress |
| BQ516960 | Biological process | response to cold and salt stress |
| BQ515215 | Response to light stimulus | response to stress |
| BQ518795 | Biological process | response to cold and salt stress |
| BQ516985 | Biological process | response to cold and salt stress |
| BQ512282 | Response to light stimulus | response to stress |
| BQ516986 | Biological process | response to cold and salt stress |
4. Discussion
In this study, we have examined the distribution of genes showing differential expression under salt stress. All four algorithms resulted in similar patterns of clustering of genes. An earlier study have used HCL algorithm to list the genes of barley, expressed under salinity stress. The identified groups of genes with similar expression patterns were up and downregulated separately. The group of upregulated genes were mainly responsible for heat shock protein (HSP), calmodulin and arginine/serine rich protein [23]. Earlier studies demonstrated that the genes responsible for HSPs show change in expression in A. thaliana and rice under abiotic stresses including salt [26], [27]. The calmodulin genes have been earlier reported to enhance drought and salt tolerance in rice plant [28]. The objective of the clustering was to use the appropriate method for grouping the expression data having similarity in their expression values. All algorithms resulted in equal number of clusters. However, the difference was observed in the pattern of genes in the cluster. In our analysis, cluster 5 of HCL and 1 of KMC showed same set of genes having up-expression under salt stress (Fig. 7a). The genes ware mainly responsible for carbohydrate metabolism like fructose 1,6-bisphosphate 1-phosphatase activity, kinase activity, response to abscisic acid stimuli and some heat shock proteins clustered together. Cluster 1 of KMC and 2 of HCL also showed similar expression pattern (Fig. 7b). According to functional similarity, the above genes belonged mainly to stress responses viz., serine/threonine phosphatase an enzyme of MAP kinase pathway activated in stress response, calmodulin binding and peroxidase activity responsible for oxidative stress. MAP kinases play an important role in abiotic stress signaling pathway [29]. Cluster 5 of HCL and 1 of KMC were compared and revealed 19 common genes showing expression level and functional similarity (Fig. 7c). All the genes were downregulated in the presence of salt stress. The expression of these genes was lowered in the presence of salt stress. The genes of both the clusters belonged to stress responses such as zinc and cadmium ion binding. The genes had biological functions like ATP binding, transferase activity and hydrolase activity. Expression views made by cluster 2 analyzed from two algorithms HCL and SOM were found to be similar to each other (Fig. 8). The number of common genes in both the clusters were 19. These genes showed upregulation under the salt stress condition. The intensity value was observed with range from 0.58 to 1.02. The functional similarity among the genes was observed as most of the genes belonged to peroxidase activity in the presence of oxidative stress and response to abscisic acid signaling pathways due to stress condition. Comparative analysis of the results indicated that the clusters of all three algorithms showed similar patterns of genes e.g. cluster 5 of HCL, cluster 6 of SOM and cluster 1 of KMC have similar function and are downregulated under stress (Fig. 9). Among three clusters 15 genes were common having range of intensity value of − 0.58 to − 0.69 indicating that they are downregulated. The functional similarities in terms of oxidase activity in response to cold suggested that these genes were responsible both for cold and salt stress. The histone deacetylase activity showed the response of genes for abscisic acid stimulus. It was earlier demonstrated that expression of histone deacetylase gene was repressed by ABA in A. thaliana, suggesting its possible role in the ABA response [30]. Thus, clustering of the genes in accordance to the similarity in their expression profiles along six different time points was useful to find out the co-expressed genes. Some clusters obtained through different clustering algorithms were similar indicating that the genes were clustered accurately. The distribution of the genes in clusters was found to be similar in each algorithm. The functional annotation of all the genes was done. The uncharacterized genes were predicted for their function based on their similarity to the genes of participating cluster. All the genes were assigned functions for cold and salt stress responses. The expression level analysis of each uncharacterized genes at all time points was done (Fig. 10). The genes showing expression values below the mean were considered to be downregulated. Genes BQ516961 and BQ516960 showed same pattern of down expression. The mean intensity value of both the genes was − 0.6045. The expression patterns of BQ515216 and BQ515215 was similar having 0.692667 intensity value and up expression. BQ516985 and BQ516986 showed down expression of − 0.5915 intensity value.
Fig. 8.

Centroid views of similar clusters made from SOM and HCL.
One cluster of SOM and HCL is represented in a comparative manner. The pink line in each cluster is showing the pattern of mean expression values in each sample.
Fig. 9.

Centroid views of similar clusters made from SOM, KMC and HCL.
One cluster of SOM, KMC and HCL is similar and represented in a comparative manner. The pink line in each cluster is showing the pattern of mean expression values in each sample.
Fig. 10.

Expression level representation of eight genes.
The expression view of the uncharacterized genes is represented. The pink line, which lies above and below the black line, represents up and down expression respectively.
5. Conclusion
In this work, we aimed to study the clustering analysis of genes showing differential expression in salinity stress. The genes of S. tuberosum showing change in their expression in C. annuum were taken. The clustering programs are routinely run as a first step of data summary and grouping genes in a microarray data analysis [9], [10], [17]. The prediction of correct number of clusters is a critical problem in unsupervised algorithms like KMC and SOM. HCL, an agglomerative method is used for pre-specification of the number of clusters for other algorithms. Use of multiple clustering algorithms is required for accuracy of the results. In our study, the application of four clustering algorithms, HCL, KMC, PCA and SOM resulted in almost similar clusters. The similarity in the clusters from multiple algorithms indicates that the co-expressed genes are clustered efficiently. Earlier reports have shown that application of more than one clustering technique may facilitate the generation of more accurate and reliable results [31]. The gene ontology annotation from A. thaliana helped to find out the functions for the genes, which were found to be up- and down-regulated in C. annuum, under salt stress. Functional annotation revealed that most of the genes were related to stress response. The functions of eight unknown genes were predicted based on their occurrence in the cluster. Out of these eight genes, 3 were up and 5 downregulated. Thus, clustering of the co-expressed genes is also helpful in getting information about the uncharacterized genes. The functional information of uncharacterized genes can be predicted based on other participating genes in particular cluster. The efficiency of the clustering algorithm can be measured by validating the analyzed clusters. The validation of the clusters will be our future endeavor to find the co-expressed genes.
The following are the supplementary data related to this article.
Gene expression data of S. tuberosum showing differential expression in C. annuum at different time periods of salt.
Conflict of interest
Authors declare no conflict of interest.
Acknowledgments
We would like to thank C Robin Buell who has submitted her experiments in GEO database of NCBI and make them freely available to the scientific community. S is thankful to CSIR, New Delhi, India for CSIR-SRF fellowship.
References
- 1.Krasensky J., Jonak C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012;63:1593–1608. doi: 10.1093/jxb/err460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu J.K. Encyclopedia of Life Science. Wiley Online Library; 2007. Plant salt stress. [Google Scholar]
- 3.Babu M.A., Singh D., Gothandam K.M. The effect of salinity on growth, hormones and mineral elements in leaf and fruit of tomato cultivar Pkm1. J. Anim. Plant Sci. 2012;22:159–164. [Google Scholar]
- 4.Shuji Y., Bressan R.A., Hasegawa P.M. JIRCAS Working Report. 2002. Salt stress tolerance of plants; pp. 25–33. [Google Scholar]
- 5.Hazen S.P., Wu Y., Kreps J.A. Gene expression profiling of plant responses to abiotic stress. Funct. Integr. Genomics. 2003;3:105–111. doi: 10.1007/s10142-003-0088-4. [DOI] [PubMed] [Google Scholar]
- 6.Eisen M.B., Brown P.O. DNA arrays for analysis of gene expression. Methods Enzymol. 1999;303:179–205. doi: 10.1016/s0076-6879(99)03014-1. [DOI] [PubMed] [Google Scholar]
- 7.Rensink W.A., Iobst S., Hart A., Stegalkina S., Liu J. Gene expression profiling of potato responses to cold, heat, and salt stress. Funct. Integr. Genomics. 2005;5:201–207. doi: 10.1007/s10142-005-0141-6. [DOI] [PubMed] [Google Scholar]
- 8.Yeung K.Y., Ruzzo W.L. Principal component analysis for clustering gene expression data. Bioinformatics. 2001;17:763–774. doi: 10.1093/bioinformatics/17.9.763. [DOI] [PubMed] [Google Scholar]
- 9.Chandrasekhar T., Thangavel K., Elayaraja E. Effective clustering algorithms for gene expression data. Int. J. Comput. Appl. 2011;32:0975–8887. [Google Scholar]
- 10.Costa I.G., de Carvalho F.A.T., de Souto M.C.P. Comparative analysis of clustering methods for gene expression time course data. Genet. Mol. Biol. 2004;27:623–631. [Google Scholar]
- 11.Eisen M.B., Spellman P.T., Brown P.O., Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U. S. A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tavazoie S., Hughes J.D., Campbell M.J., Cho R.J., Church G.M. Systematic determination of genetic network architecture. Nat. Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- 13.Raychaudhuri S., Stuart J.M., Altman R.B. Principal components analysis to summarize microarray experiments: application to sporulation time series. Pac. Symp. Biocomput. 2000;455-466 doi: 10.1142/9789814447331_0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamayo P., Slonim D., Mesirov J., Zhu Q., Kitareewan S. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc. Natl. Acad. Sci. U. S. A. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown M.P.S., Grundy W.N., Lin D., Cristianini N., Sugnet C.W. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl. Acad. Sci. U. S. A. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quackenbush J. Computational analysis of microarray data. Nat. Rev. Genet. 2001;2:418–427. doi: 10.1038/35076576. [DOI] [PubMed] [Google Scholar]
- 17.Datta S. Comparisons and validation of statistical clustering techniques for microarray gene expression data. Bioinformatics. 2003;19:459–466. doi: 10.1093/bioinformatics/btg025. [DOI] [PubMed] [Google Scholar]
- 18.Gilbert D.R., Schroeder M., van Helden J. Interactive visualization and exploration of relationships between biological objects. Trends Biotechnol. 2000;18:487–494. doi: 10.1016/s0167-7799(00)01510-9. [DOI] [PubMed] [Google Scholar]
- 19.Gibbons F.D., Roth F.P. Judging the quality of gene expression-based clustering methods using gene annotation. Genome Res. 2002;12:1574–1581. doi: 10.1101/gr.397002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sturn A., Quackenbush J., Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18:207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 21.Spellman P.T., Sherlock G., Zhang M.Q., Iyer V.R., Anders K. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edgar R., Domrachev M., Lash A E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walia H., Wilson C., Wahid A., Condamine P., Cui X. Expression analysis of barley (Hordeum vulgare L.) during salinity stress. Funct. Integr. Genomics. 2006;6:143–156. doi: 10.1007/s10142-005-0013-0. [DOI] [PubMed] [Google Scholar]
- 24.Babu M.M. Computational genomics. Horizon Press; 2004. An introduction to microarray data analysis; pp. 225–249. [Google Scholar]
- 25.Jombart T., Devillard S., Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010;11:1–15. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swindell W.R., Huebner M., Weber A.P. Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genomics. 2007;8:1–15. doi: 10.1186/1471-2164-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu W.H., Hu G.C., Han B. Genome-wide survey and expression profiling of heat shock proteins and heat shock factors revealed overlapped and stress specific response under abiotic stresses in rice. Plant Sci. 2009;176:583–590. doi: 10.1016/j.plantsci.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Xu G.Y., Rocha P.S., Wang M.L., Xu M.L., Cui Y.C. A novel rice calmodulin-like gene, OsMSR2, enhances drought and salt tolerance and increases ABA sensitivity in Arabidopsis. Planta. 2011;234:47–59. doi: 10.1007/s00425-011-1386-z. [DOI] [PubMed] [Google Scholar]
- 29.Jonak C., Ligterink W., Hirt H. MAP kinases in plant signal transduction. Cell. Mol. Life Sci. 1999;55:204–213. doi: 10.1007/s000180050285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sridha S., Wu K.Q. Identification of AtHD2C as a novel regulator of abscisic acid responses in Arabidopsis. Plant J. 2006;46:124–133. doi: 10.1111/j.1365-313X.2006.02678.x. [DOI] [PubMed] [Google Scholar]
- 31.Azuaje F. Clustering-based approaches to discovering and visualising microarray data patterns. Brief. Bioinform. 2003;4:31–42. doi: 10.1093/bib/4.1.31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene expression data of S. tuberosum showing differential expression in C. annuum at different time periods of salt.