

Abstract
Phosphatidylethanolamine (PE) is the second most abundant glycerophospholipid in eukaryotic cells. The existence of four only partially redundant biochemical pathways that produce PE, highlights the importance of this essential phospholipid. The CDP-ethanolamine and phosphatidylserine decarboxylase pathways occur in different subcellular compartments and are the main sources of PE in cells. Mammalian development fails upon ablation of either pathway. Once made, PE has diverse cellular functions that include serving as a precursor for phosphatidylcholine and a substrate for important posttranslational modifications, influencing membrane topology, and promoting cell and organelle membrane fusion, oxidative phosphorylation, mitochondrial biogenesis, and autophagy. The importance of PE metabolism in mammalian health has recently emerged following its association with Alzheimer's disease, Parkinson's disease, nonalcoholic liver disease, and the virulence of certain pathogenic organisms.
1. Introduction
Phosphatidylethanolamine (PE) is a multifunctional phospholipid required for mammalian development that is essential for a variety of cellular processes. PE is a nonbilayer forming phospholipid containing a small polar head group diameter in proportion to its fatty-acid chains. The intrinsic biophysical properties of this cone-shaped lipid induces the formation of hexagonal phases within the membrane and, in so doing, promotes membrane fusion and fission events, protein integration into membranes, and conformational changes in protein structure (Dowhan and Bogdanov, 2009; van den Brink-van der Laan et al., 2004). PE is the second most abundant phospholipid in the cell, comprising 15–25% of total phospholipids in mammalian cells (Vance, 2015). However, PE is not simply a passive membrane constituent but is functionally associated with protein biogenesis and activity (Becker et al., 2013; Bogdanov and Dowhan, 1995, 1998, 1999), oxidative phosphorylation (Bottinger et al., 2012; Tasseva et al., 2013), autophagy (Ichimura et al., 2000), membrane fusion (Verkleij et al., 1984), mitochondrial stability (Birner et al., 2001; Steenbergen et al., 2005; Storey et al., 2001), and is an important precursor of other lipids (Bremer and Greenberg, 1961; Menon and Stevens, 1992).
Four biosynthetic pathways produce PE in the cell, and notably, one of these pathways resides within the mitochondrion. The redundancy in PE biosynthetic pathways is not sufficient to allow for normal cellular function in the absence of either of the two major PE-producing pathways (Birner et al., 2001; Fullerton et al., 2007; Steenbergen et al., 2005; Storey et al., 2001). This suggests that different pools of PE are required for specified purposes in the cell. The abundance of PE varies in the membranes of different tissues and cells in mammals and organelles of both yeast and mammals (Bleijerveld et al., 2007; Colbeau et al., 1971; Nelson, 1967; Van Deenen and De Gier, 1974; Vance, 2015; Zinser et al., 1991). This review will focus on the numerous biological functions conferred by the intrinsic properties of PE. Recently, disturbances in PE metabolism have been implicated in both chronic and infectious disease (Chen et al., 2010; Deleault et al., 2012; Nesic et al., 2012; Wang et al., 2014). Phenotypic characterization of the cell biology of these diseases using a variety of model organisms collectively reveals a vital role for PE in mammalian health.
2. Heterogeneity of Biological Membranes
Biological membranes form the barriers that define cells and separate specified cellular functions into distinct but interconnected compartments. Beyond their ability to delineate different cell and organelle morphologies, cellular membranes are also multifunctional platforms involved in signaling, regulation of solute, metabolite, and protein transport; and are necessary mediums for proteins that require a hydrophobic environment for enzymatic function and stability. The wide range of biological processes mediated across membranes can be attributed to the mixture of proteins, lipids, and carbohydrates that concomitantly interact to give rise to specialized membrane environments. Greater than 1000 lipid species are present in the cell and over 30% of an organism's translated genome is dedicated to the production of alpha helical membrane proteins (Stevens and Arkin, 2000; Sud et al., 2007). With respect to carbohydrates, there are innumerable structures, conformations, and combinations of sugars that can be formed in the cell, which further add to the diversity of the membrane environment.
The major classes of lipids in the cell include phospholipids, sterols, and sphingolipids. The rigidity, thickness, hydrophobicity, and function of cellular membranes are dependent upon the presence and relative abundance of these different classes of lipid. Glycerophospholipids, sterols, and sphingolipids comprise ∼75%, 12–14%, and 8–12% of lipids in the cell, respectfully (Drin, 2014). Phospholipids are accountable for the formation of the membrane bilayer; the different classes of phospholipid in a membrane further modulate membrane identity and fluidity. Sterols, cholesterol in mammals, and ergosterol in yeast, decrease cell permeability by increasing membrane thickness and rigidity. Interestingly, the level of cholesterol is highest at the plasma membrane (20–40%), moderate in the Golgi (8%) and endoplasmic reticulum (ER) (6%), and scarcely detected in mitochondria (4%). The presence of sterols in conjunction with sphingolipids on the plasma membrane is important for cell-to-cell signaling events. As PE is the focus of this review, the biological importance of sterols and sphingolipids is beyond our scope but has been discussed in extensive detail in several fantastic reviews (Cowart and Obeid, 2007; Espenshade and Hughes, 2007; Futerman and Hannun, 2004; Hannun and Obeid, 2008; Mouritsen and Zuckermann, 2004; Ohvo-Rekila et al., 2002; Vance and Van den Bosch, 2000).
Phospholipids are the predominant lipid components of most cellular membranes and are typically characterized by a glycerol backbone containing two ester linked fatty acid chains at the sn-1 and sn-2 positions and a phosphate head group at the sn-3 position (Figure 1; Van Deenen and De Gier, 1974). The head group attached at the sn-3 position distinguishes the different classes of phospholipid while subspecies of each phospholipid class also arise from differences in their acyl chain composition. The major glycerophospholipids in the cell include phosphatidylcholine (PC), PE, phosphatidylserine (PS), phosphatidylinositol (PI), phosphatidic acid (PA), phosphatidylglycerol (PG), and cardiolipin (CL). The distribution of each phospholipid can vary on different leaflets of the membrane bilayer, between organellar membranes, and by cell type and organism (Bretscher, 1972; Colbeau et al., 1971; Van Deenen and De Gier, 1974; Zinser et al., 1991). A typical mammalian cell contains approximately 45–55% PC, 15–25% PE, 10–15% PI, 5–10% PS, 2–5% CL, and 1–2% PA (Vance, 2015). PC is found equally distributed across cellular membranes while PS and PE are primarily found on the inner but not the outer leaflet of the plasma membrane. In addition, PE and CL are particularly abundant in the inner membrane of mitochondria (Vance, 2015). Further, CL is absent in other nonmitochondrial membranes of the cell. Enrichment of lipids in different corners of the cell can be attributed to numerous factors including their different sites of synthesis, interconversion, acyl chain remodeling, trafficking mechanisms, and degradation.
Figure 1.
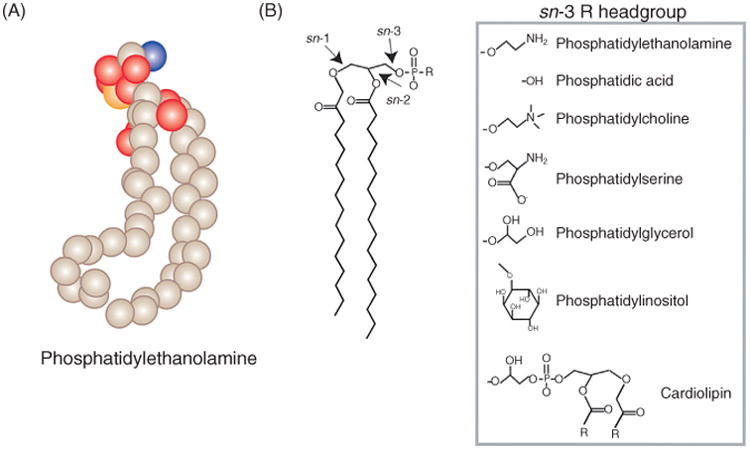
The glycerophospholipids. (A) Diagram of phosphatidylethanolamine structure. The spheres represent different atoms present in the phospholipid structure tan: carbon, red: oxygen, orange: phosphate, and blue: nitrogen (hydrogen atoms are not represented). (B) General glycerophospholipid structure. Fatty acids are linked to the glycerol backbone at the sn-1 and sn-2 positions while the phosphate headgroup is linked at the sn-3 position. Different variations of headgroups are shown (for cardiolipin, R indicates additional acyl groups attached at these positions).
The ER is the primary site of synthesis for the majority of lipids in the cell. Many essential cell processes are sequestered in the ER, and as such, this organelle has compartmentalized some of these functions into distinct domains (Vance, 2014). Initial studies on the subcellular localization of phospholipid synthesizing enzymes localized them to microsomal fractions, but some microsomal vesicles containing high PS synthase activity were not enriched for the known ER-specific marker, NADPH-cytochrome-c reductase (Dennis and Kennedy, 1972; van Golde et al., 1974; Zinser et al., 1991). Subsequently, the mitochondrial-associated membrane (MAM) of the ER was identified as a distinct site of lipid synthesis that harbors multiple phospholipid biosynthetic enzymes, including PS synthase, PI synthase, and PE methyl transferase (Cui et al., 1993; Gaigg et al., 1995; Vance, 1990). Additionally, the MAM is an important depot for the transport of substrates required for the biosynthesis of PE, PA, CDP-DAG, PG, and CL in mitochondria (transport mechanisms for PS and PE are covered in Sections 3.2.3 and 3.2.4) although CDP-DAG and PA can be synthesized in both the ER and mitochondria (Chen et al., 2006; Colbeau et al., 1971; Kuchler et al., 1986; Tamura et al., 2013; van Golde et al., 1974; Wirtz and Zilversmit, 1968; Yet et al., 1993).
3. PE Biosynthesis and Metabolism
There are four independent pathways by which PE is generated in eukaryotic cells (Figure 2). The CDP-ethanolamine pathway (Hjelmstad and Bell, 1991; Ishidate et al., 1985; Mancini et al., 1999; van Hellemond et al., 1994; Wittenberg and Kornberg, 1953), acylation of lyso-PE (Riekhof et al., 2007b), and head group base exchange reactions (Dennis and Kennedy, 1972) are sequestered in the ER while the phosphatidylserine decarboxylase (Psd) pathway is largely specific to mitochondria (Borkenhagen et al., 1961; Horvath et al., 2012; Tamura et al., 2012b; Zborowski et al., 1983). While mammals have only one Psd enzyme that is localized in the mitochondrion, yeast have both the mitochondrially localized Psd1p (Horvath et al., 2012; Tamura et al., 2012b) and the endosome localized Psd2p (Gulshan et al., 2010; Trotter and Voelker, 1995). Of these two enzymes, Psd1p is the major source of cellular decarboxylase activity in yeast (Trotter et al., 1995). The predominant pathways for PE biosynthesis are the Psd and CDP-ethanolamine pathways (Birner et al., 2001); the other two ER pathways (acylation of lyso-PE and head group base exchange) weakly contribute to the cellular pool of PE (Sundler et al., 1974; Zelinski and Choy, 1982). While the CDP-ethanolamine pathway produces a species of PE that is enriched with mono-or diunsaturated fatty acids in the sn-2 position, the mitochondrial Psd pathway generates PE species with polyunsaturated fatty acids in the sn-2 position (Bleijerveld et al., 2007). However, the functional difference between PE produced by the Psd and the CDP-ethanolamine pathways is not clear although such a difference(s) is presumed based on the fact that each pathway is required for mammalian development (Fullerton et al., 2007; Steenbergen et al., 2005). Preference for either the Psd or CDP-ethanolamine pathway varies between organisms and tissues within metazoans although both pathways have been conserved from prokaryotes to eukaryotes (Dowhan et al., 1974; Miller and Kent, 1986; Tijburg et al., 1989). There are numerous fates of newly synthesized PE. It can be integrated into membranes at its site of synthesis, targeted to other cellular compartments, used as a precursor for the production of another essential phospholipid, PC, or utilized as a substrate for the production of basic posttranslational modifications such as glycosylphosphatidylinositol (GPI) anchors (Bremer and Greenberg, 1961; Menon and Stevens, 1992).
Figure 2.
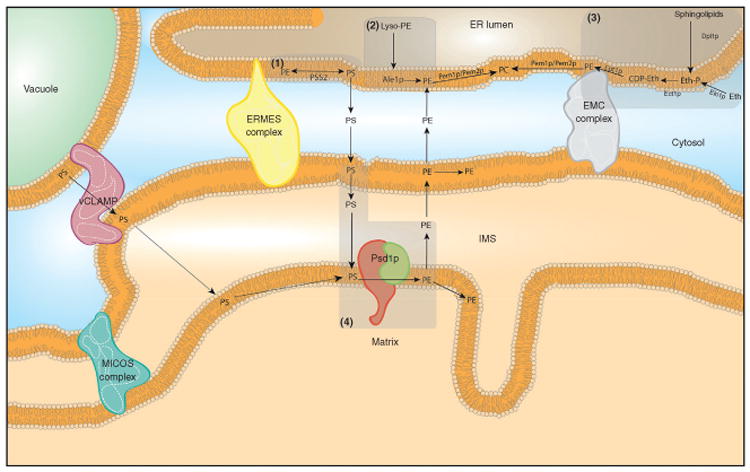
PE biosynthetic pathways at the ER–mitochondria interface in yeast. (1) Base exchange pathway. In the biosynthesis of PS, head group exchange with PE is mediated by PSS2 in mammals. The reverse reaction can also synthesize PE from PS in small amounts. In yeast, calcium mediates base exchange between PS and PE through poorly understood mechanisms. (2) Acylation of lyso-PE to PE. Ale1p is an acyl transferase that facilitates the conversion of lyso-PE to PE. (3) CDP-ethanolamine pathway or Kennedy pathway. Phosphoethanolamine (Eth-P) is generated by phosphorylation of ethanolamine (Eth) by ethanolamine kinase (Ek1p) or through degradation of sphingolipids by Dpl1p. Phosphoethanolamine and CTP are metabolized by CTP:phosphoethanolamine cytidylyltransferase (Ect1p in yeast, ET in mammals) to generate CDP-ethanolamine (CDP-Eth), which with 1,2-diacylglycerol ethanolamine phosphotransferase (Ept1p in yeast, ETP in mammals) undergoes a condensation reaction with DAG to form the final product, PE. (4) Phosphatidylserine decarboxylase pathway. Upon its synthesis, PS is transported from the MAM of the ER to the OM of mitochondria until it reaches the IM where Psd1p is located. EMC and ERMES may facilitate transfer of PS to the OM. The OM and IM of mitochondria are tethered by mitochondrial contact site and cristae organizing system (MICOS) structures (please refer to text for mammalian proteins that also serve tethering functions). Alternatively, PS can be transferred to mitochondrial membranes through the yeast vacoule facilitated by v-CLAMP membrane tethers. In the enzymatic step, Psd1p decarboxylates PS to generate PE that is integrated in mitochondrial membranes or exported to other locations in the cell. PE generated by any of these pathways can be converted to PC through the action of PE methyltransferases (Pem1p/Pem2p in yeast or PEMT in mammals).
3.1 ER Pathways
There are three distinct PE biosynthetic pathways in the ER. While the major CDP-ethanolamine pathway is in the bulk ER, the head group base exchange and lyso-PE pathways reside in the MAM subcompartment of the ER that is in close physical proximity to the mitochondrion (Stone and Vance, 2000). The PE produced by these ER pathways gains access to membranes throughout the endomembrane system via the secretory pathway. As the mitochondrion is not part of this system, transfer of any phospholipid, including PE, from the ER to the mitochondrion must occur through other mechanisms that have substrate specificity (e.g., some phospholipids such as PC move quickly in both directions (de Kroon et al., 2003), whereas others, including PE, move in one direction better than the other (Birner et al., 2001; Burgermeister et al., 2004; Vance, 1991)).
3.1.1 Kennedy or CDP-Ethanolamine Pathway
The CDP-ethanolamine pathway resides within the ER and is the preferential pathway for PE biosynthesis in hamster heart and rat liver (Miller and Kent, 1986; Tijburg et al., 1989; Zelinski and Choy, 1982). The CDP-ethanolamine pathway consists of three enzymatic steps. The first step involves the ATP-dependent phosphorylation of ethanolamine by ethanolamine kinase to form phosphoethanolamine with ADP released as a byproduct (Lykidis et al., 2001). Mice in which ethanolamine kinase has been deleted have decreased litter size and about 20% die prenatally. Phosphoethanolamine can also be generated through the action of dihydrosphingosine-1-lyase (Dpl1p), which degrades sphingosine-1-phosphate producing phosphoethanolamine and a fatty aldehyde (Gottlieb et al., 1999; Zhou and Saba, 1998). The second step of the CDP-ethanolamine pathway, which is considered to be rate-limiting, is catalyzed by the protein product of the PCYT2 gene, CTP:phosphoethanolamine cytidylyltransferase (ET; Nakashima et al., 1997). ET uses phosphoethanolamine and CTP to form the high-energy donor CDP-ethanolamine with the release of inorganic phosphate. PCYT2 mRNA is highly expressed in the heart, liver, and skeletal muscle (Fullerton et al., 2007) tissues with high specific activities for these enzymes (Miller and Kent, 1986; Tijburg et al., 1989; Zelinski and Choy, 1982). Deletion of pcyt2 is embryonically lethal and although the heterozygotes appear normal (Fullerton et al., 2007), they experience metabolic defects as adults and during ageing (Fullerton et al., 2009). In the final step of PE synthesis by the CDP-ethanolamine pathway, 1,2-diacylglycerol ethanolamine phosphotransferase (ETP) utilizes the energy provided by CDP-ethanolamine to attach ethanolamine to the membrane-embedded diacylglycerol (DAG) thus forming PE (Lykidis et al., 2001; Sundler, 1975; Sundler and Akesson, 1975a; Tijburg et al., 1987).
The CDP-ethanolamine pathway is a major PE producing pathway in eukaryotes. Mammals and yeast lack the ability to produce ethanolamine, a substrate required for PE formation via the CDP-ethanolamine pathway, de novo. Ethanolamine used for PE synthesis derives from the breakdown of existing PE, exogenously added ethanolamine, and through the action of Dpl1p, which generates phosphoethanolamine from sphingosine-1-phosphate (Gottlieb et al., 1999; Zhou and Saba, 1998). In addition, mammals acquire ethanolamine through the diet usually in the form of lipids (Gottlieb et al., 1999).
3.1.2 Acylation of Lyso-PE and Head Group Exchange
PE can also be formed via acylation of lyso-PE (Jain et al., 2007; Riekhof and Voelker, 2006; Riekhof et al., 2007a,b; Tamaki et al., 2007) and calcium-dependent head group exchange with pre-existing phospholipids (Bjerve, 1984; Sundler et al., 1974); these two forms of PE synthesis are considered minor pathways of PE production. Lyso-PE is brought in through the exogenous lysophospholipid metabolism pathway. This pathway can utilize dietary lyso-PE that is first translocated across the plasma membrane and then acylated (Riekhof and Voelker, 2006). The uptake of lyso-PE is mediated by the plasma membrane aminophospholipid translocases Dnf1p and Dnf2p and their obligate partner, Lem3p (Riekhof et al., 2007a). Following its uptake, lyso-PE is converted to PE by Ale1p, an acyl-CoA-dependent acyltransferase (Jain et al., 2007; Riekhof et al., 2007b; Tamaki et al., 2007). Ale1p activity is enriched in the MAM; however, how lyso-PE traffics from its point of entry/ production to the MAM is not known. PE formed from the acylation of lyso-PE can substitute for the PE produced by the CDP-ethanolamine pathway and to some extent, the Psd pathway, as long as Ale1p is functional and lyso-PE is present (Riekhof et al., 2007b). Thus, the PE made by this minor pathway can access the same membrane compartments as normally supplied by the CDP-ethanolamine pathway. Also, the PE made by both of these ER-based pathways has a limited capacity to replace PE made in the mitochondrion (Tasseva et al., 2013).
3.2 Mitochondrial Phosphatidylserine Decarboxylase (Psd) Pathway
In contrast to the three aforementioned PE biosynthetic pathways, the second major route for PE production, the Psd pathway, resides in mitochondria. In the mitochondrion, PS decarboxylation to PE is performed by a single protein, Psd1p, that is embedded in the mitochondrial inner membrane (IM; Borkenhagen et al., 1961; Horvath et al., 2012; Tamura et al., 2012b; Trotter et al., 1993; Zborowski et al., 1983). Approximately 90% of PE in yeast cells is produced by Psd1p (Birner et al., 2001). The Psd pathway is also the major source of PE in baby hamster kidney and CHO1 cell lines in vitro (Miller and Kent, 1986; Voelker, 1984). In mammals, the Psd pathway is essential for viability since deletion of the gene encoding PSD1, PISD, is lethal between murine embryonic days 8 and 10 (Steenbergen et al., 2005). Although PISD−/+ heterozygote brain, testes, and liver tissues have similar PE levels to wild type embryos, this reflects a compensatory increase in PE produced by the CDP-ethanolamine pathway. Still, this increase fails to substitute for the lack of mitochondrial PSD1 as evidenced by the embryonic lethality of pisd−/− mice. Consistent with the inability of the CDP-ethanolamine pathway to fully complement the mitochondrial need for PE, mitochondria in pisd−/− mouse embryonic fibroblasts are aberrantly shaped and fragmented (Steenbergen et al., 2005) and RNAi silencing of PISD in CHO1 cells impairs oxidative phosphorylation (Tasseva et al., 2013). Moreover, psd1Δ yeast have numerous mitochondrial defects even though they retain the ability to synthesize PE via a second Psd enzyme (Psd2p), the CDP-ethanolamine pathway, and Ale1p (Birner et al., 2001; Storey et al., 2001; Trotter and Voelker, 1995). The failure of PE made in the ER to fully support mitochondrial functions in the absence of Psd1p likely reflects its inefficient trafficking into mitochondrial membranes (Birner et al., 2001; Burgermeister et al., 2004; Riekhof et al., 2007b; Shiao et al., 1995) although analysis of the acyl chain composition of PE by mass spectrometry has shown that PE species synthesized by the CDP-ethanolamine pathway are incorporated into the IM in mammalian cells (Bleijerveld et al., 2007; Kainu et al., 2013). Whether the acyl chain composition of PE affects mitochondrial function or if there is a required role in the mitochondrion for Psd1p independent of PE synthesis remain to be investigated.
3.2.1 Biogenesis of Phosphatidylserine Decarboxylase 1
Phosphatidylserine decarboxylase 1 (Psd1p) belongs to a family of decarboxylases that contain pyruvoyl prosthetic groups necessary for the decarboxylation of their substrate (van Poelje and Snell, 1990). PS decarboxylases and their role in phospholipid metabolism have been evolutionarily conserved from bacteria to humans (Schuiki and Daum, 2009). Yeast contain two Psd enzymes, the mitochondrial Psd1p (Clancey et al., 1993; Trotter et al., 1993) and Psd2p; Psd2p localizes to endosomes and is only a minor source of cellular PE (Birner et al., 2001; Gulshan et al., 2010; Trotter and Voelker, 1995). Importantly, the combined deletion of PSD1 and PSD2 produces a yeast strain that is auxotrophic for ethanolamine or lyso-PE, which allow PE to be produced by the CDP-ethanolamine pathway or through the reacylation of lyso-PE by Ale1p, respectively (Atkinson et al., 1980; Riekhof et al., 2007b).
The gene encoding Psd1p, PSD1 in yeast or PISD in mammals, is nuclear-encoded and synthesized as a proenzyme by cytosolic ribosomes (Clancey et al., 1993; Kuge et al., 1991; Trotter et al., 1993). In yeast, Psd1p is targeted to the mitochondrial IM via its N-terminal mitochondrial targeting sequence. Upon its import into the IM, two matrix peptidases, matrix processing peptidase (MPP) and the octapeptidase, Oct1p, act to sequentially remove the mitochondrial targeting sequence (Horvath et al., 2012; Nebauer et al., 2007). A final autocatalytic processing step separates the enzyme into two subunits, α and β, generates a pyruvoyl prosthetic group at the N-terminus of the smaller α subunit, and is required to generate an active enzyme (Choi et al., 2012, 2015; Dowhan et al., 1974; Horvath et al., 2012; Kuge et al., 1996; Li and Dowhan, 1988, 1990; Satre and Kennedy, 1978). The α subunit is anchored to the membrane by remaining noncovalently associated with the β subunit that is integrated in the inner mitochondrial membrane facing the intermembrane space (IMS; Horvath et al., 2012; Li and Dowhan, 1988; Tamura et al., 2012b).
Pyruvoyl groups are not encoded by the genome and arise as a consequence of a posttranslational modification of the inactive proenzyme. The prosthetic group is a free carbonyl of pyruvate that is covalently linked to the modified protein (Satre and Kennedy, 1978). Studies on the biochemistry of autocatalytic processing in PS decarboxylases have mostly been done with bacterial Psd and show that following synthesis of the proenzyme, it undergoes an unusual processing event called nonhydrolytic serinolysis. Serinolysis not only separates the α and β subunits but also additionally generates the pyruvoyl prosthetic group on the N-terminus of the α subunit. This processing event and the pyruvoyl group that it makes is essential for PS decarboxylation from bacteria to humans (Choi et al., 2012, 2015; Dowhan et al., 1974; Horvath et al., 2012; Kuge et al., 1996; Li and Dowhan, 1988, 1990; Onguka et al., 2015; Satre and Kennedy, 1978).
Interestingly, a catalytic triad typical of serine proteases is required for Psd1p autocatalysis (Choi et al., 2015). In addition to a conserved serine residue (Choi et al., 2012; Horvath et al., 2012; Kuge et al., 1996; Li and Dowhan, 1988), there are evolutionarily conserved aspartic acid and histidine residues that when individually mutated, prevent Psd1p autocatalysis (Choi et al., 2015). Consequently, prior to becoming a decarboxylase, Psd1p must first function as a serine protease. Yeast Psd1p redirected to the secretory pathway is autocatalytically competent, enzymatically active, and fully capable of supporting growth of psd1Δpsd2Δ yeast in the absence of ethanolamine (Onguka et al., 2015). Thus, as long as it is anchored in a membrane, Psd1p itself contains everything else needed for autocatalysis and does not require the assistance of a mitochondrial-specific component.
3.2.2 PS Synthesis
In yeast and mammals, PS is synthesized in the MAM by phosphatidylserine synthase-1 (PSS1; Stone et al., 1998; Voelker, 1985). Mammalian cells also contain a second PS synthase enzyme, PSS2 (Stone and Vance 1999). PSS1 is ubiquitously expressed in all tissues and is enriched in brain and skeletal muscle (Nishijima et al., 1986; Sturbois-Balcerzak et al., 2001). Predominantly expressed in the testes, PSS2 is only a minor contributor of PS in other cell types (Bergo et al., 2002; Sturbois-Balcerzak et al., 2001). In yeast (and prokaryotes), PS is synthesized by the condensation of CDP-diacylglycerol and L-serine (Bae-Lee and Carman, 1984; DeChavigny et al., 1991). In contrast, mammals produce PS from a base-exchange reaction of L-serine with either PC or PE. PSS1 catalyzes a choline for serine head group exchange while PSS2 has been shown to have specificity for PE (Kuge et al., 1985; Suzuki and Kanfer, 1985). Overexpression of PSS1 but not PSS2 in rat hepatoma cells increases PS synthase activity suggesting that PSS1 activity is rate-limiting (Stone and Vance, 1999). Importantly, an increase in PS synthesis by PSS1 subsequently leads to an increase in the flux through the mitochondrial Psd pathway (Stone et al., 1998). Thus, the cell may harbor mechanisms to sense the relative ratio of phospholipids in different cellular membranes and respond in order to maintain this ratio. Alternatively, the increased flux through the Psd pathway could simply reflect an increase in substrate availability for PSD1 upon PSS1 overexpression. Deletion of PSS1 or PSS2 in mammals results in a compensatory increase of PS synthesis by the remaining enzyme (Arikketh et al., 2008; Borkenhagen et al., 1961). In a pss2−/− mouse model, the absence of PSS2 does not result in any changes in the PS/PE ratio across all tissues tested although some mice are infertile (Bergo et al., 2002). pss1−/− mice have a slightly decreased PS/PE ratio in various tissues but for the most part the absence of PSS1 is compensated for by PSS2. However, simultaneous deletion of pss1 and pss2 is incompatible with life (Arikketh et al., 2008). This may be an indication of the fundamental importance of PS for cell function and viability. Or instead, it may reflect the subsequent importance of PS as a substrate for PE production via PSD1 whose ablation is also embryonically lethal (Steenbergen et al., 2005).
3.2.3 PS Transport
Since biosynthesis of PS occurs in the MAM of the ER and Psd1p is anchored to the IM with its catalytic site facing the IMS, biosynthesis of PE involves obligate inter- and intra-organellar lipid trafficking steps (Voelker, 1984). What is known about these transport processes is discussed next.
3.2.3.1. Into Mitochondria
The mechanistic details of how PS travels between ER and mitochondrial membranes remains to be fully characterized although this process has been heavily investigated in yeast and mammals (Achleitner et al., 1995; Daum et al., 1986; Hovius et al., 1992; Kuge et al., 1986; Vance, 1991; Voelker, 1984, 1985, 1989a,b, 1990; Wirtz and Zilversmit, 1968). It was postulated that transport of PS between MAM and the mitochondrial outer membrane (OM) could be mediated by vesicular transport, lipid carrier(s), or passive diffusion facilitated by proximal contact sites (Voelker, 1985, 1989b). Vesicular transport was presumed unlikely since mitochondria are not part of the classical secretory pathway. To test the involvement of lipid carriers, PS transport was analyzed in detergent solubilized cells, which effectively removes the cytosol. Results from this experimental paradigm indicate that in mammalian cells, the import of PS from MAM to the OM is an ATP-dependent process whereas PS transport in yeast does not require ATP (Achleitner et al., 1995; Daum et al., 1986; Voelker, 1985, 1989a,b, 1990). Collectively, these results strongly suggest that soluble lipid carriers are not required for ER to mitochondria PS transport (they would be removed with the bulk cytosol in detergent permeabilized cells) and that the mechanism of PS transport between these organelles has a different energetic requirement in mammals and yeast.
In contrast, there is an abundance of evidence to support contact-site facilitated transport of PS between the ER and mitochondria. The possibility that transport could utilize interorganelle contact sites was initially provided by electron micrographs, which showed regions in which the ER and mitochondria are closely juxtaposed (∼ 30 nm; Csordas et al., 2006; Mannella et al., 1998; Robertson, 1960). Subsequent studies identified MAM as an ER hub enriched in lipid-synthesizing enzymes (e.g., PSS1) whose presence is important for the mitochondrial synthesis of PE (Vance, 1990). In yeast, a physical multiprotein complex termed the ER–mitochondria encounter structure (ERMES) bridges the small space between these organelles at proximal junctions (Kornmann et al., 2009). ERMES is composed of a complex of proteins that includes the outer mitochondrial membrane proteins, Mdm10p (Meisinger et al., 2004; Sogo and Yaffe, 1994) and Mdm34p (Dimmer et al., 2002; Youngman et al., 2004), an ER-membrane protein, Mmm1p (Burgess et al., 1994; Kornmann et al., 2009), and cytosolic Mdm12p (Berger et al., 1997; Kornmann et al., 2009). Furthermore, the ERMES complex is regulated by Gem1p, a mitochondrial GTPase (Kornmann et al., 2011). Although disruption of ERMES components decreases the number of contact sites between the ER and mitochondria and results in aberrant mitochondrial morphology (Berger et al., 1997; Burgess et al., 1994; Dimmer et al., 2002; Sogo and Yaffe, 1994; Youngman et al., 2004), the cellular and mitochondrial levels of PE in the cell is unaffected in its absence (Kornmann et al., 2009; Nguyen et al., 2012). However, there is an increase in cellular and mitochondrial PS in ERMES mutants (Tamura et al., 2012a). Beyond its function as an interorganelle tether, it is unclear if the ERMES complex has a direct biochemical role in translocating PS from the MAM to the mitochondrial OM (Nguyen et al., 2012; Voss et al., 2012). Indeed, the ERMES complex can be replaced by the synthetic ER–mitochondria tether, ChiMERA, which restores the phenotypic consequences that result when the ERMES complex is missing (Kornmann et al., 2009; Nguyen et al., 2012).
Recent evidence has shown that a second protein complex named the ER–membrane protein complex (EMC) contributes to phospholipid transfer between mitochondrial and ER membranes and importantly, is conserved across species (Lahiri et al., 2014). The EMC is composed of six different proteins, Emc1-6p. Localization of EMC proteins across ER membranes is dispersed as opposed to forming clear puncta as expected for a protein tether. Deletion of a single protein within the complex is not sufficient to disrupt ER–mitochondria contact site formation, and a decrease in PE synthesis is only observed upon the combinatorial deletion of multiple EMC subunits. Deletion of the EMC is synthetically lethal with the absence of the ERMES complex, suggesting that these two ER–mitochondria tethers serve overlapping essential roles in maintaining cellular and mitochondrial function. Although a marked decrease in PE synthesis by Psd1p occurs in the absence of the EMC, such production can be rescued by expression of the synthetic tether CHiMERA suggesting that like ERMES, EMC has no direct biochemical role in phospholipid transport and may simply function as a biological tether between organelles. The EMC associates with a component of the translocase of the outer membrane, Tom5p; however, as PS transport into the OM is normal in tom5Δ yeast, other mitochondrial factors must also be involved in bridging this interaction (Lahiri et al., 2014).
Orthologs of ERMES-like proteins have yet to be identified in mammals although contact site formation is still observed. Further, whether the conserved EMC functions in a similar manner in mammals as in yeast has not been determined. However, numerous other proteins are enriched at ER–mitochondria junctions in mammals that may collectively serve redundant functions linking these two organelles. These candidate ER–mitochondria tethers include GTPases and proteins with roles in regulating mitochondrial morphology. Mitofusin 2 (MFN2), a mitochondrial dynamin-related GTPase, forms ER–mitochondria tethers by virtue of an ER-localized MFN2 in a homo- or heterotypic complex with MFN2 or MFN1 on the outer membrane of mitochondria, respectfully (de Brito and Scorrano, 2008). Phosphofurin acid cluster sorting-protein (PACS-2) is a multifunctional protein that is important for organelle morphology, apoptotic signaling, ER homeostasis, and calcium signaling. PACS-2 depletion causes mitochondrial fragmentation, dissociates ER from mitochondria, and decreases the level of PSS1 present in MAM (Simmen et al., 2005). Voltage-dependent anion channels (VDAC) are outer membrane proteins that allow the passive diffusion of small molecules and metabolites across the outer membrane (Rapizzi et al., 2002) and which accumulate in regions of close apposition between mitochondria and ER (Garcia-Perez et al., 2011; Shoshan-Barmatz et al., 2004). As mitochondria buffer calcium released by the ER (Hajnoczky and Thomas, 1997), known and proposed proteins that promote contact sites with mitochondria are associated with decreased calcium signaling in their absence. Indeed, the selectivity of the VDAC channel is modified through its interaction with the molecular chaperone glucose-regulated protein 75 (GRP75), which links VDAC to the ER calcium-release channel inositol 1,4,5-trisphosphate receptor (IP3R) in HeLa cells (Szabadkai et al., 2006). As both VDAC and IP3R coimmunoprecipitated with GRP75, but not with each other, GRP75 forms a bridge that juxtaposes ER-based calcium release with mitochondria thus supporting the latter organelle's calcium buffering capacity. Consistent with the notion of tethering redundancy, knock down of MFN2 in HeLa cells does not diminish PS import to mitochondria or PE synthesis via the Psd pathway as measured by mass spectrometry (Kainu et al., 2013). Clearly, the mechanism of PS transport between the ER and mitochondria remains nebulous.
Lipid trafficking in and out of mitochondria is expected to be critical for cell viability. Since the ERMES complex is nonessential (Kornmann et al., 2009; Nguyen et al., 2012; Tamura et al., 2012a; Voss et al., 2012), the possibility of an alternative pathway for PS import was suggested. Recently, a yeast vacuole–mitochondria tether termed v-CLAMP was shown to be essential for cell survival in the absence of ERMES components (Elbaz-Alon et al., 2014; Honscher et al., 2014). In the absence of the ERMES complex, vacuoles surround mitochondria and the contacts between these two organelles increase. When v-CLAMP expression is repressed in a yeast background unable to form ERMES complexes, PE levels and synthesis are significantly reduced (Elbaz-Alon et al., 2014). The extent of mitochondria– vacuole contacts versus ER–mitochondria contacts varies depending on the available carbon source; ERMES-mediated ER–mitochondria contacts dominate in nonfermentative media whereas mitochondria–vacuole contacts generated by v-CLAMP predominate in fermentable media (Honscher et al., 2014). The synthetic lethality between ERMES and v-CLAMP demonstrates the obligate need of mitochondria to obtain lipid precursors from neighboring organelles for the biosynthesis of PE (and CL) as well as the acquisition of phospholipids not made in the cell's powerhouse.
3.2.3.2. Within Mitochondria
In mammals, dinitrophenol but not carbonyl cyanide m-chlorophenyl hydrazone inhibits PS import from the OM to the IM (Hovius et al., 1992). Since both compounds are uncouplers that disrupt oxidative phosphorylation, this suggests that dinitrophenol impairs PS uptake by either disturbing IM/OM contacts or increasing the distance between these two membranes (Knoll and Brdiczka, 1983). Similarly, PS import from the OM to the IM in yeast is ATP-independent and does not require either a functional membrane potential or oxidative phosphorylation machinery (Achleitner et al., 1995, 1999; Gnamusch et al., 1992). Akin to the numerous tethers between the outer membrane and other organelles, there is an IM/OM scaffold termed the mitochondrial contact site and cristae organizing system (MICOS); this scaffold is also crucial for cristae junction formation (Alkhaja et al., 2012; Harner et al., 2011; Hoppins et al., 2011; Itoh et al., 2013; Jans et al., 2013; von der Malsburg et al., 2011). MICOS is a hetero-oligomeric protein complex that is conserved from yeast to mammals and is important for mitochondrial biogenesis, morphology, and inheritance. Whether transport of PS between OM and IM leaflets utilizes MICOS-based IM/OM scaffolds has yet to be elucidated.
Lipid carriers may also be involved in shuttling incoming PS to the IM although a potential PS carrier has not been identified. The role of the Ups family of proteins (Ups1-3p) in mitochondrial phospholipid metabolism has been investigated and evidence supports their involvement in both PE and CL synthesis (Osman et al., 2009; Potting et al., 2010; Tamura et al., 2009). The unique dimeric phospholipid CL is specific to mitochondrial and bacterial membranes. In eukaryotes, CL biosynthesis requires that its substrate PA is trafficked to the matrix side of the IM (Schlame and Haldar, 1993). Ups (unprocessed) proteins are putative lipid carriers that were identified in yeast and are homologous with the mammalian MSF1/PRELI protein families (Sesaki et al., 2006). The stability of Ups proteins within the intermembrane space is dependent upon their productive association with the IMS protein Mdm35p (Potting et al., 2010; Tamura et al., 2010). As the Ups1p/Mdm35p complex transports PA across the IMS, deletion of UPS1 results in a decrease in CL (Connerth et al., 2012; Potting et al., 2013; Tamura et al., 2009). Interestingly, UPS2 deletion in ups1Δ yeast prevents the decrease in CL (Tamura et al., 2009). Further, overexpression of Ups2p results in a decrease in CL suggesting that it regulates Ups1p function (Osman et al., 2009). In addition to their antagonistic roles in CL metabolism, the absence of Ups2p results in a decrease in PE that is associated with a faster rate of PE to PC conversion (Tamura et al., 2012a). As such, Ups1p is suggested to accelerate export of PE from mitochondria and increase its subsequent conversion to PC in the MAM. In turn, the presence of Ups2p somehow regulates this process. The exact role of Ups1p and Ups2p in PE synthesis and export is presently unclear as is the molecular function of Ups3p, whose deletion does not alter mitochondrial phospholipid profiles (Tamura et al., 2009).
Given its lack of known energy requirements, the directionality of PS transport is likely provided through its conversion to PE by Psd1p in the inner membrane. The capacity to import phospholipids irrespective of mitochondrial function may allow for maintenance of mitochondrial membranes in situations where oxidative phosphorylation is disrupted. If the oxidative phosphorylation dysfunction is secondary to defects in its membranes, then this could allow the membranes to be repaired thus restoring mitochondrial energy production.
3.2.4 Fate of PE Produced by Psd1p
PE produced by Psd1p in the context of the IMS-leaflet of the IM has at least two fates. It can either stay in mitochondria (associated with the IM or OM) and support mitochondrial functions therein (discussed in Section 4.3). Or alternatively, it can exit mitochondria and contribute to the cellular pool of PE and lipid derivatives of PE. PE export from mitochondria is stimulated in cases where substrates for PE production via the CDP-ethanolamine pathway are lacking (Kainu et al., 2013). In contrast, robust levels of PE in ER membranes discourage PE export as this process may be more energetically unfavorable. Shuttling of PE from the IM to other parts of the cell is an additional phospholipid trafficking process that remains poorly characterized. As previously mentioned, the absence of Ups2p in yeast results in a decrease in mitochondrial PE suggesting that Ups1p plays a role in expediting PE export from the IM. Presumably, Ups2p regulates this export. However, in the absence of Ups1p, no accumulation of PE is observed (Osman et al., 2009; Tamura et al., 2012a). The role of inter- and intraorganelle contact sites or additional mitochondrial lipid carriers needs to be interrogated in this process.
3.3 PE as Precursor for Other Lipids and Substrate for Posttranslational Modifications
The cell is a sophisticated factory of proteins, lipids, and carbohydrates where each of these components can be modified or recycled to accommodate the cell's metabolic state. An interesting aspect of lipid biology is that phospholipids can be interconverted between distinct classes. For example, PE produced by the mitochondrial Psd pathway derives from the decarboxylation of PS. Further, PE produced by either the CDP-ethanolamine or decarboxylation pathways can be converted to PC (Kennedy and Weiss, 1956; Ridgway and Vance, 1987). PE is also a critical substrate for at least two fundamental posttranslational modifications, GPI anchors (discussed later; Menon and Stevens, 1992; Wilson-Zbinden et al., 2015) and lipidation of Atg8p/LC-3 (discussed in Section 4.4; Ichimura et al., 2000).
3.3.1 Methylation of PE to Form PC
Constituting 40–50% of total phospholipids in most organelles, phosphatidylcholine is the most abundant phospholipid in eukaryotes (Vance, 2015). PC has a cylindrical shape in which its phospholipid head group and fatty acyl tails are of equal diameter. This property allows PC to become tightly packed within the membrane, which promotes the formation of membrane bilayers.PC is predominantly produced by the CDP-choline pathway, where CDP-choline condenses with a DAG moiety to generate PC (Kennedy and Weiss, 1956). However, a minor pathway involving trimethylation of PE to PC also exists and can account for 20–30% of PC in liver cells (Sundler and Akesson, 1975b). PC made by this pathway, which is performed in the MAM (Cui et al., 1993; Vance, 1990), can utilize PE produced by any of the four routes of PE biosynthesis. A single enzyme is responsible for PC synthesis from PE in mammals, PE N-methyl transferase (PEMT), while yeast contains two enzymes, PE methyltransferase (Pem1p) and phospholipid methyltransferase (Pem2p). PE undergoes three successive methylation reactions by PEMT for its full conversion to PC (Ridgway and Vance, 1987). In yeast, Pem1p converts PE to phosphatidyl-N-monomethylethanolamine (PMME) in the first methylation reaction; Pem2p has a low affinity for this reaction as well. Pem2p then successively methylates PMME to phosphatidyl-N, N-dimethylethanolamine (PDME) and PDME to PC (Kodaki and Yamashita, 1987).
3.3.2 Phosphoethanolamine for GPI Anchor Formation
GPI anchoring is a posttranslational modification that is conserved among all eukaryotes. A GPI anchor is a glycolipid structure that is added posttranslationally to the C-terminus of many eukaryotic proteins. GPIs are synthesized in the ER by at least 10 enzymes and up to 20 genes are involved (Fujita and Kinoshita, 2010; Kinoshita et al., 2008). GPI biogenesis is essential for embryogenesis (Nozaki et al., 1999), neurogenesis (Ueda et al., 2007), immune responses, and fertility (Ueda et al., 2007). GPI-anchored proteins selectively associate with lipid rafts, membrane domains that serve as platforms for signaling and protein trafficking. Proteins that are linked to the plasma membrane by GPI anchors can remain thusly associated or are instead cleaved and released from the membrane.
The GPI core consists of phosphatidylinositol, a glucosamine moiety, three mannoses, and phosphoethanolamine (Ferguson, 1999; Ikezawa, 2002; Nosjean et al., 1997). A complete GPI precursor is transferred to proteins containing a GPI anchor signal sequence at the C-terminus. GPI anchors form on the lumenal side of the ER where a glycan core is assembled from a complex of mannose residues linked to the inositol head group of PI. A phosphoethanolamine linker is then attached to the C-terminus of the target protein and a mannose residue of the glycan core, which anchors the protein to the ER membrane facing the lumen (Kinoshita et al., 2008). PE provides the ethanolamine group used to make the phosphoethanolamine bridge between the glycan and C-terminal amino acid of the GPI-anchored protein (Menon and Stevens, 1992). There are three proteins involved in transferring additional phosphoethanolamine groups to mannose residues on newly formed GPI anchors. The first mannose residue is modified by Mcd4p in yeast and PIG-N in mammals, which localize to the ER (Gaynor et al., 1999; Hong et al., 1999). Deletion of MCD4 is lethal in yeast as GPI anchor biosynthesis is critical for cell wall integrity (Gaynor et al., 1999). Addition of phosphoethanolamine to mannose 2 and 3 is carried out by Gpi7p and the protein product of yeast YLL031C (PIG-O in mammals), respectively (Flury et al., 2000). As GPI-anchor formation is a multistep process, defects and/or mutations that impair any of a number of its intermediate reactions, including formation of the phosphoethanolamine bridge, can cause disease (Maydan et al., 2011; Takeda et al., 1993). The impact of PE depletion on GPI anchor formation and its potential relevance to Parkinson's disease is discussed in Section 5.2.
4. Cellular and Molecular Functions of PE
The existence of multiple PE-producing pathways combined with the absolute requirement of both the CDP-ethanolamine pathway in the ER and the Psd pathway in the mitochondrion for mammalian development highlights the importance of PE in performing specified functions in the cell (Fullerton et al., 2007; Steenbergen et al., 2005). As a nonbilayer forming phospholipid, a high PE content in cellular membranes promotes the formation of hexagonal phase structures, which antagonize the tight packing of membrane bilayer forming phospholipids such as PC (Osman et al., 2011). Hexagonal phase structures in the membrane have been shown to induce bilateral membrane stress that can be relieved by membrane bending events, protein insertion at sites containing poor membrane packing, or conformational changes within membrane proteins that rearrange the lipid distribution (van den Brink-van der Laan et al., 2004). It is no surprise then that PE associates with and modulates the behavior of a variety of proteins and protein complexes with diverse roles in a range of cellular processes.
4.1 PE as a Determinant of Protein Topology
The final topology of membrane proteins is determined by several intrinsic features including the presence of hydrophobic alpha helical stretches, which are often flanked by charged amino acid residues. Of particular importance are flanking positively charged residues, which orient a membrane anchor such that these reside in the cytosol (the positive-inside rule; Nilsson and von Heijne, 1990). In addition to such intrinsic topological information, the presence or absence of certain phospholipids in a membrane can interact with membrane proteins and introduce dynamic or steady state effects on its topology, structure, and function (Bogdanov et al., 2002, 2009; DeChavigny et al., 1991; Seto-Young et al., 1985). One such lipid is PE (Bogdanov and Dowhan, 1995). Importantly, the topologies that are sensitive to PE can be reversible (occur post-biosynthesis and membrane integration) and switch the membrane protein between active and inactive states (Bogdanov and Dowhan, 1998, 1999; Bogdanov et al., 2002). Work in bacteria and in reconstituted liposomes demonstrated that PE is required for energy-dependent uphill substrate accumulation but not energy-independent downhill substrate equilibration mediated by lactose permease (LacY; Bogdanov and Dowhan, 1995). The lack of PE induces topological inversion of the N-terminal six transmembrane (TM) domains of LacY, exposure of the seventh TM domain to the periplasm, and misfolding of a periplasmically exposed domain (Bogdanov et al., 2008); proper folding of this domain is linked to the ability of LacY to mediate secondary active transport (Bogdanov and Dowhan, 1999).
Interestingly, if PE is made after LacY is improperly membrane-integrated, the seventh TM domain in the periplasm reinserts across the membrane, which causes the preceding four TM domains to change their topology; the first TM domain remains in the opposite orientation as observed when PE is present during the biogenesis of LacY while the second TM domain adopts an interfacial conformation that goes into but not through the membrane (Bogdanov et al., 2008; Zhang et al., 2003). All of these changes induced by PE on existing LacY restore its ability to utilize a proton gradient to transport sugars uphill. How does PE have such a major impact? Presently, it is speculated that uncharged phospholipids such as PE shield negatively charged amino acid residues adjacent to TM domains (Bogdanov et al., 2008, 2014). This both diminishes their potential to serve as topological determinants and increases the strength of surrounding positively charged amino acids to act in this capacity. In the absence of PE, negatively charged TM-flanking residues regain topogenic strength. If revealed in the context of TM domains that are not very hydrophobic, such as TM domain 7 of LacY, this can provoke significant topological changes with functional consequences.
The importance of PE in topological orientation has also been determined for other bacterial transporters, including the 12 TM domain-containing phenylalanine and γ-aminobutyrate permeases (Bogdanov et al., 2008; Zhang et al., 2003, 2005). The ability of PE to influence the topology and function of eukaryotic proteins has not been demonstrated. Still, these elegant studies in bacteria suggest that this mode of regulation may be relevant in eukaryotic cells especially since many organelles are ensheathed by membranes of distinct lipid compositions.
4.2 Membrane Fusion
During the late stages of cell division, daughter cells separate into individual cells at cytokinesis. The contractile ring marks the boundary to separate the dividing cell and is composed of actin filaments that create tension at the plasma membrane to promote fusion (Schroeder, 1990). This generates what is widely known as the “cleavage furrow” at the membrane of the dividing cell, which becomes more pronounced and fuses at the end of telophase creating two individual daughter cells. PE, which is normally enriched on the inner leaflet of the plasma membrane, accumulates on the external leaflet specifically at the cleavage furrow during a late stage of telophase (Emoto et al., 1996). Interestingly, a streptavidin-conjugated fluorescent probe that specifically binds to PE inhibits cytokinesis at telophase by trapping PE on the external surface. Importantly, the block in cytokinesis is reversed by incubating cells with PE-loaded liposomes (Emoto and Umeda, 2000). Further, a cell line defective in the mitochondrial Psd pathway of PE biosynthesis has a defect in cytokinesis that is rescued if provided supplemental PE or ethanolamine, the latter of which stimulates PE production via the CDP-ethanolamine pathway. Mechanistically, retention of PE on the cell surface does not disturb the formation of cytoskeletal components (microtubules and actin) important for cytokinesis, membrane furrowing, or separation of chromosomes but does prevent actin filament disassembly and membrane fusion (Emoto and Umeda, 2000). Thus, the transient surface appearance of PE along the cleavage furrow and its subsequent reinternalization to the inner leaflet of the plasma membrane are temporally regulated events that are of fundamental importance at a late stage in cell division. Moreover, PE produced by either the Psd or CDP-ethanolamine pathways can fulfill this cellular function.
Additionally, PE has a pivotal role in the fusion of Golgi membranes that occurs after cell division. Fusion of Golgi membranes involves the association of cytosolic factors with components of the Golgi membrane fusion machinery. During vesicle fusion, p97, a cytosolic ATPase of the fusion machinery, associates with the t-SNARE associated factor, p47 (Otter-Nilsson et al., 1999). The activity of the p97/p47 complex, which is sufficient for mediating fusion, is influenced by the presence of PE in Golgi membranes. PE increases the head group spacing between lipids present in Golgi membranes allowing conformational changes that are required for membrane fusion to be induced within the p97/p47 complex (Pecheur et al., 2002).
4.3 PE in Mitochondrial Function
CL and PE make up about 35–50% of the total phospholipids within the mitochondrial inner membrane (Daum, 1985). CL is a dimeric phospholipid that contains two phosphatidic acids bridged by a glycerol group and four fatty acyl chains. Mitochondria are the sole manufacturers of CL in eukaryotic cells and generate CL on the matrix-side of the IM (Baile et al., 2014; Claypool and Koehler, 2012; Schlame and Haldar, 1993). Ablating mitochondrial PE (psd1Δ) or CL (crd1Δ; CRD1 encodes cardiolipin synthase) biosynthesis in yeast yields viable cells (Chang et al., 1998; Clancey et al., 1993; Jiang et al., 1997; Trotter et al., 1993; Tuller et al., 1998). psd1Δ and crd1Δ yeast share several phenotypes including reduced growth in nonfermentable carbon sources, increased frequency of petite formation (the mitochondrial genome is absent or defective), and sensitivity to increased temperature (Birner et al., 2001; Jiang et al., 1997; Storey et al., 2001; Zhong et al., 2004). While the single mutants are viable, the combined crd1Δpsd1Δ mutant is not (Gohil et al., 2005). Only deletion of PSD1, and not the CDP-ethanolamine pathway or PSD2, is synthetically lethal with crd1Δ (Gohil et al., 2005). This likely reflects the fact that PE produced by either of these pathways is poorly integrated in mitochondrial membranes (Birner et al., 2001; Burgermeister et al., 2004) and that the pool of PE in the IM of mitochondria is predominantly produced by Psd1p (Shiao et al., 1995).
In the membranes of prokaryotic cells, which lack PC but are enriched in both PE and CL, the absence of PE results in a compensatory increase in CL (Rietveld et al., 1993). This reciprocal relationship is also observed in yeast when pools of PE or CL are depleted through deletion of genes involved in their biosynthesis and/or regulation (Osman et al., 2009; Zhong et al., 2004). psd1Δ and crd1Δ yeast also share synthetic genetic interactions with genes that encode similar mitochondrial proteins; namely the prohibitin lipid scaffolds and the lipid transport proteins, Ups1p and Ups2p (Hoppins et al., 2011; Kornmann et al., 2009; Osman et al., 2009). Thus, PE and CL have converging functions in mitochondria that collectively highlight their individual and combined importance for mitochondrial function (Gohil et al., 2005). The specific roles that CL plays in mitochondrial biology, health, and disease merits more detailed attention, which has been provided by several excellent reviews (Claypool and Koehler, 2012; Joshi et al., 2009; Lu and Claypool, 2015; Schlame and Ren, 2009).
4.3.1 Oxidative Phosphorylation
psd1Δ yeast have impaired growth and increased petite formation when grown in respiratory media, characteristic of yeast with disrupted oxidative phosphorylation capacities (Birner et al., 2001). When mitochondrial PE production is reduced in CHO-K1 cell lines by RNAi inhibition of the mammalian PISD gene, mitochondrial membrane potential is increased, complex I and complex IV activity of the respiratory chain is reduced, and ATP production is significantly decreased (Tasseva et al., 2013). Individual electron transport chain complexes assemble in higher order structures known as supercomplexes (Cruciat et al., 2000; Schagger and Pfeiffer, 2000). Supercomplex formation of the different electron transport chain components maximizes respiratory capacity in mitochondria (Acín-Pérez et al., 2008; Lapuente-Brun et al., 2013). The role of PE in supercomplex formation is species-specific as results between yeast and mammals differ. In mammals, depletion of mitochondrial PE disrupts the formation of higher order complexes associated with complex IV (Tasseva et al., 2013). In contrast, in yeast, supercomplexes between respiratory complexes III and IV (yeast lack complex I) are stabilized in the absence of PE (Bottinger et al., 2012). While the impact of PE on supercomplex stability is different, its absence in yeast and mammals alike decreases respiratory function.
Cytochrome c oxidase (complex IV) oxidizes reduced cytochrome c by reducing divalent oxygen to water. The crystal structure of complex IV from bovine heart shows that there are three PE monomers associated with different components of this respiratory complex (Shinzawa-Itoh et al., 2007). Importantly, two PE monomers are at the interface where complex IV dimerizes. Inpsd1Δ yeast, complex IV activity is significantly decreased (Bottinger et al., 2012). This suggests that PE plays an indispensable and unique role in complex IV activity. Interestingly, even though ubiquinol: cytochrome c oxidoreductase (complex III) from yeast cocrystallizes with PE (Lange et al., 2001), complex III function is normal in psd1Δ yeast (Bottinger et al., 2012).
An interesting role for PE in hepatocyte mitochondrial membranes has been proposed for glucose metabolism and oxidative phosphorylation in mice. As previously mentioned, about 20–30% of PC in hepatocytes is made by the trimethylation of PE to PC (Sundler and Akesson, 1975b). pemt−/− mice are protected against insulin resistance when supplemented with a high fat diet (more on the characteristics of pemt−/− mice in Section 5.3; van der Veen et al., 2014). Elimination of PE methylation increases mitochondrial PE levels as well as pyruvate flux through the tricarboxylic acid cycle for ATP production. Abolishing PEMT did not affect the enzymatic activity of proteins involved in gluconeogenesis, but nonetheless mice were protected against insulin resistance. Instead, PEMT ablation appears to lower hepatic glucose production by increasing the activity of the electron transport chain. This increase in respiratory function correlates with a decrease in the mitochondrial PC:PE ratio (van der Veen et al., 2014). The increase in hepatocyte mitochondrial PE levels in pemt−/− mice indicates that the mitochondrial Psd pathway is an important source of PE used by PEMT, at least in the liver. Whether and how the altered mitochondrial PC:PE ratio causes the observed changes in mitochondrial substrate utilization is unclear. In sum, these results suggest that in the liver, this dynamic pathway of PC production utilizes PE made in mitochondria, is important for normal hepatocyte physiology, and potentially, whole-body glucose homeostasis.
4.3.2 Mitochondrial Protein Biogenesis and Activity
Mitochondrial protein import at the OM and IM is defective when PE is limiting. The import of preproteins into and across the IM through the translocases of the inner membrane (TIM) complexes (TIM23 and TIM22) is driven by the electrochemical gradient across the IM, which is generated by the respiratory chain and reduced in psd1Δ and psd1Δpsd2Δ yeast (Bottinger et al., 2012; Chacinska et al., 2009). Thus, the defect in IM biogenesis in psd1Δ and psd1Δpsd2Δ yeast is secondary to a decrease in the functionality of the respiratory chain. PE is additionally important for the biogenesis of β-barrel proteins of the mitochondrial OM (Becker et al., 2013). Biogenesis of β-barrel proteins begins with specific recognition of the imported precursor by the translocase of the outer membrane (TOM) translocon (Chacinska et al., 2009). Next, β-barrel precursors are translocated into the IMS before being passed to the sorting and assembly machinery (SAM) complex in a process that requires the small TIM complexes, IMS resident chaperones. Proper membrane insertion and folding of β-barrel proteins is mediated by the SAM complex. In psd1Δ and psd1Δpsd2Δ yeast, biogenesis of β-barrel proteins is impaired early in the process and at multiple steps (Becker et al., 2013). First, precursor binding by the TOM complex is reduced in the absence of normal levels of mitochondrial PE. And second, translocation of β-barrel precursors through the TOM complex into the IMS, as determined by resistance to protease digestion, is also reduced in psd1Δ and psd1Δpsd2Δ yeast. Interestingly, this decrease in activity is independent of the stability of the TOM complex since assembly by blue-native PAGE electrophoresis and protein–protein interactions by coimmunoprecipitation show that the TOM complex remains intact (Becker et al., 2013). Additionally, this defect is specific for β-barrel proteins, as the biogenesis of alpha helical OM proteins is not influenced by the presence or absence or PE.
4.3.3 Mitochondrial Fusion
Mitochondria are dynamic organelles that frequently fuse and divide to maintain their function and morphology (Friedman and Nunnari, 2014; van der Bliek et al., 2013). Mitochondrial fission and fusion is mediated by members of the dynamin-like GTPase protein family. Fission is dependent on DRP1 (Dnm1p in yeast; Bleazard et al., 1999; Smirnova et al., 2001). OM fusion is executed by the mitofusins (MFN1 and MFN2 in mammals and Fzo1p in yeast; Chen et al., 2003; Hermann et al., 1998; Santel and Fuller, 2001) and IM fusion involves OPA1 in mammals and Mgm1p in yeast (Alexander et al., 2000; Delettre et al., 2000; Jones and Fangman, 1992). OPA1 and Mgm1p are processed into long and short forms that differ with respect to their membrane association and which are both required for IM fusion (Griparic et al., 2007; Song et al., 2007; Zick et al., 2009). Fragmented and aggregated mitochondria accumulate in psd1Δ yeast, which also have a skewed long:short Mgm1p ratio (less short than normal; Chan and McQuibban, 2012; Osman et al., 2009). This suggests that PE produced by Psd1p influences the production of short Mgm1p likely at an ATP-dependent step that is required upstream of its cleavage by the rhomboid protease, Pcp1p (Herlan et al., 2003, 2004; McQuibban et al., 2003). In addition to the involvement of these GTPases in mitochondrial fission and fusion, the biophysical properties of certain lipids, including PE, may also influence these dynamic processes. In vitro fusion experiments using protein-free liposomes that mimic mitochondrial membranes with reduced PE levels have decreased rates of lipid mixing postfusion, which could decrease the efficiency of this process (Chan and McQuibban, 2012). When CL levels become limiting in psd1Δ yeast, the membrane potential is reduced, mitochondrial DNA is lost, expression of Mgm1p (long and short) is reduced, and mitochondria become highly fragmented (Joshi et al., 2012). These results suggest that PE has an important role in mitochondrial fusion that becomes essential in the absence of CL.
4.4 Autophagy
Autophagy is an important catabolic process that recycles cytosolic proteins and membranes through their degradation within lytic compartments in the cell (lysosomes in mammals or vacuoles in yeast; Mizushima et al., 2011; Ohsumi, 2014). Macroautophagy (for other forms of autophagy please refer to Mizushima et al. (2011); Reggiori and Klionsky (2013)) involves formation of the autophagosome, a double membrane-bound structure that encases cargo destined for degradation and delivers it to the lysosome/ vacoule (Tooze and Yoshimori, 2010). Autophagosome biogenesis begins with the formation of a cup-like membrane sac referred to as the isolation membrane or phagophore. To date, the origin of the phagophore membrane remains controversial, as various organelles have been speculated to be the responsible membrane donors. However, various links have implicated mitochondria and mitochondrial-derived PE in the formation and expansion of at least some autophagosomes.
Over 30 proteins are involved in the various forms of autophagy in yeast, most of which are conserved in mammals (Reggiori and Klionsky, 2013). One of these components, Atg8p, and its functional mammalian ortholog LC3 (microtubule-associated protein light chain 3; Tanida et al., 2001), is covalently attached to the headgroup of PE, anchoring it to the developing autophagosomal membrane (Kabeya et al., 2000; Kirisako et al., 1999). Lipidation of Atg8p affects membrane dynamics during different stages of autophagosome formation since the association and disassociation of Atg8p/ LC3 with autophagosomal membranes plays an important role in determining the ultimate size of the autophagosome formed (Nair et al., 2012; Xie et al., 2008).
The process by which Atg8p is modified by PE occurs in an ubiquitin-like fashion where Atg8p is transferred and activated between cysteine residues on Atg proteins prior to being anchored to the phagophore membrane (Figure 3; Mizushima et al., 2011; Reggiori and Klionsky, 2013). Atg8p is synthesized in the cytosol where Atg4p, a cysteine protease, cleaves the C-terminal arginine of Atg8p exposing an essential N-terminal glycine residue (Kirisako et al., 2000). Atg7p recognizes the exposed glycine residue and binds Atg8p in an ATP-dependent manner prior to transferring it Atg3p (Ichimura et al., 2000). In mouse embryonic stem cells, membrane association of LC3 is dependent upon formation of the ATG12-ATG5 autophagy conjugation system (Mizushima et al., 2001). Indeed, further studies in yeast (Hanada et al., 2007; Sakoh-Nakatogawa et al., 2013) and structural characterization of the yeast and mammalian ATG12-ATG5 conjugate (Noda et al., 2013; Otomo et al., 2013) verified the activity of this complex in functioning as a platform that directly interacts with ATG3 and enhances/ specifies attachment of the N-terminal glycine residue of Atg8p/LC3 to the primary amine group of PE. Although the process of autophagosome formation involves the coordinated activity of various ATG proteins, the importance of conjugating PE to Atg8p is evidenced by the fact that autophagic activity decreases when the C-terminal glycine of Atg8p is removed (Hemelaar et al., 2003; Kim et al., 2001; Kirisako et al., 2000; Noda et al., 1995). In its final modification step, Atg8p is cleaved from the autophagosomal membrane at its C-terminal glycine residue by Atg4p (the second time Atg8p is cleaved by Atg4p), an event that controls the size of the autophagosome (Nair et al., 2012; Xie et al., 2008).
Figure 3.
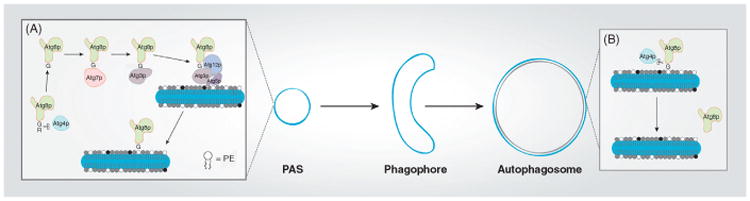
Atg8p lipidation in yeast. (A) Atg8p is proteolytically processed by the cysteine-protease Atg4p, which removes the C-terminal arginine residue and exposes a critical glycine that is recognized by Atg7p. Atg7p transfers Atg8p to Atg3p that together with the Atg12p–Atg5p protein complex conjugates Atg8p to PE. Atg8p is tethered via PE to membranes on the preautophagosomal structure (PAS) as it expands to the phagophore membrane. The phagophore membrane increases in size as it surrounds its cellular cargo to form a mature autophagosome. (B) When the autophagosome has reached its target size, Atg8p is cleaved at its terminal glycine residue by Atg4p. Similar mechanisms have been observed for LC3 lipidation using the ATG machinery in mammals.
Under nutrient starving conditions, the mitochondrial outer membrane supplies lipids for autophagosome biogenesis in a manner that is independent of mitophagy (Hailey et al., 2010), a form of macroautophagy that removes damaged or excess mitochondria (Twig and Shirihai, 2010). This process is dependent on ER–mitochondria contact sites as deletion of MFN2, which encodes a previously mentioned protein involved in mitochondrial fusion and ER–mitochondria tethering (de Brito and Scorrano, 2008), ablates induction of autophagy in normal rat kidney cells (Hailey et al., 2010). Interestingly, upon its addition to cells, the fluorescent lipid probe, NBD–PS, is initially present in the ER and accumulates in mitochondria over time, which is consistent with its subsequent conversion to NBD–PE by PSD1. Upon serum starvation, the mitochondrial NBD signal shifts to autophagosomes suggesting that mitochondrial-derived PE is used for autophagosome membrane formation (Hailey et al., 2010). However, whether the NBD signal detected in autophagosomes upon starvation was in fact NBD–PE was not experimentally verified. The origin of phagophore membranes is debated in the autophagy field (Mizushima et al., 2011; Tooze and Yoshimori, 2010) although electron microscope 3D tomography and cofractionation studies support a role for the MAM as also contributing to this process (Hamasaki et al., 2013).
A recent study has shown that PE-lipidation of Atg8p competes for the same pool of PE that is required for GPI anchor formation and PC synthesis. Growth of a yeast strain harboring a temperature sensitive (ts) allele of Mcd4p (Gaynor et al., 1999), a protein that appends a phosphoethanolamine group to the GPI anchor glycan core (Hong et al., 1999), is rescued at the nonpermissive temperature when genes encoding the PE-methylation enzyme, Pem1p, or either Atg7p or Atg14p, are deleted (Wilson-Zbinden et al., 2015). A decrease in Mcd4p activity results in accumulation of GPI-anchored proteins in the ER. By disrupting autophagy (atg7Δ or atg14Δ) or methylation of PC (pem1Δ), the localization defect of GPI-anchored proteins is restored. Overexpression of Pem1p is lethal to the mcd4-ts yeast strain presumably by increasing the conversion of PE to PC, thus reducing the pool of PE available for GPI anchor formation. Consistent with this idea, reducing cellular PE levels by deleting PSD2 or disrupting the CDP-ethanolamine pathway also impairs growth of the mcd4-ts yeast strain (Wilson-Zbinden et al., 2015). The availability of PE as a substrate for these processes is a potentially limiting factor that may regulate flux through each pathway.
High PE levels also positively regulate longevity in yeast and mammalian cells (Rockenfeller et al., 2015). This effect correlates with induction of autophagy as measured by levels of Atg8p and LC3 lipidation in cells supplemented with ethanolamine. Additionally, fly lifespan is extended upon ethanolamine supplementation although the role of autophagy in these latter findings remains to be validated. Nonetheless, these studies illustrate the interplay between PE metabolism and processes that require PE as a substrate, suggesting that further research may reveal the coregulation of these functions.
5. PE and Diseases
PE influences a variety of cellular processes and the stability and function of numerous membrane proteins. Mice lacking either of the two major PE-producing pathways are inviable (Fullerton et al., 2009; Steenbergen et al., 2005). Consequently, impairments in the major PE biosynthetic pathways are incompatible with life and thus significant defects in the metabolism of PE are presumably eliminated during mammalian development. However, slight impairments that result in a change in PE abundance are associated with Alzheimer's and Parkinson's disease. A critical PC:PE ratio in the liver is also implicated in glucose metabolism (van der Veen et al., 2014) and liver disease (Li et al., 2006). Finally, PE is an important cofactor and membrane component that is required for the pathogenicity of a variety of infectious organisms (Chen et al., 2010; Deleault et al., 2012). In the following sections, we will highlight the emerging roles of PE in mammalian disease.
5.1 Alzheimer's Disease
Alzheimer's disease (AD) is a late onset neurodegenerative disorder characterized by abundant intracellular neurofibrillary tangles rich in hyperphosphorylated microtubule-associated TAU and extracellular plaques of amyloid-β (Aβ) peptide derived from the amyloid precursor protein (APP; Goedert and Spillantini, 2006; Pimplikar, 2009; Selkoe, 2001). Pathophysiologically, AD is characterized by progressive neuronal loss, especially in the cortex and the hippocampus, which results in a decline in memory and cognition (Goedert and Spillantini, 2006). Aβ peptides are derived from the sequential proteolytic cleavage of APP by β- and γ-secretase (Haass and Selkoe, 2007). APP is a type I integral membrane protein with extracellular domains that are released upon cleavage by α- or β- secretases, which cut APP at different positions (Figure 4(A); Buxbaum et al., 1998; Vassar et al., 1999). Both processes occur normally, precede cleavage by γ-secretase, and have beneficial physiological roles (Mattson et al., 1993; Ring et al., 2007). β-secretase cleavage of APP leaves a 99 amino acid C-terminal portion (C99) in the membrane (Selkoe, 2001; Vassar et al., 1999). γ-secretase cleaves within the TM domain of C99 (Edbauer et al., 2003; Qi-Takahara et al., 2005). This results in the liberation of the APP intracellular domain, which may have functions within the cell, and the release of Aβ. As α- secretase cleaves at a position more proximal to the transmembrane domain of APP than β-secretase, its activity prevents generation of Aβ (Lammich et al., 1999) There are two factors that influence the propensity of Aβ to form extracellular plaques: its abundance and its self-aggregation potential (Haass and Selkoe, 2007). The aggregation potential of Aβ is dictated by the position within the TM domain of C99 that is cleaved by γ-secretase (Jung et al., 2014; Munter et al., 2007). Further, certain AD-associated mutations in APP enhance Aβ aggregation without increasing its rate of production (Hutton et al., 1998; Mullan et al., 1992).
Figure 4.
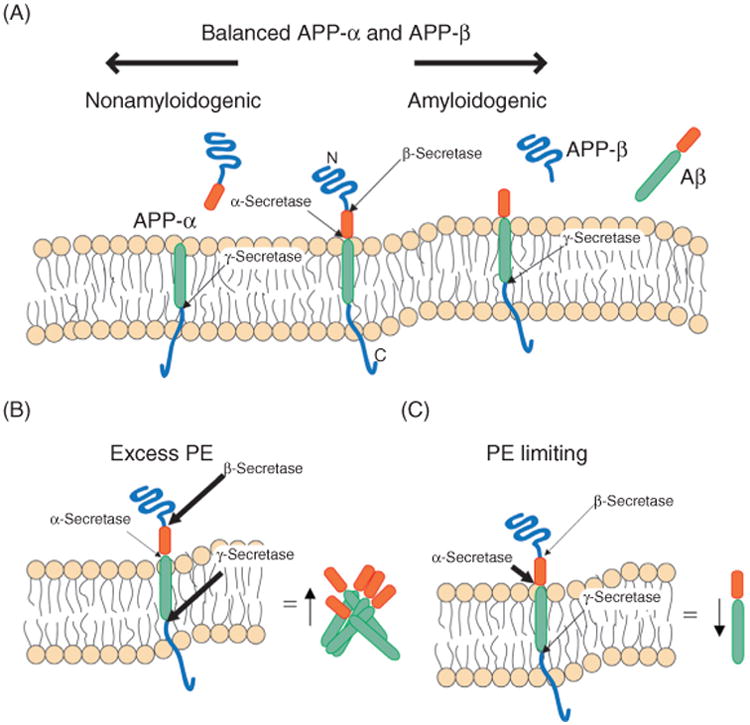
Alzheimer's disease. Schematic representation of amyloidogenic and nonamyloidogenic processing of APP. (A) Under physiologic conditions, APP is processed by both the α- and β-secretases, each event followed by cleavage by γ-secretase, and an equilibrium exists in which Aβ aggregates do not accumulate. (B) However, when there is excess PE, β- and γ-secretase activity is increased, which shifts the equilibrium toward the amyloidogenic pathway thus driving the accumulation and aggregation of Aβ. (C) In contrast, when PE is limiting (i.e., psd1Δ yeast), α-secretase activity is increased thus promoting the nonamyloidogenic pathway. How PE mechanistically alters the activity of the assorted secretases is presently unclear. The thickness of the arrows indicates the relative activity of a particular enzyme. APP-α is a cleavage product of α-secretase; APP-β is a cleavage product of β-secretase.
There are two types of AD, namely, sporadic AD, which constitute the majority of AD cases, and familial AD, which is an early onset form of AD that has been genetically linked to various mutations. The familial form is caused by mutations in genes encoding APP or the presenilins (PS; Tanzi and Bertram, 2005). In humans, there are two homologs of PS, PS1 and PS2. PS (either PS1 or PS2) is a required component of the catalytic core of the γ-secretase complex and is thus essential for its activity (Wolfe et al., 1999). The actual course of the sporadic form of AD is not clearly understood. Nevertheless, the two forms of AD are both characterized by hyperphosphorylation of intracellular TAU and an increase in the abundance of Aβ peptides.
There has been substantial debate concerning the subcellular localization of the γ-secretase complex and where pathogenic APP cleavage in fact occurs (Ankarcrona and Hultenby, 2002; Annaert and De Strooper, 1999; Kimura et al., 2001; Marambaud et al., 2002; Pasternak et al., 2003; Vetrivel et al., 2004). However, PS1, PS2, and γ-secretase activity, as well as APP have been recently shown to localize to the MAM whose membranes are characterized as being detergent resistant (Area-Gomez et al., 2009; Browman et al., 2006; Poston et al., 2011). AD-associated mutations in PS1 or PS2 result in a closer apposition of the MAM with mitochondria, which correlates with an increase in PE synthesis and γ-secretase activity (Area-Gomez et al., 2009, 2012). This tighter association between the MAM and mitochondria increases the activity of resident enzymes in both compartments, including PSD1 (Area-Gomez et al., 2012). Overall, mutations in PS1 and PS2 lead to an increase in PE levels in mitochondria, MAM, and the plasma membrane (Area-Gomez et al., 2012). Subsequently, the rise in PE levels increases γ-secretase activity and generation of Aβ (Figure 4(B)).
When PE levels are decreased, γ-secretase activity is reduced in mammalian cells and flies, and at least in the former model, α-secretase activity is increased (Figure 4(C); Nesic et al., 2012). Importantly, less Aβ accumulates in PE-depleted mammalian cells and flies. Clearly, an alteration in lipid metabolism correlates with AD. Whether the changes in lipid composition affect the accessibility of the γ-secretase cleavage site of Aβ is not known. Potentially, an increase in PE in the membrane obscures the α-secretase cleavage site thus shifting the balance in favor of the sequential action of β- and γ-secretases. Alternatively, perhaps the topology of one of the secretases (β or γ) is affected by PE, since PE has also been shown to influence certain membrane proteins in this manner (Section 4.1; Dowhan and Bogdanov, 2009). In summary, mutations in PS1 and PS2 increase MAM-mitochondria apposition although the underlying mechanism is not known (Area-Gomez et al., 2009, 2012). The tighter association between MAM and mitochondria increases production of PE and the amount of PE in membranes, which augments production of Aβ. How changes in the abundance of PE in membranes modulates the activity of α- and γ-secretase, is currently not known.
5.2 Parkinson's Disease
Parkinson's disease (PD) is morphologically characterized by the presence of intracellular proteinaceous inclusions called Lewy Bodies, which are mainly composed of cytosolic aggregates of α-synuclein in the affected neurons (Klein and Westenberger, 2012; Spillantini et al., 1997). Accumulation of these aggregates results in the progressive loss of dopaminergic neurons in the midbrain region called the substantia nigra pars compacta (Dawson and Dawson, 2003; Polymeropoulos et al., 1997). The majority (∼90%) of PD cases are sporadic; only ∼10% of PD cases have a familial history.
Even though over 28 chromosomal regions have either been directly or loosely associated with PD, to date there are only six genes that have been conclusively linked to PD (e.g., a mutation is sufficient to cause disease). Mutations in SNCA (PARK4) and LRRK2 (PARK8) result in autosomal dominant PD (Polymeropoulos et al., 1996, 1997; Singleton et al., 2003), while Parkin (PARK2), PINK1 (PARK6), DJ-1 (PARK7), and ATP13A2 (PARK9) mutations are linked with autosomal recessive PD (Abbas et al., 1999; Bonifati et al., 2003; Kitada et al., 1998). Here, we will only focus on mutations in the α-synuclein encoding gene, SNCA, which are associated with alterations in cellular PE levels. Patients with mutations in SNCA have early onset PD (age <50 years; Polymeropoulos et al., 1996). Currently there are five different missense mutations in α-synuclein as well as SNCA duplications and triplications that have been associated with PD (Kara et al., 2013; Klein and Schlossmacher, 2006; Kruger et al., 1998; Polymeropoulos et al., 1997; Singleton et al., 2003; Uchiyama et al., 2008; Zarranz et al., 2004).
The mechanism(s) by which α-synuclein confers selective death of dopaminergic neurons is currently unclear. α-synuclein is found in presynaptic neurons where it is thought to participate in neurotransmitter vesicle release by promoting assembly of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes (Burré et al., 2010). Overexpression of α-synuclein increases the pool of docked but unfused secretory vesicles with the end result being a decrease in neurotransmitter release (Nemani et al., 2010). While the majority of α-synuclein is cytosolic, it also associates with membranes including lipid rafts (Fortin et al., 2004). In fact, when the ability to interact with lipid rafts is disturbed, either through pharmacologic inhibition of lipid raft formation or introduction of a PD-associated mutation A30P, α-synuclein (mutant or wild-type) does not properly localize to synapses (Fortin et al., 2004). Interestingly, the MAM subcompartment of the ER has properties that are reminiscent of lipid rafts (Hayashi and Fujimoto, 2010) and a fraction of α-synuclein, but not the A30P mutant unable to bind lipid rafts, cofractionates with this compartment (Figure 5(A); Guardia-Laguarta et al., 2014). When the association of α-synuclein and MAM is disturbed or diminished, mitochondria–ER contacts are reduced, mitochondria become fragmented, PS synthesis in MAM and conversion to PE in mitochondria is reduced, and mitochondrial Ca2+ buffering following release of ER-Ca2+ stores is impaired (Figure 5(B); Cali et al., 2012; Guardia-Laguarta et al., 2014). These results indicate that in addition to its synaptic function, α-synuclein serves an important role at the ER–mitochondrial interface (Figure 5(A)). Relevant to PD associated with duplications and triplications of SNCA, the severe overexpression of α-synuclein increases mitochondrial fragmentation (Guardia-Laguarta et al., 2014) and reduces mitochondrial Ca2+ accumulation (Cali et al., 2012). Whether these overexpression-based alterations result from too much, or instead not enough (e.g., at high levels of expression, cytosolic α-synuclein aggregation is stimulated), MAM-associated α-synuclein is presently unclear.
Figure 5.
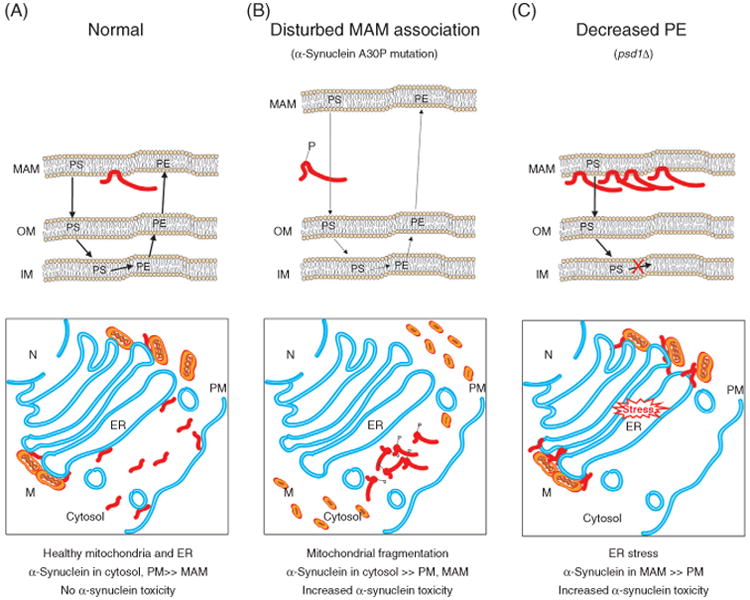
Parkinson's disease. The role of α-synuclein at the MAM and how disturbances in this association impact cell function. (A) Under physiologic conditions, α-synuclein is natively unfolded and found primarily in the cytoplasm or associated with the plasma membrane, but a small fraction associates with the MAM where it supports full mitochondrial function. (B) Mutations that affect α-synuclein membrane association prevent its association with the MAM, drive its cytosolic accumulation and aggregation, and induce mitochondrial fragmentation. (C) When PE levels become limiting, α-synuclein accumulates at the MAM leading to ER stress and cytotoxicity. Relative activity is reflected by the thickness of the arrows. N, Nucleus; M, mitochondria; PM, plasma membrane.
In psd1Δ yeast, heterologously expressed α-synuclein accumulates in the ER at the expense of its normal association with the plasma membrane; this altered subcellular distribution correlates with an increase in ER stress and yeast doubling time (Figure 5(C); Wang et al., 2014). While not improving the mitochondrial dysfunctions caused by the absence of Psd1p function, ethanolamine supplementation, which drives production of PE via the CDP-ethanolamine pathway, restores the normal α-synuclein plasma membrane localization in psd1Δ yeast and rescues the phenotypes that correlate with its accumulation at the ER. Similarly, in a worm PD model, the survival of dopaminergic neurons expressing α-synuclein is reduced in the absence of Psd1p. The addition of ethanolamine prevents this neurodegeneration in a manner that is dependent on a functional CDP-ethanolamine pathway for PE production (Wang et al., 2014). Thus, when PE is limiting, α-synuclein homeostasis is perturbed such that it is more toxic to its cellular host.
How does limiting amounts of PE disturb α-synuclein homeostasis? An intriguing possibility is centered on PE's role as a required substrate for GPI anchor formation (Menon and Stevens, 1992). The biogenesis of GPI-anchored proteins, which associate with lipid rafts, is impaired in psd1Δ yeast (Birner et al., 2001; Wang et al., 2014). α-synuclein also accumulates intracellularly in a yeast GPI anchor biosynthesis mutant (Wang et al., 2014). Interestingly, the ability of an enzyme, which is recruited to lipid rafts by GPI-anchored proteins (Okamoto et al., 2006) to access the plasma membrane is reduced in psd1Δ yeast similar to α-synuclein (Wang et al., 2014). As GPI-anchored proteins and α-synuclein, which lacks a TM domain and can bind lipids directly (Chandra et al., 2003), occupy opposite sides of the membrane, it is unlikely that α-synuclein is recruited to lipid rafts by GPI-anchored proteins. Therefore, it is tempting to speculate that in the absence of robust GPI anchor formation (e.g., in psd1Δ yeast), there is a defect in the ability to form lipid rafts that are recognized by α-synuclein, or alternatively, the ability of lipid rafts to traffic out of the ER is reduced. Regardless of the exact mechanism, the end result is a disturbance in α-synuclein homeostasis that increases its cellular toxicity.
5.3 The Balance of PE and PC in Liver Steatosis and Steatohepatitis
With over 30% of adults affected in developed countries, nonalcoholic fatty liver disease (NAFLD) is the most prevalent form of liver disease in the world (Dietrich and Hellerbrand, 2014). NAFLD encompasses a range of liver maladies that are diagnosed by assessing the level of fat deposits in and injury to hepatocytes (Tuyama and Chang, 2012). Steatosis or fatty liver, develops when excess lipids are stored as triglycerides in fat droplets or instead incorporated and secreted by the liver as very low-density lipoproteins. Increased accumulation of fat in liver cells subsequently leads to disrupted cellular functions with desensitized responses to metabolic changes making cells more susceptible to lipotoxicity and ER and oxidative stress.
Nonalcoholic steatohepatitis (NASH) involves liver steatosis, inflammation, and hepatocyte injury (Tuyama and Chang, 2012). Hepatocytes show features of “ballooning” when membrane integrity becomes compromised, which can lead to cell rupture and induction of an inflammatory response (Kleiner et al., 2005). Subsequent tissue injury leads to fibrosis in the liver, which can later mature into liver cirrhosis or cancer.
Risk factors for NAFLD include age (Frith et al., 2009), ethnicity, but most notably life style as NAFLD is often associated with obesity (Williams et al., 2011), insulin resistance (Gastaldelli et al., 2009), heart disease (Gastaldelli et al., 2009; Mirbagheri et al., 2007), and metabolic syndrome (combination of obesity, hypertension, hypertriglyceridemia, low levels of high-density lipoprotein, and/or hyperglycemia; Ryan et al., 2005; Tuyama and Chang, 2012; Williams et al., 2011). Alarmingly, it is speculated that the majority of NAFLD cases go undiagnosed, as invasive liver biopsies are the most reliable form of assessing liver damage (although some predictive tests and biomarkers for NASH exist, they may not be entirely accurate; Dietrich and Hellerbrand, 2014; Tuyama and Chang, 2012). Further, as no pharmacological treatments for NAFLD exist, patients are presently prescribed regimented diets and exercise plans designed to prevent further tissue damage and diminish the severity of steatosis.
Mice with a hepatocyte-specific deletion of PCYT2, encoding CTP: phosphoethanolamine cytidylyltransferase in the CDP-ethanolamine pathway, have normal growth and differentiation of liver cells even though there is a 50% drop in cellular PE levels (Leonardi et al., 2009). The remaining PE is made by the mitochondrial Psd pathway and is sufficient to support basic cellular function. However, mice lacking PCYT2 in hepatocytes develop steatosis that stems from a 10-fold increase in hepatocyte triglyceride synthesis. Thus, the DAG that is normally consumed to produce PE by the CDP-ethanolamine pathway is instead converted to triglycerides, which promote lipid droplet formation (Fullerton et al., 2009; Leonardi et al., 2009).
Hepatocytes generate 30% of their total PC from the trimethylation of PE by PEMT (Sundler and Akesson, 1975b). pemt−/− mice develop steatohepatitis and progress to liver failure within 3 days of being fed a choline-deficient diet (choline supplementation supports production of PC through the CDP-choline pathway; Walkey et al., 1998). The high demand for hepatic PC secretion into bile reduces levels of PC by 50% in pemt−/− mice (Li et al., 2006; Walkey et al., 1998). Secretion of PC into bile is mediated by MDR2 (multiple drug-resistant protein 2), a PC-specific flippase (Ruetz and Gros, 1994; Smit et al., 1993). Surprisingly, mdr2−/− x pemt−/− mice fed with a choline-deficient diet are resistant to the development of steatohepatitis and liver failure despite the fact that PC levels are still decreased by 50% (Li et al., 2005, 2006). The varied sensitivity of pemt−/− and mdr2−/− × pemt−/− mice to choline deprivation is due to a striking difference in their respective plasma membrane PC:PE ratios (Li et al., 2006). Interestingly, in mdr2−/− × pemt−/− mice in which the ability to secrete PC into the bile is impaired, PE and PC catabolism is increased to maintain the PC:PE ratio. The decrease in the PC:PE ratio in pemt−/− mice increases membrane permeability and facilitates hepatocyte damage. Depletion of CTP:phosphoethanolamine cytidylyltransferase in pemt−/− mice increases the PC:PE ratio, restores membrane integrity, and prevents the development of steatohepatitis although intriguingly, lipid droplets still accumulate in liver tissues (Li et al., 2006). Collectively, these results indicate that the plasma membrane PC:PE ratio is critical for hepatocyte cell integrity and that PC produced by the trimethylation of PE is physiologically important in maintaining this lipid balance.
Importantly, a decrease in the PC:PE ratio was also observed in liver biopsies from patients with nonalcoholic steatohepatitis (Arendt et al., 2013; Li et al., 2006). Thus, the maintenance of the PC:PE ratio in hepatocyte membranes is critical for the integrity of the cell although each individual phospholipid plays a different role in hepatic lipid metabolism. As PC is essential for bile secretion and incorporation into lipoproteins, PC synthesis is of particular importance for liver physiology. An increase in the ratio of hepatic PE to PC makes membranes more permeable and can account for tissue damage in pemt−/− mice (Figure 6(A) and (B); Li et al., 2006). In contrast, depletion of the CDP-ethanolamine pathway in hepatocytes leads to liver steatosis due to increased triglyceride synthesis (Figure 6(C) and (D); Leonardi et al., 2009). Therefore, changes that alter the relative abundance of PE or that directly impair PE metabolism can disturb liver function by multiple mechanisms.
Figure 6.
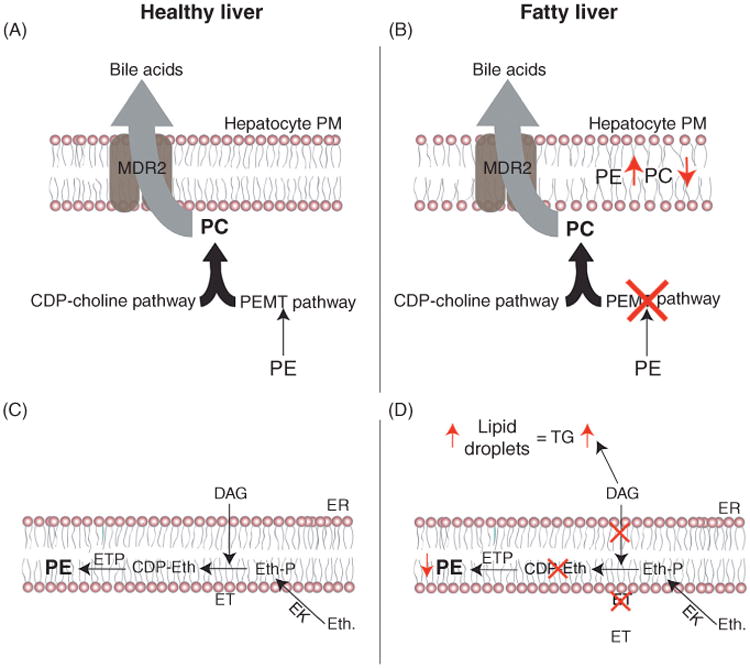
PE:PC ratio in liver disease. (A) MDR2 is a PC-specific flippase that mediates the secretion of PC into bile. (B) Depletion of PC levels by ablating its synthesis via the PEMT pathway leads to an increase in the PE:PC ratio in hepatocyte membranes due to both a decrease in its synthesis as well as continued secretion by MDR2. A decrease in the levels of PC relative to PE results in leaky hepatocyte membranes causing cell lysis and subsequent tissue damage. (C) CDP-ethanolamine pathway in hepatocytes. (D) Upon deletion of CTP:phosphoethanolamine cytidylyltransferase in the CDP-ethanolamine pathway, the DAG that is normally used to generate CDP-ethanolamine accumulates and is instead consumed to form triglyceride (TG). Increased accumulation of triglycerides leads to the development of steatosis. EK, ethanolamine kinase.
5.4 Infectious Disease
5.4.1 Pathogenic Prion Generation
Prions are infectious proteins that contain a pathogenic conformer of the mammalian prion protein (PrPsc), which can be produced by the noninfective mammalian conformer (PrPc), RNA, and lipid molecules (Deleault et al., 2003, 2012). Prion infectivity and strain properties are encoded by specific protein conformations (Kretzschmar and Tatzelt, 2013). However, neither the tertiary structure of infectious PrPsc nor the molecular mechanism(s) responsible for generating this infectious form are known. Prpsc molecules exist as insoluble aggregates, which do not allow for the determination of their crystal structure. Prion propagation occurs independent of the presence of nucleic acid (Deleault et al., 2012). NMR and mass spectrometry identified PE as a cofactor that associates with recombinant PrP. Moreover, the association of PE transforms recombinant PrP into a form capable of infecting mice. Astonishingly, treatment with phospholipase C, which removes the headgroups from phospholipids including PE, ablates the ability of PrPsc to infect brain tissues (Deleault et al., 2012). This suggests that the PE is a required cofactor necessary for the propagation of at least some infective prions.
5.4.2 Candida Virulence
Candidiasis (or candidemia) infection is the leading cause of opportunistic fungal infections in immunocompromised individuals where it has a 33.9% mortality rate (Pfaller and Diekema, 2007). Additionally, candida infections are becoming increasingly resistant to existing therapeutic treatments (Lortholary et al., 2014). Thus, studies to find ways to specifically target these infections are of critical biomedical importance (Thomas et al., 2013). Cell wall stability is essential for the virulence of Candida albicans (Ruiz-Herrera et al., 2006). Depletion of PE by targeting PSD1, PSD2, or PSS1 in C. albicans reduces its infectivity in an immunocompromised mouse model (Chen et al., 2010). Growth of pss1Δ and psd1Δpsd2Δ strains of C. albicans is decreased on dextrose-based agar plates in the absence of osmotic stabilizers (sorbitol, CaCl2, or NaCl). In addition, pss1Δ and psd1Δpsd2Δ strains are more sensitive to caspofungin treatment suggesting that cell wall integrity is affected in the absence of these lipid-synthesizing proteins. This is complemented by studies in psd1Δ yeast where the lower levels of PE impair GPI-anchor formation, which compromises cell wall integrity (Wang et al., 2014). Thus, inhibiting the Psd pathway in a manner that specifically targets proteins conserved in fungi but not humans (e.g., PSS1) represents a candidate therapeutic approach to combat fungal infections.
6. Concluding Remarks
PE is an abundant membrane phospholipid that is essential for membrane integrity, cell division, maximum mitochondrial respiratory function, membrane protein topology, and function, an important precursor for PC, and can be used to modify certain proteins. These modifications can be necessary for proper localization and/or function. One such example includes GPI-anchored proteins, which are retained in the ER when PE is limiting. Traditionally, the main function attributed to PE other than as a precursor to PC, has been associated with its nonbilayer forming structural capacity that is implicated in membrane fusion and fission events, cytokinesis, and vacuolar delivery. However, as illustrated in the present review, the functions attributed to PE go well beyond tradition.
Eukaryotes have multiple PE biosynthetic pathways. The existence of a multitude of pathways reflects the importance of PE in a cell. However, that both major pathways of PE synthesis, the CDP-ethanolamine pathway and the phosphatidylserine decarboxylation pathway, are required during mammalian development indicates that not all PE is made the same. A systematic assessment of the relative importance of each PE-producing pathway in different cells and tissues has not been provided. Such information is expected to identify functions and processes that are uniquely dependent on a given pathway.
While most studies have focused on the roles of different proteins in the development of pathologies, less has been done from a lipid perspective. Recent advances in research highlight the need to incorporate the study of lipids, as they are critical determinants of protein structure and function. The link between PE metabolism and disease is emerging and here we have highlighted those disease states in which PE is associated. Interestingly, PE can contribute to disease pathogenesis by being either increased or decreased (Alzheimer's and Parkinson's diseases). Our understanding of the role of PE in AD, PD, liver disease, fungal pathogenicity, and prion infectivity is just in its nascent stage. As such, defining the mechanistic role of PE in these and likely additional human diseases will be a main research objective in the coming years.
Acknowledgments
The authors' work was supported by National Institutes of Health Grant R01GM111548 (S.M.C.).
References
- Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Bohme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet. 1999;8:567–574. doi: 10.1093/hmg/8.4.567. [DOI] [PubMed] [Google Scholar]
- Achleitner G, Gaigg B, Krasser A, Kainersdorfer E, Kohlwein SD, Perktold A, Zellnig G, Daum G. Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur J Biochem. 1999;264:545–553. doi: 10.1046/j.1432-1327.1999.00658.x. [DOI] [PubMed] [Google Scholar]
- Achleitner G, Zweytick D, Trotter PJ, Voelker DR, Daum G. Synthesis and intracellular transport of aminoglycerophospholipids in permeabilized cells of the yeast, Saccharomycescerevisiae. J Biol Chem. 1995;270:29836–29842. doi: 10.1074/jbc.270.50.29836. [DOI] [PubMed] [Google Scholar]
- Acín-Pérez R, Fernández-Silva P, Peleato ML, Pérez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Mol Cell. 2008;32:529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- Alkhaja AK, Jans DC, Nikolov M, Vukotic M, Lytovchenko O, Ludewig F, Schliebs W, Riedel D, Urlaub H, Jakobs S, Deckers M. MINOS1 is a conserved component of mitofilin complexes and required for mitochondrial function and cristae organization. Mol Biol Cell. 2012;23:247–257. doi: 10.1091/mbc.E11-09-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankarcrona M, Hultenby K. Presenilin-1 is located in rat mitochondria. Biochem Biophys Res Commun. 2002;295:766–770. doi: 10.1016/s0006-291x(02)00735-0. [DOI] [PubMed] [Google Scholar]
- Annaert W, De Strooper B. Presenilins: molecular switches between proteolysis and signal transduction. Trends Neurosci. 1999;22:439–443. doi: 10.1016/s0166-2236(99)01455-1. [DOI] [PubMed] [Google Scholar]
- Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, Yu WH, Duff KE, Yaffe MP, Pon LA, Schon EA. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol. 2009;175:1810–1816. doi: 10.2353/ajpath.2009.090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL, Schon EA. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012;31:4106–4123. doi: 10.1038/emboj.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt BM, Ma DW, Simons B, Noureldin SA, Therapondos G, Guindi M, Sherman M, Allard JP. Nonalcoholic fatty liver disease is associated with lower hepatic and erythrocyte ratios of phosphatidylcholine to phosphatidylethanolamine. Appl Physiol Nutr Metab. 2013;38:334–340. doi: 10.1139/apnm-2012-0261. [DOI] [PubMed] [Google Scholar]
- Arikketh D, Nelson R, Vance JE. Defining the importance of phosphatidylserine synthase-1 (PSS1): unexpected viability of PSS1-deficient mice. J Biol Chem. 2008;283:12888–12897. doi: 10.1074/jbc.M800714200. [DOI] [PubMed] [Google Scholar]
- Atkinson KD, Jensen B, Kolat AI, Storm EM, Henry SA, Fogel S. Yeast mutants auxotrophic for choline or ethanolamine. J Bacteriol. 1980;141:558–564. doi: 10.1128/jb.141.2.558-564.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae-Lee MS, Carman GM. Phosphatidylserine synthesis in Saccharomycescerevisiae. Purification and characterization of membrane-associated phosphatidylserine synthase. J Biol Chem. 1984;259:10857–10862. [PubMed] [Google Scholar]
- Baile MG, Lu YW, Claypool SM. The topology and regulation of cardiolipin biosynthesis and remodeling in yeast. Chem Phys Lipids. 2014;179:25–31. doi: 10.1016/j.chemphyslip.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Horvath SE, Bottinger L, Gebert N, Daum G, Pfanner N. Role of phosphatidylethanolamine in the biogenesis of mitochondrial outer membrane proteins. J Biol Chem. 2013;288:16451–16459. doi: 10.1074/jbc.M112.442392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger KH, Sogo LF, Yaffe MP. Mdm12p, a component required for mitochondrial inheritance that is conserved between budding and fission yeast. J Cell Biol. 1997;136:545–553. doi: 10.1083/jcb.136.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergo MO, Gavino BJ, Steenbergen R, Sturbois B, Parlow AF, Sanan DA, Skarnes WC, Vance JE, Young SG. Defining the importance of phosphatidylserine synthase 2 in mice. J Biol Chem. 2002;277:47701–47708. doi: 10.1074/jbc.M207734200. [DOI] [PubMed] [Google Scholar]
- Birner R, Burgermeister M, Schneiter R, Daum G. Roles of phosphatidylethanolamine and of its several biosynthetic pathways in Saccharomyces cerevisiae. Mol Biol Cell. 2001;12:997–1007. doi: 10.1091/mbc.12.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerve KS. Phospholipid substrate-specificity of the L-serine base-exchange enzyme in rat liver microsomal fraction. Biochem J. 1984;219:781–784. doi: 10.1042/bj2190781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleijerveld OB, Brouwers JF, Vaandrager AB, Helms JB, Houweling M. The CDP-ethanolamine pathway and phosphatidylserine decarboxylation generate different phosphatidylethanolamine molecular species. J Biol Chem. 2007;282:28362–28372. doi: 10.1074/jbc.M703786200. [DOI] [PubMed] [Google Scholar]
- Bogdanov M, Dowhan W. Phosphatidylethanolamine is required for invivo function of the membrane-associated lactose permease of Escherichia coli. J Biol Chem. 1995;270:732–739. doi: 10.1074/jbc.270.2.732. [DOI] [PubMed] [Google Scholar]
- Bogdanov M, Dowhan W. Phospholipid-assisted protein folding: phosphatidylethanolamine is required at a late step of the conformational maturation of the polytopic membrane protein lactose permease. EMBO J. 1998;17:5255–5264. doi: 10.1093/emboj/17.18.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Dowhan W. Lipid-assisted protein folding. J Biol Chem. 1999;274:36827–36830. doi: 10.1074/jbc.274.52.36827. [DOI] [PubMed] [Google Scholar]
- Bogdanov M, Dowhan W, Vitrac H. Lipids and topological rules governing membrane protein assembly. Biochim Biophys Acta. 2014;1843:1475–1488. doi: 10.1016/j.bbamcr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Heacock PN, Dowhan W. A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J. 2002;21:2107–2116. doi: 10.1093/emboj/21.9.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Xie J, Dowhan W. Lipid-protein interactions drive membrane protein topogenesis in accordance with the positive inside rule. J Biol Chem. 2009;284:9637–9641. doi: 10.1074/jbc.R800081200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Xie J, Heacock P, Dowhan W. To flip or not to flip: lipid-protein charge interactions are a determinant of final membrane protein topology. J Cell Biol. 2008;182:925–935. doi: 10.1083/jcb.200803097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Borkenhagen LF, Kennedy EP, Fielding L. Enzymatic formation and decarboxylation of phosphatidylserine. J Biol Chem. 1961;236:PC28–PC30. [Google Scholar]
- Bottinger L, Horvath SE, Kleinschroth T, Hunte C, Daum G, Pfanner N, Becker T. Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J Mol Biol. 2012;423:677–686. doi: 10.1016/j.jmb.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer J, Greenberg DM. Methyl transfering enzyme system of microsomes in the biosynthesis of lecithin (phosphatidylcholine) Biochim Biophys Acta. 1961;46:205–216. [Google Scholar]
- Bretscher MS. Asymmetrical lipid bilayer structure for biological membranes. Nat New Biol. 1972;236:11–12. doi: 10.1038/newbio236011a0. [DOI] [PubMed] [Google Scholar]
- Browman DT, Resek ME, Zajchowski LD, Robbins SM. Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J Cell Sci. 2006;119:3149–3160. doi: 10.1242/jcs.03060. [DOI] [PubMed] [Google Scholar]
- Burgermeister M, Birner-Grunberger R, Nebauer R, Daum G. Contribution of different pathways to the supply of phosphatidylethanolamine and phosphatidylcholine to mitochondrial membranes of the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2004;1686:161–168. doi: 10.1016/j.bbalip.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Burgess SM, Delannoy M, Jensen RE. MMM1 encodes a mitochondrial outer membrane protein essential for establishing and maintaining the structure of yeast mitochondria. J Cell Biol. 1994;126:1375–1391. doi: 10.1083/jcb.126.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- Cali T, Ottolini D, Negro A, Brini M. Alpha-synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum–mitochondria interactions. J Biol Chem. 2012;287:17914–17929. doi: 10.1074/jbc.M111.302794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EY, McQuibban GA. Phosphatidylserine decarboxylase 1 (Psd1) promotes mitochondrial fusion by regulating the biophysical properties of the mitochondrial membrane and alternative topogenesis of mitochondrial genome maintenance protein 1 (Mgm1) J Biol Chem. 2012;287:40131–40139. doi: 10.1074/jbc.M112.399428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Chen X, Rizo J, Jahn R, Südhof TC. A broken α-helix in folded α-synuclein. J Biol Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- Chang SC, Heacock PN, Mileykovskaya E, Voelker DR, Dowhan W. Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae. J Biol Chem. 1998;273:14933–14941. doi: 10.1074/jbc.273.24.14933. [DOI] [PubMed] [Google Scholar]
- Chen D, Zhang XY, Shi Y. Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem J. 2006;398:169–176. doi: 10.1042/BJ20060303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YL, Montedonico AE, Kauffman S, Dunlap JR, Menn FM, Reynolds TB. Phosphatidylserine synthase and phosphatidylserine decarboxylase are essential for cell wall integrity and virulence in Candida albicans. Mol Microbiol. 2010;75:1112–1132. doi: 10.1111/j.1365-2958.2009.07018.x. [DOI] [PubMed] [Google Scholar]
- Choi JY, Augagneur Y, Ben Mamoun C, Voelker DR. Identification of gene encoding Plasmodium knowlesi phosphatidylserine decarboxylase by genetic complementation in yeast and characterization of in vitro maturation of encoded enzyme. J Biol Chem. 2012;287:222–232. doi: 10.1074/jbc.M111.313676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, Duraisingh MT, Marti M, Ben Mamoun C, Voelker DR. From protease to decarboxylase: The molecular metamorphosis of phosphatidylserine decarboxylase. J Biol Chem. 2015;290:10972–10980. doi: 10.1074/jbc.M115.642413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancey CJ, Chang SC, Dowhan W. Cloning of a gene (PSD1) encoding phosphatidylserine decarboxylase from Saccharomyces cerevisiae by complementation of an Escherichiacoli mutant. J Biol Chem. 1993;268:24580–24590. [PubMed] [Google Scholar]
- Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends Biochem Sci. 2012;37:32–41. doi: 10.1016/j.tibs.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbeau A, Nachbaur J, Vignais PM. Enzymic characterization and lipid composition of rat liver subcellular membranes. Biochim Biophys Acta. 1971;249:462–492. doi: 10.1016/0005-2736(71)90123-4. [DOI] [PubMed] [Google Scholar]
- Connerth M, Tatsuta T, Haag M, Klecker T, Westermann B, Langer T. Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science. 2012;338:815–818. doi: 10.1126/science.1225625. [DOI] [PubMed] [Google Scholar]
- Cowart LA, Obeid LM. Yeast sphingolipids: recent developments in understanding biosynthesis, regulation, and function. Biochim Biophys Acta. 2007;1771:421–431. doi: 10.1016/j.bbalip.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciat CM, Brunner S, Baumann F, Neupert W, Stuart RA. The cytochrome bc1 and cytochrome c oxidase complexes associate to form a single supracomplex in yeast mitochondria. J Biol Chem. 2000;275:18093–18098. doi: 10.1074/jbc.M001901200. [DOI] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Vance JE, Chen MH, Voelker DR, Vance DE. Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. A specific biochemical and cytological marker for a unique membrane fraction in rat liver. J Biol Chem. 1993;268:16655–16663. [PubMed] [Google Scholar]
- Daum G. Lipids of mitochondria. Biochim Biophys Acta. 1985;822:1–42. doi: 10.1016/0304-4157(85)90002-4. [DOI] [PubMed] [Google Scholar]
- Daum G, Heidorn E, Paltauf F. Intracellular transfer of phospholipids in the yeast Saccharomycescerevisiae. Biochim Biophys Acta. 1986;878:93–101. doi: 10.1016/0005-2760(86)90347-4. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- de Kroon AI, Koorengevel MC, Vromans TA, de Kruijff B. Continuous equilibration of phosphatidylcholine and its precursors between endoplasmic reticulum and mitochondria in yeast. Mol Biol Cell. 2003;14:2142–2150. doi: 10.1091/mbc.E02-08-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChavigny A, Heacock PN, Dowhan W. Sequence and inactivation of the pss gene of Escherichiacoli. Phosphatidylethanolamine may not be essential for cell viability. J Biol Chem. 1991;266:10710. [PubMed] [Google Scholar]
- Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature. 2003;425:717–720. doi: 10.1038/nature01979. [DOI] [PubMed] [Google Scholar]
- Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci USA. 2012;109:8546–8551. doi: 10.1073/pnas.1204498109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- Dennis EA, Kennedy EP. Intracellular sites of lipid synthesis and the biogenesis of mitochondria. J Lipid Res. 1972;13:263–267. [PubMed] [Google Scholar]
- Dietrich P, Hellerbrand C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract Res Clin Gastroenterol. 2014;28:637–653. doi: 10.1016/j.bpg.2014.07.008. [DOI] [PubMed] [Google Scholar]
- Dimmer KS, Fritz S, Fuchs F, Messerschmitt M, Weinbach N, Neupert W, Westermann B. Genetic basis of mitochondrial function and morphology in Saccharomycescerevisiae. Mol Biol Cell. 2002;13:847–853. doi: 10.1091/mbc.01-12-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowhan W, Bogdanov M. Lipid-dependent membrane protein topogenesis. Annu Rev Biochem. 2009;78:515–540. doi: 10.1146/annurev.biochem.77.060806.091251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowhan W, Wickner WT, Kennedy EP. Purification and properties of phosphatidylserine decarboxylase from Escherichia coli. J Biol Chem. 1974;249:3079–3084. [PubMed] [Google Scholar]
- Drin G. Topological regulation of lipid balance in cells. Annu Rev Biochem. 2014;83:51–77. doi: 10.1146/annurev-biochem-060713-035307. [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V, Futerman AH, Geiger T, Schuldiner M. A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell. 2014;30:95–102. doi: 10.1016/j.devcel.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Emoto K, Kobayashi T, Yamaji A, Aizawa H, Yahara I, Inoue K, Umeda M. Redistribution of phosphatidylethanolamine at the cleavage furrow of dividing cells during cytokinesis. Proc Natl Acad Sci USA. 1996;93:12867–12872. doi: 10.1073/pnas.93.23.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto K, Umeda M. An essential role for a membrane lipid in cytokinesis. Regulation of contractile ring disassembly by redistribution of phosphatidylethanolamine. J Cell Biol. 2000;149:1215–1224. doi: 10.1083/jcb.149.6.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet. 2007;41:401–427. doi: 10.1146/annurev.genet.41.110306.130315. [DOI] [PubMed] [Google Scholar]
- Ferguson MA. The structure, biosynthesis and functions of glycosylphosphatidylinositol anchors, and the contributions of trypanosome research. J Cell Sci. 1999;112(Pt 17):2799–2809. doi: 10.1242/jcs.112.17.2799. [DOI] [PubMed] [Google Scholar]
- Flury I, Benachour A, Conzelmann A. YLL031c belongs to a novel family of membrane proteins involved in the transfer of ethanolaminephosphate onto the core structure of glycosylphosphatidylinositol anchors in yeast. J Biol Chem. 2000;275:24458–24465. doi: 10.1074/jbc.M003844200. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505:335–343. doi: 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith J, Day CP, Henderson E, Burt AD, Newton JL. Non-alcoholic fatty liver disease in older people. Gerontology. 2009;55:607–613. doi: 10.1159/000235677. [DOI] [PubMed] [Google Scholar]
- Fujita M, Kinoshita T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Lett. 2010;584:1670–1677. doi: 10.1016/j.febslet.2009.10.079. [DOI] [PubMed] [Google Scholar]
- Fullerton MD, Hakimuddin F, Bakovic M. Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2) Mol Cell Biol. 2007;27:3327–3336. doi: 10.1128/MCB.01527-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton MD, Hakimuddin F, Bonen A, Bakovic M. The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J Biol Chem. 2009;284:25704–25713. doi: 10.1074/jbc.M109.023846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, Hannun YA. The complex life of simple sphingolipids. EMBO Rep. 2004;5:777–782. doi: 10.1038/sj.embor.7400208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaigg B, Simbeni R, Hrastnik C, Paltauf F, Daum G. Characterization of a microsomal subfraction associated with mitochondria of the yeast, Saccharomycescerevisiae. Involvement in synthesis and import of phospholipids into mitochondria. Biochim Biophys Acta. 1995;1234:214–220. doi: 10.1016/0005-2736(94)00287-y. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez C, Schneider TG, Hajnoczky G, Csordas G. Alignment of sarcoplasmic reticulum-mitochondrial junctions with mitochondrial contact points. Am J Physiol Heart Circ Physiol. 2011;301:H1907–H1915. doi: 10.1152/ajpheart.00397.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaldelli A, Kozakova M, Hojlund K, Flyvbjerg A, Favuzzi A, Mitrakou A, Balkau B. Fatty liver is associated with insulin resistance, risk of coronary heart disease, and early atherosclerosis in a large European population. Hepatology. 2009;49:1537–1544. doi: 10.1002/hep.22845. [DOI] [PubMed] [Google Scholar]
- Gaynor EC, Mondésert G, Grimme SJ, Reed SI, Orlean P, Emr SD. MCD4 encodes a conserved endoplasmic reticulum membrane protein essential for glycosylphosphatidylinositol anchor synthesis in yeast. Mol Biol Cell. 1999;10:627–648. doi: 10.1091/mbc.10.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnamusch E, Kalaus C, Hrastnik C, Paltauf F, Daum G. Transport of phospholipids between subcellular membranes of wild-type yeast cells and of the phosphatidylinositol transfer protein-deficient strain Saccharomycescerevisiae sec 14. Biochim Biophys Acta. 1992;1111:120–126. doi: 10.1016/0005-2736(92)90281-p. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Gohil VM, Thompson MN, Greenberg ML. Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine and cardiolipin biosynthetic pathways in Saccharomyces cerevisiae. J Biol Chem. 2005;280:35410–35416. doi: 10.1074/jbc.M505478200. [DOI] [PubMed] [Google Scholar]
- Gottlieb D, Heideman W, Saba JD. The DPL1 gene is involved in mediating the response to nutrient deprivation in Saccharomycescerevisiae. Mol Cell Biol Res Commun. 1999;1:66–71. doi: 10.1006/mcbr.1999.0109. [DOI] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, Voos W, Schon EA, Przedborski S. Alpha-synuclein is localized to mitochondria-associated ER membranes. J Neurosci. 2014;34:249–259. doi: 10.1523/JNEUROSCI.2507-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulshan K, Shahi P, Moye-Rowley WS. Compartment-specific synthesis of phosphatidylethanolamine is required for normal heavy metal resistance. Mol Biol Cell. 2010;21:443–455. doi: 10.1091/mbc.E09-06-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G, Thomas AP. Minimal requirements for calcium oscillations driven by the IP3 receptor. EMBO J. 1997;16:3533–3543. doi: 10.1093/emboj/16.12.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T. Autophagosomes form at ER–mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Harner M, Korner C, Walther D, Mokranjac D, Kaesmacher J, Welsch U, Griffith J, Mann M, Reggiori F, Neupert W. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 2011;30:4356–4370. doi: 10.1038/emboj.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Fujimoto M. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol Pharmacol. 2010;77:517–528. doi: 10.1124/mol.109.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemelaar J, Lelyveld VS, Kessler BM, Ploegh HL. A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J Biol Chem. 2003;278:51841–51850. doi: 10.1074/jbc.M308762200. [DOI] [PubMed] [Google Scholar]
- Herlan M, Bornhövd C, Hell K, Neupert W, Reichert AS. Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J Cell Biol. 2004;165:167–173. doi: 10.1083/jcb.200403022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlan M, Vogel F, Bornhövd C, Neupert W, Reichert AS. Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J Biol Chem. 2003;278:27781–27788. doi: 10.1074/jbc.M211311200. [DOI] [PubMed] [Google Scholar]
- Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, Shaw JM. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol. 1998;143:359–373. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmstad RH, Bell RM. sn-1,2-diacylglycerol choline- and ethanolaminephosphotransferases in Saccharomycescerevisiae. Mixed micellar analysis of the CPT1 and EPT1 gene products. J Biol Chem. 1991;266:4357–4365. [PubMed] [Google Scholar]
- Hong Y, Maeda Y, Watanabe R, Ohishi K, Mishkind M, Riezman H, Kinoshita T. Pig-n, a mammalian homologue of yeast Mcd4p, is involved in transferring phosphoethanolamine to the first mannose of the glycosylphosphatidylinositol. J Biol Chem. 1999;274:35099–35106. doi: 10.1074/jbc.274.49.35099. [DOI] [PubMed] [Google Scholar]
- Honscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, van der Laan M, Cabrera M, Reggiori F, Ungermann C. Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell. 2014;30:86–94. doi: 10.1016/j.devcel.2014.06.006. [DOI] [PubMed] [Google Scholar]
- Hoppins S, Collins SR, Cassidy-Stone A, Hummel E, Devay RM, Lackner LL, Westermann B, Schuldiner M, Weissman JS, Nunnari J. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol. 2011;195:323–340. doi: 10.1083/jcb.201107053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath SE, Bottinger L, Vogtle FN, Wiedemann N, Meisinger C, Becker T, Daum G. Processing and topology of the yeast mitochondrial phosphatidylserine decarboxylase 1. J Biol Chem. 2012;287:36744–36755. doi: 10.1074/jbc.M112.398107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovius R, Faber B, Brigot B, Nicolay K, de Kruijff B. On the mechanism of the mitochondrial decarboxylation of phosphatidylserine. J Biol Chem. 1992;267:16790–16795. [PubMed] [Google Scholar]
- Hutton M, Perez-Tur J, Hardy J. Genetics of Alzheimer's disease. Essays Biochem. 1998;33:117–131. doi: 10.1042/bse0330117. [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- Ikezawa H. Glycosylphosphatidylinositol (GPI)-anchored proteins. Biol Pharm Bull. 2002;25:409–417. doi: 10.1248/bpb.25.409. [DOI] [PubMed] [Google Scholar]
- Ishidate K, Furusawa K, Nakazawa Y. Complete co-purification of choline kinase and ethanolamine kinase from rat kidney and immunological evidence for both kinase activities residing on the same enzyme protein(s) in rat tissues. Biochim Biophys Acta. 1985;836:119–124. [PubMed] [Google Scholar]
- Itoh K, Tamura Y, Iijima M, Sesaki H. Effects of Fcj1-Mos1 and mitochondrial division on aggregation of mitochondrial DNA nucleoids and organelle morphology. Mol Biol Cell. 2013;24:1842–1851. doi: 10.1091/mbc.E13-03-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Stanford N, Bhagwat N, Seiler B, Costanzo M, Boone C, Oelkers P. Identification of a novel lysophospholipid acyltransferase in Saccharomycescerevisiae. J Biol Chem. 2007;282:30562–30569. doi: 10.1074/jbc.M706326200. [DOI] [PubMed] [Google Scholar]
- Jans DC, Wurm CA, Riedel D, Wenzel D, Stagge F, Deckers M, Rehling Ps, Jakobs S. STED super-resolution microscopy reveals an array of MINOS clusters along human mitochondria. Proc Natl Acad Sci USA. 2013;110:8936–8941. doi: 10.1073/pnas.1301820110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Rizavi HS, Greenberg ML. Cardiolipin is not essential for the growth of Saccharomyces cerevisiae on fermentable or non-fermentable carbon sources. Mol Microbiol. 1997;26:481–491. doi: 10.1046/j.1365-2958.1997.5841950.x. [DOI] [PubMed] [Google Scholar]
- Jones BA, Fangman WL. Mitochondrial DNA maintenance in yeast requires a protein containing a region related to the GTP-binding domain of dynamin. Genes Dev. 1992;6:380–389. doi: 10.1101/gad.6.3.380. [DOI] [PubMed] [Google Scholar]
- Joshi AS, Thompson MN, Fei N, Huttemann M, Greenberg ML. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J Biol Chem. 2012;287:17589–17597. doi: 10.1074/jbc.M111.330167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AS, Zhou J, Gohil VM, Chen S, Greenberg ML. Cellular functions of cardiolipin in yeast. Biochim Biophys Acta. 2009;1793:212–218. doi: 10.1016/j.bbamcr.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JI, Premraj S, Cruz PE, Ladd TB, Kwak Y, Koo EH, Felsenstein KM, Golde TE, Ran Y. Independent relationship between amyloid precursor protein (APP) dimerization and gamma-secretase processivity. PLoS One. 2014;9:e111553. doi: 10.1371/journal.pone.0111553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kainu V, Hermansson M, Hanninen S, Hokynar K, Somerharju P. Import of phosphatidylserine to and export of phosphatidylethanolamine molecular species from mitochondria. Biochim Biophys Acta. 2013;1831:429–437. doi: 10.1016/j.bbalip.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Kara E, Lewis PA, Ling H, Proukakis C, Houlden H, Hardy J. Alpha-synuclein mutations cluster around a putative protein loop. Neurosci Lett. 2013;546:67–70. doi: 10.1016/j.neulet.2013.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EP, Weiss SB. The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem. 1956;222:193–214. [PubMed] [Google Scholar]
- Kim J, Huang WP, Klionsky DJ. Membrane recruitment of Aut7p in the autophagy and cytoplasm to vacuole targeting pathways requires Aut1p, Aut2p, and the autophagy conjugation complex. J Cell Biol. 2001;152:51–64. doi: 10.1083/jcb.152.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura N, Nakamura SI, Honda T, Takashima A, Nakayama H, Ono F, Sakakibara I, Doi K, Kawamura S, Yoshikawa Y. Age-related changes in the localization of presenilin-1 in cynomolgus monkey brain. Brain Res. 2001;922:30–41. doi: 10.1016/s0006-8993(01)03146-8. [DOI] [PubMed] [Google Scholar]
- Kinoshita T, Fujita M, Maeda Y. Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: recent progress. J Biochem. 2008;144:287–294. doi: 10.1093/jb/mvn090. [DOI] [PubMed] [Google Scholar]
- Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147:435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, Ohsumi Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–276. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Klein C, Schlossmacher MG. The genetics of Parkinson disease: Implications for neurological care. Nat Clin Pract Neurol. 2006;2:136–146. doi: 10.1038/ncpneuro0126. [DOI] [PubMed] [Google Scholar]
- Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- Knoll G, Brdiczka D. Changes in freeze-fractured mitochondrial membranes correlated to their energetic state: Dynamic interactions of the boundary membranes. Biochim Biophys Acta. 1983;733:102–110. doi: 10.1016/0005-2736(83)90095-0. [DOI] [PubMed] [Google Scholar]
- Kodaki T, Yamashita S. Yeast phosphatidylethanolamine methylation pathway. Cloning and characterization of two distinct methyltransferase genes. J Biol Chem. 1987;262:15428–15435. [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER–mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Osman C, Walter P. The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc Natl Acad Sci USA. 2011;108:14151–14156. doi: 10.1073/pnas.1111314108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar H, Tatzelt J. Prion disease: a tale of folds and strains. Brain Pathol. 2013;23:321–332. doi: 10.1111/bpa.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Kuchler K, Daum G, Paltauf F. Subcellular and submitochondrial localization of phospholipid-synthesizing enzymes in Saccharomycescerevisiae. J Bacteriol. 1986;165:901–910. doi: 10.1128/jb.165.3.901-910.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuge O, Nishijima M, Akamatsu Y. Isolation of a somatic-cell mutant defective in phosphatidylserine biosynthesis. Proc Natl Acad Sci USA. 1985;82:1926–1930. doi: 10.1073/pnas.82.7.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuge O, Nishijima M, Akamatsu Y. Phosphatidylserine biosynthesis in cultured Chinese hamster ovary cells. III. Genetic evidence for utilization of phosphatidylcholine and phosphatidylethanolamine as precursors. J Biol Chem. 1986;261:5795–5798. [PubMed] [Google Scholar]
- Kuge O, Nishijima M, Akamatsu Y. A cloned gene encoding phosphatidylserine decarboxylase complements the phosphatidylserine biosynthetic defect of a Chinese hamster ovary cell mutant. J Biol Chem. 1991;266:6370–6376. [PubMed] [Google Scholar]
- Kuge O, Saito K, Kojima M, Akamatsu Y, Nishijima M. Post-translational processing of the phosphatidylserine decarboxylase gene product in Chinese hamster ovary cells. Biochem J. 1996;319(Pt 1):33–38. doi: 10.1042/bj3190033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S, Chao JT, Tavassoli S, Wong AK, Choudhary V, Young BP, Loewen CJ, Prinz WA. A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 2014;12:e1001969. doi: 10.1371/journal.pbio.1001969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Nett JH, Trumpower BL, Hunte C. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J. 2001;20:6591–6600. doi: 10.1093/emboj/20.23.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapuente-Brun E, Moreno-Loshuertos R, Acin-Perez R, Latorre-Pellicer A, Colas C, Balsa E, Perales-Clemente E, Quiros PM, Calvo E, Rodriguez-Hernandez MA, Navas P, Cruz R, Carracedo A, Lopez-Otin C, Perez-Martos A, Fernandez-Silva P, Fernandez-Vizarra E, Enriquez JA. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340:1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- Leonardi R, Frank MW, Jackson PD, Rock CO, Jackowski S. Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis. J Biol Chem. 2009;284:27077–27089. doi: 10.1074/jbc.M109.031336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QX, Dowhan W. Structural characterization of Escherichiacoli phosphatidylserine decarboxylase. J Biol Chem. 1988;263:11516–11522. [PubMed] [Google Scholar]
- Li QX, Dowhan W. Studies on the mechanism of formation of the pyruvate prosthetic group of phosphatidylserine decarboxylase from Escherichia coli. J Biol Chem. 1990;265:4111–4115. [PubMed] [Google Scholar]
- Li Z, Agellon LB, Allen TM, Umeda M, Jewell L, Mason A, Vance DE. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006;3:321–331. doi: 10.1016/j.cmet.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Li Z, Agellon LB, Vance DE. Phosphatidylcholine homeostasis and liver failure. J Biol Chem. 2005;280:37798–37802. doi: 10.1074/jbc.M508575200. [DOI] [PubMed] [Google Scholar]
- Lortholary O, Renaudat C, Sitbon K, Madec Y, Denoeud-Ndam L, Wolff M, Fontanet A, Bretagne S, Dromer F. Worrisome trends in incidence and mortality of candidemia in intensive care units (Paris area, 2002-2010) Intensive Care Med. 2014;40:1303–1312. doi: 10.1007/s00134-014-3408-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YW, Claypool SM. Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front Genet. 2015;6:3. doi: 10.3389/fgene.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykidis A, Jackson P, Jackowski S. Lipid activation of CTP: phosphocholine cytidylyltransferase alpha: characterization and identification of a second activation domain. Biochemistry. 2001;40:494–503. doi: 10.1021/bi002140r. [DOI] [PubMed] [Google Scholar]
- Mancini A, Del Rosso F, Roberti R, Orvietani P, Coletti L, Binaglia L. Purification of ethanolaminephosphotransferase from bovine liver microsomes. Biochim Biophys Acta. 1999;1437:80–92. doi: 10.1016/s1388-1981(98)00011-0. [DOI] [PubMed] [Google Scholar]
- Mannella CA, Buttle K, Rath BK, Marko M. Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. Biofactors. 1998;8:225–228. doi: 10.1002/biof.5520080309. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK. A presenilin-1/ gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21:1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- Maydan G, Noyman I, Har-Zahav A, Neriah ZB, Pasmanik-Chor M, Yeheskel A, Albin-Kaplanski A, Maya I, Magal N, Birk E, Simon AJ, Halevy A, Rechavi G, Shohat M, Straussberg R, Basel-Vanagaite L. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet. 2011;48:383–389. doi: 10.1136/jmg.2010.087114. [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Saurya S, Freeman M. Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature. 2003;423:537–541. doi: 10.1038/nature01633. [DOI] [PubMed] [Google Scholar]
- Meisinger C, Rissler M, Chacinska A, Szklarz LK, Milenkovic D, Kozjak V, Schonfisch B, Lohaus C, Meyer HE, Yaffe MP, Guiard B, Wiedemann N, Pfanner N. The mitochondrial morphology protein Mdm10 functions in assembly of the preprotein translocase of the outer membrane. Dev Cell. 2004;7:61–71. doi: 10.1016/j.devcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Menon AK, Stevens VL. Phosphatidylethanolamine is the donor of the ethanolamine residue linking a glycosylphosphatidylinositol anchor to protein. J Biol Chem. 1992;267:15277–15280. [PubMed] [Google Scholar]
- Miller MA, Kent C. Characterization of the pathways for phosphatidylethanolamine biosynthesis in Chinese hamster ovary mutant and parental cell lines. J Biol Chem. 1986;261:9753–9761. [PubMed] [Google Scholar]
- Mirbagheri SA, Rashidi A, Abdi S, Saedi D, Abouzari M. Liver: an alarm for the heart? Liver Int. 2007;27:891–894. doi: 10.1111/j.1478-3231.2007.01531.x. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- Mouritsen OG, Zuckermann MJ. What's so special about cholesterol? Lipids. 2004;39:1101–1113. doi: 10.1007/s11745-004-1336-x. [DOI] [PubMed] [Google Scholar]
- Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair U, Yen WL, Mari M, Cao Y, Xie Z, Baba M, Reggiori F, Klionsky DJ. A role for Atg8-PE deconjugation in autophagosome biogenesis. Autophagy. 2012;8:780–793. doi: 10.4161/auto.19385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima A, Hosaka K, Nikawa J. Cloning of a human cDNA for CTP-phosphoethanolamine cytidylyltransferase by complementation in vivo of a yeast mutant. J Biol Chem. 1997;272:9567–9572. doi: 10.1074/jbc.272.14.9567. [DOI] [PubMed] [Google Scholar]
- Nebauer R, Schuiki I, Kulterer B, Trajanoski Z, Daum G. The phosphatidylethanolamine level of yeast mitochondria is affected by the mitochondrial components Oxa1p and Yme1p. FEBS J. 2007;274:6180–6190. doi: 10.1111/j.1742-4658.2007.06138.x. [DOI] [PubMed] [Google Scholar]
- Nelson GJ. Lipid composition of erythrocytes in various mammalian species. Biochim Biophys Acta. 1967;144:221–232. doi: 10.1016/0005-2760(67)90152-x. [DOI] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesic I, Guix FX, Vennekens K, Michaki V, Van Veldhoven PP, Feiguin F, De Strooper B, Dotti CG, Wahle T. Alterations in phosphatidylethanolamine levels affect the generation of Abeta. Aging Cell. 2012;11:63–72. doi: 10.1111/j.1474-9726.2011.00760.x. [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Lewandowska A, Choi JY, Markgraf DF, Junker M, Bilgin M, Ejsing CS, Voelker DR, Rapoport TA, Shaw JM. Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic. 2012;13:880–890. doi: 10.1111/j.1600-0854.2012.01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson I, von Heijne G. Fine-tuning the topology of a polytopic membrane protein: role of positively and negatively charged amino acids. Cell. 1990;62:1135–1141. doi: 10.1016/0092-8674(90)90390-z. [DOI] [PubMed] [Google Scholar]
- Nishijima M, Kuge O, Akamatsu Y. Phosphatidylserine biosynthesis in cultured Chinese hamster ovary cells. I. Inhibition of de novo phosphatidylserine biosynthesis by exogenous phosphatidylserine and its efficient incorporation. J Biol Chem. 1986;261:5784–5789. [PubMed] [Google Scholar]
- Noda NN, Fujioka Y, Hanada T, Ohsumi Y, Inagaki F. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep. 2013;14:206–211. doi: 10.1038/embor.2012.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Matsuura A, Wada Y, Ohsumi Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1995;210:126–132. doi: 10.1006/bbrc.1995.1636. [DOI] [PubMed] [Google Scholar]
- Nosjean O, Briolay A, Roux B. Mammalian GPI proteins: sorting, membrane residence and functions. Biochim Biophys Acta. 1997;1331:153–186. doi: 10.1016/s0304-4157(97)00005-1. [DOI] [PubMed] [Google Scholar]
- Nozaki M, Ohishi K, Yamada N, Kinoshita T, Nagy A, Takeda J. Developmental abnormalities of glycosylphosphatidylinositol-anchor-deficient embryos revealed by Cre/loxP system. Lab Invest. 1999;79:293–299. [PubMed] [Google Scholar]
- Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24:9–23. doi: 10.1038/cr.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohvo-Rekila H, Ramstedt B, Leppimaki P, Slotte JP. Cholesterol interactions with phospholipids in membranes. Prog Lipid Res. 2002;41:66–97. doi: 10.1016/s0163-7827(01)00020-0. [DOI] [PubMed] [Google Scholar]
- Okamoto M, Yoko-o T, Umemura M, Nakayama Ki, Jigami Y. glycosylphosphatidylinositol-anchored proteins are required for the transport of detergent-resistant microdomain-associated membrane proteins Tat2p and Fur4p. J Biol Chem. 2006;281:4013–4023. doi: 10.1074/jbc.M504684200. [DOI] [PubMed] [Google Scholar]
- Onguka O, Calzada E, Ogunbona OB, Claypool SM. Phosphatidylserine decarboxylase 1 autocatalysis and function does not require a mitochondrial-specific factor. J Biol Chem. 2015 doi: 10.1074/jbc.M115.641118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, Brugger B, Westermann B, Langer T. The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J Cell Biol. 2009;184:583–596. doi: 10.1083/jcb.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12∼ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol. 2013;20:59–66. doi: 10.1038/nsmb.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otter-Nilsson M, Hendriks R, Pecheur-Huet EI, Hoekstra D, Nilsson T. Cytosolic ATPases, p97 and NSF, are sufficient to mediate rapid membrane fusion. EMBO J. 1999;18:2074–2083. doi: 10.1093/emboj/18.8.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak SH, Bagshaw RD, Guiral M, Zhang S, Ackerley CA, Pak BJ, Callahan JW, Mahuran DJ. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem. 2003;278:26687–26694. doi: 10.1074/jbc.m304009200. [DOI] [PubMed] [Google Scholar]
- Pecheur EI, Martin I, Maier O, Bakowsky U, Ruysschaert JM, Hoekstra D. Phospholipid species act as modulators in p97/p47-mediated fusion of Golgi membranes. Biochemistry. 2002;41:9813–9823. doi: 10.1021/bi0259195. [DOI] [PubMed] [Google Scholar]
- Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimplikar SW. Reassessing the amyloid cascade hypothesis of Alzheimer's disease. Int J Biochem Cell Biol. 2009;41:1261–1268. doi: 10.1016/j.biocel.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, Sanges G, Stenroos ES, Pho LT, Schaffer AA, Lazzarini AM, Nussbaum RL, Duvoisin RC. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science. 1996;274:1197–1199. doi: 10.1126/science.274.5290.1197. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Poston CN, Duong E, Cao Y, Bazemore-Walker CR. Proteomic analysis of lipid raft-enriched membranes isolated from internal organelles. Biochem Biophys Res Commun. 2011;415:355–360. doi: 10.1016/j.bbrc.2011.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potting C, Tatsuta T, König T, Haag M, Wai T, Aaltonen Mari J, Langer T. TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 2013;18:287–295. doi: 10.1016/j.cmet.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Potting C, Wilmes C, Engmann T, Osman C, Langer T. Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 2010;29:2888–2898. doi: 10.1038/emboj.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y. Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci. 2005;25:436–445. doi: 10.1523/JNEUROSCI.1575-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE, Rizzuto R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol. 2002;159:613–624. doi: 10.1083/jcb.200205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Klionsky DJ. Autophagic processes in yeast: mechanism, machinery and regulation. Genetics. 2013;194:341–361. doi: 10.1534/genetics.112.149013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgway ND, Vance DE. Purification of phosphatidylethanolamine N-methyl-transferase from rat liver. J Biol Chem. 1987;262:17231–17239. [PubMed] [Google Scholar]
- Riekhof WR, Voelker DR. Uptake and utilization of lyso-phosphatidylethanolamine by Saccharomycescerevisiae. J Biol Chem. 2006;281:36588–36596. doi: 10.1074/jbc.M608851200. [DOI] [PubMed] [Google Scholar]
- Riekhof WR, Wu J, Gijon MA, Zarini S, Murphy RC, Voelker DR. Lysophosphatidylcholine metabolism in Saccharomyces cerevisiae: the role of P-type ATPases in transport and a broad specificity acyltransferase in acylation. J Biol Chem. 2007a;282:36853–36861. doi: 10.1074/jbc.M706718200. [DOI] [PubMed] [Google Scholar]
- Riekhof WR, Wu J, Jones JL, Voelker DR. Identification and characterization of the major lysophosphatidylethanolamine acyltransferase in Saccharomyces cerevisiae. J Biol Chem. 2007b;282:28344–28352. doi: 10.1074/jbc.M705256200. [DOI] [PubMed] [Google Scholar]
- Rietveld AG, Killian JA, Dowhan W, de Kruijff B. Polymorphic regulation of membrane phospholipid composition in Escherichia coli. J Biol Chem. 1993;268:12427–12433. [PubMed] [Google Scholar]
- Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, Wolfer DP, Muller UC. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the natomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci. 2007;27:7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JD. The molecular structure and contact relationships of cell membranes. Prog Biophys Mol Biol. 1960;10:343–418. [PubMed] [Google Scholar]
- Rockenfeller P, Koska M, Pietrocola F, Minois N, Knittelfelder O, Sica V, Franz J, Carmona-Gutierrez D, Kroemer G, Madeo F. Phosphatidylethanolamine positively regulates autophagy and longevity. Cell Death Differ. 2015;22:499–508. doi: 10.1038/cdd.2014.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruetz S, Gros P. Phosphatidylcholine translocase: a physiological role for the mdr2 gene. Cell. 1994;77:1071–1081. doi: 10.1016/0092-8674(94)90446-4. [DOI] [PubMed] [Google Scholar]
- Ruiz-Herrera J, Elorza MV, Valentin E, Sentandreu R. Molecular organization of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res. 2006;6:14–29. doi: 10.1111/j.1567-1364.2005.00017.x. [DOI] [PubMed] [Google Scholar]
- Ryan MC, Wilson AM, Slavin J, Best JD, Jenkins AJ, Desmond PV. Associations between liver histology and severity of the metabolic syndrome in subjects with nonalcoholic fatty liver disease. Diabetes Care. 2005;28:1222–1224. doi: 10.2337/diacare.28.5.1222. [DOI] [PubMed] [Google Scholar]
- Sakoh-Nakatogawa M, Matoba K, Asai E, Kirisako H, Ishii J, Noda NN, Inagaki F, Nakatogawa H, Ohsumi Y. Atg12-Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat Struct Mol Biol. 2013;20:433–439. doi: 10.1038/nsmb.2527. [DOI] [PubMed] [Google Scholar]
- Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114:867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- Satre M, Kennedy EP. Identification of bound pyruvate essential for the activity of phosphatidylserine decarboxylase of Escherichia coli. J Biol Chem. 1978;253:479–483. [PubMed] [Google Scholar]
- Schagger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000;19:1777–1783. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, Haldar D. Cardiolipin is synthesized on the matrix side of the inner membrane in rat liver mitochondria. J Biol Chem. 1993;268:74–79. [PubMed] [Google Scholar]
- Schlame M, Ren M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim Biophys Acta. 2009;1788:2080–2083. doi: 10.1016/j.bbamem.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder TE. The contractile ring and furrowing in dividing cells. Ann NY Acad Sci. 1990;582:78–87. doi: 10.1111/j.1749-6632.1990.tb21669.x. [DOI] [PubMed] [Google Scholar]
- Schuiki I, Daum G. Phosphatidylserine decarboxylases, key enzymes of lipid metabolism. IUBMB Life. 2009;61:151–162. doi: 10.1002/iub.159. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Presenilin, Notch, and the genesis and treatment of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:11039–11041. doi: 10.1073/pnas.211352598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H, Dunn CD, Iijima M, Shepard KA, Yaffe MP, Machamer CE, Jensen RE. Ups1p, a conserved intermembrane space protein, regulates mitochondrial shape and alternative topogenesis of Mgm1p. J Cell Biol. 2006;173:651–658. doi: 10.1083/jcb.200603092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto-Young D, Chen CC, Wilson TH. Effect of different phospholipids on the reconstitution of two functions of the lactose carrier of Escherichiacoli. J Membr Biol. 1985;84:259–267. doi: 10.1007/BF01871389. [DOI] [PubMed] [Google Scholar]
- Shiao YJ, Lupo G, Vance JE. Evidence that phosphatidylserine is imported into mitochondria via a mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine. J Biol Chem. 1995;270:11190–11198. doi: 10.1074/jbc.270.19.11190. [DOI] [PubMed] [Google Scholar]
- Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007;26:1713–1725. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Zalk R, Gincel D, Vardi N. Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochim Biophys Acta. 2004;1657:105–114. doi: 10.1016/j.bbabio.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005;24:717–729. doi: 10.1038/sj.emboj.7600559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit JJM, Schinkel AH, Elferink RPJO, Groen AK, Wagenaar E, van Deemter L, Mol CAAM, Ottenhoff R, van der Lugt NMT, van Roon MA, van der Valk MA, Offerhaus GJA, Berns AJM, Borst P. Homozygous disruption of the murine MDR2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- Sogo LF, Yaffe MP. Regulation of mitochondrial morphology and inheritance by Mdm10p, a protein of the mitochondrial outer membrane. J Cell Biol. 1994;126:1361–1373. doi: 10.1083/jcb.126.6.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, Vance JE. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J Biol Chem. 2005;280:40032–40040. doi: 10.1074/jbc.M506510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens TJ, Arkin IT. Do more complex organisms have a greater proportion of membrane proteins in their genomes? Proteins. 2000;39:417–420. doi: 10.1002/(sici)1097-0134(20000601)39:4<417::aid-prot140>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Stone SJ, Cui Z, Vance JE. Cloning and expression of mouse liver phosphatidylserine synthase-1 cDNA. Overexpression in rat hepatoma cells inhibits the CDP-ethanolamine pathway for phosphatidylethanolamine biosynthesis. J Biol Chem. 1998;273:7293–7302. doi: 10.1074/jbc.273.13.7293. [DOI] [PubMed] [Google Scholar]
- Stone SJ, Vance JE. Cloning and expression of murine liver phosphatidylserine synthase (PSS)-2: differential regulation of phospholipid metabolism by PSS1 and PSS2. Biochem J. 1999;342(Pt 1):57–64. [PMC free article] [PubMed] [Google Scholar]
- Stone SJ, Vance JE. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem. 2000;275:34534–34540. doi: 10.1074/jbc.M002865200. [DOI] [PubMed] [Google Scholar]
- Storey MK, Clay KL, Kutateladze T, Murphy RC, Overduin M, Voelker DR. Phosphatidylethanolamine has an essential role in Saccharomycescerevisiae that is independent of its ability to form hexagonal phase structures. J Biol Chem. 2001;276:48539–48548. doi: 10.1074/jbc.M109043200. [DOI] [PubMed] [Google Scholar]
- Sturbois-Balcerzak B, Stone SJ, Sreenivas A, Vance JE. Structure and expression of the murine phosphatidylserine synthase-1 gene. J Biol Chem. 2001;276:8205–8212. doi: 10.1074/jbc.M009776200. [DOI] [PubMed] [Google Scholar]
- Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, Merrill AH, Jr, Murphy RC, Raetz CR, Russell DW, Subramaniam S. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007;35:D527–D532. doi: 10.1093/nar/gkl838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundler R. Ethanolaminephosphate cytidylyltransferase. Purification and characterization of the enzyme from rat liver. J Biol Chem. 1975;250:8585–8590. [PubMed] [Google Scholar]
- Sundler R, Akesson B. Biosynthesis of phosphatidylethanolamines and phosphatidylcholines from ethanolamine and choline in rat liver. Biochem J. 1975a;146:309–315. doi: 10.1042/bj1460309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundler R, Akesson B. Regulation of phospholipid biosynthesis in isolated rat hepatocytes. Effect of different substrates. J Biol Chem. 1975b;250:3359–3367. [PubMed] [Google Scholar]
- Sundler R, Akesson B, Nilsson A. Quantitative role of base exchange in phosphatidylethanolamine synthesis in isolated rat hepatocytes. FEBS Lett. 1974;43:303–307. doi: 10.1016/0014-5793(74)80667-8. [DOI] [PubMed] [Google Scholar]
- Suzuki TT, Kanfer JN. Purification and properties of an ethanolamine-serine base exchange enzyme of rat brain microsomes. J Biol Chem. 1985;260:1394–1399. [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, Takahashi M, Kitani T, Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- Tamaki H, Shimada A, Ito Y, Ohya M, Takase J, Miyashita M, Miyagawa H, Nozaki H, Nakayama R, Kumagai H. LPT1 encodes a membrane-bound O-acyltransferase involved in the acylation of lysophospholipids in the yeast Saccharomycescerevisiae. J Biol Chem. 2007;282:34288–34298. doi: 10.1074/jbc.M704509200. [DOI] [PubMed] [Google Scholar]
- Tamura Y, Endo T, Iijima M, Sesaki H. Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J Cell Biol. 2009;185:1029–1045. doi: 10.1083/jcb.200812018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Harada Y, Nishikawa S, Yamano K, Kamiya M, Shiota T, Kuroda T, Kuge O, Sesaki H, Imai K, Tomii K, Endo T. Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab. 2013;17:709–718. doi: 10.1016/j.cmet.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Iijima M, Sesaki H. Mdm35p imports Ups proteins into the mitochondrial intermembrane space by functional complex formation. EMBO J. 2010;29:2875–2887. doi: 10.1038/emboj.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Onguka O, Hobbs AE, Jensen RE, Iijima M, Claypool SM, Sesaki H. Role for two conserved intermembrane space proteins, Ups1p and Ups2p, [corrected] in intra-mitochondrial phospholipid trafficking. J Biol Chem. 2012a;287:15205–15218. doi: 10.1074/jbc.M111.338665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Onguka O, Itoh K, Endo T, Iijima M, Claypool SM, Sesaki H. Phosphatidylethanolamine biosynthesis in mitochondria: phosphatidylserine (PS) trafficking is independent of a PS decarboxylase and intermembrane space proteins UPS1P and UPS2P. J Biol Chem. 2012b;287:43961–43971. doi: 10.1074/jbc.M112.390997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Tanida-Miyake E, Ueno T, Kominami E. The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J Biol Chem. 2001;276:1701–1706. doi: 10.1074/jbc.C000752200. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Tasseva G, Bai HD, Davidescu M, Haromy A, Michelakis E, Vance JE. Phosphatidylethanolamine deficiency in mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J Biol Chem. 2013;288:4158–4173. doi: 10.1074/jbc.M112.434183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas E, Roman E, Claypool S, Manzoor N, Pla J, Panwar SL. Mitochondria influence CDR1 efflux pump activity, Hog1-mediated oxidative stress pathway, iron homeostasis, and ergosterol levels in Candida albicans. Antimicrob Agents Chemother. 2013;57:5580–5599. doi: 10.1128/AAC.00889-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijburg LB, Geelen MJ, Van Golde LM. Biosynthesis of phosphatidylethanolamine via the CDP-ethanolamine route is an important pathway in isolated rat hepatocytes. Biochem Biophys Res Commun. 1989;160:1275–1280. doi: 10.1016/s0006-291x(89)80141-x. [DOI] [PubMed] [Google Scholar]
- Tijburg LB, Houweling M, Geelen JH, van Golde LM. Stimulation of phosphatidylethanolamine synthesis in isolated rat hepatocytes by phorbol 12-myristate 13-acetate. Biochim Biophys Acta. 1987;922:184–190. doi: 10.1016/0005-2760(87)90153-6. [DOI] [PubMed] [Google Scholar]
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831–835. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- Trotter PJ, Pedretti J, Voelker DR. Phosphatidylserine decarboxylase from Saccharomyces cerevisiae. Isolation of mutants, cloning of the gene, and creation of a null allele. J Biol Chem. 1993;268:21416–21424. [PubMed] [Google Scholar]
- Trotter PJ, Pedretti J, Yates R, Voelker DR. Phosphatidylserine decarboxylase 2 of Saccharomycescerevisiae. Cloning and mapping of the gene, heterologous expression, and creation of the null allele. J Biol Chem. 1995;270:6071–6080. doi: 10.1074/jbc.270.11.6071. [DOI] [PubMed] [Google Scholar]
- Trotter PJ, Voelker DR. Identification of a non-mitochondrial phosphatidylserine decarboxylase activity (PSD2) in the yeast Saccharomyces cerevisiae. J Biol Chem. 1995;270:6062–6070. doi: 10.1074/jbc.270.11.6062. [DOI] [PubMed] [Google Scholar]
- Tuller G, Hrastnik C, Achleitner G, Schiefthaler U, Klein F, Daum G. YDL142c encodes cardiolipin synthase (Cls1p) and is non-essential for aerobic growth of Saccharomycescerevisiae. FEBS Lett. 1998;421:15–18. doi: 10.1016/s0014-5793(97)01525-1. [DOI] [PubMed] [Google Scholar]
- Tuyama AC, Chang CY. Non-alcoholic fatty liver disease. J Diabetes. 2012;4:266–280. doi: 10.1111/j.1753-0407.2012.00204.x. [DOI] [PubMed] [Google Scholar]
- Twig G, Shirihai OS. The Interplay Between Mitochondrial Dynamics and Mitophagy. Antioxidants Redox Signaling. 2010;14:1939–1951. doi: 10.1089/ars.2010.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama T, Ikeuchi T, Ouchi Y, Sakamoto M, Kasuga K, Shiga A, Suzuki M, Ito M, Atsumi T, Shimizu T, Ohashi T. Prominent psychiatric symptoms and glucose hypometabolism in a family with a SNCA duplication. Neurology. 2008;71:1289–1291. doi: 10.1212/01.wnl.0000327607.28928.e6. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Yamaguchi R, Ikawa M, Okabe M, Morii E, Maeda Y, Kinoshita T. PGAP1 knock-out mice show otocephaly and male infertility. J Biol Chem. 2007;282:30373–30380. doi: 10.1074/jbc.M705601200. [DOI] [PubMed] [Google Scholar]
- Van Deenen LLM, De Gier J. The Red Blood Cell. Vol. 1. Academic Press Inc.; New York, New York: 1974. Lipids of the red cell membrane; pp. 147–211. [Google Scholar]
- van den Brink-van der Laan E, Killian JA, de Kruijff B. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim Biophys Acta. 2004;1666:275–288. doi: 10.1016/j.bbamem.2004.06.010. [DOI] [PubMed] [Google Scholar]
- van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen JN, Lingrell S, da Silva RP, Jacobs RL, Vance DE. The concentration of phosphatidylethanolamine in mitochondria can modulate ATP production and glucose metabolism in mice. Diabetes. 2014;63:2620–2630. doi: 10.2337/db13-0993. [DOI] [PubMed] [Google Scholar]
- van Golde LM, Raben J, Batenburg JJ, Fleischer B, Zambrano F, Fleischer S. Biosynthesis of lipids in golgi complex and other subcellular fractions from rat liver. Biochim Biophys Acta. 1974;360:179–192. doi: 10.1016/0005-2760(74)90168-4. [DOI] [PubMed] [Google Scholar]
- van Hellemond JJ, Slot JW, Geelen MJ, van Golde LM, Vermeulen PS. Ultrastructural localization of CTP:phosphoethanolamine cytidylyltransferase in rat liver. J Biol Chem. 1994;269:15415–15418. [PubMed] [Google Scholar]
- van Poelje PD, Snell EE. Pyruvoyl-dependent enzymes. Annu Rev Biochem. 1990;59:29–59. doi: 10.1146/annurev.bi.59.070190.000333. [DOI] [PubMed] [Google Scholar]
- Vance DE, Van den Bosch H. Cholesterol in the year 2000. Biochim Biophys Acta. 2000;1529:1–8. doi: 10.1016/s1388-1981(00)00133-5. [DOI] [PubMed] [Google Scholar]
- Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- Vance JE. Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J Biol Chem. 1991;266:89–97. [PubMed] [Google Scholar]
- Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta. 2014;1841:595–609. doi: 10.1016/j.bbalip.2013.11.014. [DOI] [PubMed] [Google Scholar]
- Vance JE. Phospholipid synthesis and transport in mammalian cells. Traffic. 2015;16:1–18. doi: 10.1111/tra.12230. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Verkleij AJ, Leunissen-Bijvelt J, de Kruijff B, Hope M, Cullis PR. Non-bilayer structures in membrane fusion. Ciba Found Symp. 1984;103:45–59. doi: 10.1002/9780470720844.ch4. [DOI] [PubMed] [Google Scholar]
- Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. Association of gamma-secretase with lipid rafts in post-golgi and endosome membranes. J Biol Chem. 2004;279:44945–44954. doi: 10.1074/jbc.M407986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelker DR. Phosphatidylserine functions as the major precursor of phosphatidylethanolamine in cultured BHK-21 cells. Proc Natl Acad Sci USA. 1984;81:2669–2673. doi: 10.1073/pnas.81.9.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelker DR. Disruption of phosphatidylserine translocation to the mitochondria in baby hamster kidney cells. J Biol Chem. 1985;260:14671–14676. [PubMed] [Google Scholar]
- Voelker DR. Phosphatidylserine translocation to the mitochondrion is an ATP-dependent process in permeabilized animal cells. Proc Natl Acad Sci USA. 1989a;86:9921–9925. doi: 10.1073/pnas.86.24.9921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelker DR. Reconstitution of phosphatidylserine import into rat liver mitochondria. J Biol Chem. 1989b;264:8019–8025. [PubMed] [Google Scholar]
- Voelker DR. Characterization of phosphatidylserine synthesis and translocation in permeabilized animal cells. J Biol Chem. 1990;265:14340–14346. [PubMed] [Google Scholar]
- von der Malsburg K, Muller JM, Bohnert M, Oeljeklaus S, Kwiatkowska P, Becker T, Loniewska-Lwowska A, Wiese S, Rao S, Milenkovic D, Hutu DP, Zerbes RM, Schulze-Specking A, Meyer HE, Martinou JC, Rospert S, Rehling P, Meisinger C, Veenhuis M, Warscheid B, van der Klei IJ, Pfanner N, Chacinska A, van der Laan M. Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev Cell. 2011;21:694–707. doi: 10.1016/j.devcel.2011.08.026. [DOI] [PubMed] [Google Scholar]
- Voss C, Lahiri S, Young BP, Loewen CJ, Prinz WA. ER-shaping proteins facilitate lipid exchange between the ER and mitochondria in S. cerevisiae. J Cell Sci. 2012;125:4791–4799. doi: 10.1242/jcs.105635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkey CJ, Yu L, Agellon LB, Vance DE. Biochemical and evolutionary significance of phospholipid methylation. J Biol Chem. 1998;273:27043–27046. doi: 10.1074/jbc.273.42.27043. [DOI] [PubMed] [Google Scholar]
- Wang S, Zhang S, Liou LC, Ren Q, Zhang Z, Caldwell GA, Caldwell KA, Witt SN. Phosphatidylethanolamine deficiency disrupts alpha-synuclein homeostasis in yeast and worm models of Parkinson disease. Proc Natl Acad Sci USA. 2014;111:E3976–E3985. doi: 10.1073/pnas.1411694111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- Wilson-Zbinden C, Dos Santos AX, Stoffel-Studer I, van der Vaart A, Hofmann K, Reggiori F, Riezman H, Kraft C, Peter M. Autophagy competes for a common phosphatidylethanolamine pool with major cellular PE-consuming pathways in Saccharomycescerevisiae. Genetics. 2015;199:475–485. doi: 10.1534/genetics.114.169797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz KW, Zilversmit DB. Exchange of phospholipids between liver mitochondria and microsomes invitro. J Biol Chem. 1968;243:3596–3602. [PubMed] [Google Scholar]
- Wittenberg J, Kornberg A. Choline phosphokinase. J Biol Chem. 1953;202:431–444. [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008;19:3290–3298. doi: 10.1091/mbc.E07-12-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yet SF, Lee S, Hahm YT, Sul HS. Expression and identification of p90 as the murine mitochondrial glycerol-3-phosphate acyltransferase. Biochemistry. 1993;32:9486–9491. doi: 10.1021/bi00087a029. [DOI] [PubMed] [Google Scholar]
- Youngman MJ, Hobbs AE, Burgess SM, Srinivasan M, Jensen RE. Mmm2p, a mitochondrial outer membrane protein required for yeast mitochondrial shape and maintenance of mtDNA nucleoids. J Cell Biol. 2004;164:677–688. doi: 10.1083/jcb.200308012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zborowski J, Dygas A, Wojtczak L. Phosphatidylserine decarboxylase is located on the external side of the inner mitochondrial membrane. FEBS Lett. 1983;157:179–182. doi: 10.1016/0014-5793(83)81141-7. [DOI] [PubMed] [Google Scholar]
- Zelinski TA, Choy PC. Phosphatidylethanolamine biosynthesis in isolated hamster heart. Can J Biochem. 1982;60:817–823. doi: 10.1139/o82-102. [DOI] [PubMed] [Google Scholar]
- Zhang W, Bogdanov M, Pi J, Pittard AJ, Dowhan W. Reversible topological organization within a polytopic membrane protein is governed by a change in membrane phospholipid composition. J Biol Chem. 2003;278:50128–50135. doi: 10.1074/jbc.M309840200. [DOI] [PubMed] [Google Scholar]
- Zhang W, Campbell HA, King SC, Dowhan W. Phospholipids as determinants of membrane protein topology. Phosphatidylethanolamine is required for the proper topological organization of the gamma-aminobutyric acid permease (GabP) of Escherichia coli. J Biol Chem. 2005;280:26032–26038. doi: 10.1074/jbc.M504929200. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Gohil VM, Ma L, Greenberg ML. Absence of cardiolipin results in temperature sensitivity, respiratory defects, and mitochondrial DNA instability independent of pet56. J Biol Chem. 2004;279:32294–32300. doi: 10.1074/jbc.M403275200. [DOI] [PubMed] [Google Scholar]
- Zhou J, Saba JD. Identification of the first mammalian sphingosine phosphate lyase gene and its functional expression in yeast. Biochem Biophys Res Commun. 1998;242:502–507. doi: 10.1006/bbrc.1997.7993. [DOI] [PubMed] [Google Scholar]
- Zick M, Duvezin-Caubet S, Schäfer A, Vogel F, Neupert W, Reichert AS. Distinct roles of the two isoforms of the dynamin-like GTPase Mgm1 in mitochondrial fusion. FEBS Lett. 2009;583:2237–2243. doi: 10.1016/j.febslet.2009.05.053. [DOI] [PubMed] [Google Scholar]
- Zinser E, Sperka-Gottlieb CD, Fasch EV, Kohlwein SD, Paltauf F, Daum G. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomycescerevisiae. J Bacteriol. 1991;173:2026–2034. doi: 10.1128/jb.173.6.2026-2034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]