

Abstract
Systemic lupus erythematosus (SLE) occurs nine times more often in females than males. The purpose of this study was to determine the impact of estrogen receptor (ER) null genotypes on disease in lupus prone NZM2410 (NZM) and MRL/lpr mice, as a method to define the role of estrogen receptor signaling in lupus. ERα deficient NZM females, but not males, had significantly prolonged survival, reduced proteinuria, renal pathology scores and serum urea nitrogen levels compared to wildtype mice, despite higher serum anti-dsDNA levels. ERα deficient MRL/lpr female, but not male, mice also had significantly less proteinuria and renal pathology scores with no effect on autoantibody levels. Deficiency of ERβ had no effect on disease in either strain or sex. Taken together, these data demonstrate a key role for ERα, but not ERβ, in the development of lupus like disease, but not autoimmunity, in female NZM and MRL/lpr mice.
Keywords: Lupus, Estrogen receptors, Renal disease, Autoimmunity
Introduction
Systemic lupus erythematosus is a prototypic autoimmune disease characterized by the production of autoantibodies, formation of immune complexes (IC) and subsequent IC deposition in target tissues with resultant local inflammation and organ damage [1]. A key unexplained epidemiologic observation in lupus is the profound 9/1 female/male prevalence in the reproductive years [1]. Female predominance is much less prevalent in prepubertal or postmenopausal ages (2–3/1 female/male prevalence) suggesting a role for sex hormones in modulating disease. Significant research effort, however, has not clearly identified a mechanism by which estrogen could modulate such a profound effect on disease. Despite the higher prevalence of lupus in females, disease severity, renal disease progression and mortality are higher in males affected by the disease. This dichotomy of gender effects on the prevalence vs. severity of lupus remains an unexplained, yet highly investigated aspect of lupus.
In experiments by Talal et al. and later by Tarkowski et al., prepubertal castration, with hormonal manipulation, led to significant effects on disease expression in both NZB/NZW and MRL/lpr mice [2–6]. The mechanisms underlying these effects are unclear as castration alone had no effect on disease in either NZB/NZW or MRL/lpr mice indicating that the lack of estrogen alone is insufficient to protect against disease. Administering high dose estrogen after castration worsened disease while giving testosterone to castrated female mice improved disease. Manipulation of sex hormones/castration post-onset of sexual maturity, however, had minimal effect on disease expression except in MRL/lpr mice where high dose testosterone given to female mice led to mild improvement in disease manifestations. Thus, although clearly castration and sex hormone manipulation impacts disease, the mechanisms underlying these effects are unclear. These results further suggest that a significant effect of female sex/hormones on disease susceptibility occurs via imprinting the immune system early in life as hormonal manipulation later in life has minimal impact on disease. We thus undertook these experiments using ER deficient mice to further define gender effects on disease expression in lupus prone mice.
Estrogen classically functions by activating one of its two nuclear receptors, ERα or ERβ [7]. ERα and ERβ are encoded by separate genes and differ in promoter sequences, indicating likely differences in function. In most tissues, ERα is the predominant receptor expressed. Both receptors are expressed in most immune cells, where again, ERα is the predominant receptor expressed. To define the differential biologic functions of the two estrogen receptors, knockout mice for both ERα and ERβ were developed [8]. The ERβ knockout is complete, while the ERα knockout expresses a small truncated ERα that lacks the ligand dependent activation domain (AF1), but retains the ligand independent activation domain (AF2). Only small amounts of this aberrant receptor are made by the ERα knockout mice. The phenotype of ERα deficient mice in numerous published studies indicates a lack of estrogen mediated effects despite the presence of the truncated receptor [9]. Surprisingly, despite the expression of ERs in almost all immune cells, immune parameters measured in ER deficient mice were similar to wildtype littermates [10]. Recently, however, ERα deficiency, on the B6/129 background, resulted in development of autoantibodies and lupus like immune complex glomerulonephritis in both male and female mice, while ERβ deficiency had no effect, suggesting that the lack of ERα signaling is proautoimmune [11].
The purpose of this study was to determine the impact of ER null genotypes on lupus prone NZM2410 and MRL/lpr mice compared to controls. Our hypothesis was that ERα deficiency would accelerate disease in lupus prone mice based on development of lupus like disease in B6/129 ERα deficient mice and the overall immunosuppressive effects of estrogen in most in vitro systems. We hypothesized that ERβ deficiency would either have no effect or protect against disease given its more limited expression.
ERα and ERβ knockout mice on the C57BL/6 background were backcrossed to lupus prone NZM2410 and MRL/lpr mice for 9 generations. Female NZM2410 mice, deficient in ERα, had prolonged survival and significant protection from renal disease compared to controls as measured by proteinuria, serum urea nitrogen, renal pathology and survival. This improvement in disease parameters occurred despite increased serum levels of anti-dsDNA antibodies, estrogen and testosterone. MRL/lpr female mice that were ERα deficient had similar protection against renal disease with no effect on autoantibody production. ERα deficiency in males had minimal to no effect on disease expression in either strain. ERβ deficiency had no significant impact on disease in either sex. Taken together, these data demonstrate a key role for the ERα AF1 domain in mediating renal disease in female NZM2410 and MRL/lpr mice while not impacting development of autoimmunity.
Materials and methods
Mice
All mice were maintained at the Ralph H. Johnson VAMC Animal Care Facility (Charleston, SC) and experiments were approved by the Institutional Animal Care and Use Committee. ERα and ERβ knockout mice on the C57BL/6 background were backcrossed to NZM2410 mice (Jackson Laboratory, Bar Harbor, ME) for 9 generations. Verification of NZM2410 background was done by identification of selected micro-satellite markers within susceptibility loci for NZM2410 mice including MHC and SLE 1, 2 and 3 loci [12]. We studied 20 mice in each group of wildtype, heterozygote and homozygote knockout mice with 10 mice of each gender. The groups of mice of each genotype were sacrificed at 32 weeks of age. The final sacrifice time was determined based on proteinuria and appearance of the mice to insure presence of active disease without reaching end stage disease. All ER knockout mice were compared to wildtype and heterozygote littermate mice as controls. Groups of MRL/lpr mice (Jackson Laboratory) were derived in a similar fashion sacrificing them at 24 weeks using similar proteinuria criteria.
Urine protein excretion
NZM 2410 mice were placed in metabolic cages for 24-hour urine collections at weeks 18, 22, 26, 28, 30, and 32 vs. 12, 16, 20 and 24 weeks for the MRL/lpr mice. To prevent bacterial growth, antibiotics (ampicillin, gentamicin and chloramphenicol) were added to the collection tubes. Urinary protein excretion was determined by mouse albumin ELISA using known standards.
Renal pathology
At the time of sacrifice (32 vs. 24 weeks depending on strain), the kidneys were removed. One kidney was fixed with buffered formalin, embedded in paraffin, and then sectioned before staining with hematoxylin and eosin. The slides were read and interpreted in a masked fashion grading the kidneys for glomerular hypercellularity, segmental mesangial expansion, neutrophils/cell debris, crescent formation and necrosis. These parameters were combined for a total glomerular pathology score with a maximum score of 16. Interstitial changes and vasculitis were also noted and graded separately. Scores from 0–4 were assigned to each of the features and added together to yield a final renal score.
Immunofluorescence staining
Kidney tissue was snap frozen in liquid nitrogen and stored at −80 °C until analysis. The deposition of IgG and complement component C3 was analyzed by immunofluorescence by incubating the slides with rabbit anti-mouse IgG FITC (Sigma) and sheep anti-mouse C3 FITC (Sigma). IgG and C3 deposition was graded 0–3 for intensity of staining in a masked fashion.
Measure of autoantibodies
Serum levels of anti-dsDNA, anti-glomerular antigens (GA), total IgG, and serum IgG isotypes were measured as previously described [13]. Isotype specific anti-dsDNA antibody ELISAs were performed using isotype specific conjugates (i.e. anti-mouse IgG1, IgG2a, IgG2b and IgG3). Standard curves using IgG isotypes were used to determine the dilution of the anti-IgG antibody to use to ensure equal detection of isotypes. To measure relative affinity of anti-dsDNA antibodies, we performed competitive inhibition ELISA assays. Sera from 28-week old mice were incubated with increasing concentrations of soluble dsDNA for 24 h at 4 °C prior to performing anti-dsDNA ELISAs. Soluble dsDNA concentrations ranged from 1 μg/ml–30 μg/ml. Anti-dsDNA ELISAs were then carried out as described above. Anti-GA antibodies, serum IgG iso-types and total IgG were determined as previously described [14].
Anti-glomerular Ab injections
Methods for this experiment were previously described [14]. Briefly, five wildtype and ERα knockout female NZM 2410 mice at 8–10 weeks of age (predisease) were used in this study. Nephritis was induced using a combination of 50 μg LPS (administered i.p.) and 250 μg of anti-glomerular sera (i.v.) given simultaneously to each mouse. Following this challenge, the mice were monitored for proteinuria over a 2-week period.
Measurement of serum hormones
Serum was collected from wildtype and ERα knockout female NZM 2410 mice at 32 weeks of age. Estrogen and testosterone were both measured via ELISA (Bio-Quant Inc.) according to the manufacturer’s instructions.
Statistical analysis
Two-way ANOVA was utilized to test for significance when comparing groups in proteinuria and autoantibody experiments. Mann–Whitney nonparametric t-tests were utilized to test for significance between groups in single group comparisons as in pathology and immunofluorescence scoring. Standard error of the means was reported where applicable. p values ≤0.05 were considered significant. Logrank analysis was used to compare trends in animal survival.
Results
Effect of ER deficiency on albuminuria in NZM2410 mice
NZM2410 mice of various ER genotypes were placed in metabolic cages and 24-hour urine samples were collected. Urine was analyzed by ELISA for albumin as a measure of renal disease. Wildtype female NZM2410 mice, as expected (Fig. 1A), developed severe albuminuria (>1 mg/mouse/day) by 26 weeks of age, while ERα heterozygote littermate females developed severe albuminuria at 30 weeks of age. ERα homozygote knockout NZM2410 females displayed significant protection from development of albuminuria with 24-hour albumin excretion levels never exceeding 100 μg/mouse/day out to 32 weeks of age. There was a statistically significant decrease in albuminuria in ERα knockout female NZM2410 mice compared to both heterozygote and wildtype ERα NZM2410 knockout mice. A significant decrease in albuminuria was also observed comparing female heterozygote to wildtype mice indicating an ERα gene dosing effect on albuminuria in NZM2410 female mice. ERα deficiency resulted in a trend towards decreased albuminuria in male NZM2410 mice that did not reach statistical significance (Fig. 1B).
Figure 1.

Proteinuria in ER deficient NZM2410 lupus mice. NZM females with differing ERα genotype were compared for excretion of urine albumin at the various ages indicated. (A) ANOVA analysis demonstrated a significant decrease in proteinuria in ERα deficient females compared to wildtype and heterozygote mice p<0.05 as indicated by the * and φ respectively. (B) The decreased proteinuria in ERα deficient males did not reach statistical significance overall. (C) There was a significant decrease in ERβ deficient females compared to normal mice only at 30 weeks of age. p<0.05 as indicated by the *. (D) No significant differences were noted in males regardless of ERβ genotype. (n=10 in all groups).
Similarly, NZM2410 mice of various ERβ genotypes were analyzed for development of albuminuria. Wildtype females began to develop severe albuminuria at 28 weeks (Fig. 1C) while ERβ heterozygote females developed severe albuminuria at 30–32 weeks. ERβ homozygote KO females developed severe albuminuria at 32 weeks of age, demonstrating a trend towards delayed development of albuminuria in the KO females that did not reach statistical significance. The male mice of each ERβ genotype developed mild proteinuria by 32 weeks of age (Fig. 1D). There was no significant difference between ERβ genotypes in albuminuria despite the trends noted. The decreased proteinuria in the male ERβ wildtype vs. male ERα wildtype groups emphasizes the need for using littermates for controls.
Effect of ER deficiency on renal pathology in NZM2410 mice
Kidneys were collected at 32 weeks of age and analyzed for glomerular pathology. Female NZM2410 mice, deficient in ERα, had significantly reduced pathology scores compared to wildtype NZM2410 female mice (p =0.041) and heterozygote ERα female mice (p = 0.032) as shown in Figure 2A. There was no significant difference in renal pathology scores between heterozygote and wildtype mice. This lack of a difference may reflect that the most severely affected wildtype mice had died prior to 32 weeks, thus falsely lowering the pathology scores in this group. ERα knockout mice showed a decrease in all renal pathology parameters scored indicating a global impact on renal disease (Table 1). In male NZM2410 mice, ERα homozygote and heterozygote knockout mice had trends toward lower renal pathology scores than wildtypes, but did not reach a significant difference (p =0.08 and 0.10 respectively) when compared to wildtype (Fig. 2B).
Figure 2.

Renal pathology in ER deficient NZM2410 lupus mice. Total glomerular pathology scores were determined for female NZM mice of various ERα genotypes. (A) ERα null females had significantly lower pathology score compared to both wildtype and ERα heterozygotes. p<0.05 by Mann–Whitney analysis. (B) Male littermates of various ERα status were also compared for glomerular scores with no significant difference. (C) ERβ null females had a trend towards lower glomerular scores but did not reach statistical significance. Littermate male NZM mice had no difference in pathology regardless of ERβ genotype (D).
Table 1.
Percentage of female NZM mice with renal pathologic parameters
| ERα | HC (3+) | PMNS | EPI REAC | MEM | MES | SCLER |
|---|---|---|---|---|---|---|
| (+/+) | 30% | 30% | 60% | 60% | 40% | 10% |
| (+/−) | 0 | 30% | 30% | 30% | 60% | 0 |
| (−/−) | 0 | 0 | 20% | 0 | 0 | 0 |
Kidneys from female NZM 2410 mice of various ERα genotypes were scored 0–3+ for hypercellularity (HC), neutrophils/cell debris (PMNS), epithelial reactivity (EPI REAC), membranous changes (MEM), segmental mesangial expansion (MES), and sclerosis (SCLER). Values represent a percentage of 10 mice scored for each group.
ERβ deficient and heterozygote females displayed a non-significant trend towards decreased glomerular pathology scores compared to wildtype NZM 2410 mice (Fig. 2C). There were no differences observed in renal scores of NZM 2410 male mice, regardless of ERβ status (Fig. 2D).
IgG and C3 kidney deposition in ER deficient NZM2410 mice
We next assessed renal deposition of complement component C3 and IgG by immunofluorescence. Surprisingly, despite the significant differences in pathology and albuminuria, there was no difference in total IgG deposition between the three study groups (Fig. 3A). When C3 deposition was assessed (Fig. 3B), there was a minimal decrease in the ERα deficient female mice compared to wildtype littermates that did not reach statistical significance. Male ERα KO mice displayed no difference in either IgG or C3 glomerular deposition compared to wildtype litter-mates (Figs. 3C and D). Figures 3E and F show the diffuse glomerular staining observed in wildtype and ERα KO females respectively. There were no differences in IgG or C3 deposition in ERβ deficient females or males compared to wildtype littermates.
Figure 3.

Renal immune complex deposition in ERα deficient NZM2410 mice. Female NZM2410 kidneys were analyzed for IgG deposition (A) and C3 deposition (B) in wildtype and ERα deficient mice by immunofluorescence. There was no difference regardless of ERα genotype. Male littermates were similarly examined and compared between wildtype (C) and ERα knockout (D). No statistical difference was observed regardless of ERα. IgG stainings of glomeruli from wildtype and ERα knockout females are shown respectively in (E) and (F).
Serum urea nitrogen levels in ERα deficient NZM2410 mice
We assessed SUN levels from the NZM2410 female wildtype and ERα KO littermates. At the age of 18 weeks, there was a trend towards lower SUN levels in ERα knockout females compared to wildtypes. At 32 weeks, SUN levels were significantly higher (24.2±2.26 mg/dl) in the wildtype female mice compared to 17.5±3.4 mg/dl in the knockout females (p<0.01). The male NZM2410 ERα KO mice showed no significant difference in SUN levels compared to their wildtype littermates (data not shown).
Effect of ER deficiency on survival of NZM2410 mice
Wildtype NZM2410 females began to die at 26 weeks of age with 40% dying by 32 weeks of age. Survival in the heterozygote and wildtype groups was 100% at 32 weeks of age (Fig. 4). Logrank analysis demonstrated a statistical difference in survival between wildtype and ERα knockout and heterozygote mice. There were no deaths of male mice of any ERα status by 32 weeks of age. There was also no difference in survival of NZM2410 mice regardless of ERβ genotype in either sex (data not shown).
Figure 4.
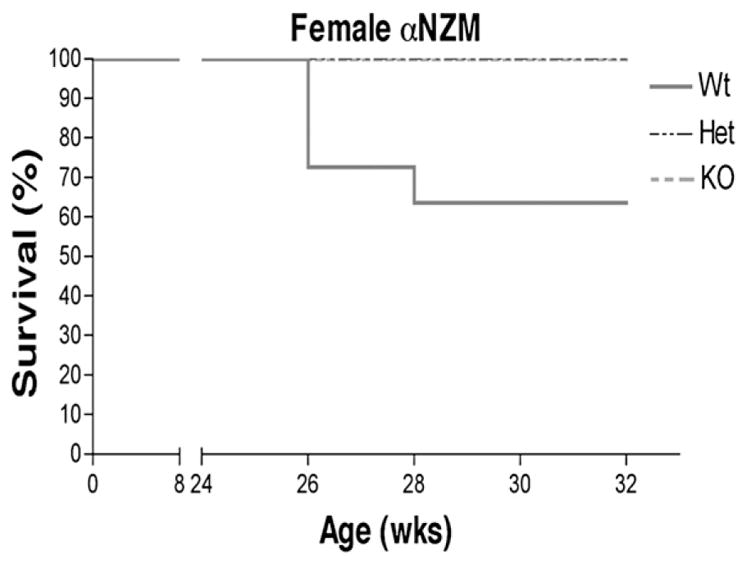
Survival of ER deficient NZM2410 lupus mice. ERα knockouts and heterozygotes had significantly prolonged survival by Logrank analysis compared to wildtypes (n=10).
Effect of ER deficiency on autoantibody levels in NZM2410 mice
The different groups of NZM2410 mice were assessed by ELISA for total serum IgG levels as they aged. We found no significant difference in serum IgG levels between groups or any difference in serum IgG isotypes (data not shown). Serum levels of anti-double stranded DNA (dsDNA) antibodies were measured by ELISA in the ERα study groups. In both sexes of NZM2410 mice, we observed an expected increase in serum anti-dsDNA antibodies as they aged (Figs. 5A and B). Surprisingly, the ERα null females had significantly higher serum levels of anti-dsDNA antibodies than wildtype mice from weeks 28 to 32 (p<0.05). The IgG isotypes of autoantibodies were measured as isotype differences could impact renal disease through differential complement fixation and FcR activation. Serum IgG1 and IgG3 anti-dsDNA antibodies were similar in the female NZM2410 mice regardless of genotype (data not shown). Serum IgG2a anti-dsDNA antibodies in the female NZM2410 mice, however, were significantly higher in ERα KOs compared to wildtype mice from 28 to 32 weeks of age (p<0.05). The difference in IgG2a anti-DNA antibodies between ERα KOs and wildtype female mice accounted for the majority of the total anti-dsDNA difference observed in Figure 5A. Heterozygote ERα KO mice were intermediate in their anti-dsDNA and IgG2a anti-dsDNA antibody production. ERα status did not impact serum anti-dsDNA antibody levels in male mice. ERβ deficient mice showed no difference in serum anti-dsDNA production compared to wildtype regardless of gender (data not shown). When we assessed serum autoantibodies against glomerular antigens (GA) in these mice, there was a trend towards increased anti-GA antibodies in the ERα knockout females that did not reach statistical significance (data not shown). There was no effect of ERα genotype on anti-GA levels in male mice or of ERβ genotype of either gender.
Figure 5.

Serum anti-dsDNA antibodies in ERα deficient NZM2410 mice. (A) Both wildtype and knockout mice demonstrated an increase in anti-double stranded DNA production that paralleled disease development (n=10). There was a significant increase in anti-dsDNA levels in ERα null females compared to wildtypes (* indicates p<0.05). Similarly, littermate male NZM mice demonstrated an increase in anti-dsDNA production that increased with disease progression (B), but, with no difference between groups (n=10).
A possible mechanism by which the ERα KO mice would have less renal disease, despite higher serum levels of anti-dsDNA antibodies, is that the affinity of the anti-dsDNA antibodies was less in the ERα KO mice. To test this possibility, we performed competitive inhibition ELISA assays on serum from 28-week old NZM2410 mice of ERα deficient and wildtype genotypes. There was no significant difference in soluble dsDNA inhibition (from 1–30 μg/ml) of plate bound dsDNA between serum from ERα KO vs. ERα wildtype mice with 50% inhibition achieved at 3 μg/ml of soluble dsDNA in both groups.
Anti-glomerular antibody injection
We next assessed if the renal protective effect seen in the female NZM2410 mice was due primarily to the response of the kidney to inflammatory insult. We determined the effect of ERα status on the renal response to injection of rabbit anti-glomerular antigen antibodies. Using the protocol developed by Mohan et al. [14], groups of five NZM2410 wildtype and ERα KO female mice at 8 weeks of age (predisease) were injected with LPS 50 μg IP followed by rabbit anti-mouse glomerular antigen antibodies intravenously. The mice were then followed weekly for the development of albuminuria. ERα genotype did not impact the development of albuminuria following anti-glomerular antibody injection (not shown). These data suggest that the effect of ERα deficiency extends beyond the acute renal response to an inflammatory insult in this complement dependent model.
Serum hormone levels in ERα deficient female NZM2410 mice
Prior reports indicate that C57Bl6 mice deficient in ERα, but not ERβ, developed hypergonadism and partial endocrine sex reversal [15]. Plasma was collected from the ERα and wildtype mice at 32 weeks of age. Wildtype female NZM2410 mice produced an average of 74.8 ± 12.5 pg/ml of estradiol compared to 149.8±27.8 pg/ml in the ERα deficient mice (p<0.01). Similarly, we analyzed serum from 32-week old female mice for testosterone. Wildtype female NZM2410 mice produced undetectable levels of testosterone as expected. ERα deficient females, however, produced significantly elevated levels of testosterone reaching 6.6±2.9 ng/ml (p<0.01). Depending on the strain, male mice have plasma testosterone levels ranging from 5–30 ng/ml.
Disease expression in MRL/lpr mice
MRL/lpr female ERα deficient mice also exhibited significant protection against renal disease as measured by proteinuria and renal pathologic scores (Figs. 6A and B). Serum anti-dsDNA antibody levels were similar (data not shown), though complement deposition was significantly decreased in the female ERα knockout mice (Fig. 6C). As in the NZM2410 mice, there was no statistically significant impact of ERα status on male MRL/lpr mice and ERβ status had no effect on MRL/lpr disease in males or females (data not shown). These results indicate very similar responses to ERα deficiency in two different strains of lupus mice.
Figure 6.

Effects of ER deficiency on disease expression in MRL/lpr mice. MRL females with differing ERα genotype were compared for excretion of urine albumin at the various ages indicated (A). Total glomerular pathology scores were determined for female MRL/lpr mice of various ERα genotypes (B). ERα null females had significantly lower pathology score compared to wildtypes. p<0.01 by Mann–Whitney analysis. Female MRL kidneys were analyzed for C3 deposition (C) in wildtype and ERα deficient mice by immunofluorescence. ERα null females had significantly lower deposition compared to wildtype mice. p<0.05 by Mann–Whitney analysis.
Discussion
Due to the profound gender difference in SLE prevalence, defining the phenotypic effects of the genetic lack of the estrogen receptors on disease provides new insight into the mechanistic role of estrogen/ERs in the pathogenesis of lupus. The purpose of this study was to determine whether the deficiency of either of the known ERs affected the development of disease in the NZM 2410 and MRL/lpr lupus prone strains. Conflicting prior studies of ovariectomy, estrogen replacement and ER deficiency on immune function and autoimmunity in normal and lupus mice suggested that ER deficiency could either improve disease, worsen disease or have no impact on disease. Of particular importance to this study was the prior finding that the lack of ERα in B6/129 mice led to the development of autoantibodies and immune complex glomerulonephritis in both male and female mice. We found, in contrast, the lack of ERα resulted in a significant decrease in disease expression occurred in both lupus prone strains in female mice. In our current study, phenotypic differences in ERα deficient NZM female mice, compared to wildtype, included decreased albuminuria, renal pathology, SUN levels and mortality. Similar decreases in albuminuria and renal pathology were seen in female ERα deficient MRL/lpr mice. All aspects of renal pathology (i.e. proliferation, inflammatory infiltrate and sclerosis) were decreased in the KO mice. This profound impact on disease of ERα deficiency occurred despite an increase in anti-dsDNA antibodies (NZM) or no difference in anti-dsDNA antibodies (MRL/lpr) indicating that the impact of ERα deficiency was at the later stage of autoimmune disease development, but not at the earlier stage of autoantibody production. Higher serum estrogen levels were present in the ERα deficient mice, which would be predicted to worsen disease based on prior studies of estrogen administration to lupus mice and castrated lupus mice, yet the ERα deficient female mice had less disease. ERα deficiency had no significant effect on total serum immunoglobulin levels, or spleen T cell/B cell subsets (data not shown) or on the response of the kidney to acute inflammatory injury. C3 deposition was significantly decreased in the ERα deficient MRL/lpr mice, while C3 deposition was not significantly decreased in the NZM2410 mice. ERα deficiency had minimal to no protective effect on disease in male NZM2410 or MRL/lpr mice, indicating a gender specific effect of ERα deficiency on lupus like disease. ERβ deficiency resulted in a trend towards less severe disease, but none of the differences in disease measures was significant in either males or females. Thus, ERα deficiency had a unique protective effect on clinical disease in female NZM 2410 and MRL/lpr mice.
This is the first report of the effect of ER deficiency in lupus mice. Prior studies of estrogen effects in murine models of lupus found that prepubertal ovariectomy and hormonal manipulation affected development of autoimmune disease [6,16]. NZB/NZW mice, specific gene knockout mice that develop lupus (i.e. DNase KOs) and some of the derived NZM strains have significant female predominance of disease and disease severity [17]. Ovariectomy alone had no effect on disease in NZB/NZW mice. Estrogen replacement in the ovariectomized mice resulted in disease expression similar or worse than unmanipulated mice. Administration of testosterone post-ovariectomy led to improvement in disease severity. When male NZB/NZW mice were castrated, prior to sexual maturity, and given estrogen, disease developed similar in severity to unmanipulated female mice. Later studies in MRL/lpr mice found similar impacts of ovariectomy and sex hormone manipulation on disease as in NZB/NZW mice [21]. Thus, these previous studies indicated that ovariectomy, and/or estrogen deficiency alone, had no impact on disease, but that ovariectomy plus sex hormone manipulation significantly impacted disease expression in lupus mice.
Our studies provide the next step towards understanding gender differences in disease. The simple lack of estrogen, as in castrated NZB/NZW female mice, did not impact disease, yet giving extraneous estrogen to these mice enhanced disease. Thus, estrogen effects alone are not sufficient to explain the gender differences in disease. The lack of ERα signaling, however, does have significant impacts on disease expression, but only in female mice. Why, then are the ERα KOs protected, while castration alone has no effect? The castrations occurred at 2 weeks, while ERα deficiency is present in utero and in the first two weeks of life. Thus, the ERα effect may occur prior to 2 weeks of age. Compensatory changes due to ERα deficiency are also a potential mechanism including the hormonal changes we noted. Perhaps high levels of estrogen signaling through ERβ, in the absence of ERα, are protective. The lack of a significant effect of ERβ deficiency on disease would appear to mitigate against this possibility.
In the only prior study of lupus susceptibility in ER deficient mice, ERα deficient C57BL/6J/129 mice spontaneously developed autoimmune glomerulonephritis at 1 year of age [11]. Nephritis occurred in ERα−/−, but not ERβ−/−mice, and unexpectedly occurred in both male and female mice. High levels of serum IgG3 autoantibodies, increased numbers of memory B cells and renal disease were noted in the ERα deficient mice. The contrasting results between this study and our findings of disease protection in two different lupus prone strains, are most likely due to the differences strains to ER deficiency. There are a number of reports of phenotypic differences in gene knockout mice depending on which genetic background the knockout is expressed. B6/129 mice develop spontaneous autoimmune disease when C1q is deleted [18]. When C1q deficiency is then expressed on a pure B6 background, the autoimmune phenotype is absent. In studies of serologic and pathologic manifestations in B6 ERα and ERβ KO, no evidence of autoimmunity was noted at any age in either gender (Korach, unpublished data). In the B6/129 study, there were effects of ERα deficiency on B cell subsets and serum IgG3 levels. Unlike this study, we did not see an impact of ERα on IgG3 or B cell subsets in either NZM2410 or MRL/lpr mice. The same knockout construct was used in both studies. Thus, the enhancement of autoimmune disease in B6/129 mice, the lack of effect in B6 mice and the protection against disease in MRL/lpr and NZM2410 mice can best be hypothesized to reflect strain differences in response to the lack of ERα signaling. It is of interest that in neither study was there any evidence of a significant effect of ERβ deficiency, though trends towards improved disease were seen in both the lupus prone strains in female mice. Thus, it appears that disease modulating effects of estrogen receptors on autoimmune disease are via ERα and more specifically the AP1 domain of ERα.
To define mechanistic impacts of genetic manipulations on lupus, it is helpful to consider SLE as a two-step process. The first manifestation of disease is “autoimmunity”, characterized by autoantibody production. Grimaldi et al. demonstrated that ERα expression increases during differentiation of B cells [19]. Earlier the same group observed that estrogen enhanced survival of autoreactive B cells, although the receptor through which this effect was mediated is unclear [20]. Thus, one impact of estrogen receptor deficiency could be on autoanti-body production. In our study, however, autoantibody production (anti-dsDNA) was either not impacted or significantly increased in ERα−/− female mice. The increased anti-dsDNA production in the NZM2410 female mice may reflect B cell effects via increased levels of estrogen signaling through ERβ or through non-genomic effects of estrogen. This explanation is consistent with the B cell and autoantibody effects of estrogen reported by Grimaldi et al. [20,21]. We also did not see any impact of ER deficiency on anti-glomerular antigen antibodies, which we and others have linked with development of pathologic renal disease.
It is important to understand that production of auto-antibodies, the first phase of “disease” does not necessarily result in development of clinical disease. Some individuals (first-degree relatives) and murine models of disease (C3H/lpr mice) do not progress beyond the production of autoantibodies, never developing significant organ involvement [21]. This is, perhaps, best illustrated in first-degree relatives of patients with lupus, who often have similar serum autoantibody profiles as the person with lupus [21]. Of importance to this line of investigation, recent studies in human lupus, first-degree relatives and unrelated controls indicate no significant difference in autoantibody production in male vs. female first-degree relatives of lupus patients or unrelated controls [22]. Thus the development of autoimmunity, as measured by a +ANA, does not appear to differ between men and women, yet a 10/1 predominance of clinically apparent lupus is reproducibly noted in women compared to men. Our data suggest that ERα exhibits contrasting effects on the two stages of lupus development. Deficiency of ERα appears to either enhance or have no effect on autoimmunity (anti-dsDNA antibodies), while inhibiting autoimmune disease (glomerulonephritis).
The lack of an effect of ERα deficiency on lupus prone male mice suggests different mechanisms of disease development in the male vs. female mice of these two strains. Clearly, ERα expression is required for full disease expression in female mice, but is not necessary for full disease expression in male mice. One potential explanation for this difference could be in the effects of ERα deficiency on hormone levels in the two sexes. In the female mice, testosterone and estrogen levels are increased, while in male mice, ERα deficiency has no effect on hormone levels. Since testosterone levels are similar in the male and female ERα KO mice, yet females are protected, while males are not, it is difficult to attribute the protective effect of ERα deficiency in the female mice to high testosterone levels alone. As noted above, the impact of ERα deficiency appears to be on renal disease development rather than development of autoimmunity. In most murine models of renal disease and human renal disease, testosterone worsens renal disease severity, thus protection against renal disease by high testosterone in lupus mice, would be contradictory to almost all other models of renal disease. As opposed to the Talal castration experiments, when testosterone was given from 2 weeks of age, in the ERα KO mice, testosterone levels do not become elevated until after puberty, when testosterone had no impact in the castration models. Thus, the improvement of renal disease in both strains is unlikely due to high testosterone alone. Similarly, estrogen administration to lupus mice either has no effect or makes disease worse and thus the higher estrogen levels in the ERα KO are also unlikely to explain the disease differences noted. No prior manipulations of lupus mice have given both testosterone and estrogen, the scenario that is present in the ERα KO mice. Thus, it is difficult to attribute the protection seen in the ERα deficient mice to these hormonal changes alone.
Another potential mechanism of disease impact of estrogen receptor deficiency is via effects on complement factor production. C3 production in the rat, human and mouse uterus is estrogen dependent, although the receptor through which this effect is mediated is unknown [23]. Decreased local production of C3 in the kidney during inflammation could impact disease expression and local C3 production is a known contributor to renal disease in MRL/lpr mice [24]. There was a significant decrease in C3 deposition in the kidneys of the MRL/lpr mice and a trend towards decreased deposition in the NZM2410 mice. Assessment of C3 deposition by IF intensity is semiquantitative at best and thus impact on local C3 production in the ERα deficient mice remains a possible mechanism for disease protection that warrants further study.
There are limitations to the study as presented. The hormonal changes in female ERα deficient mice are obvious confounders, yet prior studies suggest that these hormonal changes alone are insufficient to have the disease impact noted in the ERα deficient mice. Studies of ovariectomized, estrogen supplemented ERα deficient mice are currently in progress in our laboratory. We believe, therefore, that the effect of ERα deficiency is multifactorial impacting a number of inflammatory pathways based on the multiple genes influenced by estrogen that have ER response elements in their promoters [23]. We did not find a detectable difference in spleen cell lymphocyte subsets or in urine or serum MCP1 or IL6 levels (data not shown) suggesting that systemic inflammatory responses are not impacted by ERα deficiency effects. The lack of an effect in the induced anti-GA model suggests that the effect is not limited to a differential renal response to inflammation, though the inflammatory insult in the GA model is primarily complement mediated and may overwhelm the modulatory impacts of ERα on renal responses.
In summary, our results are the first report of the effect of ER deficiency on disease expression in a murine model of lupus. The profound impact of ERα deficiency indicates a key role for ERα in the development of renal disease in NZM2410 and MRL/lpr lupus prone mice. ERα−/− females demonstrated significant protection from renal disease while showing enhanced or no change in autoimmunity towards dsDNA and glomerular antigens. Additional studies of this intriguing finding should provide further understanding of the complex impact of female gender on lupus. Furthermore specific targeted modulation of ERα function may provide a novel approach to therapy in human lupus.
Acknowledgments
This work was supported by the Medical Research Service, Ralph H. Johnson VAMC and a REAP award from the VA Medical Research Service. The authors are indebted to Drs. John Couse and Derek Henley, NIEHS, for their expertise in estrogen receptor biology.
References
- 1.Pisetsky DS, Gilkeson G, St Clair EW. Systemic lupus erythematosus. Diagnosis and treatment. Med Clin North Am. 1997;81:113–128. doi: 10.1016/s0025-7125(05)70507-1. [DOI] [PubMed] [Google Scholar]
- 2.Talal N. Sex hormones and modulation of immune response in SLE. Clin Rheum Dis. 1982;8:23–28. [PubMed] [Google Scholar]
- 3.Talal N. Sex steroid hormones and systemic lupus erythematosus. Arthritis Rheum. 1981;24:1054–1056. doi: 10.1002/art.1780240811. [DOI] [PubMed] [Google Scholar]
- 4.Talal N, Ahmed SA, Dauphinee M. Hormonal approaches to immunotherapy of autoimmune disease. Ann NY Acad Sci. 1986;475:320–328. doi: 10.1111/j.1749-6632.1986.tb20880.x. [DOI] [PubMed] [Google Scholar]
- 5.Ansar Ahmed S, Penhale WJ, Talal N. Sex hormones, immune responses, and autoimmune diseases. Mechanisms of sex hormone action. Am J Pathol. 1985;121:531–551. [PMC free article] [PubMed] [Google Scholar]
- 6.Carlsten H, Tarkowski A, Holmdahl R, Nilsson LA. Oestrogen is a potent disease accelerator in SLE-prone MRL lpr/lpr mice. Clin Exp Immunol. 1990;80:467–473. doi: 10.1111/j.1365-2249.1990.tb03311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 8.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 9.Couse JF, Korach KS. Contrasting phenotypes in reproductive tissues of female estrogen receptor null mice. Ann NY Acad Sci. 2001;948:1–8. doi: 10.1111/j.1749-6632.2001.tb03981.x. [DOI] [PubMed] [Google Scholar]
- 10.Smithson G, Couse JF, Lubahn DB, Korach KS, Kincade PW. The role of estrogen receptors and androgen receptors in sex steroid regulation of B lymphopoiesis. J Immunol. 1998;161:27–34. [PubMed] [Google Scholar]
- 11.Shim GJ, Kis LL, Warner M, Gustafsson JA. Autoimmune glomerulonephritis with spontaneous formation of splenic germinal centers in mice lacking the estrogen receptor alpha gene. Proc Natl Acad Sci U S A. 2004;101:1720–1724. doi: 10.1073/pnas.0307915100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morel L, Yu Y, Blenman KR, Caldwell RA, Wakeland EK. Production of congenic mouse strains carrying genomic intervals containing SLE-susceptibility genes derived from the SLE-prone NZM2410 strain. Mamm Genome. 1996;7:335–339. doi: 10.1007/s003359900098. [DOI] [PubMed] [Google Scholar]
- 13.Gilkeson GS, Grudier JP, Karounos DG, Pisetsky DS. Induction of anti-double stranded DNA antibodies in normal mice by immunization with bacterial DNA. J Immunol. 1989;142:1482–1486. [PubMed] [Google Scholar]
- 14.Fu Y, Xie C, Chen J, Zhu J, Zhou H, Thomas J, Zhou XJ, Mohan C. Innate stimuli accentuate end-organ damage by nephrotoxic antibodies via Fc receptor and TLR stimulation and IL-1/TNF-alpha production. J Immunol. 2006;176:632–639. doi: 10.4049/jimmunol.176.1.632. [DOI] [PubMed] [Google Scholar]
- 15.Couse JF, Yates MM, Walker VR, Korach KS. Characterization of the hypothalamic–pituitary–gonadal axis in estrogen receptor (ER) null mice reveals hypergonadism and endocrine sex reversal in females lacking ERalpha but not ERbeta. Mol Endocrinol. 2003;17:1039–1053. doi: 10.1210/me.2002-0398. [DOI] [PubMed] [Google Scholar]
- 16.Shear HL, Wofsy D, Talal N. Effects of castration and sex hormones on immune clearance and autoimmune disease in MRL/Mp-lpr/lpr and MRL/Mp-+/+ mice. Clin Immunol Immunopathol. 1983;26:361–369. doi: 10.1016/0090-1229(83)90120-4. [DOI] [PubMed] [Google Scholar]
- 17.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 18.Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiology. 2002;205:395–406. doi: 10.1078/0171-2985-00141. [DOI] [PubMed] [Google Scholar]
- 19.Grimaldi CM, Cleary J, Dagtas AS, Moussai D, Diamond B. Estrogen alters thresholds for B cell apoptosis and activation. J Clin Invest. 2002;109:1625–1633. doi: 10.1172/JCI14873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bynoe MS, Grimaldi CM, Diamond B. Estrogen up-regulates Bcl-2 and blocks tolerance induction of naive B cells. Proc Natl Acad Sci U S A. 2000;97:2703–2708. doi: 10.1073/pnas.040577497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon C, Salmon M. Update on systemic lupus erythematosus: autoantibodies and apoptosis. Clin Med. 2001;1:10–14. doi: 10.7861/clinmedicine.1-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramos PS, Kelly JA, Gray-McGuire C, Bruner GR, Leiran AN, Meyer CM, Namjou B, Espe KJ, Ortmann WA, Reichlin M, Langefeld CD, James JA, Gaffney PM, Behrens TW, Harley JB, Moser KL. Familial aggregation and linkage analysis of autoantibody traits in pedigrees multiplex for systemic lupus erythematosus. Genes Immun. 2006;7:417–432. doi: 10.1038/sj.gene.6364316. [DOI] [PubMed] [Google Scholar]
- 23.Roper RJ, Griffith JS, Lyttle CR, Doerge RW, McNabb AW, Broadbent RE, Teuscher C. Interacting quantitative trait loci control phenotypic variation in murine estradiol-regulated responses. Endocrinology. 1999;140:556–561. doi: 10.1210/endo.140.2.6521. [DOI] [PubMed] [Google Scholar]
- 24.Passwell J, Schreiner GF, Nonaka M, Beuscher HU, Colten HR. Local extrahepatic expression of complement genes C3, factor B, C2, and C4 is increased in murine lupus nephritis. J Clin Invest. 1988;82:1676–1684. doi: 10.1172/JCI113780. [DOI] [PMC free article] [PubMed] [Google Scholar]