

Abstract
Human reticulocyte 12/15-Lipoxygenase (h12/15-LOX) is a lipid-oxidizing enzyme that can directly oxidize lipid membranes in the absence of a phospholipase, leading to a direct attack on organelles, such as the mitochondria. This cytotoxic activity of h12/15-LOX is up-regulated in neurons and endothelial cells after a stroke and thought to contribute to both neuronal cell death and blood-brain barrier leakage. The discovery of inhibitors that selectively target recombinant h12/15-LOX in vitro, as well as possessing activity against the murine ortholog ex vivo, could potentially support a novel therapeutic strategy for the treatment of stroke. Herein, we report a new family of inhibitors discovered in a High Throughput Screen (HTS) that are selective and potent against recombinant h12/15-LOX and cellular mouse 12/15-LOX (m12/15-LOX). MLS000099089 (compound 99089), the parent molecule, exhibits an IC50 potency of 3.4 ± 0.5 µM against h12/15-LOX in vitro and an ex vivo IC50 potency of approximately 10 µM in a mouse neuronal cell line, HT-22. Compound 99089 displays greater than 30-fold selectivity versus h5-LOX and COX-2, 15-fold versus h15-LOX-2 and 10-fold versus h12-LOX, when tested at 20 µM inhibitor concentration. Steady-state inhibition kinetics reveals that the mode of inhibition of 99089 against h12/15-LOX is that of a mixed inhibitor with a Kic of 1.0 ± 0.08 µM and a Kiu of 6.0 ± 3.3 µM. These data indicate that 99089 and related derivatives may serve as a starting point for the development of anti-stroke therapeutics due to their ability to selectively target h12/15-LOX in vitro and m12/15-LOX ex vivo.
Keywords: inhibitor, selective, high-throughput, lipoxygenase, human, murine
Graphical abstract

1.1 Introduction
Lipoxygenases are a family of non-heme iron containing enzymes that oxidize lipids, generating eicosanoids that are important mediators in inflammation and numerous diseases.1,2 The human genome contains six functional LOX genes (ALOX3, ALOX5, ALOX12, ALOX12B, ALOX15B, ALOX15) which encode for six distinct LOX-isoforms.3 The ALOX3 and ALOX12B genes encode for two different epidermis type LOX enzymes that are co-expressed in the skin, a hydroperoxide isomerase (eLOX-3) and a 12R-lipoxygenase (h12R-LOX), respectively.3 These two isoforms have been implicated in epidermal differentiation as well as epidermal water barrier function.3 The ALOX5 gene encodes for a 5-lipoxygenating enzyme (h5-LOX) found in leukocytes as well as macrophages and plays an extensive role in leukotriene biosynthesis.3 The ALOX12 gene encodes for a 12-lipoxygenating enzyme (h12S-LOX) that is highly expressed in blood platelets and implicated as a key player in regulating platelet function and thrombosis.3 The ALOX15B gene encodes for a 15-lipoxygenating enzyme (h15-LOX-2) found to be abundant in epithelial cells and linked to the pathogenesis of atherosclerosis.3 Human reticulocyte 12/15-LOX (h12/15-LOX and/or h15-LOX-1), encoded by the ALOX15 gene, preferentially reacts with free polyunsaturated fatty acids (PUFAs) but also can directly oxidize lipid membranes containing PUFAs.3,4,5 These oxidized lipids within the lipid bilayer lead to the formation of hydrophilic pores, which affect the barrier function of the membrane and thus lead to cellular dysfunction, particularly in stroke.6
Stroke kills approximately 130,000 Americans each year and is the fifth leading cause of death in the United States.7 Two types of stroke can occur in the brain, ischemic and hemorrhagic.7 Ischemic stroke makes up approximately 85% of all strokes and is associated with thrombosis (clots), while hemorrhagic stroke results in excessive bleeding due to the bursting or leaking of the blood vessels.7 There is currently only one FDA approved drug, tissue plasminogen activator (tPA). tPA is a protease that dissolves blood clots but, due to its propensity to initiate a hemorrhagic response, it is given to less than 5% of patients and therefore other therapeutic targets would be beneficial.8
A target strongly linked to stroke is h12/15-LOX. The activity of m12/15-LOX is increased in the mouse brain following ischemic induction and m12/15-LOX co-localizes with a marker for oxidized lipids, MDA2.4 In addition, a similar co-localization was detected in the brain of human stroke patients, as well as increased levels of apoptosis-inducing factor, AIF.4,6 Finally, animal studies by van Leyen et al. demonstrated a 40% reduction of infarct size in a m12/15-LOX knockout (K/O) mouse relative to wild-type (WT).9 These data strongly suggest that h12/15-LOX is involved in stroke and might be an attractive therapeutic target.
Discovery of potent and selective inhibitors that target h12/15-LOX for the treatment of human disorders has proved to be a challenge. Although many inhibitors have been reported targeting 12/15-LOX, most of them target the non-human species like soybean lipoxygenase, which lack clinical significance.10–12 Another factor is the lack of an X-ray crystal structure of h12/15-LOX and a bound inhibitor, which hampers rational inhibitor design. In addition, the murine ortholog of 12/15-LOX (m12/15-LOX), which is involved in mouse stroke, exhibits 12-lipoxygenating activity, while h12/15-LOX exhibits 15-lipoxygenating activity, which is reflected in a difference in their inhibitor profiles.13–15 This is problematic because mice are effective/inexpensive models for stroke, making an inhibitor that targets both h12/15-LOX and m12/15-LOX critical for developing a stroke therapeutic.
Recently, Pelcman et al. identified BLX-2477 (N-(2-chloro-4-fluorophenyl)triazole-4-carboxamide) (Figure 1), as a potential therapeutic due to its potency (IC50 = 99 nM) and selectivity for h12/15-LOX, but unfortunately it had adverse off-target effects in mini-pigs and did not exhibit potency against the dog or rat orthologs.16,17 Bristol-Myers Squibb synthesized a series of sulfonamide derivatives, which inhibited rabbit reticulocyte 12/15-LOX in vitro (IC50 = 19 nM) and were later tested in a Chinese hamster ovary (CHO) cell-based assay that over expressed human recombinant 12/15-LOX.18 However, these analogs exhibited poor pharmacokinetics in rodent models and were deemed not suitable for further evaluation.18 Parke-Davis/Warner-Lambert (Pfizer) discovered an indole-based inhibitor, PD-146176 (IC50 = 810 nM), which had in vivo activity against rabbit reticulocyte 12/15-LOX, for atherosclerosis.19 Eleftheriadis et. al. published a novel class of 6-benzyloxysalicylates inhibitors that selectively target h15-LOX-1 (N206, IC50 = 7100 nM).20 Our collaborative team has discovered two different families of inhibitors, ML094 (4-(5-(Naphthalen-1-yl)-1,3,4-oxadiazol-2-ylthio)but-2-ynylthiophene-2-carboxylate) and ML351 (5-(methylamino)-2-(naphthalene-1-yl)oxazole-4-carbonitrile) (Figure 1). ML094 has low nanomolar potency against recombinant h12/15-LOX in vitro (IC50 = < 10 nM) but lacks activity in cell-based assays, possibly due to the hydrolyzable nature of the ester moiety.21 In comparison, ML351 has sub-micromolar potency (IC50 = 200 nM) and exhibits in vivo activity in mice.13 In the current work, we expand on our initial h12/15-LOX screen and present another unique chemical scaffold for h12/15-LOX inhibition. Utilizing HTS screening, cellular screening, SAR, and kinetic studies, we report a new family of inhibitors that are both selective and potent against h12/15-LOX, as well as being active in mouse neuronal cells.
Figure 1.


Relevant h12/15-LOX inhibitors.
1.2 Materials and Methods
1.2.1 Materials
All commercial fatty acids were purchased from Nu Chek Prep, Inc. (MN, USA). BWb70c was purchased from Sigma/Aldrich Chemicals. The inhibitors were obtained from the NIH Molecular Libraries Small Molecule Repository (MLSMR): (https://mli.nih.gov/mli/compound-repository/). All other chemicals were reagent grade or better and were used without further purification.
1.2.2 Protein Expression
All the LOX isozymes used in this publication were expressed and purified as previously published (h5-LOX22, h12-LOX23, h12/15-LOX23 and h15-LOX-224). Human leukocyte 5-lipoxygenase was expressed as a non-tagged protein and used as a crude ammonium sulfate precipitated protein. The remaining enzymes: h12/15-LOX, h15-LOX-2 and h12-LOX were expressed as N-terminal His6-tagged proteins and purified via immobilized metal affinity chromatography (IMAC) using Ni-NTA resin. The purity of each protein was analyzed by SDS-PAGE and found to be greater than 90% purity, except for h5-LOX. The iron content of each protein, except for h5-LOX, were determined with a Thermo Element XR inductively coupled plasma mass spectrometer (ICP-MS), using cobalt (EDTA) as an internal standard. Iron concentrations were compared to standard iron solutions. The protein concentrations were determined using the Bradford assay, with bovine serum albumin (BSA) as the protein standard. The iron loading was found to range from 20 – 50% for all proteins except h5-LOX.
1.2.3 Lipoxygenase UV-Vis-based IC50 Assay
One-point inhibition percentages of approximately 100 derivatives of MLS000099089 (99089), the parent compound, were determined by following the formation of the conjugated diene product at 234 nm (ε = 27,000 M−1cm−1) with a Perkin-Elmer Lambda 40 UV/Vis spectrophotometer at 20 µM inhibitor concentration. Twenty-nine selective derivatives were investigated further to determine their IC50 values. The full IC50 experiments were done with at least four different inhibitor concentrations. All reactions mixtures were 2 mL in volume and constantly stirred using a magnetic stir bar at room temperature (23°C) with the appropriate amount of LOX isozyme h12/15-LOX (~ 30 nM). Reactions were carried out in 25 mM HEPES buffer (pH 7.5), 0.01% Triton X-100 and 10 µM AA. The concentration of AA was quantitated by allowing the enzymatic reaction to proceed to completion in the presence of s12/15-LOX. IC50 values were obtained by determining the enzymatic rate at various inhibitor concentrations and plotted against inhibitor concentration, followed by a hyperbolic saturation curve fit. The data used for the saturation curves were performed in duplicate or triplicate, depending on the quality of the data. All inhibitors were stored at −20°C in DMSO.
1.2.4 HT-22 Cellular Assay
Oxidative stress leading to 12/15-LOX dependent cell death was induced in HT-22 cells by glutamate treatment as described.13,25,26 Briefly, HT-22 cells were cultured in DMEM containing 10% fetal bovine serum and penicillin / streptomycin (all media from Invitrogen). For initial screening of compounds, cells were seeded at 1 × 104 cells/well in 96-well plates (Corning) and treated 18h later, when the cells were approximately 50–70% confluent. Treatment consisted of exchanging the medium to 100 µL fresh culturing medium and adding 5 mM glutamate (stock solution 0.5 M in PBS) in the presence or absence of DMSO (maximum 0.1% final concentration) as control or 5, 10, or 20 µM concentration of the indicated compound. Titration of 99089 over a broader concentration range was then carried out in the same way in 24-well plates, with the cells seeded at 5 × 104 cells/well. In all cases, lactate dehydrogenase (LDH) content was determined separately for the cell extracts and corresponding media using a Cytotoxicity Detection Kit (Roche), and the percentage of LDH released to the medium calculated after subtracting the corresponding background value. The Z' values for the 96-well plate format are typically in the range of 0.6–0.9, indicating an excellent assay.27
1.2.5 Selectivity Assay
99089 was screened to determine its selectivity towards h12/15-LOX. Reactions with h12-LOX were carried out in 25 mM HEPES (pH 8.0) 0.01% Triton X-100 and 10 µM AA. Reactions with the crude, ammonium sulfate precipitated h5-LOX were carried out in 25 mM HEPES (pH 7.3), 0.3 mM CaCl2, 0.1 mM EDTA, 0.2 mM ATP, 0.01% Triton X100 and 10 µM AA. Reactions with h15-LOX-2 were carried out in 25 mM HEPES buffer (pH 7.5), 0.01% Triton X-100 and 10 µM AA. Human 12-LOX and h15-LOX-2 were tested with 20 µM, 25 µM, and 35 µM inhibitor concentration and all reactions were performed in duplicate. Concentrations above 35 µM for 99089 were not possible due to solubility problems with the inhibitor. Human 5-LOX and COX-2 was screened only at 20 µM inhibitor concentration of 99089, due to their low inhibition. Zileuton (20 µM) was utilized as a positive control with h5-LOX and shown to produce 97% inhibition.
1.2.6 Steady-State Inhibition Kinetics
The steady-state equilibrium constants of inhibition for 99089 and h12/15-LOX rates were determined the same way as the UV-Vis-Based Assay (Section 1.2.5). Reactions were initiated by adding h12/15-LOX to a constantly stirring 2 mL reaction mixture containing 2 µM – 14 µM AA in 25 mM HEPES buffer (pH 7.5), in the presence of 0.01% Triton X-100. Kinetic data were obtained by recording initial enzymatic rates, at varied inhibitor concentrations, and subsequently fitted to the Henri-Michaelis-Menten equation, using KaleidaGraph (Synergy) to determine the microscopic rate constants, Vmax (µmol/min/mg) and Vmax/Km (µmol/min/mg/µM). These rate constants were subsequently replotted as 1/Vmax or Km/Vmax versus inhibitor concentration, to yield Kiu and Kic, respectively, which are defined as the equilibrium constant of dissociation from the secondary and catalytic sites. The primary data were also plotted in the Dixon format, graphing 1/v vs. [I] µM at the chosen substrate concentrations. From the Dixon plots, the slope at each substrate concentration was extracted and plotted against 1/[S] µM to produce the Dixon parameters, Kic and Kiu.
1.2.7 Pseudoperoxidase Assay
The pseudo-peroxidase activity of 99089 was determined with h12/15-LOX enzyme utilizing 13-HpODE as the oxidant and BWb70c as the positive control on a Perkin-Elmer Lambda 40 UV/Vis spectrophotometer, as described previously published.28 The reaction was initiated by addition of 20 µM 13-HpODE to 2 mL buffer (50 mM Sodium Phosphate (pH 7.4), 0.3 mM CaCl2, 0.1 mM EDTA, 0.01% Triton X-100) containing 20 µM 99089 and 60 nM h12/15-LOX. The reactions were constantly mixed with a stir bar at 23 °C. Activity was determined by monitoring the decrease at 234 nm (13-HpODE consumption) and the percent consumption of 13-HpODE was recorded. More than 25% 13-HpODE degradation indicates redox activity of that particular inhibitor. The negative controls used were; enzyme alone with product, enzyme alone with inhibitor, and inhibitor alone with product. These formed a baseline for the assay, reflecting non-pseudo-peroxidase dependent hydroperoxide product decomposition. To rule out the auto-inactivation of the enzyme from pseudo-peroxidase cycling, the h12/15-LOX residual activity was observed after the addition of 20 µM AA at the end of each reaction. In addition, initial rates of inhibitor and 13-HpODE were compared to initial rates of inhibitor alone because the inhibitor by itself inherently lowers the rate of oxygenation. Activity is characterized by direct measurement of the product formation with the increase of absorbance at 234 nm.
1.2.8 Computational Modeling
A homology model of human reticulocyte 12/15-lipoxygenase protein (Uniprot ID P16050) was built using the software PRIME Version 3.9 (Schrodinger Inc)29 from the rabbit reticulocyte 15-Lipoxygenase-1 crystal structure (PDB ID 2p0m, chain B). The two LOX isozymes have 90% similarity and 81% identity between the two. Both the co-crystallized ligand and the metal ion were retained during the homology modeling. After the model was built, it was subjected to a protein preparation step using Protein Preparation Wizard (Schrodinger Inc). During this step hydrogen atoms were added, proper bond-orders and atom-types were set and the protein structure was minimized such that heavy-atoms were not allowed to move beyond 0.3Å. Iron was treated as ferric ion (Fe3+). The inhibitor, 99089, structure was built using Maestro’s Edit/Build panel. We minimized the structure using LigPrep software (Schrodinger Inc), and enumerated plausible protonation states of the inhibitor by applying the empirical pKa prediction software Epik (Schrodinger Inc). We docked the inhibitor to h12/15-LOX active site using Glide software with the standard-precision docking scoring function (Schrodinger Inc).
1.3 Results and Discussion
1.3.1 HTS Discovery
A HTS of approximately 74,000 compounds was performed with h12/15-LOX, as previously reported,21 and the top 1000 compounds were manually screened using the UV-Vis assay with h12/15-LOX. All molecules were screened at 20 µM inhibitor concentration, with 99089 inhibiting h12/15-LOX greater than 90% at this concentration. A full IC50 was performed on h12/15-LOX with five different 99089 concentrations and an IC50 of 3.4 ± 0.5 µM was determined (Figure 2).
Figure 2.

Structure and potency of the novel h12/15-LOX inhibitor (error in parentheses).
1.3.2 HT-22 Cellular Assay
As mentioned above, in order for an h12/15-LOX inhibitor to be developed into a therapeutic, it needs to be effective in the mouse stroke model. Therefore, we have modified our workflow such that, as soon as a potent h12/15-LOX inhibitor is discovered, it is tested against m12/15-LOX in a mouse neuronal cell line, HT-22, with 10 µM ML351 used as reference standard. In this cell line, glutathione depletion is induced by adding exogenous glutamate. This leads to oxidative stress and subsequent cell death, termed oxytosis or oxidative glutamate toxicity, which is dependent on 12/15-LOX activity and thus reversed by inhibition of 12/15-LOX.13,25 In the current work, we screened 99089 against HT-22 and found it to be potent, with an approximate cellular EC50 of 10 µM (Figure 3).
Figure 3.

Titration of 99089 in HT-22 cells, with increasing cell survival. ML351 was used as a positive control at 10 µM.
1.3.3 Compound Selectivity
After determining that 99089 was active against both h12/15-LOX and m12/15-LOX, we sought to establish whether our new inhibitor scaffold is selective to h12/15-LOX over other human LOX isozymes. Therefore, 99089 was screened against human lipoxygenases for its selectivity towards h12/15-LOX, utilizing the UV-Vis assay (Figure 4). 99089 exhibited approximately 15-fold selectivity over h12-LOX and h15-LOX-2 and greater than 30-fold selectivity over h5-LOX. The potency of 99089 was also investigated against COX-2, with no inhibitor potency being observed (extrapolated IC50 > 100 µM).
Figure 4.

Selectivity potency (µM) against h12-LOX, h15-LOX-2 and 5-LOX with 99089. Three inhibitor concentrations (20 µM, 25 µM, 35 µM) were screened against h12-LOX and h15-LOX-2, and the IC50 value averaged. For h5-LOX and COX-2 we only screened at 20 µM, due to their low potency and the solubility problems of 99089 above 35 µM.
1.3.4 Structure-Activity Relationship (SAR) Study
After the identification of the parent analogue, 28 derivatives were screened to investigate structure-activity relationships (Table 1–3). The compounds were subjected to IC50 determination, using 4 or more inhibitor concentrations (ranging from 0.5 µM to 30 µM), depending on the potency of the molecule.
Table 1.
IC50 values of h12/15-LOX inhibitors selected for the structure-activity relationship study, errors are in parentheses when available. Experiments were conducted in the presence of 10 µM AA and 0.01% TritonX-100. Each experiment was performed in duplicate with at least four different inhibitor concentrations.
| Compound | Structure | %Inhibition (20 µM) |
IC50 (±SD) (µM) |
HT-22 Survival % (5 µM) |
|---|---|---|---|---|
| 99089 | 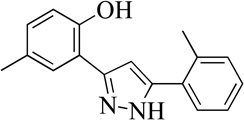 |
94% | 3.4 (0.5) | 20 |
| 1MLS000068545 | 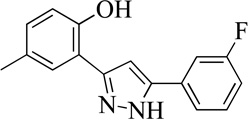 |
80 | 10 (1) | 49 |
| 2MLS000856489 | 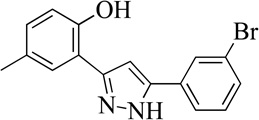 |
75 | 9.1 (2) | 18 |
| 3MLS000551826 | 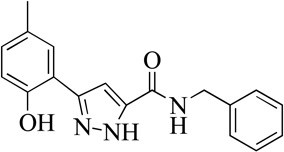 |
0% | >50 | |
| 4MLS0001163892 | 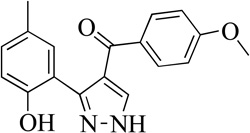 |
15% | >50 | |
| 5MLS000066802 | 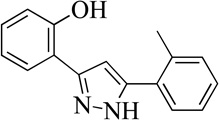 |
92 | 7.0 (0.4) | 76 |
| 6MLS000060847 | 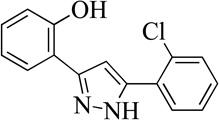 |
74 | 4.0 (0.4) | |
| 7MLS000551030 | 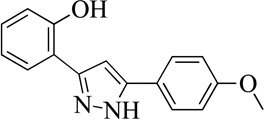 |
75 | 10 (1) | 7 |
| 8MLS000122998 | 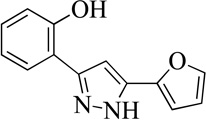 |
66 | 7.0 (2) | 34 |
| 9MLS002473404 | 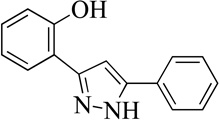 |
59 | 20 (4) |
Table 3.
IC50 values of h12/15-LOX inhibitors selected for the structure-activity relationship study, errors are in parentheses when available. Experiments were conducted in the presence of 10 µM AA and 0.01% TritonX-100. Each experiment was performed in duplicate with at least four different inhibitor concentrations.
| Compound | Structure | %Inhibition (20 µM) |
IC50 (±SD) (µM) |
HT-22 Survival % (5 µM) |
|---|---|---|---|---|
| 99089 | 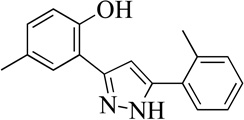 |
94% | 3.4 (0.5) | 20 |
| 20MLS0001194968 | 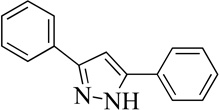 |
14% | >50 | |
| 21MLS0002473395 |  |
4% | >50 | |
| 22MLS000035498 | 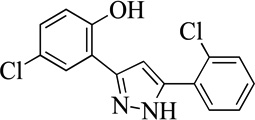 |
29 | > 50 | |
| 23MLS000547918 | 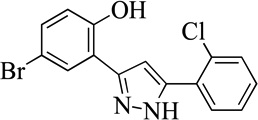 |
32% | >50 | |
| 24MLS000547918 | 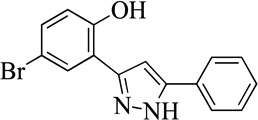 |
35% | >50 |
Structurally the parent molecule, 99089 contains a pyrazole ring, with phenyl substituents at C-3 and C-5. The 3-position of the pyrazole is substituted with a 2-OH, 5-Me-phenyl group, while the 5-position is substituted with a 2-Me-phenyl group (Figure 2). As observed in Table 1, modification of the 5-position with either a 3-F-phenyl or 3-Br-phenyl had little effect on the observed inhibition (1 and 2, respectively). However, replacing the 5-phenyl group with an N-benzylcarboxamide (3) or a 4-methoxyphenylmethanone (4) lowered activity over 10-fold, suggesting larger substituents on this side of the molecule are not well tolerated. It was also observed that removing the 5-Me on the 3-phenyl group of the parent molecule (5) had little effect on its potency. Subsequent changes to the 5-phenyl group of compound 5 had minor effects on potency, such as replacement of the 2-Me with a 2-Cl (6), or a 4-OMe (7), or complete replacement of the 5-phenyl moiety with a furan (8). Interestingly, removal of the 2-Me decreased the potency approximately 6-fold (9). Replacement of the 5-Me on the other phenyl group (C-3) with a 5-OMe also had minimal effect on potency (10–13) as shown in Table 2. Moreover, repositioning of the 5-Me on the 3-phenyl to the 4-position (14), also had no effect. The additional change of the 2-Me on the 5-phenyl group to a halogen had varied effects (15–17), but in general did not affect potency dramatically. However, converting the 2-Me to a 4-Me (18) lowered the potency greater than 15-fold, suggesting that adding steric bulk on opposite ends of the molecule possibly induces steric clashes with active site residues. This hypothesis is supported by the lack of potency of 19, which has a 4-OMe on the 3-phenyl group and a 4-Me on the 5-phenyl. As seen in Table 3, the most sensitive moiety of the parent molecule was the 2-OH on the 3-phenyl group, whose removal abolishes all potency (20), as compared to compound 9. Potency was not regained if the 2-OH group was restored to a minimal structure (21). These data suggest that the 2-OH moiety binds directly to the active-site iron. This is a reasonable hypothesis since phenols are known to be strong ligands to ferric ions. However, the fact that compound 21 did not inhibit h12/15-LOX indicates that there are other structural interactions that contribute to inhibitor potency, besides metal ligation. This hypothesis of metal ligation is supported by the fact that electron withdrawing groups opposite the 2-OH group eliminated all potency (22, 23 & 24). The electron withdrawing substituents lower the phenolate’s ability to bind the ferric iron because the electrons are not readily available to ligate the metal. It should be noted that although we propose that these inhibitors ligate the iron center, preventing the catalytically essential ferric-hydroxyl moiety being formed, these inhibitors do not exhibit redox activity (vide infra), indicating a mismatch in their reduction potentials. In summary, the collective data indicates that bulky substituents on both the phenyl substituents of the pyrazole can have deleterious effects, as does lowering the ability of the molecule to ligate the active site iron.
Table 2.
IC50 values of h12/15-LOX inhibitors selected for the structure-activity relationship study, errors are in parentheses when available. Experiments were conducted in the presence of 10 µM AA and 0.01% TritonX-100. Each experiment was performed in duplicate with at least four different inhibitor concentrations.
| Compound | Structure | %Inhibition (20 µM) |
IC50 (±SD) (µM) |
HT-22 Survival % (5 µM) |
|---|---|---|---|---|
| 99089 | 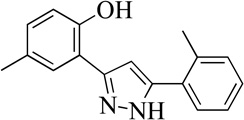 |
94% | 3.4 (0.5) | 20 |
| 10MLS000068611 | 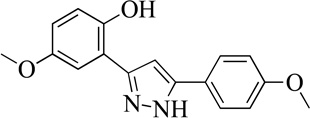 |
82 | 6.0 (0.5) | 96 |
| 11MLS000108293 | 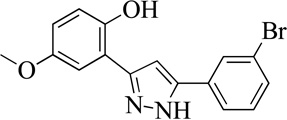 |
78 | 7.0 (1) | 70 |
| 12MLS001204578 | 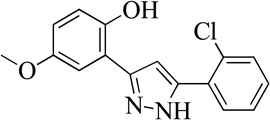 |
82 | 6.6 (0.4) | 83 |
| 13MLS001163964 | 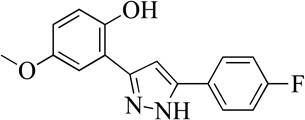 |
74 | 11 (2) | 78 |
| 14MLS000547497 | 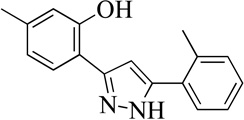 |
66 | 10 (1) | 71 |
| 15MLS000106958 | 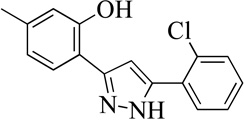 |
79 | 17 (2) | 77 |
| 16MLS001207693 | 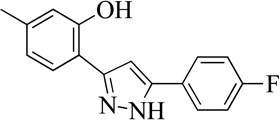 |
69 | 11 (1) | 25 |
| 17MLS000066801 | 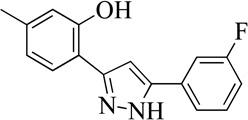 |
65 | 18 (2) | 66 |
| 18MLS001209799 | 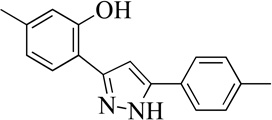 |
32 | > 50 | |
| 19MLS0001197234 | 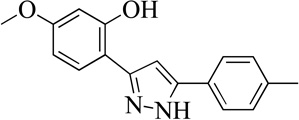 |
24% | >50 |
As mentioned above, the mouse stroke model is essential for developing a human stroke therapeutic; however, the mouse analogue to the human LOX (m12/15-LOX) preferentially generates 12-HpETE, suggesting a difference in their active sites. This hypothesis is supported by our HT-22 mouse neuronal cell assay, which models the stress induced by stroke. Effective 12/15-LOX inhibitors increase the survival percentage of the HT-22 cells by lowering the activity of the enzyme. As seen in Tables 1–3, there is a poor correlation between h12/15-LOX and HT-22 potency, indicating a difference in the active sites of h12/15-LOX and m12/15-LOX. For example, compounds 7 and 10 have comparable potency against h12/15-LOX, but exhibit dramatically different potency against m12/15-LOX. It should be noted that the HT-22 assay is a cellular assay and thus other factors could be at play, such as cell permeability, which was not investigated in the current work.
1.3.5 Steady-State Inhibition Kinetics
The mode of inhibition by which 99089 inhibits h12/15-LOX was investigated utilizing steady-state inhibition kinetics. The formation of hydroperoxide product was monitored in a UV-Vis assay as a function of substrate and inhibitor concentration in the presence of 0.01% Triton-X-100. The reaction was performed with substrate concentration ranging from 2 µM to 14 µM and three inhibitor concentrations of 2.5 µM, 5 µM and 10 µM. A Dixon plot of the primary data for 99089 is shown in Figure 5A, while a Dixon replot of the secondary data is shown in Figure 5B. Fitting the data yielded a Kic of 1.0 ± 0.08 µM and a Kiu of 6.0 ± 3.3 µM, which are defined as the equilibrium constants of dissociation from the enzyme and enzyme substrate complex, respectively. These numbers are slightly lower than the IC50 values and are indicative of a mixed type inhibitor, which is a common trait among LOX inhibitors.
Figure 5.

Steady-state inhibition kinetics for the determination of Kic and Kiu of h12/15-LOX and 99089. (A) Dixon plot of the primary data of h12/15-LOX and 99089. The substrate concentrations are 2 µM (open triangles), 4 µM (closed squares), 9 µM (open circles), 15 µM (closed diamonds). (B) The Dixon replot of slope versus [Inhibitor] in µM yielded a Kic of 1.0 (0.08) µM and a Kiu of 6.0 (3) µM.
1.3.6 Pseudoperoxidase Activity Assay
Some LOX inhibitors in the literature inactivate these enzymes by reduction of the iron center.24,30–32 Because redox inhibitors can cause off-target reactions in the cell, the mechanism of action of a particular inhibitor is important.32,33 A UV-Vis pseudoperoxidase assay was utilized to determine the mechanism of inhibition of 99089.28 In the pseudoperoxidase assay, the only fatty acid present is 13-HpODE, which oxidizes the inactive Fe2+ enzyme to an active Fe3+ enzyme. If the inhibitor (99089) were a redox inhibitor, it would reduce the ferric iron to a ferrous iron and lead to the consumption of more 13-HpODE. The consumption of 13-HpODE would be seen in the degradation of the 234 nm signal on the UV-Vis spectrophotometer. The degradation of the hydroperoxide product, 13-HpODE, was not observed, demonstrating that 99089 does not inhibit h12/15-LOX via a redox mechanism.
1.3.7 Computational Modeling
99089 was docked to a homology model of h12/15-LOX model, and the standard-precision Glide score for the docked pose is −7.77 kcal/mol (Figure 6). In this pose, the inhibitor directly interacts with the metal ion and hydrogen bonds with the side chain of Glu356. The predicted pose in which the phenolate anion directly interacts with the metal ion may explain why removing the 2-OH group of 3-phenyl ring (as in 20) abolishes all potency. In addition, the ortho-methyl 5-phenyl ring nicely packs into the hydrophobic pocket created by Phe352, Phe414 and Met418 and appears to have a π-π stacking interaction with Phe414. The close proximity (3.5–4.5Å) of the 5-phenyl ring to these hydrophobic residues may further explain why larger substitution to the 5-phenyl ring is not well tolerated. Thus, the docking results are consistent with the key aspects of the SAR.
Figure 6.

99089 docking pose in the active site of h12/15-LOX. Carbon atoms of the protein and ligand are shown in grey and green, respectively. Nitrogen, oxygen, hydrogen atoms are blue, red and white, respectively. The ferric ion is shown as an orange sphere. 99089 is shown in ball-and-stick representation.
1.4 Conclusion
In summary, we have identified a novel chemical scaffold that selectively inhibits h12/15-LOX versus other human LOX isozymes, but still inhibits m12/15-LOX. This property is critical for development of an anti-stroke therapeutic which will require the use of a mouse animal model. The SAR studies indicate that this class of inhibitors ligate the metal center and are constrained by sterics on either end of the molecule. We are currently investigating its potency and efficacy in our MCAO mouse stroke model.
Acknowledgments
This work was supported by the NIH NS081180.
Abbreviations
- LOX
lipoxygenase
- hLOX
human lipoxygenase
- h15-LOX-2
human epithelial 15-lipoxygenase-2
- h12/15-LOX, h15-LOX-1
human reticulocyte 15-lipoxygenase-1
- h12-LOX
human platelet 12-lipoxygenase
- s15-LOX-1
soybean 15-lipoxygenase-1
- h5-LOX
human leukocyte 5-lipoxygenase
- AA
arachidonic acid
- LA
linoleic acid
- 12-HETE
12-hydroxy-5,8,10-14-eicosatetraenoic acid
- 12-HpETE
12-hydroperoxyeicosatetraenoic acid
- 15-HpETE
15-hydroperoxyeicosatetraenoic acid
- 13-HpODE
13-(S)-hydroperoxyoctadecadienoic acid
- tPA
tissue plasminogen activator
- AIF
apoptosis-inducing factor
- HTS
High Throughput Screen
- CHO
Chinese hamster ovary
- MLSMR
Molecular Libraries Small Molecule Repository
- 99089
MLS000099089
- NIH
National Institutes of Health
- ICP-MS
Inductively Coupled Plasma Mass Spectrometer
- BSA
Bovine Serum Albumin
- EDTA
Ethylenediaminetetraacetic acid
- DMSO
Dimethyl Sulfoxide
- SAR
Structure-Activity Relationship
- MCAO
Middle Cerebral Artery Occlusion
- SDS-PAGE
Sodium Dodecyl Sulphate-Polyacrylamide Gel Electrophoresis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brash AR. J. Biol. Chem. 1999;274:23679. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- 2.Funk CD. Arterioscle. Thromb. Vasc. Biol. 2006;26:1204. doi: 10.1161/01.ATV.0000222960.43792.ff. [DOI] [PubMed] [Google Scholar]
- 3.Kuhn H, et al. Biochim. Biophys. Acta. 2014;4C:1. [Google Scholar]
- 4.van Leyen K. CNS Neurol. Disord. Drug Targets. 2013;12:191. doi: 10.2174/18715273112119990053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Leyen K, Holman TR, Maloney DJ. Future Med. Chem. 2014;6:1853. doi: 10.4155/fmc.14.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhn H, Banthiya S, van Leyen K. Biochim. Biophys. Acta. 2015;1851:308. doi: 10.1016/j.bbalip.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mozaffarian D, Benjamin EJ, Go AS, et al. Circulation. 2015:e29. [Google Scholar]
- 8.Marler JR. N. Engl. J. Med. 1995;333:1581. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 9.van Leyen K, Kim HY, Lee SR, et al. Stroke. 2006;37:3014. doi: 10.1161/01.STR.0000249004.25444.a5. [DOI] [PubMed] [Google Scholar]
- 10.Mahdavi M, Taherkhani R, Saeedi M, Alipour E, Moradi A, Nadri H, Emami S, Firoozpour L, Shafiee A, Foroumadi A. Eur. J. Med. Chem. 2014;82:308. doi: 10.1016/j.ejmech.2014.05.054. [DOI] [PubMed] [Google Scholar]
- 11.Tehrani MB, Emami S, Asadi M, Saeedi M, Mirzahekmati M, Ebrahimi SM, Mahdavi M, Nadri H, Moradi A, Moghadam FH, Farzipour S, Vosooghi M, Foroumadi A, Shafiee A. Eur. J. Med. Chem. 2014;87:759. doi: 10.1016/j.ejmech.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 12.Iranshahi M, Jabbari A, Orafaie A, Mehri R, Zeraatkar S, Ahmadi T, Alimardani M, Sadeghian H. Eur. J. Med. Chem. 2012;57:134. doi: 10.1016/j.ejmech.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 13.Rai G, Joshi N, Perry S, Yasgar A, Schultz L, Jung JE, Liu Y, Terasaki Y, Diaz G, Kenyon V, Jadhav A, Simeonov A, van Leyen K, Holman TR, Maloney DJ. Probe Reports from the NIH Molecular Libraries Program. 2010 [Google Scholar]
- 14.Rai G, Joshi N, Jung EJ, Liu Y, Schultz L, Yasgar A, Perry S, Diaz G, Zhang Q, Kenyon V, Jadhav A, Simeonov A, Lo EH, van Leyen K, Maloney DJ, Holman TR. J. Med. Chem. 2014;57:4035. doi: 10.1021/jm401915r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haeggstrom JZ, Funk CD. Chem. Rev. 2011;111:5866. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 16.Pelcman B, Sanin A, Nilsson P, No K, Schaal W, Ohrman S, et al. Bioorg. Med. Chem. Lett. 2015;25:3024. doi: 10.1016/j.bmcl.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Pelcman B, Sanin A, Nilsson P, Schaal W, Olofsson K, et al. Bioorg. Med. Chem. Lett. 2015;25:3017. doi: 10.1016/j.bmcl.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Weinstein DS, Liu W, Gu Z, Langevine C, Ngu K, et al. Bioorg. Med. Chem. Lett. 2005;15:1435. doi: 10.1016/j.bmcl.2004.12.081. [DOI] [PubMed] [Google Scholar]
- 19.Nair DG, Funk CD. Prostaglandins Other Lipid Mediat. 2009;90:98. doi: 10.1016/j.prostaglandins.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Eleftheriadis N, et al. Eur. J. Med. Chem. 2015;94:265. doi: 10.1016/j.ejmech.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 21.Rai G, Kenyon V, Jadhav A, Schultz L, Armstrong M, Jameson B, Leister W, Simeonov A, Holman TR, Maloney DJ. J. Med. Chem. 2010;53:7392. doi: 10.1021/jm1008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson SJ, Riener M, Loveridge ST, Tenney K, Valeriote FA, Holman TR, Crews P. J. Nat. Prod. 2009;72:1857. doi: 10.1021/np900465e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amagata T, Whitman S, Johnson TA, Stessman CC, Loo CP, Lobkovsky E, Clardy J, Crews P, Holman TR. J. Nat. Prod. 2003;66:230. doi: 10.1021/np020462l. [DOI] [PubMed] [Google Scholar]
- 24.Vasquez-Martinez Y, Kenyon V, Holman TR, Sepulveda-Boza S. Bioorg. Med. Chem. 2007;15:7408. doi: 10.1016/j.bmc.2007.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Maher P, Schubert D. Neuron. 1997;19:453. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 26.van Leyen K, Arai K, Jin G, Kenyon V, Gerstner B, Rosenberg PA, Holman TR, Lo EH. J. Neurosci. Res. 2008;86:904. doi: 10.1002/jnr.21543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang JH, Chung TD, Oldenburg KR. J. Biomol. Screen. 1999;4:67. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 28.Hoobler EK, Holz C, Holman TR. Bioorg. Med. Chem. 2013;21:3894. doi: 10.1016/j.bmc.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prime, version 3.9., Glide, version 6.6, LigPrep, version 3.3., Maestro, version 10.1. New York, NY: Schrodinger, LLC; 2015. [Google Scholar]
- 30.Carter GW, Young PR, Albert DH, Bouska J, Dyer R, Bell RL, Summers JB, Brooks DW. J. Pharmacol. Exp. Ther. 1991;256:929. [PubMed] [Google Scholar]
- 31.Cichewicz RH, Kenyon VA, Whitman S, Morales NM, Arguello JF, Holman TR, Crews P. J. Am. Chem. Soc. 2004;126:14910. doi: 10.1021/ja046082z. [DOI] [PubMed] [Google Scholar]
- 32.Pergola C, Werz O. Expert Opin. Ther. Pat. 2010;20:355. doi: 10.1517/13543771003602012. [DOI] [PubMed] [Google Scholar]
- 33.McMillan RM, Walker ER. Trends Pharmacol. Sci. 1992;13:323. doi: 10.1016/0165-6147(92)90100-k. [DOI] [PubMed] [Google Scholar]