

Abstract
We have previously demonstrated a crucial role of NUPR1 in tumor development and progression. In this work we report the functional characterization of a novel Nupr1-like isoform (NUPR1L) and its functional interaction with the protumoral factor NUPR1. Through the use of primary sequence analysis, threading, and homology-based molecular modeling as well as expression and immunolocalization, studies reveal that NUPR1L displays properties which are similar to member of the HMG-like family of chromatin regulators including its ability to translocate to the cell nucleus and bind to DNA. Analysis of the NUPR1L promoter showed the presence of two p53-response elements at positions -37 and -7 respectively. Experiments using reporter assays combined with site-directed mutagenesis and using cells with controllable p53 expression demonstrate that both of these sequences are responsible for the regulation of NUPR1L expression by p53. Congruently, NUPR1L gene expression is activated in response to DNA damage induced by Oxaliplatin treatment or cell cycle arrest induced by serum starvation, two well-validated methods to achieve p53 activation. Interestingly, expression of NUPR1L down-regulates the expression of NUPR1, its closely related protumoral isoform, by a mechanism that involves the inhibition of its promoter activity. At the cellular level, overexpression of NUPR1L induces G1 cell cycle arrest and a decrease in their cell viability, an effect that is mediated, at least in part, by down-regulating NUPR1 expression. Combined these experiments constitute the first functional characterization of NUPR1L as a new p53-induced gene which negatively regulates the protumoral factor NUPR1.
Keywords: p53-dependent gene, stress gene, DNA damage, Nupr1L, Nupr1, p53
Introduction
The Nuclear Protein 1 (NUPR1), also known as p8 and Com1, is a stress-induced gene that was first identified in pancreas because its activation during the acute phase of pancreatitis (Mallo et al., 1997). This gene encodes an 82 amino acid polypeptide with a theoretical molecular mass of 8872.7 Da and a pI of 9.98. NUPR1 is an evolutionary conserved gene present in Drosophila, Xenopus and mammals but not in yeast. Despite NUPR1 being structurally related to the HMG (high-mobility group) transcriptional regulators (Encinar et al., 2001), it currently does not share significant homology with any other protein. NUPR1 is a basic helix-loop-helix molecule that contains a canonical bipartite nuclear localization signal (NLS) (Vasseur et al., 1999) and an N-terminal PEST (Pro/Glu/Ser/Thr-rich) region indicating a possible regulation by the ubiquitin/proteasome system (Goruppi and Kyriakis, 2004) and suggesting a role in transcriptional regulation (Goruppi and Iovanna, 2010; Urrutia et al., 2014). Nupr1is considered as a stress protein because it is induced in response to several injurious stimuli, such as hypoxia, apoptosis inducers, glucose starvation and several anticancer agents. Likewise, NUPR1 is overexpressed in several types of human cancers, including pancreatic ductal adenocarcinoma (PDAC) (Su et al., 2001) and its metastasis (Ree et al., 2000), suggesting a crucial role in cancer biology (Cano et al., 2014; Vasseur et al., 2002). Therefore, during the last decade we have focused our studies on the role of NUPR1 on cancer, especially on PDAC which remain one the most lethal tumor diseases. In this regard, we have demonstrated in our recent studies the crucial function of NUPR1 as a cooperator factor with the oncogenic form of KrasG12D to promote Pancreas Intraepithelial Neoplasias (PanIN) in vivo and in vitro (Cano et al., 2014; Grasso et al., 2014; Hamidi et al., 2012a; Vasseur et al., 2002).
The recent detailed analysis of databases reveals the presence of a NUPR1-related sequence in human genome and consequently denominated NUPR1-Like (NUPR1L). NUPR1 and NUPR1L have been recently proposed as members of a new family of small chromatin regulators based on theoretical dynamic analysis of their spatial structure (Urrutia et al., 2014). Due to the critical role of NUPR1 in cancer development and progression, our aim was to study the function of NUPR1L, as well as the possible interaction with its homologous, NUPR1, in order to shed additional light to the NUPR1-associated cancer processes which remains poorly understood at the mechanistic level. To this end, we have cloned both the cDNA and the promoter region of human NUPR1L into vectors that have allowed us to analyze the function and regulation of this protein. In this paper, we demonstrated that NUPR1L is a new direct p53 target gene which down-regulates the tumorigenic gene NUPR1 at the transcriptional level by repressing the activity of its promoter. In addition, the NUPR1L-induced decrease in pancreatic cancer cells viability is rescued by the forced expression of NUPR1 showing a functional interaction between them. Thus, because of the key role of NUPR1 in cancer, the new information emerging from this study has biomedical relevance that aid to a better understanding on the pathobiology of cancer.
Material and Methods
Primary structure analysis and bioinformatics tools
NUPR1L sequence was obtained using the NCBI database (Reference Sequence: NP_001139184.1). NLS motif was determined by ELM (Eukaryotic Linear Motifs) server (http://elm.eu.org/)(Dinkel et al., 2012). Putative p53-sites binding of NUPR1L promoter were evaluated using web-based tools to identify conserved patterns in sequences (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) (Farre et al., 2003). NUPR1 and NUPR1L sequences were compared by pairwise alignment (www.ebi.ac.uk/Tools/msa/clustalw2/).
Cell culture
The immortalized Mouse Embryonic Fibroblasts (MEFs), 3T3, 10.1 and (10.1)Val5, were a kind gift from Levine AJ (Harvey and Levine, 1991; Wu and Levine, 1994). MEFs, HEK-293T (human embryonic kidney cells), MCF-7 (breast cancer cells), Hela (cervical cancer cells) and pancreatic cancer-derived cell lines, Capan-2, Panc-1 and MiaPaca-2, parental and transduced, were maintained in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS at 37°C with 5% CO2 in a humidified atmosphere and manipulated following ATCC's recommendations. To induce DNA damage, MCF-7, Capan-2 and Hela cells were treated with increasing concentrations of Oxaliplatin (10, 20 and 40 μM) for 18 and 24 h. For FBS starvation, cells were maintained in serum-free DMEM for 12, 24 and 60 h. Glucose starvation was obtained by cultivating cells with DMEM (no glucose) (Invitrogen, Ref# 11966) supplemented with 10% FBS for 24 h.
DNA constructs
According to the predicted cDNA sequence of human (Accession No. ENSG00000185290) deposited in GenBank, two gene-specific primers were designed to amplify 5′-cDNA and 3′-cDNA ends of Nupr1L mRNA from human embryonic kidney 293T cells using Trizol reagent (Invitrogen) and ImProm-II Reverse Transcription System (Promega). Nupr1L mRNA was obtained from HEK-293T cells because are normal human cells. PCR was performed under the following conditions: 5 min at 95°C for denaturation, followed by 25 cycles (10 sec at 98°C, 1 min at 65°C and 1 min at 72°C) of reactions, ending with a 5-min extension at 72°C (GoTaq® DNA Polymerase, Promega). The primers used are listed in Table S2. The full-length cDNA for human NUPR1L was subcloned into pCCL-WPS-PGK vector modified to express 6-histidines and a Flag at N-terminus of the expressed protein (6His-Flag-Nupr1L), using BamHI and XhoI restriction sites. Genomic DNA fragment (-159/+179) corresponding to the NUPR1L promoter was cloned into luciferase pGL3-basic (Promega) reported plasmid using MluI and BglII restriccion sites (pGL3-Nupr1L-luciferase). Human genomic DNA from mononuclear cells was used as a PCR template. PCR conditions are reported above. The potential p53-binding sequences of NUPR1L were mutated using the QuikChange®II-E Site-Directed Mutagenesis Kit (Agilent Technologies). The primers used are listed in Table S2. Plasmids were verified by DNA sequencing. The Nupr1 promoter (-2111/+229) which is cloned into the luciferase pGL3-basic reported plasmid (pGL3-Nupr1L-luciferase) was a kind gift from Bartholin L. (INSERM U1052, France) (Pommier et al., 2012).
Transductions
Lentiviral particles were generated by transfecting HEK-293T cells as reported previously (Bonacci et al., 2014). Briefly, HEK-293T cells were transfected with a mix of pCCL construct (Nupr1L, Nupr1 or GFP), Helper (carries sequence necessary for viral assembly of lentivirus) and pVsVg (expresses the vesicular stomatitis virus envelop glycoprotein G pseudotype), using Lipofectamine reagent (Invitrogen) and following manufacturer's recommendations. Viruses containing medium was collected and filtered through a 0.2 μm filter. MiaPaCa-2 and Hela cells were grown at 50%-60% confluence and infected with the supernatant containing viruses. Cells were used for experiments 72-96 hours after transduction. Expression of GFP was verified by fluorescence microscopy and NUPR1L expression controlled by western blot.
siRNA transfection
Cells were plated at 70% confluence. Nupr1 and Nupr1L were knockeddown using 140 ng of specific siRNA. INTERFERin reagent (Polyplus-transfection, New York, NY, USA) was used to perform siRNA transfections according to the manufacturer's protocol. Scrambled siRNA targeting no known gene sequence was used as negative control. The sequences of the siRNA used are shown in Table S2.
Immunofluorescence
Transduced cells with the lentiviral vector 6His-Flag-Nupr1L, as previously described, were cultured on glass coverslips, fixed, permeabilized, and incubated with mouse monoclonal anti-Flag (M2, Sigma) followed by Alexa Fluor antimouse IgG 488 (Invitrogen) secondary antibody. Samples were mounted in ProLongAntifade Reagent with DAPI (Invitrogen) and examined in an Eclipse 90i Nikon microscope (Nikon Instruments Europe B.V., Champigny-sur-Marne, France).
Reporter gene assay
The reporter assays were performed with Luciferase Assay System (Promega) according to the manufacturer's instructions. Cells were plated in 24-well plates and the following day co-transfected with 200 ng of pGL3-Nupr1L-luciferase, pGL3-Nupr1-luciferase or 6His-Flag-Nupr1L, and 20 ng of pRL-SV40 plasmids. The concentrations of pcDNA3-p53 were 200, 400 and 800 ng, under the needs of each experiment. Transient transfections were performed using Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer's protocol. The luciferase reporter activity of each sample was normalized against the internal control activity of Renilla luciferase developed with coelenterazine (Sigma-Aldrich). Each sample was determined in triplicate. The results represent means ± S.D. from three experiment runs.
RT-qPCR
RNA from cells was prepared using Trizol reagent (Invitrogen). cDNA was obtained using the ImProm-II Reverse Transcription System (Promega). Real-time quantitative PCR was performed in a Stratagene Cycler (La Jolla, CA, USA) using GoTaq® qPCR Master Mix and ROX reference dye (Promega). Sequences of the primers used to amplify human genes are indicated in Supplementary Table 2.
Immunobloting
Protein extraction was performed on ice using total protein extraction buffer: 50 mM HEPES (pH 7.5), 150 mM NaCl, 20% SDS, 1mM EDTA,1 mM EGTA, 10% glycerol, 1% Triton, 25 mM NaF, 10 mM ZnCl2 and 50 mM DTT. Before lysis, protease inhibitor cocktail at 1:200 (Roche Applied Science, Meylan, France), 500 mM PMSF, 1 mM sodium orthovanadate and 1 mM β-glycerophosphate were added. Protein concentration was measured using a BCA Protein Assay Kit (Pierce Biotechnology). Protein samples (95 μg) were denatured at 95°C and subsequently separated by SDS-PAGE gel electrophoresis. After being transferred to PVDF, the membrane was blocked with 1% BSA, and then the samples were probed with primary antibody, followed by a horseradish peroxidase-coupled secondary antibody. Primary antibodies used were mouse monoclonal anti-Flag (M2) and anti-β-tubulin polyclonal from Sigma-Aldrich. Image acquisition was made in Fusion FX image acquisition system (Vilber Lourmat, VWR, Fontenay-sous-Bois, France).
Flow cytometry
Cell cycle analysis was performed by standard propidium iodide (PI) staining protocol (Grasso et al., 2014) MiaPaCa-2 cells were seeded at a density of 500,000 cells per well in a 6-well tissue culture plate. Then, cells were trypsinized, fixed in 70% ethanol, and stained with PI. Acquisition of 50,000 events per sample was made in a MACSQuant-VYB flow cytometer (Miltenyi Biotec, Surrey, UK). Data analysis was performed using the FlowJo (Treestar, Ashland, OR, USA) software.
Cell viability
Transduced cells were seeded at a density of 5,000 cells per well in a 96-well opaque-walled tissue culture plates compatible with fluorometer. Cell were allow to attach overnight and the following day plate was removed from the 37°C incubator, and 20 μL/well CellTiter-Blue™ (Promega, USA) were added. Cells were incubated with the reactive using standard cell-culture conditions for 24 h. Then, the plate was shaken for 10 seconds, and fluorescence was recorded at 560/590 nm (TriStar LB 941 Multimode Microplate Reader, Berthold Technologies). Cell viability in GFP infected cells was normalized to 100%.
Proliferation assay using iCELLigence System
A real-time cell proliferation assay was conducted using the iCELLigence System, (ACEA Biosciences, San Diego, CA). This system uses measurement of electrical impedance, created by cells attached to the microelectrode-integrated cell culture plated to measure cell proliferation in real time. A dimensionless parameter termed cell index (CI) was derived to represent cell status based on the measured relative change in electrical impedance that occurs in the presence and absence of cells in the wells (Irelan et al., 2011; Urcan et al., 2010). Prior to experiment, transduced cells were trypsinized and counted with Luna Counter Cells (Logos Biosystems) for getting exactly 40,000 cells in each well with 300 μl of DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS. Then, proliferation was followed using iPad and the specific application which was able to synchronize with the device and measure the impedance minutes after minutes for a total of 72 h.
Statistical analysis
Results presented in the text, tables and figures are means and standard deviation (S.D.). Before mean comparison, normality of data was checked by using the Kolmogorov-Smirnov test. For variables with a normal distribution, Student-T Test and one-way ANOVA with post hoc LSD test were used to compare the significance of differences between experimental groups. For variables lacking a normal distribution, either Mann-Whitney U test or Kruskal-Wallis one-way analysis of variance were applied. SPSS 20.0 software was used (SPSS Inc., Chicago, IL, USA). Only values of P < 0.05 were considered significant. RT-qPCR data are representative of at least three independent experiments with technical duplicated completed.
Results
NUPR1L is a protein with nuclear localization that displays homology with NUPR1
Our laboratory has extensively characterized the structure and function of NUPR1, a chromatin factor with similar biochemical properties to HMG proteins (Encinar JA, 2001). Interestingly, analyses of RNA-Seq data derived from pancreatic cancer cells as well as the EST database (NCBI) revealed the existence of a NUPR1-like protein (NUPR1L), which shares sequence homology, predicted molecular, mass, and isoelectric point with NUPR1. NUPR1L is found located on chromosome 7 (7p11.2) and contains 2 coding exons that encode a 97 amino acids polypeptide (NM_001145712). The Nupr1 gene is positioned on chromosome 16 (Vasseur et al., 1999). Similar to NUPR1, this previously uncharacterized protein also shares many properties with members of the HMG family of transcriptional regulators which are composed by intrinsically-disordered non-histone chromosomal proteins containing DNA-binding domains, called AT-hooks (DBD), which bind to the minor groove of short stretches of AT-rich DNA (Fonfría-Subirós E, 2012). These AT hooks (DBDs) are formed by a conserved sequence, which is rich in Glycine, Arginine, and Lysine (Fonfría-Subirós E, 2012). The first HMG AT-hook, DBD1, differs from DBD2 and DBD3 by the absence of single Proline residues that flank the G/R/K-rich core of this domain. Interestingly, we found that, similar to NUPR1, NUPR1L contains a single AT-hook domain, which is similar to the HMGA1 DBD1 but lacks significant homology to this protein outside of this region (Figure 1a). Based upon the high homology of NUPR1 and NUPR1L (Figure 2a), we built a model of NUPR1L using threading algorithms, a method that has been ranked by the structural biology community as one of the top systems for protein structure prediction in the CASP7 (Zhou et al., 2007), CASP8 (Zhang, 2009), CASP9 (Xu et al., 2011), and CASP10 (Zhang, 2012) experiments. The NUPR1L model was generated using as input the FASTA file corresponding to the NCBI-deposited primary structure and the I-TASSER software. According to this model, NUPR1L has the propensity to adopt a helix-loop-helix fold, a domain evolutionarily associated with DNA-binding proteins (Figure 1b). To estimate this model quality, we subsequently generated Ramachandran plots (Psi vs. Phi angles plot) using PROCHECK (Laskowski R A, 1993). The NUPR1L model had the best overall geometry with 97% of residues in favored and allowed regions (Supplementary Figure 1a). The properties of this energy minimized model, display several regions within NUPR1L which the propensity to form three α helices. Helix 1 contains 15 residues and spans from 19 to 34. Helix 2 contains 9 residues and spans from 52 to 61 while Helix 3 contains 20 residues and spans from 67 to 87 (Supplementary Figure 1b and c). Comparisons with experimentally solved structures deposited in the PDB bank indicated that NUPR1L not only shares structural properties with the previously identified most related protein NUPR1 but also with HMGA1 (PDB 2E6O). Thus, the two proteins share the propensity to form H-L-H motifs and share AT-hook-like DNA binding motif. Next, we utilized the previously reported structural features of both NUPR1 and HMGA1 to infer insights into the biochemical properties of NUPR1L by performing comparative linear motif analyses. Linear motif analyses by Psort II predicts typical NLS signal with a pattern of K (K/R) X (K/R) between residues 70 to 86 (Figure 2b) (Nakai K, 1999). In fact, the spacing of these sequences in 3D, as shown in Figure 1c, has ligand properties expected of a bipartite receptor for α-importins, which transports proteins to the nucleus. These results are congruent with the nuclear localization of Nurp1L determined experimentally by immunofluorescence (Figure2b). For the identification of residues involved in DNA binding by NUPR1, we used DP-Bind (Hwang S, 2007). This software implements three machine learning methods, namely support vector machine (SVM), kernel logistic regression (KLR), and penalized logistic regression (PLR) to predict DNA and RNA-binding residues from primary structure features, including the side-chain pKa value, hydrophobicity index and molecular mass of an amino acid (Hwang S, 2007). Figures 1d and 1e provides a graphic representation of the results obtained with this approach, which predicted that the sequences GRSKGRTRR have the potential to bind to DNA. Interestingly, this sequence is similar to the DNA binding residues found in the AT-hook-like motifs of both NUPR1 (GRKGRTKR) and HGMA1 (GRPKGSKNK). Our prediction of a DNA-binding domain within the sequence of NUPR1L prompted us to generate a model of this protein bound to DNA. For this, we applied two well-validated methods. We developed a homology-based model, which relies upon the previously solved NMR structure of the first hook of the HMGA1 bound to DNA (PDB: 3UXW), as the first 3D approach to characterize the NUPR1L DNA binding domain. Because of its simplicity, this model lent itself to the simplicity of using manual docking to superimpose the corresponding region of NUPR1L to the highly homologous HMGA1 AT-hook (Figure 1d and 1e). The NUPR1-DNA complex obtained through this homology-based approach is shown in Figure 1d and 1e. This complex was maintained through the bonding interactions, listed in Supplementary Table 1a. The second method, DP-Dock uses a nonspecific B-DNA to probe the binding site on a 3D model of a protein, which is known to bind DNA, but for which the specific amino-acid-to-nucleic-acid-base-contacts are unknown. Given the structure of a DNA-binding protein, the method first generates, automatically, an ensemble of protein-DNA complexes obtained by rigid-body docking with a nonspecific canonical B-DNA molecules with the sequence A10-T10 (Gao M, 2009). Models are subsequently selected through clustering and ranking by their DNA-protein interfacial energy (Gao M, 2009). Figure 1d and 1e shows that this method was successful in generating a NUPR1L-DNA complex where the amino acid-to-base contacts were primarily given by the same GRSKGRTRR sequence identified through DP-bind, as shown in Figure 1d and 1e. The ionic and hydrogen-bonding interactions that define the protein-DNA binding interphase are listed in Supplementary Table 1a. In addition, analyses of the DNA bound NUPR1L complex confirmed that this protein prefers to recognize the minor groove of DNA. Together, the results of these analyses support the existence of a high level of similarity between Nurp1 and Nurp1L and HMGA1. In addition, the models generated through these comparative modeling approaches constitute a step forward toward the understanding how Nurp1L binds to α-importin for localizing to the nucleus where it can use its distinct AT-hook to bind to the minor groove of DNA (Supplementary Table 1b), a characteristic previously reported for the structurally related HMGA protein to facilitate transcription initiation by several regulators of gene expression.
Figure 1. Similar to Nupr1, Nurp1L displays properties congruent with their role in gene expression regulation.
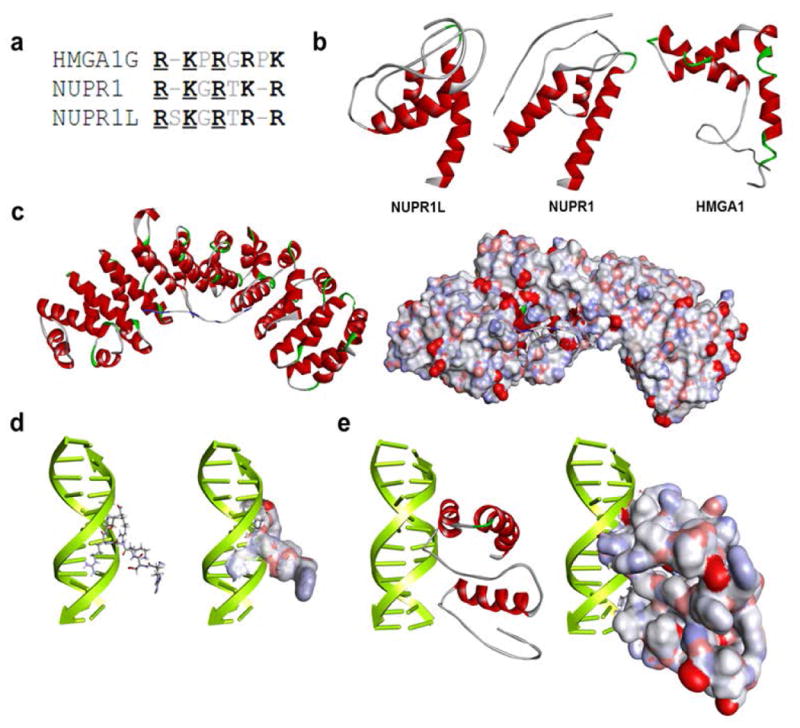
(a) Comparison between DNA-binding domains of NUPR1L and NUPR1 with the AT-hook of HMGA1.(b) NUPR1L and its closest human homolog NUPR1 share their ability to adopt a helix-loop-helix fold that is characteristic of HMGA1. Ribbon representation of their models with helical regions denoted in red and loops in white. (c) Homology-based models derived from the PDB complex 1EJY. The models show that a C-terminal peptide sequence of NUPR1L (KRVAQKLLRGQRKRR) kinds to alpha-importin which mediates the nuclear localization of most eukaryotic proteins. The ribbon model shows the C-terminal peptide from NUPR1L with blue coloring n basic regions that form the most bonds with the main chain of alpha-importin. A space-filling model displays the cavity within alpha-importin which easily accommodates the C-terminal peptide of NUPR1L. (d) Two different molecular modeling approaches were used to gain insight into the DNA-binding properties of NUPR1L. Both a homology-based approach and a DP-DOCK-based docking approach and scoring method reveal that NUPR1L binds to the minor groove of DNA using a DNA binding sequence (GRSKGRTRR) that is highly similar to the AT-hook containing transcriptional activator HMGA1. HMGA1-based homology models in a ribbon and space-filling representation are shown. (e) DP-DOCK-based model of full-length NUPRL are shown.
Figure 2. Primary structure analysis and nuclear localization of Nup1L protein.
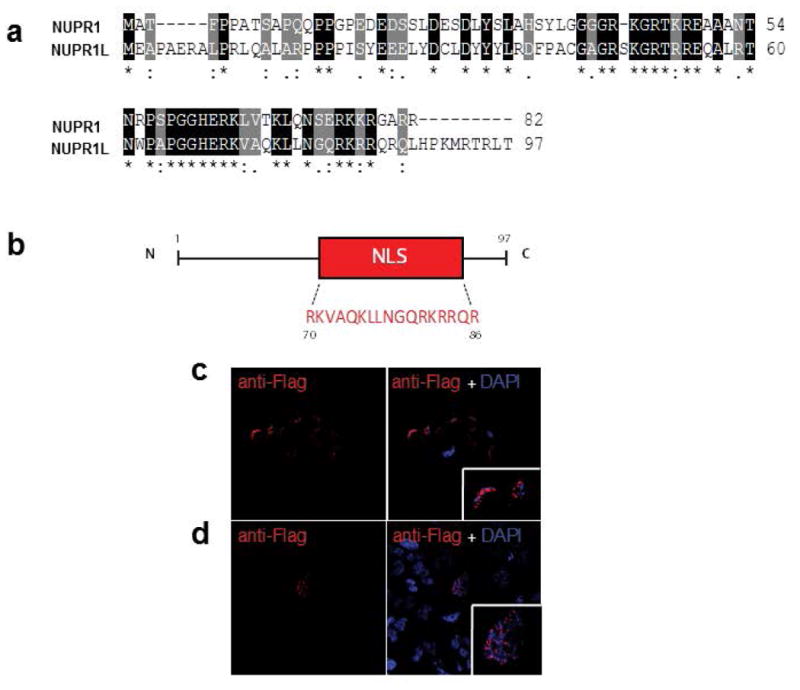
(a) Pairwise alignment of the Nupr1 and Nupr1L proteins displaying a 60 % of homology. Residues labeled with a (*) indicate 100% identity, residues labeled with (:) indicate positions with conservative substitutions, residues labeled with (.) indicate substitutions less conservative. (b) Schematic representation of Nupr1L protein, which contains a Nuclear Localization Signal (NLS). The Flag-Nupr1L immunofluorescence in MiaPaca-2 (c) and Hela cells (d) reveals its nuclear localization. The cells were transduced with a lentiviral vector (6His-Flag-Nupr1L) and were plated on coverslips. The mouse Flag was revelated using Alexa fluorantimouse IgG488. Nuclei are stained by blue fluorescent DAPI.
NUPR1L is a new p53 target gene that is regulated by two functional binding sites in its promoter region
We have previously shown that NURP1 mediates pancreatic cancer initiation and promotion by mediating downstream signal from mutated tumor suppressor such as p53. This knowledge led us to begin studies on the regulation of NUPR1L gene expression by first analyzing its promoter region using bioinformatics-based transcription factor binding site analyses (Farre et al., 2003). Analysis of the 643 bp core promoter region for the NUPR1L gene identified consensus cis-regulatory sites for E2F-1, C/EBPα, C/EBPβ, AP-1, GATA-1, NFκB, ATF-1 as well as two p53-binding sites (see Figure 3a). These sites, which conform to the typical p53 binding sequence (GGGCAGG) are located at positions -37 and -7 of the promoter, respectively (Figure3a). Thus, we generated the pGL3-Nupr1L-Luc constructs, containing these p53-binding sites to be used in reporter transcriptional studies which we performed in three different cancer cell lines by co-transfection with a wild-type p53 expression vector. Notably, these experiments demonstrate that the Nupr1L promoter is activated by p53 in a dose-dependent manner reaching a maximal upregulation of 4.78 (± 0.37) fold using 800 ng of the p53 encoding plasmid (Figure3b). A similar response was observed in Hela cells (Figure3d). In breast cancer cells, the Nupr1L promoter activity was increased by 5.87 (± 0.65) fold in response to only 200 ng of the p53 construct (Figure3c). Subsequently, we use site directed mutagenesis to inactivate the p53 binding site at both the position -37 (ΔS1 mutation) and -7 (ΔS2 mutation) (Figure4a) and repeated the reporter assays under the same conditions described above (Figure4b-d). Interestingly, mutation of one of these p53-binding sites independently did not eliminate the induction effect of the p53 expression suggesting that both sites are necessary for achieving the upregulation of Nupr1L promoter activity by p53. Consequently, inactivation of both p53-binding sites (ΔS1+ΔS2 mutation) (Figure 4a) completely abolished the activating effect of p53 in the three different cell lines studied. These results demonstrate that Nupr1L is regulated by p53 at the promoter level and defines the key cis-regulatory sequences responsible for this effect.
Figure 3. Nupr1L gene is regulated by p53 protein.
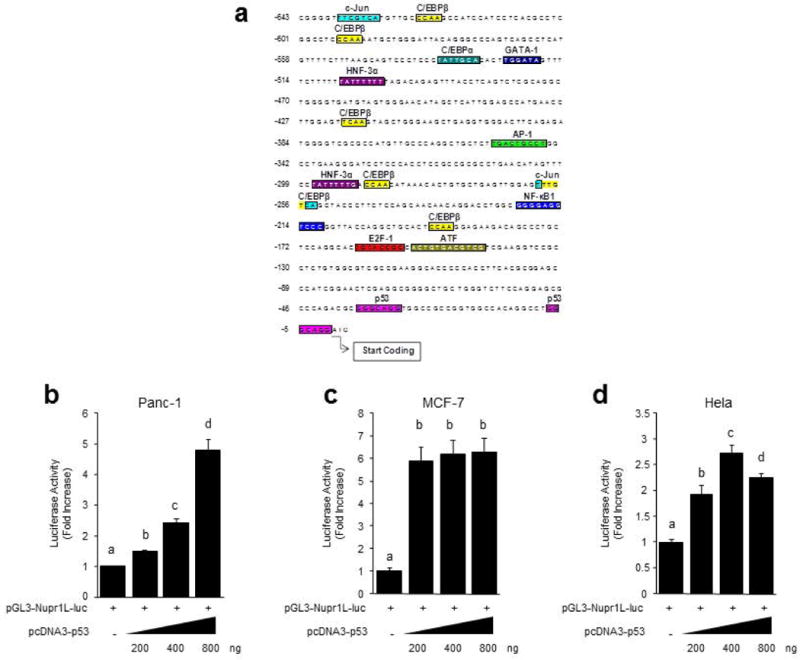
(a) Diagram of the possible binding sites of different transcription factors in the Nupr1L promoter whose sequence is illustrated since the position -643 to +3 considering +1 the translation start site. Reporter gene assay for Nupr1L promoter in response to increasing concentration of pcDNA3-p53 in Panc-1 (b), MCF-7 (c) and Hela cells (d). The region of the Nupr1L promoter that was cloned into pGL3-luciferase vector (pGL3-Nupr1L-Luc) was comprised by the sequence since -245 to +93 position. Values are expressed as mean ± S.D. of triplicate, from 2 independent experiments. Means sharing the same superscript letter are not significantly different from one another (P< 0.05).
Figure 4. Nupr1L promoter contains two p53 regulation functional sites.
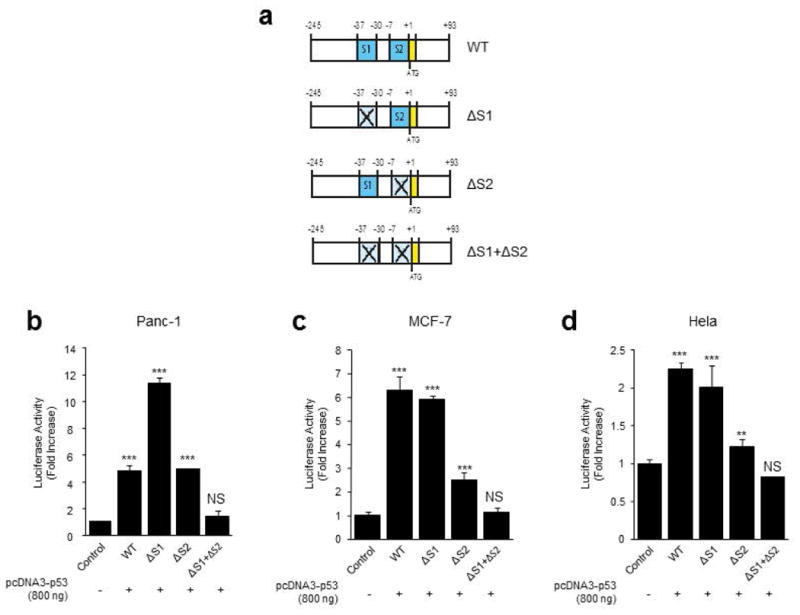
(a) Schematic representation of the different constructions of Nupr1L promoter that were cloned into pGL3-luciferase vector. WT, Nupr1L promoter sequence wild type; ΔS1, Nupr1L promoter sequence with the deleted sequence1 by directed mutagenesis; ΔS2 Nupr1L promoter sequence with the deleted sequence2 by directed mutagenesis; ΔS1+ΔS2, Nupr1L promoter sequence with both deleted sequences (1+2) by directed mutagenesis. The yellow band indicates the start codon. (b) Reporter gene assay for the different constructions of Nupr1L promoter (WT, ΔS1, ΔS2 and ΔS1+ΔS2) in Panc-1 cells, MCF-7 (c), and Hela cells (d). The bar denominated as control is a representation of the different controls corresponding to each construction. Values are expressed as mean ± S.D. of triplicate, from 2 independent experiments. Differences are expressed respect to the control; **P<0.01 and ***P<0.001; (NS) not significant differences.
NUPR1L expression is highly dependent of p53
Through promoter studies described above, we demonstrated that NUPR1L is regulated by p53. However, we needed to know and to quantify in a more elegant manner the p53 dependence in the expression of NUPR1L. For this, three cell lines characterized by a different p53 status were transfected with the pGL3-Nupr1L-Luc construct and the promoter activity was evaluated by luciferase assay. We used immortalized fibroblasts p53+/+, p53-/- (10.1 cell line) and a termosensible (ts) mutant, p53-/ts named (10.1)Val5 cell line. In fact, 10.1 cell line is an established Balb/c 3T3 line that has lost both p53 alleles during immortalization (Harvey and Levine, 1991). (10.1)Val5 is a clonal cell line derived from the 10.1 cells by stable transfection of the temperature-sensitive p53 mutant (val135) driven by the Harvey sarcoma virus long terminal repeat (Michalovitz et al., 1990). p53 exists in a mutant (inactive) conformation in this cell line at 39°C; temperature shift to 32°C results in wild-type p53 conformation and activity. At 37°C, both mutant and wild-type p53 conformational forms co-exist in the cells as previously demonstrated (Martinez et al., 1991; Wu et al., 1993). We therefore analyzed the activity of NUPR1L promoter in p53+/+, p53-/-, and p53-/ts cells where both wild-type p53 and inactive conformational forms were present (Figure 5). p53+/+ cells showed a significant increase in the promoter activity compared with the control vector (6.8 ± 1.1 folds). In the same way, luciferase assay displayed a 9.2-fold (± 0.9) increase in termosensible p53 cells, and only 2.3-fold (± 0.4) when growth at 32°C. In fact, we found that NUPR1L promoter activity was similar in both p53+/+ and p53-/ts cell line (p = 0.05). Altogether these results demonstrate a high and significant dependence of p53 activation in the NUPR1L regulation.
Figure 5. p53-dependent expression of Nupr1L.
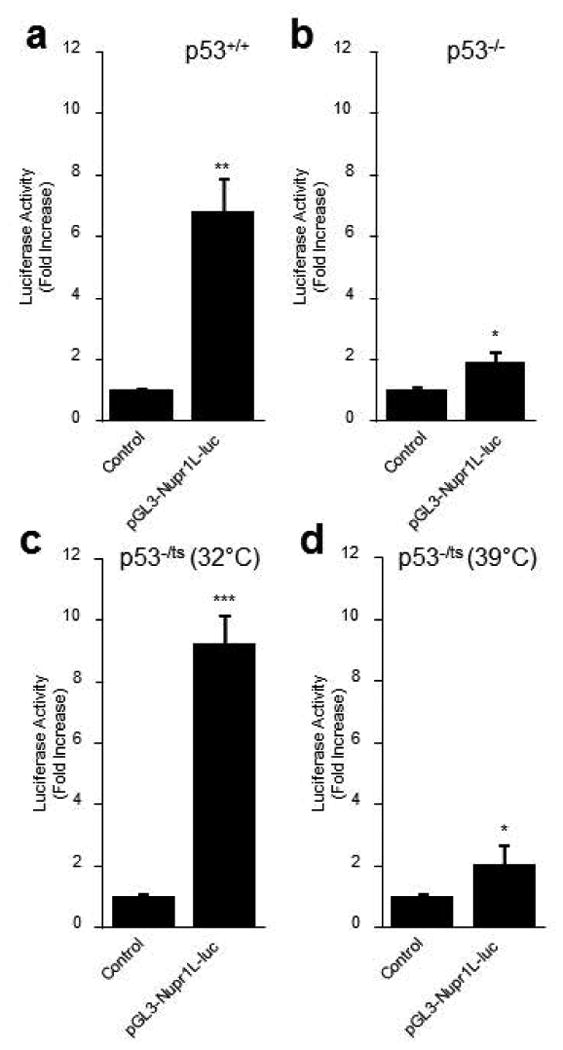
Reporter gene assay for Nupr1L promotor in (a) 3T3 fibroblastes (p53+/+), (b) 10.1 cell line (p53-/-) and (c and d) (10.1)Val5 cell line grown at 39°C and 32°C. Results are expressed as fold increase compared with the pGL3 empty vector control. Values are expressed as mean ± S.D. of triplicate, from 2 independent experiments. *p<0.05, **P<0.01 and ***P<0.001.
p53-inducing classical stimuli increase NUPR1L expression in cancer cells
The p53 tumor suppressor protein is a key regulator of cell-cycle arrest, survival, DNA repair, programmed cell death, and genetic stability. The expression and function of p53 is activated in response to several cellular stresses including serum starvation, DNA damage, and a wide range of chemotherapeutic drugs (Levine et al., 2006; Vazquez et al., 2008). Thus, having demonstrated that the Nupr1L promoter is responsive to p53, we next aim at studying whether or not classic treatments that trigger p53 activation, such as DNA damage by Oxaliplatin or cell cycle arrest induced by serum starvation, ultimately impact on the expression of the NUPR1L gene. For this purpose, we treated three cancer cell lines, which have preserved p53 wild-type, with increasing concentrations of Oxaliplatin for 18 or 24 h and measured NUPR1L mRNA expression by RT-qPCR. The results of these experiments depicted in Figure 6 demonstrate that Nupr1L mRNA is induced in a dose-dependent manner. The levels of NUPR1L mRNA were increased by almost 12 fold in MCF-7 cells upon the treatment with Oxaliplatin at 40 μM for 24 h (11.7 ± 2.9 folds) (Figure6a). A significant, although less robust increase in Nupr1L mRNA was also obtained in Capan-2 and Hela cells after of 18 h of treatment with 20 μM of Oxaliplatin (Figure6b and6c). Changes in Nupr1L mRNA levels were also evaluated following serum starvation after of 12, 24 and 60 h, a treatment that induces cell cycle arrest and p53 activation (Del Sal et al., 1995; Schneider et al., 1988). Congruently, as shown in Figure 6 Nupr1L mRNA levels are also induced by this stimulus in the three studied cell lines. Hence, we demonstrated that classical treatments that induce p53 activation by different intracellular pathways also increase NUPR1L expression, a finding that is consistent with our promoter studies.
Figure 6. DNA damage treatments and serum starvation induce the Nupr1L gene overexpression.
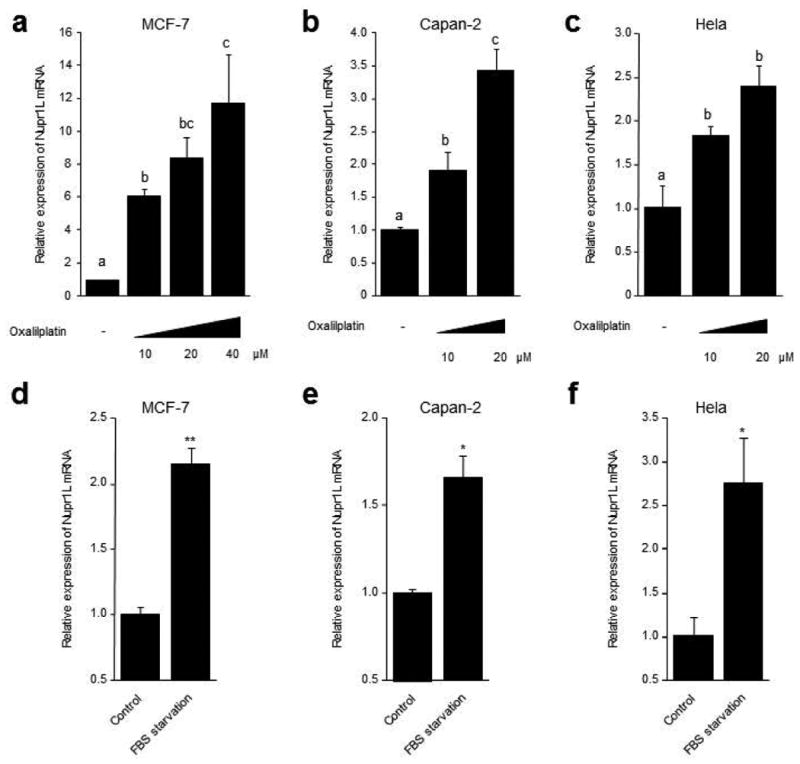
(a) MCF-7 cells were treated with increasing concentrations of Oxaliplatin (10, 20 and 40 μM) for 24 h. Capan-2 (b) and Hela cells (c) were treated with increasing concentrations of Oxaliplatin (10 and 20 μM) for 18 h. MCF-7 (d), Capan-2 (e) and Hela cells (f) were cultured in FBS starvation conditions for 24 h, 60 h and 12 h, respectively. mRNA levels of Nupr1L were analyzed by RT-qPCR. The housekeeping gene Cyclophilin was used as internal control in order to the normalizing of values. Results are expressed as fold change compared with the control ± S.D. Means sharing the same superscript letter are not significantly different from one another (P< 0.05). *P<0.05 and **P<0.01.
NUPR1L down regulates the transcriptional activity of the NUPR1 promoter
The close homology between Nupr1 and Nupr1L as well as its ability of being regulated by p53 led us to elucidate functional properties of this new p53-target gene, initially as it related to its potential interactions with Nupr1. Accordingly, we transfected pancreatic cancer cells with a 6His-Flag-Nupr1L lentiviral vector and measured its effects on NUPR1 mRNA expression by RT-qPCR. We find that viral delivery of NUPR1L significantly decreased NUPR1 mRNA levels as showed in Figure 7a. To further characterize this effect, we used reporter assay using a pGL3-Nupr1-Luc vector containing the previously characterized promoter of NUPR1 (-2111 to +229) (Pommier et al., 2012). As shown in Figure 7b, the expression of 6His-Flag-Nupr1L results in a 44 ± 9% reduction in NUPR1 promoter activity. Similarly, we observed that NUPR1 promoter activity decrease (72 ± 1%) in the presence of p53. Because p53 up-regulates expression of NUPR1L which in turn down-regulates expression of NUPR1 and moreover, NUPR1 is down-regulated by p53, we carried on luciferase assay in the presence of p53 and silencing NUPR1L by siRNA, in order to know whether the regulation exerted by p53 on NUPR1 promoter is or not NUPR1L-dependent. NUPR1L-silenced cells showed close values to siControl cells in presence of p53, revealing that the decrease of NUPR1L expression (Figure 7d) does not interfere on p53-depending regulation of NUPR1 promoter (Figure 7c). These results reveal a coordinated trans-regulation of p53, NUPR1, and NUPR1L into a gene pathway which has an impact on the regulation of cell cycle mediated processes.
Figure 7. Nupr1L down-regulates Nupr1 expression.
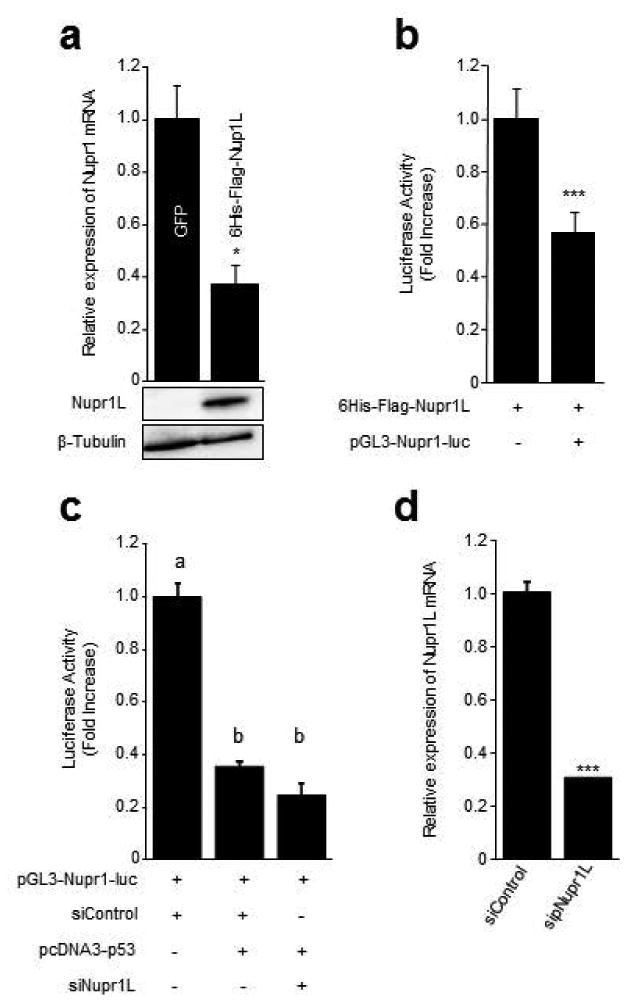
MiaPaca-2 cells were infected by 6His-Flag-Nupr1L lentiviral vector. (a) By RT-qPCR, decrease of Nupr1 transcript is observed in cells overexpressing Nupr1L. Western Blot shows the increase of NUPR1 protein in transduced cells with the lentiviral 6His-Flag-Nupr1L vector. (b) Reporter gene assay for Nupr1. Significant decrease in the activity was observed when cells were co-transfected with the 6His-Flag-Nupr1L and pGL3-Nupr1-Luc. (c) Similar results were showed at level of NUPR1 promoter in presence of p53, displaying a significant decrease in the luciferase activity. Nupr1L depletion did not modify the effect of p53 on Nupr1 promoter activity. (d) RT-qPCR of Nupr1L showing the effectiveness of specific siRNA. Values are expressed as mean ± S.D. Means sharing the same superscript letter are not significantly different from one another. *p<0.05 and ***p<0.001.
Cell cycle arrest induced by NUPR1L is rescued by NUPR1 expression
FACS-assisted cell cycle analyses carried out in the MiaPaCa-2 pancreatic cancer cell line demonstrate that NUPR1L causes an arrest at G1 (58.24 ± 2.95% in cells transduced with GFP- vs. 65.8 ± 3.71% in cells transduced with the 6His-Flag-Nupr1L expressing lentivirus, respectively). In addition, Figure 8a shows that this effect is accompanied by a decrease in S phase (25.41 ± 2.08% vs. 17.3 ± 1.28% in cells transduced under the same conditions). Concomitantly, at the cellular level, we observed that NUPR1L expression induces a significant decrease in the viability of MiaPaCa-2 cells (100 ± 1.3% vs. 59.7 ± 0.8% in cells transduced by the same set of lentivirus), as shown in Figure 8b. Likewise, if we observe the real-time cell proliferation curves we found that NUPR1L-overexpressing cells showed a significant stop in cell proliferation compared with GFP-transduced cells from 38 h (Figure 8c). These modifications in the cell cycle, viability and cell proliferation are reminiscent of those changes previously observed upon treatment of MiaPaCa-2 cells with Nupr1 siRNAs (Grasso et al., 2014; Hamidi et al., 2012b). Thus, taken together, these results led us to hypothesize that the effect of NUPR1L on the cell cycle and viability was mediated, at least in part, by down regulation of NUPR1. To test this hypothesis we sought to reestablish NUPR1 expression levels in NUPR1L overexpressing cells by concomitant lentivirus expression and measured cell cycle progression, cell viability and cell proliferation. As shown in Figure 8, NUPR1 expression does not change significantly neither cell cycle nor cell viability. Regarding to cell proliferation, NUPR1-overexpressing cells showed a proliferation rate higher than GFP-transduced cells. However, the co-expression of NUPR1 with NUPR1L almost completely reduces the effects of the latter. In the case of NUPR1 and NUPR1L co-expressing cells, proliferation curve was higher than in GFP-transduced cells, reaching values close to NUPR1-overexpressing cells. Together, these results suggest that a NUPR1L effect on cell growth is mediated, at least in part, by its ability to down-regulate NUPR1.
Figure 8. Nupr1L decreases the pancreatic cancer cells viability.
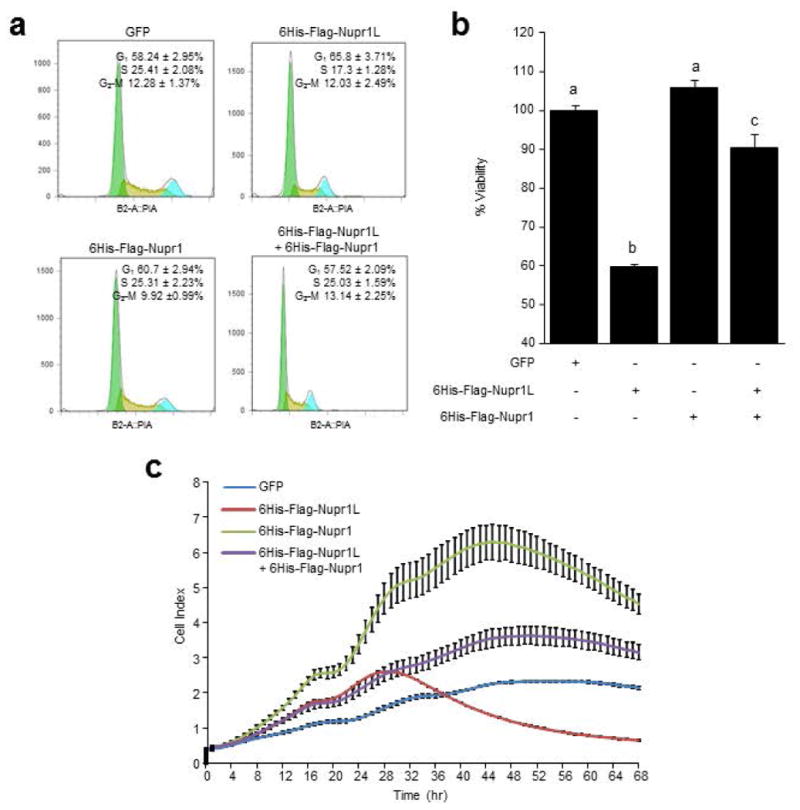
MiaPaca-2 cells were infected by 6His-Flag-Nupr1L and 6His-Flag-Nupr1 lentiviral vector, individually and together. GFP overexpressing cells were used like control (e) Cytometric cell cycle analysis showing a G1 cell cycle arrest with a significant decrease of S phase induced by Nupr1L overexpression and abolished by the concomitant overexpression of Nupr1. (f) Cell viability measured by Cell Titer-Blue assay. Decrease of viability is showed in cells infected by 6His-Flag-Nupr1L vector but not in infected cells byboth lentiviral vectors, 6His-Flag-Nupr1 and 6His-Flag-Nupr1L. Cell viability in GFP infected cells was normalized to 100%. Values are expressed as mean ± S.D. Means sharing the same superscript letter are not significantly different from one another (P< 0.05). (c) Real-time cell proliferation assay using electric impedance as a measure of cell proliferation. Electrical impedance was normalized according to the background measurement at time point 0. Impedance measurements were carried out for 72 h. Data points represent mean values ± S.D. (n = 3). Results show lower rate of proliferation in NUPR1L-overexpressing cells than GFP-transduced cells. The concomitant overexpression of Nupr1 abolished the effect induced by Nupr1L on the proliferation cell, reaching intermediate values to both infected cells (6His-Flag-Nupr1L and 6His-Flag-Nupr1 cells).
Reciprocal regulation of NUPR1L by its homologue NUPR1
In this work we describe a significant regulation of NUPR1L on the expression of NUPR1 associated to a diminution of proliferation in pancreatic cancer cell. The protumoral factor NUPR1 has been subject of study for our group during the last decades having demonstrated a crucial role in cancer development and progression (Cano et al., 2014; Grasso et al., 2014; Hamidi et al., 2012a; Vasseur et al., 2002). For this reason, our next aim was to know if this regulation is reciprocal in order to shed light on the relationship between both homologues and the cancer biology. Accordingly, MiaPaca-2 cells were transduced with the 6His-Flag-Nupr1 lentiviral vector and the NUPR1L expression was evaluated by RT-qPCR using GFP-transduced cells as control. Cells were cultured in conditioned medium as mock condition and under glucose starvation as a metabolic stress condition. No significant difference was found in basal conditions (Figure 9a). The induction of a metabolic stress increased sustantialy expression of the NUPR1L (4.5 ± 0.4 folds). However, this increment was reduced in the presence of NUPR1 (2.7 ± 0.2 folds). Indeed, NUPR1 silencing by siRNA increased slightly the expression of NUPR1L compared to siControl cells under glucose starvation conditions (Figure 9b). Effectiveness of specific siRNA was confirmed by RT-qPCR, displaying a reduction in the NUPR1 expression of 84% (Figure 9c). Finally, as previously published (Hamidi et al., 2012b), we observed that the glucose starvation increased considerably the expression of NUPR1 (352.4 ± 20.7 folds) (Figure 9d). Altogether, results revealed that the regulation of NUPR1L by its homologue the NUPR1 tumor factor only takes place under stress conditions where the expression of NUPR1 is strongly augmented.
Figure 9. Nupr1 decrease the Nupr1L expression under metabolic stress conditions.
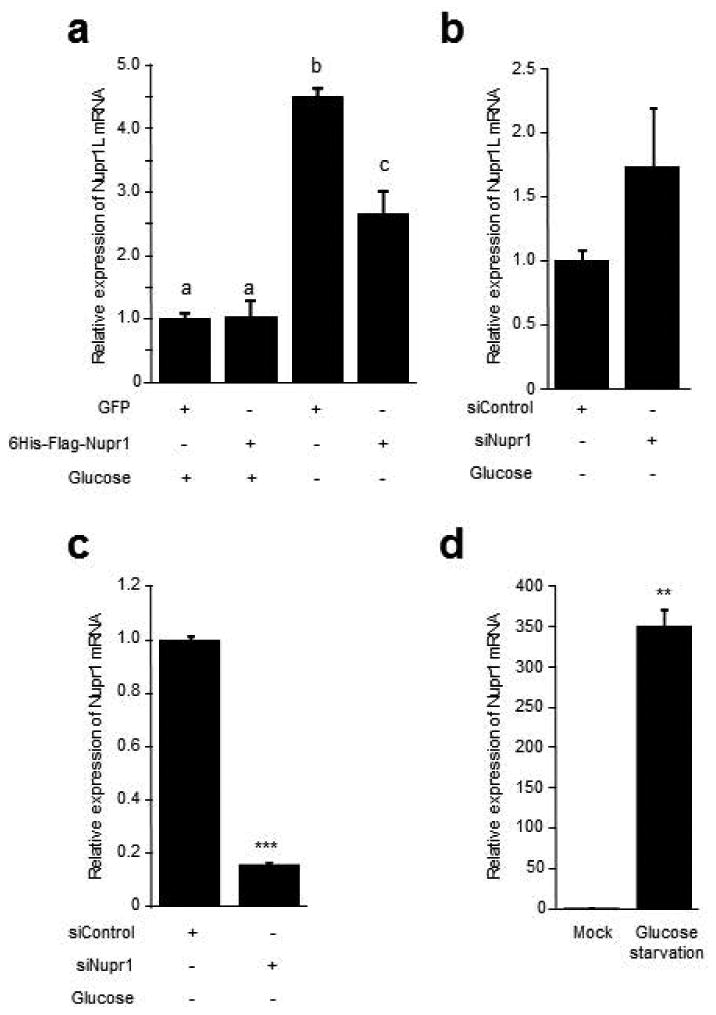
Expression of Nupr1L or Nupr1 by RT-qPCR in MiaPaca-2 cells (a) Nupr1L expression in transduced cells with 6His-Flag-Nupr1 lentiviral vector and starved of glucose for 24 h. GFP-overexpressing cells and cultured in conditioned medium were used as control. (b) Nupr1L expression in cells treated with siNupr1 or siControl and cultured under glucose starvation conditions for 24 h. (c) RT-qPCR of Nupr1 showing the effectiveness of specific siRNA. (d) Increase of Nupr1 expression under glucose starvation conditions. Conditioned medium was used as mock treatment. Values are expressed as mean ± S.D. Means sharing the same superscript letter are not significantly different from one another. *p<0.05 and ***p<0.001.
In summary, we find that in addition to the tumor promoting chromatin protein NUPR1, the human genome contains another gene encoding for a highly related isoform. At the molecular level, these two proteins share properties between themselves and with members of the HMG family of proteins. Functionally, both proteins appear to work downstream of the p53 pathway though playing antagonistic roles on the regulation of cell cycle progression, including in the presence of chemotherapeutic drugs. Together, this data expand the knowledge on the repertoire of chromatin proteins, which can mediate p53 signals in pancreatic cancer cells. This new knowledge, therefore, has both mechanistic value and biomedical relevance.
Discussion
In this work we demonstrate that NUPR1L is a new p53-inducible gene that is directed to decrease the expression of the NUPR1 tumorigenic gene through the negative regulation of its promoter activity. In addition, we also found that overexpression of NUPR1L induces G1 cell cycle arrest and a decrease in cell viability and cell proliferation, whereas concomitant overexpression of NUPR1 is able to counter arrest this effect indicating that the effect of NUPR1L on cell growth is mediated at least in part by down-regulating NUPR1 expression.
Human NUPR1L gene is located on chromosome 7 (7p11.2) and encodes a 97 aminoacid polypeptide with a theoretical molecular mass of 10177.94 Da and a pI of 10.81. Regarding the primary structure, recent in silico studies have identified a bipartite nuclear localization signal and a conserved domain which is found in several DNA-Binding proteins (Urrutia et al., 2014). In agreement with these predictions, we confirmed its nuclear localization using an immunofluorescence analysis of the 6His-Flag-Nupr1L transduced in two cell types (Figure2c and 2d). Interestingly, both NUPR1 and NUPR1L proteins are of nuclear localization. Since both proteins share subcellular localization and are highly homologous we speculated that they can have a functional relationship, both as having common functions or, on the contrary, as acting as a negative dominant.
One of the most interesting observations obtained from this work was the p53-dependency of NUPR1L gene expression but most exciting is the fact that we found two functional p53-response elements on the proximal region of the NUPR1L promoter, and that both are redundant, suggesting that during the evolution process of this gene the p53-dependency was an important criteria of selection. In the same way, we found that cellular processes inducing activation of the p53, such as DNA damage induced by anticancer drugs or cell cycle arrest induced by serum starvation, activates NUPR1L expression (Figure6), which is in agreement with the transactivation experiments (Figure 3). We also found other transcription factors that seem to bind over the proximal region of the NUPR1L promoter (Figure3a) however their functionality remains to be defined.
An apparent contradiction was acted during this study because, on one hand, NUPR1 which is a protumoral factor with antiapoptotic activity (Malicet et al., 2006), facilitating the EMT process induced by TGFβ (Cano et al., 2014; Cano et al., 2012; Pommier et al., 2012; Sandi et al., 2011), increasing resistance to anticancer treatments (Giroux et al., 2006), facilitating metastasis (Ree et al., 2000) and having a crucial role during oncogenic Kras-dependent transformation in pancreatic cancer (Garcia et al., 2014; Hamidi et al., 2012a) by bypassing the senescence process (Grasso et al., 2014), but on the other hand, on the contrary, NUPR1L was found to be activated by one of the more powerful gene suppressor, p53. In this way, overexpression of NUPR1L induces cell cycle arrest and decreases cell viability. Our studies found that in fact the effect we observed for NUPR1L was at least in part mediated since NUPR1L clearly down regulates expression of NUPR1 and because forced expression of the NUPR1 counter act the effect of NUPR1L overexpression and therefore suggesting a very fine regulatory feedback between both structurally-related molecules. Interestingly, the high homology, the functional results, as well as the fact of that both factors have been proposed to belong to the same protein family (Urrutia et al., 2014), overall, suggests that NUPR1 and NUPR1L could be possible paralogous genes arisen by a duplication phenomenon. There are many cases in the genome of most organisms, e.g. the hemoglobin genes (Nehrt et al., 2011). In addition, both homologues are regulated by p53 but in an opposite manner, suggesting a system of large selective pressure in order to maintain an anticancer phenotype through the repression of NUPR1.
Finally, we aimed to know the effect of the NUPR1 tumor factor on the expression of NUPR1L in order to complete this feedback. Results revealed that NUPR1 does not interfere in NUPR1L expression in normal conditions. It was necessary to increase the expression of the tumoral factor through metabolic stress to observe a brake in the expression of NUPR1L (Figure 9). Thus, here, we describe a system of two homologues with opposite functions where one of them inhibits the expression of the other, which is in turn a tumoral factor with a key role in cancer. However, this down-regulation is only reciprocal when the expression of NUPR1 is surprisingly high, confirming this anticancer evolutionary pressure above mentioned.
Nowadays, human cancers represent one of the biggest challenges for modern societies. In fact, it has been estimated that by 2020, cancer deaths worldwide could reach 10 million (Szychot et al., 2013). Therefore, one of the important aims of science research is the improvement of anti-malignant treatment options to mitigate cancer-related morbidity and mortality. Cancer development involves tumor suppression gene inactivation and oncogene activation, leading to uncontrolled clonal proliferation. Despite of the efforts to find an effective treatment targeting oncogenes or their protein products, the appearance of genetic mechanisms of drug resistance has become the principal issue of therapy (Szychot et al., 2013). On the one hand, the tumor suppressor p53 is considered one of the major objectives to combat cancer. In fact, several in vivo studies have demonstrated an efficient suppression of established tumor after to reinstatement of p53 (Ventura et al., 2007; Xue et al., 2007). On the other hand, NUPR1, as commented above, is a protumoral factor involved in the resistance to the therapy and whose silencing displays an anticancer phenotype. Here, we described a new factor that is regulated by the most important tumor suppressor, p53, and represses the expression of a protumorogenic factor, NUPR1, displaying an antitumoral phenotype. Thereby, a better understanding of the NUPR1L network could be important for the application of p53-based therapies.
In conclusion, we demonstrated that NUPR1L is a new p53-induced gene which negatively regulates the protumoral factor NUPR1. Thus, because of the key role of NUPR1 in cancer, the new information emerging from this study has biomedical relevance that aid to a better understanding on the pathobiology of cancer.
Supplementary Material
Figure S1. (a) More than 98% of the residues are found in the allowed regions (b). NUPR1L, display several regions within Nurp1L which the propensity to form three α helices. Helix 1 contains 15 residues and spans from 19 to 34. Helix 2 contains 9 residues and spans from 52 to 61 while Helix 3 contains 20 residues and spans from 67 to 87. (c) Topology diagram of the NUPR1L model.
Table S1a. Bonding pattern between NUPR1L and B-DNA.
Table S1a. Bonding pattern between Nurp1L and alpha-importin.
Table S2. (a) siRNA sequences used for silencing Nupr1 and Nupr1L mRNA. (b)Primer sequences used to clone and directed mutagenesis. (c) Primer sequences used to amplify human genes.
Acknowledgments
This work was supported by La Ligue Contre le Cancer, INCa, Canceropole PACA, DGOS (labelization SIRIC) and INSERM (to JLI), National Institutes of Health Grants DK52913, the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567) and the Mayo Foundation (to RU), and the Spanish Ministerio de Ciencia e Innovacion (MCINN) (CTQ2011-24393 to JLN, CSD2008-00005 to JLN, intramural BIFI 2011 projects (to JLN). BJ is supported byINCa. MIM is supported by La LigueContre le Cancer. DG and MNG are supported by l'Association pour la Recherchesur le Cancer. LMB are supported by Fundacion Alfonso Martin Escudero.
Footnotes
The authors have declared that no conflict of interest exists.
References
- Bonacci T, Audebert S, Camoin L, Baudelet E, Bidaut G, Garcia M, Witzel II, Perkins ND, Borg JP, Iovanna JL, Soubeyran P. Identification of new mechanisms of cellular response to chemotherapy by tracking changes in post-translational modifications by ubiquitin and ubiquitin-like proteins. Journal of proteome research. 2014;13(5):2478–2494. doi: 10.1021/pr401258d. [DOI] [PubMed] [Google Scholar]
- Cano CE, Hamidi T, Garcia MN, Grasso D, Loncle C, Garcia S, Calvo E, Lomberk G, Dusetti N, Bartholin L, Urrutia R, Iovanna JL. Genetic inactivation of Nupr1 acts as a dominant suppressor event in a two-hit model of pancreatic carcinogenesis. Gut. 2014;63(6):984–995. doi: 10.1136/gutjnl-2013-305221. [DOI] [PubMed] [Google Scholar]
- Cano CE, Sandi MJ, Hamidi T, Calvo EL, Turrini O, Bartholin L, Loncle C, Secq V, Garcia S, Lomberk G, Kroemer G, Urrutia R, Iovanna JL. Homotypic cell cannibalism, a cell-death process regulated by the nuclear protein 1, opposes to metastasis in pancreatic cancer. EMBO molecular medicine. 2012;4(9):964–979. doi: 10.1002/emmm.201201255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Sal G, Ruaro EM, Utrera R, Cole CN, Levine AJ, Schneider C. Gas1-induced growth suppression requires a transactivation-independent p53 function. Molecular and cellular biology. 1995;15(12):7152–7160. doi: 10.1128/mcb.15.12.7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel H, Michael S, Weatheritt RJ, Davey NE, Van Roey K, Altenberg B, Toedt G, Uyar B, Seiler M, Budd A, Jodicke L, Dammert MA, Schroeter C, Hammer M, Schmidt T, Jehl P, McGuigan C, Dymecka M, Chica C, Luck K, Via A, Chatr-Aryamontri A, Haslam N, Grebnev G, Edwards RJ, Steinmetz MO, Meiselbach H, Diella F, Gibson TJ. ELM--the database of eukaryotic linear motifs. Nucleic acids research. 2012;40(Database issue):D242–251. doi: 10.1093/nar/gkr1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinar JA, Mallo GV, Mizyrycki C, Giono L, Gonzalez-Ros JM, Rico M, Canepa E, Moreno S, Neira JL, Iovanna JL. Human p8 is a HMG-I/Y-like protein with DNA binding activity enhanced by phosphorylation. The Journal of biological chemistry. 2001;276(4):2742–2751. doi: 10.1074/jbc.M008594200. [DOI] [PubMed] [Google Scholar]
- Encinar JAMG, Mizyrycki C, Giono L, Gonzalez-Ros JM, Rico M, Cánepa E, Moreno S, Neira JL, Iovanna JL. Human p8 is a HMG-I/Y-like protein with DNA binding activity enhanced by phosphorylation. J Biol Chem. 2001;276(4):2742–2751. doi: 10.1074/jbc.M008594200. [DOI] [PubMed] [Google Scholar]
- Farre D, Roset R, Huerta M, Adsuara JE, Rosello L, Alba MM, Messeguer X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic acids research. 2003;31(13):3651–3653. doi: 10.1093/nar/gkg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfría-Subirós EA-RF, Saperas N, Pous J, Subirana JA, Campos JL. Crystal Structure of a Complex of DNA with One AT-Hook of HMGA1. PLoS ONE. 2012;7(5):e37120. doi: 10.1371/journal.pone.0037120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao MSJ. A threading-based method for the prediction of DNA-binding proteins with application to the human genome. PLoS Comput Biol. 2009;5(11):e1000567. doi: 10.1371/journal.pcbi.1000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MN, Grasso D, Lopez-Millan MB, Hamidi T, Loncle C, Tomasini R, Lomberk G, Porteu F, Urrutia R, Iovanna JL. IER3 supports KRASG12D-dependent pancreatic cancer development by sustaining ERK1/2 phosphorylation. The Journal of clinical investigation. 2014 doi: 10.1172/JCI76037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroux V, Malicet C, Barthet M, Gironella M, Archange C, Dagorn JC, Vasseur S, Iovanna JL. p8 is a new target of gemcitabine in pancreatic cancer cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(1):235–241. doi: 10.1158/1078-0432.CCR-05-1700. [DOI] [PubMed] [Google Scholar]
- Goruppi S, Iovanna JL. Stress-inducible protein p8 is involved in several physiological and pathological processes. The Journal of biological chemistry. 2010;285(3):1577–1581. doi: 10.1074/jbc.R109.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goruppi S, Kyriakis JM. The pro-hypertrophic basic helix-loop-helix protein p8 is degraded by the ubiquitin/proteasome system in a protein kinase B/Akt- and glycogen synthase kinase-3-dependent manner, whereas endothelin induction of p8 mRNA and renal mesangial cell hypertrophy require NFAT4. The Journal of biological chemistry. 2004;279(20):20950–20958. doi: 10.1074/jbc.M312401200. [DOI] [PubMed] [Google Scholar]
- Grasso D, Garcia MN, Hamidi T, Cano C, Calvo E, Lomberk G, Urrutia R, Iovanna JL. Genetic inactivation of the pancreatitis-inducible gene Nupr1 impairs PanIN formation by modulating Kras-induced senescence. Cell death and differentiation. 2014 doi: 10.1038/cdd.2014.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi T, Algul H, Cano CE, Sandi MJ, Molejon MI, Riemann M, Calvo EL, Lomberk G, Dagorn JC, Weih F, Urrutia R, Schmid RM, Iovanna JL. Nuclear protein 1 promotes pancreatic cancer development and protects cells from stress by inhibiting apoptosis. The Journal of clinical investigation. 2012a;122(6):2092–2103. doi: 10.1172/JCI60144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi T, Cano CE, Grasso D, Garcia MN, Sandi MJ, Calvo EL, Dagorn JC, Lomberk G, Urrutia R, Goruppi S, Carracedo A, Velasco G, Iovanna JL. Nupr1-aurora kinase A pathway provides protection against metabolic stress-mediated autophagic-associated cell death. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012b;18(19):5234–5246. doi: 10.1158/1078-0432.CCR-12-0026. [DOI] [PubMed] [Google Scholar]
- Harvey DM, Levine AJ. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes & development. 1991;5(12B):2375–2385. doi: 10.1101/gad.5.12b.2375. [DOI] [PubMed] [Google Scholar]
- Hwang SGZ, Kuznetsov IB. DP-Bind: a web server for sequence-based prediction of DNA-binding residues in DNA-binding proteins. Bioinformatics. 2007;23(5):634–636. doi: 10.1093/bioinformatics/btl672. [DOI] [PubMed] [Google Scholar]
- Irelan JT, Wu MJ, Morgan J, Ke N, Xi B, Wang X, Xu X, Abassi YA. Rapid and quantitative assessment of cell quality, identity, and functionality for cell-based assays using real-time cellular analysis. Journal of biomolecular screening. 2011;16(3):313–322. doi: 10.1177/1087057110397359. [DOI] [PubMed] [Google Scholar]
- Laskowski RAMMW, Moss DS, Thornton JM. PROCHECK - a program to check the stereochemical quality of protein structures. J App Cryst. 1993;26:283–291. [Google Scholar]
- Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell death and differentiation. 2006;13(6):1027–1036. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- Malicet C, Giroux V, Vasseur S, Dagorn JC, Neira JL, Iovanna JL. Regulation of apoptosis by the p8/prothymosin alpha complex. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(8):2671–2676. doi: 10.1073/pnas.0508955103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo GV, Fiedler F, Calvo EL, Ortiz EM, Vasseur S, Keim V, Morisset J, Iovanna JL. Cloning and expression of the rat p8 cDNA, a new gene activated in pancreas during the acute phase of pancreatitis, pancreatic development, and regeneration, and which promotes cellular growth. The Journal of biological chemistry. 1997;272(51):32360–32369. doi: 10.1074/jbc.272.51.32360. [DOI] [PubMed] [Google Scholar]
- Martinez J, Georgoff I, Martinez J, Levine AJ. Cellular localization and cell cycle regulation by a temperature-sensitive p53 protein. Genes & development. 1991;5(2):151–159. doi: 10.1101/gad.5.2.151. [DOI] [PubMed] [Google Scholar]
- Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell. 1990;62(4):671–680. doi: 10.1016/0092-8674(90)90113-s. [DOI] [PubMed] [Google Scholar]
- Nakai KHP. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci. 1999;24(1):34–36. doi: 10.1016/s0968-0004(98)01336-x. [DOI] [PubMed] [Google Scholar]
- Nehrt NL, Clark WT, Radivojac P, Hahn MW. Testing the ortholog conjecture with comparative functional genomic data from mammals. PLoS computational biology. 2011;7(6):e1002073. doi: 10.1371/journal.pcbi.1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier RM, Gout J, Vincent DF, Cano CE, Kaniewski B, Martel S, Rodriguez J, Fourel G, Valcourt U, Marie JC, Iovanna JL, Bartholin L. The human NUPR1/P8 gene is transcriptionally activated by transforming growth factor beta via the SMAD signalling pathway. The Biochemical journal. 2012;445(2):285–293. doi: 10.1042/BJ20120368. [DOI] [PubMed] [Google Scholar]
- Ree AH, Pacheco MM, Tvermyr M, Fodstad O, Brentani MM. Expression of a novel factor, com1, in early tumor progression of breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000;6(5):1778–1783. [PubMed] [Google Scholar]
- Sandi MJ, Hamidi T, Malicet C, Cano C, Loncle C, Pierres A, Dagorn JC, Iovanna JL. p8 expression controls pancreatic cancer cell migration, invasion, adhesion, and tumorigenesis. Journal of cellular physiology. 2011;226(12):3442–3451. doi: 10.1002/jcp.22702. [DOI] [PubMed] [Google Scholar]
- Schneider C, King RM, Philipson L. Genes specifically expressed at growth arrest of mammalian cells. Cell. 1988;54(6):787–793. doi: 10.1016/s0092-8674(88)91065-3. [DOI] [PubMed] [Google Scholar]
- Su SB, Motoo Y, Iovanna JL, Xie MJ, Mouri H, Ohtsubo K, Yamaguchi Y, Watanabe H, Okai T, Matsubara F, Sawabu N. Expression of p8 in human pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2001;7(2):309–313. [PubMed] [Google Scholar]
- Szychot E, Brodkiewicz A, Peregud-Pogorzelski J. Will therapies that target tumour suppressor genes be useful in cancer treatment? Advances in clinical and experimental medicine : official organ Wroclaw Medical University. 2013;22(6):861–864. [PubMed] [Google Scholar]
- Urcan E, Haertel U, Styllou M, Hickel R, Scherthan H, Reichl FX. Real-time xCELLigence impedance analysis of the cytotoxicity of dental composite components on human gingival fibroblasts. Dental materials : official publication of the Academy of Dental Materials. 2010;26(1):51–58. doi: 10.1016/j.dental.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Urrutia R, Velez G, Lin M, Lomberk G, Neira JL, Iovanna J. Evidence supporting the existence of a NUPR1-like family of helix-loop-helix chromatin proteins related to, yet distinct from, AT hook-containing HMG proteins. Journal of molecular modeling. 2014;20(8):2357. doi: 10.1007/s00894-014-2357-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur S, Hoffmeister A, Garcia S, Bagnis C, Dagorn JC, Iovanna JL. p8 is critical for tumour development induced by rasV12 mutated protein and E1A oncogene. EMBO reports. 2002;3(2):165–170. doi: 10.1093/embo-reports/kvf023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur S, Vidal Mallo G, Fiedler F, Bodeker H, Canepa E, Moreno S, Iovanna JL. Cloning and expression of the human p8, a nuclear protein with mitogenic activity. European journal of biochemistry / FEBS. 1999;259(3):670–675. doi: 10.1046/j.1432-1327.1999.00092.x. [DOI] [PubMed] [Google Scholar]
- Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nature reviews Drug discovery. 2008;7(12):979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes & development. 1993;7(7A):1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- Wu X, Levine AJ. p53 and E2F-1 cooperate to mediate apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(9):3602–3606. doi: 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a) More than 98% of the residues are found in the allowed regions (b). NUPR1L, display several regions within Nurp1L which the propensity to form three α helices. Helix 1 contains 15 residues and spans from 19 to 34. Helix 2 contains 9 residues and spans from 52 to 61 while Helix 3 contains 20 residues and spans from 67 to 87. (c) Topology diagram of the NUPR1L model.
Table S1a. Bonding pattern between NUPR1L and B-DNA.
Table S1a. Bonding pattern between Nurp1L and alpha-importin.
Table S2. (a) siRNA sequences used for silencing Nupr1 and Nupr1L mRNA. (b)Primer sequences used to clone and directed mutagenesis. (c) Primer sequences used to amplify human genes.