

Abstract
The molecular mechanisms underlying the differentiation of interleukin 17–producing T helper cells (TH-17 cells) are still poorly understood. Here we show that optimal transcription of the gene encoding interleukin 17 (Il17) required a 2-kilobase promoter and at least one conserved noncoding (enhancer) sequence, CNS-5. Both cis-regulatory elements contained regions that bound the transcription factors RORγt and Runx1. Runx1 influenced TH-17 differentiation by inducing RORγt expression and by binding to and acting together with RORγt during Il17 transcription. However, Runx1 also interacts with the transcription factor Foxp3, and this interaction was necessary for the negative effect of Foxp3 on TH-17 differentiation. Thus, our data support a model in which the differential association of Runx1 with Foxp3 and with RORγt regulates TH-17 differentiation.
Introduction
The differentiation of interleukin 17 (IL-17)–producing T helper cells (TH-17 cells) has been the subject of much attention, mainly because IL-17 and other cytokines released from TH-17 cells are important to the pathogenesis of autoimmune inflammation in both mice and humans1, 2, 3, 4, 5. In mice, TH-17 differentiation is induced by the combined activity of transforming growth factor-β (TGF-β) and IL-6 (refs. 6,7), whereas in humans, TGF-β and IL-21 (naive cells) or IL-1β (memory cells)8, 9 have a predominant function. In both species, however, these cytokines affect IL-17 production by inducing the expression of key lineage-specific transcription factors: the orphan nuclear receptor RORγt (A002302) and a related factor, RORα1, 8, 10.
Studies of the transcription of genes encoding T helper type 1 (TH1) or TH2 cytokine such as interferon-γ and IL-4, respectively, have shown that such transcription is controlled in part by cis-regulatory elements consisting of evolutionarily conserved noncoding sequences (CNS) elements located in the vicinity of the gene encoding the cytokine11, 12, 13. One such CNS element is also present in the Il17 locus and enhances the transcriptional activity of a minimal Il17 promoter14. However, the actual location of the Il17 promoter and the function of the CNS in Il17 transcription is still mostly undefined.
It has been shown that Foxp3 (A002750), the TGF-β-induced, lineage-specific transcription factor of regulatory T cells (Treg cells), also influences TH-17 differentiation. Specifically, Foxp3 physically interacts with RORγt, and this interaction considerably inhibits Il17 transcription15. This ‘yin-yang’ relationship of RORγt and Foxp3 is the probable basis of the observation that the differentiation of TH-17 cells and Treg cells is often reciprocal. The function of Foxp3 in TH-17 differentiation also suggests that one or more Runx transcription factors may influence Il17 transcription. This follows from the finding that Foxp3 also interacts with Runx1 (A000523) and such interaction is required for the negative regulation of Il2 transcription16. Runx proteins can regulate transcription in a context-dependent way by binding to other transcription factors to form coactivator or corepressor complexes; for example, Runx3 acts cooperatively with T-bet to promote expression of the gene encoding interferon-γ and to silence Il4 in TH1 cells17. In this study, we marshal evidence indicating that Il17 transcription is dependent on RORγt as well as the second factor Runx1 (and possibly Runx2), and that both factors act at promoter and enhancer regions to regulate transcription. In addition, we show that Runx1 binds to RORγt and to Foxp3 to bring about positive and negative effects on Il17 transcription.
Results
The cis elements regulating Il17 transcription
We initially did studies to define the promoter elements that participate in the regulation of Il17 transcription. Accordingly, we generated a series of reporter constructs containing regions upstream of the Il17 transcription start site and assessed the activity of these constructs by transfecting them into Jurkat cells, which we then stimulated with the phorbol ester PMA and ionomycin. A reporter consisting of a 2-kilobase (kb) promoter fragment containing a binding site for the TH-17 lineage–specific transcription factor RORγ showed substantial transcriptional activity, but a 1.1-kb fragment, which also contained a RORγt-binding site, showed only minimal transcriptional activity equivalent to that of a 0.6-kb fragment lacking a RORγt-binding site (Fig. 1a). To specifically determine the function of RORγt in the regulating the Il17 promoter, we then assessed the reporter activity of Il17 promoter fragments in cells cotransfected with RORγt-expressing constructs. In the presence of RORγt, the 2-kb promoter fragment had about twofold higher luciferase activity, but the 1.1-kb fragment that contained the RORγt-binding site showed no greater luciferase activity relative to that of the 0.6-kb fragment (Fig. 1b). To further assess the importance of RORγt in Il17 promoter activity, we assessed the reporter activity of a 2-kb promoter with a mutated RORγt-binding site. This mutated construct had much lower luciferase activity that was not reversed by cotransfection of exogenous RORγt (Fig. 1c). These results collectively indicate that whereas RORγt regulates Il17 transcription through its promoter, other factors in addition to RORγt may also influence Il17 transcription.
Figure 1. Mouse Il17 promoter activity.

(a) Luciferase activity of Jurkat cells transfected with fragments (0.6 kb, 1.1 kb and 2 kb) of the mouse Il17 promoter linked to firefly luciferase reporter constructs along with a renillla luciferase vector (transfection efficiency control), allowed to ‘rest’ overnight, then left unstimulated (None) or stimulated for 6 h with PMA and ionomycin (PMA + iono). Results are presented relative to renilla luciferase activity. (b) Luciferase activity of Jurkat cells transfected with luciferase reporter constructs plus RORγt-expressing vector (+RORγt) or empty vector (−RORγt), allowed to ‘rest’ overnight, then stimulated for 6 h with PMA and ionomycin and assessed as described in a. Below, immunoblot analysis of RORγt expression in transfected Jurkat cells, detected with anti-RORγ (α-RORγ). (c) Luciferase activity of Jurkat cells transfected with luciferase reporter constructs containing an intact 2-kb promoter or a 2-kb promoter with a mutated RORγt-binding site, plus RORγt-expressing or empty vector, then stimulated and assessed as described in b. Data are representative of at least four (a,b) or three (c) independent experiments (mean and s.d. of triplicate transfections).
We also searched for CNS elements, similar to those that act as distal upstream enhancers regulating Il4 and the gene encoding IFN-γ1, 12, 18, 19, that regulate Il17 transcription. For this, we compared approximately 200 kb of DNA in the mouse Il17 locus with a similar sequence in the human IL17 locus using the VISTA global alignment program. We identified several CNS sites, including CNS-5 (also called CNS-2; ref. 14), which was approximately 5 kb upstream of Il17 and contained a perfect consensus binding sequence for RORγt, TGACCT (Fig. 2a and Supplementary Fig. 1a online). Another site, CNS-19, was approximately 19 kb upstream of Il17 and lacked a RORγt-binding sequence. To determine the function of these CNS elements in Il17 transcriptional regulation, we used a promoter-reporter strategy similar to that described above but in this case with constructs containing promoter fragments linked to CNS sequences. The reporter activity of the 2-kb promoter was much greater when linked to CNS-5, particularly in the presence of exogenous RORγt; in contrast, the reporter activity of the 2-kb promoter fragment was not enhanced when linked to CNS-19 (Fig. 2b). In addition, in a way dependent on RORγt expression, CNS-5 also amplified the luciferase activity induced by the shorter IL-17 promoter fragments (Fig. 2c). However, this luciferase activity was still lower than that noted with the 2.0-kb promoter (data not shown).
Figure 2. CNS-5 is a RORγt-dependent enhancer element for Il17 transcription.
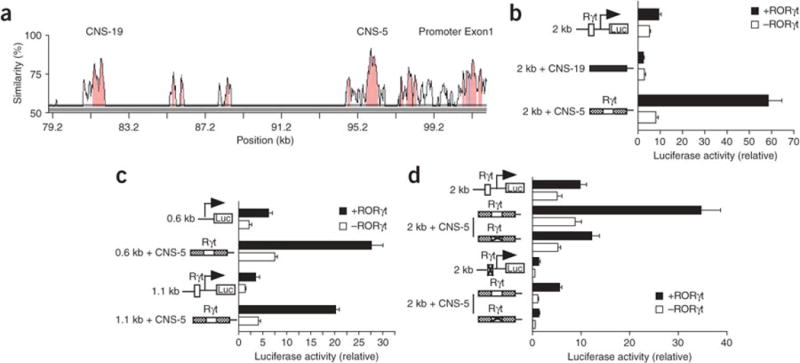
(a) VISTA plot of sequence similarity of CNS sites (pink) in human IL17 and mouse Il17 DNA, presented relative to their positions in the mouse genome (horizontal axis); exons are blue. CNS-19 and CNS-5 (also called CNS2; ref. 14) are located -19 kb and -5 kb, respectively, from the transcriptional start site of mouse Il17. (b) Luciferase activity of Jurkat cells transfected with reporter constructs (left margin) containing the 2-kb promoter alone or linked upstream to CNS-19 (filled rectangles) or CNS-5 (crosshatched rectangles), plus RORγt-expressing or empty vector, stimulated and assessed as described in Figure 1b. Open boxes, RORγt-binding site (Rγt); Luc, luciferase. (c) Luciferase activity of Jurkat cells transfected with luciferase reporter constructs containing 0.6-kb or 1.1-kb promoter fragments alone or linked to CNS-5, plus RORγt-expressing or empty vector, stimulated and assessed as described in Figure 1b. (d) Luciferase activity of Jurkat cells transfected with reporter constructs containing the 2-kb promoter fragment alone or linked to CNS-5, with (‘X’ in box) or without (open box) mutation of RORγt-binding sites, plus RORγt-expressing or empty vector, stimulated and assessed as described in Figure 1b. Data are representative of experiments repeated at least three times (mean and s.d. of triplicate transfections).
To determine if the CNS-5 enhancer activity depended on RORγt, we mutated the RORγt-binding site in CNS-5. The mutated CNS-5 construct induced much less luciferase activity than its wild-type counterpart did when cotransfected with RORγt into Jurkat cells (Fig. 2d). Moreover, luciferase activity was almost completely abolished by mutation of the RORγt-binding sites in both CNS-5 and the 2-kb promoter (Fig. 2d). These results suggested that both the 2-kb promoter and the enhancer element CNS-5 are required for maximum Il17 transcription.
To further confirm that CNS-5 is an enhancer element, we cloned CNS-5 and CNS-19 separately into a PGL4.23 reporter vector containing a minimal irrelevant promoter. Cotransfection of the CNS-5-containing reporter and an RORγt construct into Jurkat cells resulted in a strong luciferase signal, and this signal was much lower when the RORγt-binding site in CNS-5 was mutated (Supplementary Fig. 1b). In contrast, the CNS-19-containing construct showed no luciferase signal in any circumstances (Supplementary Fig. 1b). These results collectively provide evidence that CNS-5, but not CNS-19, is an RORγt-dependent enhancer element for Il17 transcription.
Regulation of Il17 transcription by Runx1
As noted above, the 1.1-kb promoter fragment containing the RORγt-binding site showed low reporter activity, whereas the 2-kb fragment showed much higher promoter activity. Because of this discrepancy, we analyzed the sequence located between 1.1 kb and 2 kb of the Il17 promoter and identified consensus binding sequences for several transcription factors, including STAT, Runx and Foxp factors (Supplementary Fig. 2a online). Of those, we focused on Runx, given published evidence that Runx family members are involved in TGF- signaling20, 21, 22, TH1 differentiation and Treg cell function16, 17. Accordingly, we did reporter assays with Jurkat cells cotransfected with constructs expressing Runx1 and the 2-kb Il17 promoter reporter. The presence of exogenous Runx1 amplified the luciferase signal induced by the 2-kb promoter fragment (Fig. 3a). Cotransfection of constructs expressing Runx2 or Runx3 also amplified the luciferase activity driven by the 2-kb promoter fragment, albeit to a lesser extent than Runx1 did (Supplementary Fig. 2b).
Figure 3. Runx1 upregulates Il17 transcription.

(a) Luciferase activity of Jurkat cells transfected with a reporter construct containing the 2-kb Il17 promoter fragment, plus Flag-tagged Runx1-expressing or empty vector and a renilla luciferase plasmid, stimulated and assessed as described in Figure 1a. Below, immunoblot analysis of Runx1 expression in transfected Jurkat cells. (b) Luciferase activity of Jurkat cells transfected with reporter constructs containing the 2-kb Il17 promoter fragment with or without mutation of the RORγt-binding site or Runx1-binding sites (Rx), plus RORγt-expressing or empty vector, stimulated and assessed as described in Figure 1b. (c) Luciferase activity of Jurkat cells transfected with reporter constructs containing the 2-kb Il17 promoter fragment with or without mutation of the RORγt-binding site, plus Runx1-expressing vector (+) or empty vector (−), stimulated and assessed as described in Figure 1a. Data are representative of three independent experiments (mean and s.d. of triplicate transfections).
In further studies, we did reporter assays with Jurkat cells transfected with promoter reporter constructs in which Runx-binding sites were mutated. Mutation of either one or both of the Runx-binding sites in the 2-kb promoter fragment led to less reporter activity and this effect was not reversed by cotransfection of an RORγt-expressing construct (Fig. 3b). In addition, Runx1 did not increase the luciferase activity of a 2-kb promoter fragment with a mutated RORγt-binding site (Fig. 3c). These studies thus indicate that Runx1 is important in the transcription of Il17 and that the effect of Runx1 is dependent on RORγt.
Runx1 is required for TH-17 differentiation
To define the requirement of Runx proteins in the differentiation of primary TH-17 cells, we measured the expression of Runx mRNA and protein in CD4 cells cultured in various T helper cell–polarizing conditions. Because all Runx family members were expressed in TH-17 cells (Supplementary Fig. 3a–c online), we further evaluated the influence of each Runx protein on Il17 transcription by measuring IL-17 expression in primary T cells. We examined IL-17 expression in primary T cells ectopically expressing individual Runx proteins. Overexpression of Runx1 induced the greatest increase in IL-17 expression, and overexpression of Runx2 led to lesser but still substantial increase; in contrast, overexpression Runx3 led to only a marginal increase (Fig. 4a). All three Runx proteins were transcriptionally active in these cells, as judged by their ability to promote the production of interferon-γ (Supplementary Fig. 4 online).
Figure 4. Runx1 is required for IL-17 expression in CD4+ T cells.
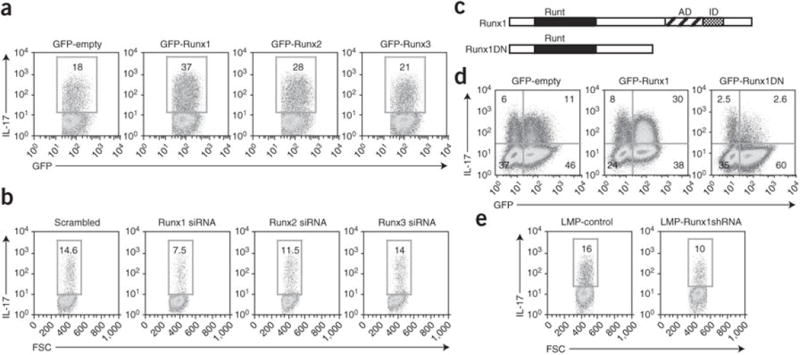
(a,b) Flow cytometry of IL-17 expression in purified CD4+ cells transduced with the MSCV-IRES-GFP retroviral vector (MIG) encoding GFP alone, GFP-Runx1, GFP-Runx2 or GFP-Runx3 constructs (a) or transfected by nucleofection with Runx1-, Runx2- or Runx3-specific siRNA or control (‘scrambled’) siRNA (b), then cultured in TH-17-polarizing conditions, followed by intracellular staining 4 d after the first transduction (5 d after activation). Plots are gated on GFP+ cells. Numbers in outlined areas indicate percent IL-17+ cells. FSC, forward scatter. Data are representative of three independent experiments. (c) Runx1 and the Runx1DN construct, including the Runt domain, activation domain (AD) and inhibition domain (ID). (d) Flow cytometry of IL-17 production by purified CD4+ T cells transduced with retroviral vectors encoding GFP alone, GFP-Runx1 or GFP-Runx1DN, then cultured in TH-17-polarizing conditions and stained 4 d after the first transduction (5 d after activation). Numbers in quadrants indicate percent cells in each. Data are representative of at least three independent experiments. (e) Flow cytometry of IL-17 in purified naive CD4+ cells transduced with retroviral vector containing control shRNA (LMP control) or Runx1-specific shRNA (LMP Runx1-shRNA), then cultured in TH-17-polarizing conditions and stained on day 5. Plots are gated on GFP+ cells. Numbers in outlined areas indicate percent IL-17+ cells. Data are representative of three independent experiments.
As Runx1, Runx2 and Runx3 are similar in structure, it is possible that overexpression studies may not be the best way to measure their specific functions. Thus, we attenuated expression of the individual Runx proteins with small interfering RNA (siRNA). First, we confirmed that each of the designed siRNA duplexes specifically affected its target Runx transcript (Supplementary Fig. 5a online). We then transfected the individual Runx-specific siRNA duplexes into T cells cultured in TH-17 conditions and evaluated IL-17 expression. Silencing of Runx1 led to a much lower percentage of IL-17+ cells, whereas silencing of Runx2 had only a modest effect and silencing of Runx3 had no effect (Fig. 4b). These results collectively indicate that although all Runx proteins are expressed in TH-17 cells, Runx1 is probably the main Runx family member involved in IL-17 expression. These results are also consistent with our reporter experiments in which Runx1 maximally enhanced the transcription mediated by the Il17 2-kb promoter fragment (Supplementary Fig. 2b).
To further confirm the function of Runx1 in the differentiation of primary TH-17 cells, we cultured CD4+ T cells in TH-17 polarizing conditions and transduced them with Runx1 or dominant negative Runx1 (Runx1DN) constructs tagged with green fluorescent protein (GFP) and containing an internal ribosomal entry site (IRES); the Runx1DN construct encoded a Runx1 protein lacking the carboxy-terminal transcriptional activation and inhibition domains16 (Fig. 4c). Consistent with our siRNA data, a much higher percentage of Runx1-transduced cells produced IL-17 whereas the percentage of IL17-producing cells among Runx1DN-transduced cells was much lower (Fig. 4d).
Finally, we ‘knocked down’ endogenous Runx1 expression with a short hairpin RNA (shRNA) retroviral vector. Runx1 expression was decreased specifically by Runx1 shRNA but not by the vector containing a control hairpin; in addition, this Runx1-specific shRNA did not influence the expression of Runx2 or Runx3 (Supplementary Fig. 5b). The proportion of IL17+ cells was lower among cells transduced with Runx1-specific shRNA than among cells transduced with the control shRNA vector (Fig. 4e). Collectively, these results demonstrate that Runx1 is required for TH-17 differentiation.
Runx1 acts together with RORγt to activate Il17
We next sought to understand the interaction between Runx1 and RORγt in TH-17 differentiation. We transduced CD4+ T cells with Runx1 or Runx1DN and cultured them with no cytokine or with IL-6 or TGF-β alone or together. In cells cultured without cytokine or with IL-6 (conditions in which endogenous RORγt is not induced), overexpression of Runx1 had no effect on IL-17 production (Fig. 5a). However, in cells cultured with TGF-β, which can induce RORγt expression23, overexpression of Runx1 lead to enhanced IL-17 expression (Fig. 5a). Thus, the greater number of IL-17-producing cells among Runx1-transduced cells cultured with TGF-β may have been due to ‘collaboration’ between Runx1 and RORγt. When we cultured cells in TH-17-polarizing conditions of IL-6 and TGF-β, in which large amounts of endogenous RORγt were produced, ectopic expression of Runx resulted in a much higher fraction of cells producing IL-17. To further demonstrate that Runx1 acts together with RORγt to induce IL-17 production, we overexpressed RORγt and Runx1 in T cells cultured in both TH0 conditions (no cytokines) and TH-17 conditions. In each condition, RORγt-induced IL-17 production was further enhanced when Runx1 was coexpressed (Fig. 5b). In contrast, RORγt-induced IL-17 production was lower when Runx1DN was coexpressed. These results collectively indicated that Runx1 acts together with RORγt to potentiate IL-17 expression and is required for the full effect of RORγt on IL-17 expression. In addition, RORγt-induced IL-17 production was also enhanced when RORγt was coexpressed with Runx2, albeit to a lesser extent than when it was coexpressed with Runx1, whereas we noted only marginal enhancement with coexpression of Runx3 (Supplementary Fig. 6 online).
Figure 5. Runx1 acts together with RORγt to induce IL-17 production.

(a) Flow cytometry of IL-17 production by purified naive CD4+ T cells transduced with retroviruses as described in Figure 4d and then cultured in the presence of various cytokines (above plots), followed by intracellular staining on day 4 after the first transduction (day 5 after activation). Numbers in quadrants indicate percent cells in each. (b) Flow cytometry of IL-17 expression in purified naive CD4+ T cells transduced with various combinations of retrovirus expressing GFP alone (MIG), Thy-1.1 alone (MIT), Thy-1.1-RORγt, GFP-Runx1 or GFP-Runx1DN, then cultured in TH0- or TH-17-polarizing conditions and analyzed as described in Figure 4a. Plots are gated on GFP+Thy-1.1+ cells. Numbers in outlined areas indicate percent IL-17+ cells. Data are representative of at least three independent experiments.
To gain further insight into how Runx1 and RORγt regulate IL17 transcription, we did chromatin immunoprecipitation (ChIP) assays to determine if these factors bind to the Il17 promoter and/or enhancer during TH-17 differentiation. In an initial study, we used ectopically expressed Flag-tagged Runx1 and Myc-tagged RORγt and detected the binding of these two transcription factors to the Il17 promoter and enhancer with antibody to Flag (anti-Flag) or anti-Myc, respectively. Both RORγt and Runx1 showed a positive ChIP signal for the Il17 promoter and the CNS-5 enhancer region but no substantial binding to the CNS-19 region (Fig. 6a,b). We also used anti-Runx1 and anti-RORγ to examine the binding of endogenous Runx1 and RORγt to the Il17 promoter and enhancers in TH-17 cells. The Runx1-specific antibody did not cross-react with either Runx2 or Runx3 (Supplementary Fig. 3a). As expected, we detected binding of RORγt and Runx1 to the Il17 promoter and CNS-5 enhancer but not to CNS-19 (Fig. 6c,d). These results indicated that the recruitment of both RORγt and Runx1 to both promoter and enhancer regions of Il17 might be necessary for optimal activation of IL-17 expression.
Figure 6. RORγt and Runx1 bind to the Il17 promoter and enhancer.

(a,b) Transcription factor binding in purified CD4+ T cells transduced with retrovirus expressing GFP-Myc-RORγt (a) or GFP-Flag-Runx1 (b) as described in Figure 4a, then cultured for 4 d in TH-17-polarizing conditions and restimulated for 4 h with PMA and ionomycin, and then assessed by ChIP with anti-Myc (a) or anti-Flag (b). (c,d) Binding of endogenous transcription factors in purified CD4+ T cells activated and cultured for 4 d in TH-17-polarizing conditions and restimulated for 4 h with PMA and ionomycin, then assessed by ChIP with anti-RORγ (c) or anti-Runx1 (d). Results are presented as the copy number of genomic DNA detected by real-time PCR in each immunoprecipitation relative to a standard dilution of input DNA. Data are representative of three independent experiments (mean and s.d. of triplicate immunoprecipitations for each antibody).
An alternative mechanism by which Runx1 may regulate IL-17 expression is by inducing RORγt expression. To investigate that possibility, we transduced CD4+ T cells with empty GFP vector or the GFP-tagged Runx1 or Runx1DN retrovirus construct and cultured the cells in TH-17-polarizing conditions. We sorted GFP+ cells and restimulated them with anti-CD3 and anti-CD28, then measured the expression of RORγt and IL-17 by real-time RT-PCR. The expression of RORγt and IL-17 was much higher in cells transduced with Runx1 and lower in cells transduced with Runx1DN than in cells transduced with empty GFP vector (Fig. 7a). To further confirm that finding, we transfected purified CD4+ cells with Runx1-specific siRNA or ‘scrambled’ siRNA (as in Fig. 4b) and measured the expression of Runx1, RORγt and IL-17 by real-time RT-PCR at 48 h after transfection. The expression of Runx1, RORγt and IL-17 was lower, but the expression of Runx2 and Runx3 remained the same in cells transfected with Runx1-specific siRNA (Fig. 7b and data not shown). These results provide evidence that an additional mechanism by which Runx1 regulates TH-17 differentiation is by inducing RORγt expression.
Figure 7. Runx1 promotes RORγt expression.

(a) Real-time RT-PCR of the expression of RORγt and IL-17 in purified CD4+ T cells activated and transduced with retrovirus expressing GFP alone, GFP-Runx1 or GFP-Runx1DN, then cultured in TH-17-polarizing conditions; GFP+ cells were then purified by flow cytometry, recultured overnight in TH-17-polarizing conditions, washed and then restimulated for 6 h with anti-CD3 plus anti-CD28. (b) Real-time RT-PCR of the expression of Runx1, RORγt and IL-17 in purified CD4+ cells transfected with scrambled siRNA or Runx1-specific siRNA as described in Figure 4b, then cultured for 48 h in TH-17-polarizing conditions. Results are presented relative to HPRT expression. Data are representative of three independent experiments (mean and s.d. of triplicates).
Foxp3 represses RORγt- and Runx1-induced IL-17 expression
Studies have shown that TGF-β induces the expression of RORγt23 and Foxp3 (refs. 23,24). RORγt enhances IL-17 expression, whereas Foxp3 inhibits IL-17 expression15. Other studies have shown that Foxp3 physically interacts with RORγt during the regulation of IL-17 induction15 and with Runx1 during the inhibition of IL-2 expression16. We therefore hypothesized that Foxp3 controls the ability of Runx1 to functionally interact with RORγt during TH-17 differentiation. To investigate that possibility, we first assessed the effect of Foxp3 on RORγt-induced IL-17 expression. We cotransduced CD4+ T cells with RORγt and Foxp3 or one of four mutant Foxp3 constructs; three Foxp3 mutants were carboxy-terminal truncations and the fourth mutant (Foxp3 329VHL) contained three point substitutions in the Runx1-binding site16 (Fig. 8a). We cultured the transduced cells in TH-17 or TH0 conditions and assessed IL-17 expression. Wild-type Foxp3 suppressed RORγt-induced IL-17 expression, but the suppressive activity of Foxp3 was abrogated by substitution of its Runx1-binding site (Foxp3 329VHL) or by removal of the carboxy-terminal forkhead DNA-binding domain (Fig. 8b). Next, we determined the effect of Foxp3 on Runx1-induced IL-17 expression. We transduced CD4+ T cells with Runx1 alone or Runx1 in combination with wild-type or mutant Foxp3 and cultured the cells in TH-17-polarizing conditions. Wild-type Foxp3 inhibited Runx1-induced IL-17 expression, but this inhibitory effect of Foxp3 was also abrogated by substitution of the Runx1-binding domain or by truncation of the carboxy-terminal forkhead DNA-binding domain (Fig. 8c). These results suggested that both the Runx1-binding site and forkhead DNA-binding domain are required for the suppressive effect of Foxp3 on IL-17 expression.
Figure 8. Interaction of Runx1, RORγt and Foxp3 regulates TH-17 cell differentiation.
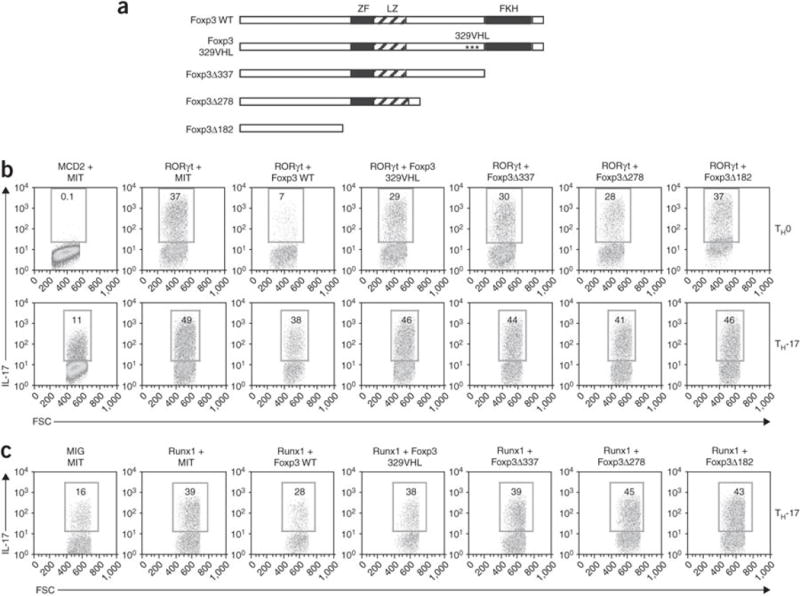
(a) Intact Foxp3 (wild-type (WT)) and mutant Foxp3 constructs, including the location of the zinc finger (ZF), leucine zipper (LZ) and forkhead (FKH) domains. Foxp3 329VHL contains the substitutions D329V, Y330H and K332L; Δ337, Δ278 and Δ182 indicate carboxy-terminal deletions (number indicates position at which deletion ends). (b) Flow cytometry of purified naive CD4+ cells activated and transduced as described in Figure 5b with various combinations of the MSCV-IRES-human CD2 (MCD2) retrovirus encoding human CD2 alone, MSCV–IRES–Thy-1.1 (MIT) encoding Thy-1.1 alone, human CD2–RORγt, Thy-1.1–Foxp3 or Thy-1.1–mutant Foxp3, then cultured in TH-17- or TH0-polarizing conditions and analyzed 4 d after transduction. Plots are gated on human CD2–positive Thy-1.1+ cells. (c) Flow cytometry of purified naive CD4+ T cells activated and transduced as described in Figure 5b with various combinations of retrovirus encoding GFP alone (MIG), Thy-1.1 alone (MIT), GFP-Runx1, Thy-1.1–Foxp3 or Thy-1.1–mutant Foxp3, then cultured in TH-17-polarizing conditions and analyzed 4 d after transduction. Plots are gated on GFP+Thy-1.1+ cells. Numbers in outlined areas (b,c) indicate percent IL-17+ cells. Data are representative of at least three independent experiments.
Having noted a functional interaction between RORγt and Runx1 and also between Foxp3 and RORγt, we next determined if RORγt and Runx1 physically interact with each other. We cotransfected 293T cells with Flag-tagged Runx1 (or Runx2 or Runx3) and Myc-tagged RORγt and immunoprecipitated cell lysates with anti-Myc or anti-Flag. Flag-tagged Runx1 (or Runx2 and Runx3) and Myc-tagged RORγt were coimmunoprecipitated, even in the presence of the DNA intercalator ethidium bromide (Fig. 9a and Supplementary Fig. 7 online), which suggested that these two proteins interact in the absence of DNA. We then examined the interaction between endogenous RORγt and Runx1 in TH-17 cells. In the presence of ethidium bromide, anti-Runx1 coimmunoprecipitated RORγt from lysates of CD4+ cells cultured in TH-17-polarizing conditions (Fig. 9b).
Figure 9. RORγt binds to both Runx1 and Foxp3.

(a) Immunoblot (IB) analysis of Runx1 and RORγt in 293T cells transfected with Flag-Runx1 and Myc-RORγt constructs with or without ethidium bromide (EtBr), detected in lysates immunoprecipitated (IP) with anti-Flag or anti-Myc. (b) Immunoassay of purified CD4+ T cells cultured for 4 d in TH-17-polarizing conditions; lysates prepared in the presence of ethidium bromide were immunoprecipitated with IgG isotype-matched control antibody or anti-Runx1, then analyzed by immunoblot with anti-RORγt or anti-Runx1. (c,d) Immunoassay of 293T cells transfected with Myc-tagged RORγt and Flag-tagged wild-type Foxp3 (c,d) or mutant Foxp3 (d); lysates immunoprecipitated with anti-Flag or anti-Myc were analyzed by immunoblot with anti-Flag or anti-Myc. Results are representative of three independent experiments.
We next assessed the interaction between RORγt and wild-type versus mutant Foxp3. We cotransfected 293T cells with Flag-tagged Foxp3 and Myc-tagged RORγt then did reciprocal coimmunoprecipitation of cell lysates, followed by immunoblot analysis with anti-Flag and anti-Myc. RORγt and wild-type Foxp3 were coimmunoprecipitated (Fig. 9c), which indicated a biochemical association between these two proteins. With a similar approach, we found that Flag-Foxp3 329VHL, as well as Flag-Foxp3Δ337 (a forkhead-truncated deletion mutant), still interacted with RORγt despite their substitutions and deletions (Fig. 9d), even though these constructs did not inhibit IL-17 transcription (Fig. 8b). This correlated with the functional data reported above suggesting that the ability of Foxp3 to inhibit IL-17 transcription is related to its interaction with Runx1 as well as RORγt (Supplementary Fig. 8 online)
Discussion
Here we have reported that Il17 transcription is governed by sequences present in the 2-kb promoter fragment upstream of the transcription start site, including sequences upstream of the RORγt-binding site. In addition, we have shown that Il17 transcription also depends on at least one conserved noncoding (enhancer) sequence (CNS-5) in the Il17 locus. These transcriptional control regions are involved in Il17 transcription, as they contain binding sites not only for RORγt but also for Runx1, which we found had a considerable potentiating effect on Il17 transcription. We found that Runx1 exerted no influence on Il17 transcription in the absence of RORγt and that Runx1 bound to RORγt during the course of TH-17 differentiation. In addition, we demonstrated that RORγt- and Runx1-induced IL-17 expression was inhibited by Foxp3 and that such inhibition required the binding of Foxp3 to Runx1. These findings allow us to propose that a complex three-way interaction among RORγt, Foxp3 and Runx1 is a chief regulator of the immune responses of both proinflammatory TH-17 cells and anti-inflammatory Treg cells.
As mentioned above, extensive data from reporter assays showed that transcriptional control through the 2-kb promoter required the influence of at least one distant enhancer site for maximum IL-17 expression. It is well known that such promoter-enhancer communication could occur through the interaction of proteins binding to each of these elements, which then leads to the ‘looping out’ of intervening DNA and the creation of a promoter-enhancer ‘holocomplex’. This ‘holocomplex’ could then maintain a high local concentration of transcription factors and establish a favorable environment at the promoter that facilitates maximum gene transcription25. This seemed to be true in the case of Il17 transcription, as mutation of the RORγt-binding site in the promoter or the enhancer resulted in much less Il17 transcription and because RORγt and Runx1, which bind to promoter, also bound to the enhancer; in addition, coimmmunoprecipitation studies indicated that RORγt and Runx1 underwent direct physical interaction in developing TH-17 cells.
One of the important aspects of Runx1 transcriptional activity in terms of IL-17 expression is its absolute dependence on the presence RORγt. Thus, in studies of CD4+ T cells, ectopic Runx1 did not enhance IL-17 expression in TH0 conditions (in the absence of TGF-β and IL-6 and thus in the absence of RORγt); in contrast, ectopic Runx1 did potentiate IL-17 expression in TH0 cells that also expressed ectopic RORγt. However, such ‘collaboration’ between Runx1 and RORγt at the time of Il17 transcription is probably not the only way that Runx1 affects IL-17 expression, as ectopic Runx1 resulted in higher IL-17 expression in cells exposed only to TGF-β, which suggests that Runx1 not only acts together with previously synthesized RORγt but also induces new RORγt synthesis. That possibility was supported by studies showing that overexpression of Runx1 resulted in considerably more RORγt synthesis and overexpression of Runx1DN resulted in less RORγt synthesis in developing TH-17 cells; furthermore, RORγt production was much lower in TH-17 cells subjected to siRNA-mediated depletion of Runx1. These findings indicate that Runx1 promotes transcription of the gene encoding RORγt in TH-17 conditions, and they are consistent with the presence of a Runx1-binding site in the promoter of the gene encoding RORγt26 and with the finding that transcription of the gene encoding RORγt is induced substantially by TGF-β23, a cytokine whose signaling has been shown to be related to Runx family members20, 21, 22.
The Runx family of transcription factors consists of three highly homologous proteins, each containing a very similar ‘runt’ DNA-binding site27, 28, 29, 30. Given that all three family members are expressed in TH-17 cells, we did several studies to determine which members were involved in Il17 transcription. We found that silencing of Runx1 expression strongly suppressed IL-17 expression, whereas silencing of Runx2 had a marginal effect and silencing of Runx3 almost no effect. Those results correlated with the results of studies in which we overexpressed the various Runx family members in TH-17 or TH0 cells either alone or with RORγt. In these studies, Runx2 had a considerable capacity to upregulate RORγt-induced IL-17 expression, whereas Runx3 had only a marginal effect most evident when RORγt was also overexpressed in TH17 conditions. We therefore conclude that Runx1 is the main Runx family member involved in TH-17 development and that Runx2 may also be involved but to a considerably lesser extent than Runx1; Runx3, in contrast, seems to have little or no involvement in IL-17 expression. In cells in which Runx and RORγt were overexpressed, we found that Runx2 and Runx3, as well as Runx1, physically interacted with RORγt in coimmunoprecipitation studies; thus, either such binding is nonphysiological or Runx activity in terms of IL-17 transcription involves more than Runx1 binding. Further studies of the function of Runx family members in Il17 transcription should use gene-targeted mice deficient in one or more Runx family member(s).
Our data add greater complexity to the understanding of the molecular basis of the reciprocal relation between TH-17 and Treg differentiation. It is now apparent that Runx1 must be included in the ‘mix’ of factors that regulate such differentiation. We have shown here that RORγt bound to both Foxp3 and Runx1, and published work has demonstrated that Foxp3 and Runx1 interact16. These interactions ‘set the stage’ to allow shifting of the direction of T cell differentiation according to extracellular and environmental conditions. Thus, in cells stimulated only by TGF-β (as in Treg cell–polarizing conditions), Foxp3 is produced in relatively large amounts and the main pathway is probably characterized by the interaction of Foxp3 with both Runx1 and RORγt. In these conditions, Foxp3 inhibits RORγt-mediated IL-17 transcription either directly in the form of a Foxp3-Runx1 complex or indirectly by blocking the ability of Runx1 to enhance RORγt-mediated Il17 transcription. That view is supported by the finding that overexpression of Foxp3 329VHL, the Foxp3 mutant with an impaired ability to interact with Runx1, did not inhibit RORγt-induced IL-17 expression, even though it was still able to bind to RORγt. In contrast, in cells stimulated in the presence of TGF-β and IL-6 or IL-21 (as in TH-17 conditions), Foxp3 synthesis is inhibited24, 31, and the main pathway is probably the binding of Runx1 to RORγt and subsequent enhancement of Il17 transcription. Finally, Runx-mediated transcription has been shown to be context dependent, which means that Runx factors recruit other transcription factors and these complexes act together to induce promoter activation or repression. Given published work showing that Foxp3 functions in tandem with the transcription factor NFAT27, 32, it is possible that interactions between Runx1 and Foxp3 and those between Runx1 and RORγt also involve a higher order of transcription factor assembly including NFAT.
The essential role of Runx1 in Foxp3 function16 and Treg cell development, as well as in RORγt function and IL-17 expression, as shown here, provides a possible basis for the finding that single-nucleotide polymorphisms affecting the consensus binding site for Runx1, or Runx1 itself, are associated with susceptibility to several autoimmune diseases, including systemic lupus erythematosus, rheumatoid arthritis and psoriasis33, 34, 35. It seems reasonable to suspect these genetic abnormalities influence susceptibility because they might cause changes in Runx1 function that ‘tip the balance’ between regulatory and effector cells in favor of effector cells. Thus, understanding the molecular basis of the interaction of Runx1, RORγt and Foxp3 in the development of Treg and TH-17 cells may provide insight into clinical immune pathologies.
Methods
Mice
BALB/c mice 8–12 weeks of age (Jackson Laboratories) were maintained in specific pathogen–free conditions. Studies followed a protocol approved by the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases.
Cell lines
Jurkat cells (American Type Culture Collection) were maintained in RPMI-1640 medium supplemented with 10% (vol/vol) FCS and antibiotics (penicillin and streptomycin; Gibco). HEK293 cells and Phoenix cells (American Type Culture Collection) for retroviral packaging were maintained in DMEM with 10% (vol/vol) FCS and antibiotics.
Plasmids
The Flag–Runx1–mouse stem cell virus (MSCV) retroviral vector and pCMV-Tag2-Foxp3 plasmid were provided by M. Ono16. The Myc-RORγt-GFP-S-003 plasmid was provided by S. Kersh26. Full-length wild-type Foxp3 cDNA and the Foxp3 mutant constructs were amplified by PCR and were cloned into MSCV–IRES–Thy-1.1 (MIT) retroviral vector. The Foxp3 329VHL construct was made with the Quickchange II site-directed mutagenesis kit (Stratagene). Thy-1.1–RORγt was made by cloning of the RORγt fragment released from RORγt–human CD2-MSCV provided by Y. He36 into the MSCV–Thy-1.1 vector.
Promoter and CNS promoter reporter constructs
The 0.6-kb, 1.1-kb and 2-kb fragments of the Il17 proximal promoter were obtained by PCR with mouse genomic DNA as template. The PCR products were cloned into the pGL4.10 basic luciferase vector (Promega) through the use of the NheI and BglII entry sites. CNS elements were generated by PCR, then were ligated into upstream of the 2-kb, 1.1-kb or 0.6-kb promoter fragments in the pGL4.10 luciferase vector. Mutations in the promoter and CNS were introduced with the Quickchange II site-directed mutagenesis kit (Stratagene; PCR primers, Supplementary Table 1 online). All constructs were verified by sequencing.
Luciferase reporter assay
Jurkat cells were transfected with a Nucleofector II (Amaxa). In a typical study, 3 × 106 cells in 100 μl Amaxa solution V were transfected with 2 μg luciferase reporter construct and 0.1 μg pRL-TK plasmid along with 2 μg plasmid encoding RORγt or Runx1 or control plasmid. Transfected cells were incubated overnight, then were stimulated for 6 h with 20 nM PMA (phorbol 12-myristate 13-acetate) and 2 μM ionomycin. Cells were lysed and luciferase activity was measured with a dual luciferase assay system (Promega). Each transfection was done in triplicate.
Retroviral transduction and intracellular staining
For the production of ecotropic retroviruses, Phoenix cells were transfected by Lipofectamine 2000 (52887; Invitrogen) with retroviral constructs (20 μg for single-plasmid transfection and 10 μg each for double-plasmid transfection, per 10-cm plate), and 48 h after transfection, virus supernatants were collected and were filtered through 0.45-μm low-protein-binding membranes. For transduction of virus, naive CD4+ cells were first isolated by negative selection, then were positively selected with CD62L magnetic beads (Miltenyi) and were cultured for 16–24 h at a density of 1 × 106 cells per well in nonpolarizing conditions (no added cytokines) in wells precoated with anti-CD3 (2C11; 2 μg/ml; BD Biosciences) and anti-CD28 (37.51; 1 μg/ml; BD Biosciences). These activated cells were then transduced with retrovirus supernatant by centrifugation for 1 h at 2,000g in the presence of polybrene (8 μg/ml; Sigma). After removal of the virus-containing supernatants, cells were recultured in TH0 conditions (no cytokines and no antibody) or TH-17-polarizing conditions (10 ng/ml of IL-6 (406ML) plus 2.5 ng/ml of TGF-β (240-B); R&D systems) or other conditions. The next day, cells were again transduced with retrovirus supernatant and recultured as before. After 4 d of culture, cells were restimulated for 5 h with 20 nM PMA (Sigma) and 1 μM ionomycin with the addition of GolgiStop (BD Bioscience) during the final 3 h. Intracellular staining and flow cytometry were then done as described37.
RNA-mediated interference
For knockdown of Runx1, Runx2 or Runx3, CD4+ T cells were purified with the CD4 Pan T Cell Isolation kit (Miltenyi) and were directly transfected by nucleofection with siRNA specific for Runx1, Runx2 or Runx3, respectively (all from Dharmacon; predesigned ON-TARGETplus SMARTpool siRNA) or ‘scrambled’ control siRNA (Dhamacon). For transfection, 2 × 106 cells in 100 μl mouse T cell Nucleofector solution (Amaxa) were transfected with 300 pmol total of siRNA with the X-001 program (Amaxa). After transfection, cells were incubated for 4 h at 37 °C, then were activated with anti-CD3 (2 μg/ml) and anti-CD28 (1 μg/ml) and were cultured in TH-17-polarizing condition as described above. After 48 h of activation, some transfected cells were collected and RNA was made; RNA was then analyzed by real-time RT-PCR. After a total of 72 h of activation, the remaining cells were allowed to ‘rest’ for another 48 h and then were restimulated with PMA and ionomycin, and IL-17 production was measured as described above. For Runx1 shRNA knockdown, double-stranded DNA short hairpin sequence targeting the coding region of Runx1 (5′-ATCACTGGCGCTGCAACAAGAC-3′) or control shRNA (5′-AATGAAGATCAAGATCATTGCG-3′) was cloned into the MSCV-LTRmiR30-PIG (LMP) retroviral vector (Openbiosystems), which encodes GFP and a puromycin selection marker, according to the manufacturer’s instruction. Retroviral production and transduction was done as described above. After 4 d of culture, cells were restimulated for 5 h with PMA and ionomycin with GolgiStop added during the final 3 h. Intracellular staining was then done as described37. For transduction of the mouse lymphoma EL4 cell line, 1 × 106 cells were transduced with retroviral supernatant containing either control shRNA or Runx1-specific shRNA as described above, then puromycin (5 μg/ml) was added to the culture medium immediately after transduction. GFP+ cells were identified by flow cytometry, cells were collected and RNA was made after over 90% of the cells were GFP+.
ChIP
These assays were done as described37. Cells were fixed for 20 min on ice with 1% (vol/vol) fomaldehyde, and glycine was added to a final concentration of 0.125 M. Cells were washed with ice-cold PBS and were resuspended for 10 min in lysis buffer. Nuclei were washed with MNase digestion buffer without CaCl2, then were digested with MNase (Roche) and sonicated to obtain chromatin fragments 200–500 base pairs in length. These chromatin fragments were diluted with ChIP dilution buffer and the chromatin was precleared with protein A or G beads (Millipore) in the presence of immunoglobulin G (IgG). Chromatin was then incubated overnight at 4 °C with anti-Runx1 (39000; Active Motif), anti-Flag (F316; Sigma), anti-Myc (05-724; Millipore) or anti-RORγ (28559; Santa Cruz), after which protein A or G agarose beads were added, followed by incubation for another 2 h at 4 °C. Beads were washed extensively with wash buffer, and bead-protein-DNA complexes were then digested overnight at 56 °C with proteinase K. DNA was extracted with phenol-chloroform and ethanol precipitation in the presence of 20 μg glycogen. Finally, extracted DNA was analyzed by real-time PCR (primer pairs and probes, Supplementary Table 1 online).
Coimmunoprecipitation and immunoblot analysis
HEK293 cells in a 10-cm plate were transfected with pCMV-Tag2-Runx1 (10 μg), Myc-RORγt-S003 (10 μg), pCMV-Tag2-Foxp3 (5 μg) or with the appropriate empty vector. For each plate, at 24 h after transfection, cells (10 × 106) were lysed in 1 ml lysis buffer (80 mM NaCl and 0.5% (vol/vol) Nonidet-P40) in the presence or absence of ethidium bromide (10 μg/ml). For immunoprecipitation, whole-cell lysates were precleared with normal IgG and then were incubated overnight at 4 °C with anti-Flag or anti-Myc, then protein G beads were added, followed by incubation for additional 1 h. Immunocomplexes were extensively washed and then were resuspended in SDS loading buffer. Immunoblot analysis was done as described37. For coimmunoprecipitation of endogenous Runx1 and RORγt, CD4+ cells were purified and polarized for 4 d, lysates were made in the presence of ethidium bromide (10 μg/ml) in lysis buffer, then 1.5 mg of protein was precleared with normal IgG and incubated overnight at 4 °C as described above with anti-Runx1 (5 μg; 39000; Active Motif). Monoclonal anti-RORγt (a gift from D. R. Littman) or monoclonal anti-Runx1 (255019; MAB2399; R&D Systems) was used for immunoblot analysis. For immunoblot of Runx1, Runx2 and Runx3, cell lysates made as described above were analyzed with anti-Runx1 (39000 (Active Motif) or MAB2399 (R&D Systems)), anti-Runx2 (sc-10758; Santa Cruz Biotechnology) or anti-Runx3 (39301; Active Motif).
Isolation of mRNA and quantitative real-time PCR
For Runx RT-PCR, CD4+ cells were purified and were cultured for 4 d in various conditions, then RNA was isolated with TRIzol (Gibco). For transduced cells, CD4+ T cells were transduced with GFP-tagged Runx1, Runx1DN or empty vector and were cultured in TH-17-polarizing conditions, then GFP+ cells were sorted by flow cytometry and were restimulated for 6 h with anti-CD3 and anti-CD28. Total RNA was isolated with TRIzol. For RT-PCR, total RNA was reverse-transcribed with the oligo(dT) primer Superscript III (Invitrogen) and the resultant cDNA was analyzed by quantitative Taqman real-time PCR on an iCycler (Bio-Rad). Samples were amplified in triplicate for 40 cycles. The starting quantity of the initial cDNA sample was calculated from primer-specific standard curves with iCycler data-analysis software. The expression of Runx, RORγt and IL-17 was normalized to the expression of HPRT (hypoxanthine guanine phosphoribosyl transferase). Primer sets and probes for RORγt, IL-17 and HPRT were used as described1, 16. Primer sets and probes for mouse Runx1 (Mm00486762), Runx2 (Mm00490666) and Runx3 (Mm00501580) were from ABI.
Acknowledgments
We thank S. Goenka for reading the manuscript and for technical suggestions for the reporter assay; M. Ono (Kyoto University) for Runx1 and Foxp3 expression plasmids; Y. He (Duke University) for the MSCV-hCD2-RORγt plasmid; G.J. Kersh (Emory University) for the Myc-RORγt-GFP plasmid; G.P. Nolan (Stanford University) for Phoenix cells; D.R. Littman (New York University) for monoclonal anti-RORγt; Y. Huang and Z.Y. Hu for technical support in real time PCR experiments; and X.Y. Yan for help with making reporter constructs.
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
Author contributions
F.Z. and W.S. designed experiments; F.Z. did all of the experiments and wrote the manuscript; G.M. contributed Supplementary Fig. 3 and was involved in coimmunoprecipitation experiments; and W.S. directed the project and helped to write the manuscript.
NOTE: In the version of this article initially published, two panels in Figure 9a were horizontally inverted. The error has been corrected in the HTML and PDF versions of the article.
References
- 1.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 2.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 3.Komiyama Y, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 4.Wong CK, et al. Proinflammatory cytokines (IL-17, IL-6, IL-18 and IL-12) and Th cytokines (IFN-γ, IL-4, IL-10 and IL-13) in patients with allergic asthma. Clin Exp Immunol. 2001;125:177–183. doi: 10.1046/j.1365-2249.2001.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang XO, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Mangan PR, et al. Transforming growth factor-β induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 8.Manel N, Unutmaz D, Littman DR. The differentiation of human TH-17 cells requires transforming growth factor- and induction of the nuclear receptor RORγt. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, et al. IL-21 and TGF-β are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang XO, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR and RORγ. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ansel KM, et al. Deletion of a conserved Il4 silencer impairs T helper type 1-mediated immunity. Nat Immunol. 2004;5:1251–1259. doi: 10.1038/ni1135. [DOI] [PubMed] [Google Scholar]
- 12.Lee DU, Avni O, Chen L, Rao A. A distal enhancer in the interferon-γ (IFN-γ) locus revealed by genome sequence comparison. J Biol Chem. 2004;279:4802–4810. doi: 10.1074/jbc.M307904200. [DOI] [PubMed] [Google Scholar]
- 13.Agarwal S, Rao A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity. 1998;9:765–775. doi: 10.1016/s1074-7613(00)80642-1. [DOI] [PubMed] [Google Scholar]
- 14.Akimzhanov AM, Yang XO, Dong C. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem. 2007;282:5969–5972. doi: 10.1074/jbc.C600322200. [DOI] [PubMed] [Google Scholar]
- 15.Zhou L, et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ono M, et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- 17.Djuretic IM, et al. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. 2007;8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 18.Solymar DC, Agarwal S, Bassing CH, Alt FW, Rao A. A 3′ enhancer in the IL-4 gene regulates cytokine production by Th2 cells and mast cells. Immunity. 2002;17:41–50. doi: 10.1016/s1074-7613(02)00334-5. [DOI] [PubMed] [Google Scholar]
- 19.Hatton RD, et al. A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity. 2006;25:717–729. doi: 10.1016/j.immuni.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Ito Y. Oncogenic potential of the RUNX gene family: ‘overview’. Oncogene. 2004;23:4198–4208. doi: 10.1038/sj.onc.1207755. [DOI] [PubMed] [Google Scholar]
- 21.Ito Y, Miyazono K. RUNX transcription factors as key targets of TGF-β superfamily signaling. Curr Opin Genet Dev. 2003;13:43–47. doi: 10.1016/s0959-437x(03)00007-8. [DOI] [PubMed] [Google Scholar]
- 22.Ito Y. RUNX genes in development and cancer: regulation of viral gene expression and the discovery of RUNX family genes. Adv Cancer Res. 2008;99:33–76. doi: 10.1016/S0065-230X(07)99002-8. [DOI] [PubMed] [Google Scholar]
- 23.Zhou L, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 24.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 25.Oestreich KJ, et al. Regulation of TCRβ gene assembly by a promoter/enhancer holocomplex. Immunity. 2006;24:381–391. doi: 10.1016/j.immuni.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 26.Xi H, Schwartz R, Engel I, Murre C, Kersh GJ. Interplay between RORγt, Egr3, and E proteins controls proliferation in response to pre-TCR signals. Immunity. 2006;24:813–826. doi: 10.1016/j.immuni.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 27.Hu H, Djuretic I, Sundrud MS, Rao A. Transcriptional partners in regulatory T cells: Foxp3, Runx and NFAT. Trends Immunol. 2007;28:329–332. doi: 10.1016/j.it.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 28.Taniuchi I, Littman DR. Epigenetic gene silencing by Runx proteins. Oncogene. 2004;23:4341–4345. doi: 10.1038/sj.onc.1207671. [DOI] [PubMed] [Google Scholar]
- 29.Taniuchi I, et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- 30.Levanon D, Groner Y. Structure and regulated expression of mammalian RUNX genes. Oncogene. 2004;23:4211–4219. doi: 10.1038/sj.onc.1207670. [DOI] [PubMed] [Google Scholar]
- 31.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 32.Wu Y, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 33.Prokunina L, et al. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat Genet. 2002;32:666–669. doi: 10.1038/ng1020. [DOI] [PubMed] [Google Scholar]
- 34.Helms C, et al. A putative RUNX1 binding site variant between SLC9A3R1 and NAT9 is associated with susceptibility to psoriasis. Nat Genet. 2003;35:349–356. doi: 10.1038/ng1268. [DOI] [PubMed] [Google Scholar]
- 35.Tokuhiro S, et al. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat Genet. 2003;35:341–348. doi: 10.1038/ng1267. [DOI] [PubMed] [Google Scholar]
- 36.He YW, Deftos ML, Ojala EW, Bevan MJ. RORγt, a novel isoform of an orphan receptor, negatively regulates Fas ligand expression and IL-2 production in T cells. Immunity. 1998;9:797–806. doi: 10.1016/s1074-7613(00)80645-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang F, Boothby M. T helper type 1-specific Brg1 recruitment and remodeling of nucleosomes positioned at the IFN-γ promoter are Stat4 dependent. J Exp Med. 2006;203:1493–1505. doi: 10.1084/jem.20060066. [DOI] [PMC free article] [PubMed] [Google Scholar]