

Abstract
Plants and plant pathogens are subject to continuous co-evolutionary pressure for dominance, and the outcomes of these interactions can substantially impact agriculture and food security1–3. In virus– plant interactions, one of the major mechanisms for plant antiviral immunity relies on RNA silencing, which is often suppressed by co-evolving virus suppressors, thus enhancing viral pathogenicity in susceptible hosts1. In addition, plants use the nucleotide-binding and leucine-rich repeat (NB-LRR) domain-containing resistance proteins, which recognize viral effectors to activate effector-triggered immunity in a defence mechanism similar to that employed in non-viral infections2,3. Unlike most eukaryotic organisms, plants are not known to activate mechanisms of host global translation suppression to fight viruses1,2. Here we demonstrate in Arabidopsis that the constitutive activation of NIK1, a leucine-rich repeat receptor-like kinase (LRR-RLK) identified as a virulence target of the begomovirus nuclear shuttle protein (NSP)4–6, leads to global translation suppression and translocation of the downstream component RPL10 to the nucleus, where it interacts with a newly identified MYB-like protein, L10-INTERACTING MYB DOMAIN-CONTAINING PROTEIN (LIMYB), to downregulate translational machinery genes fully. LIMYB overexpression represses ribosomal protein genes at the transcriptional level, resulting in protein synthesis inhibition, decreased viral messenger RNA association with polysome fractions and enhanced tolerance to begomovirus. By contrast, the loss of LIMYB function releases the repression of translation-related genes and increases susceptibility to virus infection. Therefore, LIMYB links immune receptor LRR-RLK activation to global translation suppression as an antiviral immunity strategy in plants.
NIK1 was first identified as a virulence target of the begomovirus NSP5,6. For begomoviruses, a group of single-stranded DNA viruses that infect major crops, the success of infection relies not only on viral suppressors of RNA silencing4 but also on the viral inhibitor, NSP, of the immune receptor, NIK1 (ref. 5). The NIK1 protein belongs to the same LRR-RLK subfamily as the well-characterized PAMP recognition co-receptor BRI1-ASSOCIATED RECEPTOR KINASE 1 (BAK1)7,8. NIK1 is involved in plant antiviral immunity5, whereas BAK1is required for plant immunity against bacteria, fungi and oomycetes through its interactions with multiple PAMP-recognition LRR-RLKs9. We have previously demonstrated that the activation of NIK1 kinase is induced by the phosphorylation of Thr 474 within the activation (A)-loop10,11 (Supplementary Discussion 1). Apart from the identification of RPL10 as a downstream effector in NIK1-mediated antiviral immunity12,13, mechanistic knowledge of the signalling pathway is lacking, and the molecular nature of the defence response remains unclear. In this study, we replaced the normal NIK1 receptor with the NIK1 phosphomimetic gain-of-function mutant T474D11 in transgenic Arabidopsis lines to understand the molecular basis of the NIK1-mediated defence mechanism (Extended Data Fig. 1a–c). Transgenic lines possessing the gain-of-function mutant T474D in the nik1 knockout background10 were challenged with infectious clones of the Arabidopsis-infecting begomovirus cabbage leaf curl virus (CaLCuV)10,11. Then, we compared the virus-induced and T474D-induced transcriptomes at 10 days post-inoculation (dpi). A global cluster analysis of the expressed sequences among the mock-treated and infected wild-type (Col-0), NIK1 and T474D lines (Supplementary Table 1) revealed that the transcriptomes of the infected wild-type and mock-inoculated T474D lines were most closely related; these samples clustered together with a high bootstrap probability and a high approximately unbiased P value (Fig. 1a), which suggests that the NIK1-mediated response and the response to begomovirus infection share similar mechanisms. These transcriptomes differed greatly from the NIK1 mock-inoculated transcriptome, indicating that virus infection activates the NIK1-mediated response. Moreover, the gain-of-function T474D mutant might be activated in a constitutive manner that allows it to support a sustained NIK1-mediated response, in contrast with the expression of the intact NIK1 receptor in the nik1 genetic background. The transcriptome from NIK1-complemented lines clustered with the Col-0 mock-inoculated transcriptome.
Figure 1. Constitutive activation of the NIK1 receptor suppresses global host translation and confers tolerance to begomovirus.
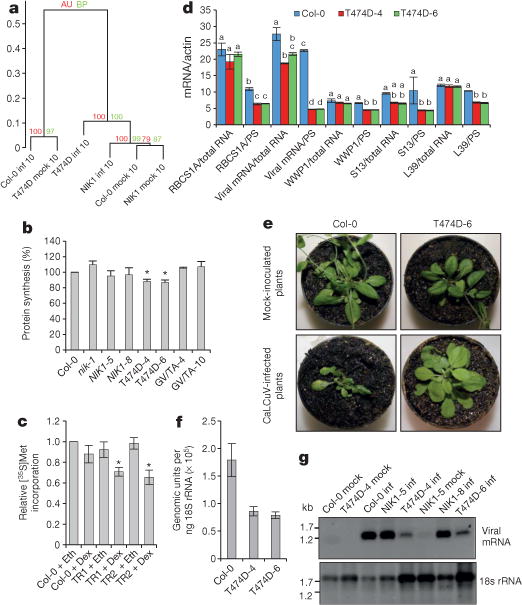
a, Ward hierarchical clustering of the gene expression data from the Arabidopsis infection experiments at 10 dpi. The dendrogram provides two types of P values: approximately unbiased (AU; red) and bootstrap probability (BP; green). inf, infected plants; mock, mock-inoculated plants. b, Global translation suppression by T474D expression. In vivo labelling of leaf proteins with [35S]Met was performed in Col-0 and transgenic seedlings. The labelling percentage was also normalized to the leaf chlorophyll content (Extended Data Fig. 1d). Estimated proportion (in %) with respective error bars showing standard error of the mean (s.e.m.) from three independent experiments (n = 3). Asterisks indicate that the proportion is significantly (P < 0.05) different from 100% using the one-sided (less) chi-squared test. GV/TA, G473V/T474A double-mutant lines. c, Inhibition of global translation upon induction of T474D expression. An oestrogen-receptor-based chemical-inducible system was used to induce T474D expression in Arabidopsis seedlings, which were treated with 10 μM dexamethasone (Dex) or ethanol (Eth) for 8 h, and then were pulse labelled with L-[35S]Met for 1 h. Tricarboxylic acid (TCA)-precipitable radioactivity in each sample was normalized to Col-0 + ethanol. Estimated proportion with respective error bars showing s.e.m. from three independent experiments (n = 3). Asterisks indicate that the proportion is significantly (*P < 0.05) different from 1 using the one-sided (less) chi-squared test. TR1 and TR2 are independently transformed lines harbouring T474D under the control of the dexamethasone-inducible promoter. d, Quantitation of total and PS-associated viral replication initiator protein (Rep) and host transcripts by qRT–PCR. The two-way analysis of variance (ANOVA) results were sliced by RNA fractions (total and PS). Different letters indicate significant differences among the RNA levels of the same gene by the TukeyHSD test (P < 0.01). mRNA/actin, mRNA of the gene normalized to actin mRNA. e, Symptoms associated with CaLCuV infection in wild-type and transgenic lines at 21 dpi. The figure shows representative samples from three independent experiments, each one with ten plants (biological replicates). f, Absolute quantitation of CaLCuV genomic units in infected lines at 14 dpi. An 18S ribosomal RNA (rRNA) target was run in parallel for normalizing the template load per reaction. Error bars, 95% confidence intervals based on bootstrap resampling replicates of four independent (n = 4) experiments. g, PS loading of viral mRNA in systemically infected leaves. PS-bound RNA was probed with viral Rep complementary DNA and 18S rDNA.
We also employed these transgenic lines to assess the T474D-induced global variation in gene expression. Gene enrichment analyses of immune system category genes indicated that ectopic expression of T474D did not activate typical viral defences, such as salicylic acid signalling or virus-induced gene silencing (Supplementary Table 2, Extended Data Fig. 2a, b and Supplementary Discussion 2). Among the differentially expressed genes, we observed an overrepresentation of translational-machinery-related genes, which largely predominated the downregulated gene list (Extended Data Fig. 3a, red spots; Supplementary Tables 2 and 3). Using enrichment analysis, these downregulated genes included ribosomal genes and other components of the protein synthesis machinery. Therefore, T474D ectopic expression downregulates components of the translational machinery, suggesting that the constitutive activation of NIK1 might influence translation. To confirm that protein synthesis was impaired by constitutive activation of NIK1 in the T474D lines, we labelled leaf proteins in vivo with [35S]Met in the control and nik1 plants, as well as in the NIK1- (ref. 11), G473V/T474A- (ref. 11) and T474D-expressing lines (Fig. 1b and Extended Data Fig. 3b). The one-proportion statistical test indicated a significant decrease (12.8% for T474D line 4 (T474D-4) and 13% for T474D-6; P < 0.05) in the amount of newly synthesized protein in T474D-expressing leaves compared with wild-type and NIK1-expressing leaves. We also demonstrated that dexamethasone-inducible T474D expression for 8 h led to a higher inhibition of de novo protein synthesis in the transgenic lines (Fig. 1c and Extended Data Fig. 3c–e). In the dexamethasone-inducible lines, the expression of T474D significantly reduced polysome (PS) and monosome (NPS) fractions (12% total reduction) to a similar extent as it reduced PS and NPS RNA (13% reduction; Extended Data Fig. 4a–c). The loading of host mRNA (RBCS, Arabidopsis thaliana (At)WWP1, S13 and S39 genes) in actively translating PS fractions was significantly reduced in T474D-overexpressing lines compared to the wild-type line, although to a different extent (Fig. 1d, Extended Data Fig. 4d and Supplementary Discussion 3). Therefore, the activation of NIK1 reduces global levels of translation, but the effect may not be the same for all mRNAs. This downregulation of cytosolic translation might at least partially underlie the molecular mechanisms involved in NIK1-mediated antiviral defences.
To examine whether the constitutive activation of NIK1 was effective at controlling begomovirus infection, the transgenic lines were inoculated with CaLCuV DNA-A and DNA-B. The wild-type plants displayed typical symptoms of CaLCuV infection, whereas the symptoms in the T474D-expressing lines were greatly attenuated (Fig. 1e and Extended Data Fig. 5b). The symptomless CaLCuV infections of the T474D-expressing lines were associated with a delayed course of infection (Extended Data Fig. 5a) and a lower accumulation of viral DNA in the systemically infected leaves (Fig. 1f).
Because T474D expression caused downregulation of protein synthesis, we determined whether viral RNA translation was impaired in the T474D-expressing lines. We examined viral RNA transcripts in actively translating PS fractions prepared from infected leaves at 10 dpi (Extended Data Fig. 4e–g), when the total viral mRNA accumulation in the Col-0 and T474D-expressing lines was not very dissimilar (Fig. 1d and Extended Data Fig. 5c). We observed a significant reduction in the PS loading of viral mRNA in the T474D infected leaves compared with the wild-type infected leaves (Fig. 1d, g), indicating that the begomovirus was not capable of sustaining high levels of viral mRNA translation in the T474D-expressing lines.
Our data indicate that the translational suppression induced by NIK1 constitutive activation is associated with the downregulation of translational-machinery-related genes. Hence, NIK1-mediated nucleocytoplasmic trafficking of RPL10 may be linked to the regulation of gene expression. The previously identified extraribosomal functions of RPL10 associated with transcription factor regulation14–16 may serve as potential targets for assessing this hypothesis. We used the two-hybrid system to search for RPL10 nuclear partners and isolated a MYB-domain-containing transcription factor, which was designated as LIMYB (Extended Data Fig. 6a–e). As a putative transcription factor, LIMYB localized in the nucleus of transiently or stably transformed plant cells (Extended Data Fig. 7a–i) and interacted with RPL10 in the nuclei of plant cells (Fig.2a and Extended Data Fig.6f, g). We further demonstrated that RPL10 and LIMYB interact in vivo using co-immunoprecipitation assays (Fig. 2b, c).
Figure 2. LIMYB interacts with RPL10 in the nucleus.
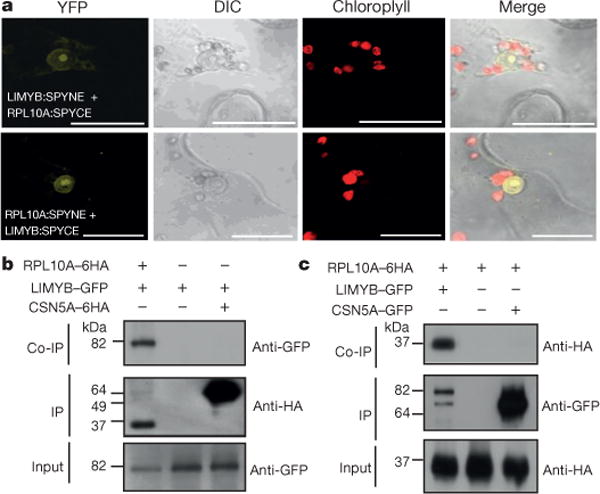
a, In vivo interaction between LIMYB and RPL10 by bimolecular fluorescence complementation (BiFC) analysis. The fluorescence (yellow fluorescent protein (YFP)) images were acquired using Nicotiana tabacum leaves co-expressing the 35S∷RPL10-SPYNE + 35S∷LIMYB-SPYCE and 35S∷RPL10-SPYCE + 35S∷LIMYB-SPYNE fusion proteins. They are representative samples from three independent biological repeats. Scale bars, 20 μm. DIC, differential interference contrast. b, LIMYB and RPL10 interaction in planta. The indicated constructs were expressed in N. tabacum leaves, and co-immunoprecipitation (Co-IP) assays were performed using an anti-haemagglutinin (HA) antibody. CSN5A is an unrelated protein used as a negative control. c, Co-immunoprecipitation assay was performed using an anti-GFP antibody.
The function of LIMYB in NIK1-mediated antiviral signalling was examined using several different approaches. We first demonstrated that LIMYB, RPL10 and NIK1 are co-expressed in several organs (Extended Data Fig. 8). Then, we identified transfer DNA (T-DNA) insertion mutants (limyb-32 and limyb-82) in the LIMYB gene (Extended Data Fig.1e, f). We also prepared LIMYB-overexpressing lines (Extended Data Fig. 1g) and determined the LIMYB-induced global variation in gene expression compared to Col-0 leaves. Remarkably, the overexpression of LIMYB resulted in a downregulation of translational-machinery-related genes similar to that induced by T474D expression (Extended Data Fig. 1h, red spots). We selected five ribosomal protein (RP) genes to confirm the deep-sequencing results for the LIMYB-overexpressing lines by quantitative polymerase chain reaction with reverse transcription (qRT–PCR) (Fig. 3a). LIMYB overexpression downregulated the expression of the selected RP genes but not of the unrelated gene (AtWWP1; Extended Data Fig. 9c). Conversely, in the limyb-32 line, the RP genes were up-regulated (Fig. 3 and Extended Data Fig. 9a, d). Expression of LIMYB in limyb lines restored the wild-type expression of the RP genes (Extended Data Fig. 9b). As in the T474D-expressing lines, protein synthesis was slightly but significantly reduced in the LIMYB-overexpressing lines (Fig. 3c and Extended Data Fig. 4h). Because RPL10 functions in NIK1-mediated antiviral signalling and interacts with LIMYB, we examined whether RPL10 also controls the expression of RP genes. In RPL10-overexpressing lines (Extended Data Fig. 1i, j), the expression of RP genes but not of the unrelated AtWWP1 gene was downregulated (Fig. 3d and Extended Data Fig.9e). We then examined whether LIMYB, a putative transcription factor, binds to an RP promoter (RPL18) in vivo. We performed chromatin immunoprecipitation (ChIP) experiments in which a 150 base pair (bp) RPL18 promoter was amplified from the precipitated DNA of LIMYB-expressing tissues but not of wild-type tissues (Fig. 3e). ChIP-qPCR showed that the 150-bp promoter fragment was significantly enriched in samples precipitated by anti-green fluorescent protein (GFP) antibody but not in samples pulled down from wild-type lines (Fig. 3f). These results suggest that LIMYB may function as a DNA-binding protein that associates with RP promoters in vivo.
Figure 3. LIMYB links NIK1 activation to the downregulation of RP genes and confers tolerance to begomovirus.
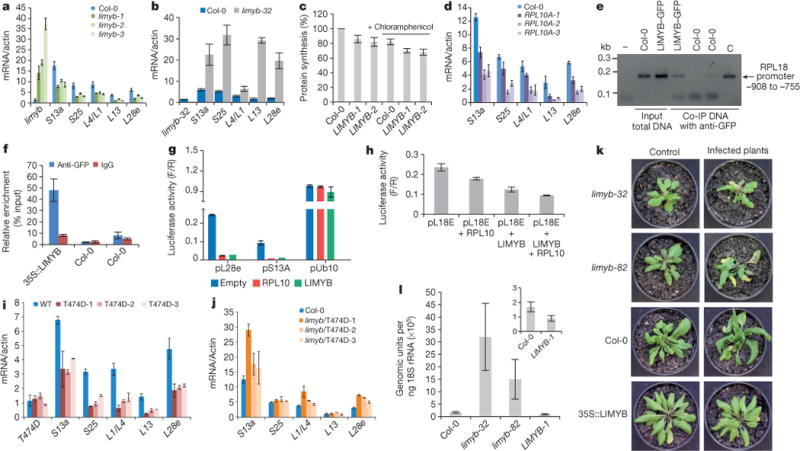
a, LIMYB overexpression downregulates RP genes. RP gene expression was monitored by qRT–PCR of RNA from LIMYB-overexpressing leaves and Col-0. b, Induction of RP genes in the limyb-32 mutant, monitored as in a. c, LIMYB overexpression suppresses global host translation. Col-0 and LIMYB-overexpressing seedlings were labelled with [35S]Met in the presence and absence of chloramphenicol, as described in Fig. 1b. d, RPL10 downregulates RP genes, monitored as in a. e, LIMYB binds to the RPL18e promoter in vivo. ChIP assay was performed with LIMYB–GFP-expressing leaves or wild type using anti-GFP antibodies. The co-immunoprecipitated (Co-IP) 150-bp fragment of the RPL18 promoter was detected by PCR. C, amplification of the control plasmid. f, ChIP-qPCR assay of 35S:LIMYB–GFP transgenic and wild-type (Col-0) seedlings. Samples were immunoprecipitated with anti-GFP antibody or IgG antibody. ChIP DNA was quantified with real-time PCR. g, LIMYB or RPL10 repress the RP promoter. N. benthamiana leaves were agro-infiltrated with plasmids carrying the prL28e-luciferase (pL28e), prS23A-luciferase (pS13A) or prUb10-luciferase (pUb10) genes in combination with either the 35S∷LIMYB or 35S:RPL10 DNA constructs. Luciferase activity was measured 48 h after agro-infiltration. F/R, firefly luciferase activity to Renilla activity ratio. h, LIMYB and RPL10 determine the full transcriptional repression of the RP promoter. N. benthamiana leaves were agro-infiltrated with the indicated combination of DNA constructs and luciferase activity was measured 48 h after infiltration. i, Downregulation of RP genes in T474D-expressing lines, monitored as in a. j, T474D requires the function of LIMYB to mediate RP gene suppression. The limyb-32 (limyb) mutant was transformed with 35S∷T474D, and the expression of RP genes was monitored as described in a. k, Symptoms associated with CaLCuV infection in Col-0 plants, the LIMYB-overexpressing line and limyb mutants at 21 dpi. Representative samples from three independent experiments, each one with ten plants (biological replicates). l, Absolute quantitation of CaLCuV genomic units in infected LIMYB-overexpressing lines and limyb mutants. Viral accumulation was determined by quantitative PCR at 14 dpi, as described in Fig. 1f. a–d, f–j, l, The respective 95% confidence interval limits were estimated based on bootstrap resampling replicates of three independent (n = 3) experiments and two technical repeats, except that in g, n = 2 with two technical repeats.
To provide further evidence for the regulation of RP target genes by LIMYB and RPL10, we performed a luciferase transactivation assay in agro-infiltrated leaves. Consistent with the gene expression profile, LIMYB and RPL10 specifically repressed the L28e, S13A and L18E promoters but not an unrelated ubiquitin promoter (Fig. 3g, h). The co-transfection of Arabidopsis protoplasts with both LIMYB and RPL10 promoted increased repression of the L18E promoter compared to regulation of the gene reporter by individual expression of the transfactors. Collectively, these results indicate that LIMYB and RPL10 function as transcriptional repressors of common RP genes and that both transfactors are required for full regulation (Fig. 3h). Therefore, LIMYB and RPL10 may coordinately regulate common target promoters.
We also demonstrated that T474D expression downregulated the RP genes (Fig. 3i) but not AtWWP1 (Extended Data Fig. 9f), confirming the RNA-sequencing data (Extended Data Fig. 3a). As an additional control, we showed that the double-mutant NIK1 G4743V/T474A12, which does not complement the enhanced susceptibility phenotype of the nik1-null alleles (Extended Data Fig. 5a), also does not affect RP genes (Extended Data Fig. 9g). Nevertheless, the loss of LIMYB function prevented the T474D-mediated downregulation of the RP genes in both the limyb-32 transgenic lines that stably expressed T474D (Fig. 3j and Extended Data Fig. 9h, i) and the limyb-32 protoplasts that transiently expressed T474D (Extended Data Fig. 10). These results genetically link LIMYB to the translation suppression portion of the NIK1-mediated antiviral response.
We predicted that if the suppression of host translation was the basis for the begomovirus-tolerant phenotype of the T474D-expressing lines, LIMYB overexpression would also be effective against CaLCuV and the loss of LIMYB function would further debilitate the plant upon begomovirus infection. Col-0, limyb-32, limyb-82 and LIMYB-overexpressing lines were inoculated with infectious clones of CaLCuV DNA-A and DNA-B (Fig. 3k). Both Col-0 and limyb lines developed typical CaLCuV symptoms, although to different extents. The disease symptoms varied in severity from extreme stunting and leaf distortion with severe chlorosis in the limyb-32 and limyb-82 leaves to mild leaf distortion and moderate chlorosis in Col-0. The course of infection in the limyb leaves was accelerated compared to that in the Col-0 plants (Extended Data Fig. 5d) and the limyb-32 and limyb-82 systemic leaves accumulated higher levels of viral DNA than Col-0 (Fig. 3l). Therefore, the loss of LIMYB function recapitulated the enhanced begomovirus susceptibility phenotype of the nik1-null alleles, as would be expected from a downstream component of the NIK1-mediated antiviral defence. By contrast, the LIMYB-overexpressing lines did not develop symptoms, displayed delayed infection and accumulated a lower level of viral DNA in their systemic leaves, resembling the tolerant phenotype of the T474D-overexpressing lines. We also found that LIMYB overexpression did not induce salicylic-acid-signalling marker genes or typical defence responses to viral infection (Extended Data Fig. 2c and Supplementary Table 2). Collectively, these results further support the notion that the inhibition of host translation observed in the T474D and LIMYB lines may be an effective mechanism exploited by plant cells to fight begomovirus infection. Therefore, the demonstration that immune-receptor-mediated defence signalling controls translation in plant cells may represent a new paradigm for antiviral defences in plants (Supplementary Discussion 4).
METHODS
Plasmid constructs
The clone pK7F-NIK1T474D was described previously11. This clone harbours a GFP gene fused in frame after the last codon of the respective mutant cDNA under the control of the CaMV 35S promoter. In the mutant cDNA T474D, the threonine residue at position 474 within the activation loop of NIK1 was replaced with an aspartate residue. The clones rpL10AST-pDONR201 (pUFV608), rpL10ANS-pDONR201 (pUFV609), rpL10AST-pDONR207 (pUFV900) and rpL10ANS-pDONR207 (pUFV901), which harbour the RPL10A coding region either with (ST) or without (NS) a translational stop codon inserted into the entry vectors pDONR201 or pDONR207, have also been previously described12. The RPL10A coding region was transferred from these entry clones to the yeast expression vectors pDEST32 and pDEST22, generating the clones pBD-RPL10A (pUFV1422) and pAD-RPL10 (pUFV785) as GAL4 binding domain (BD) or activation domain (AD) fusions. For BiFC, the RPL10A coding region was transferred to the vectors SPY-NE-GW and SPY-CE-GW. The resulting clones, pSPYNE-RPL10A (pUFV1652) and pSPYCE-RPL10A (pUFV1653), harboured the RPL10A coding region fused to either the N-terminal (NE) region of YFP or the YFP C terminus (CE), respectively, under the control of the 35S promoter. The clones pK7F-L10 and pYFP-L10, which harbour RPL10A fused in frame with GFP after its last codon or with YFP before its first codon under the control of the 35S promoter, respectively, have been previously described10.
The clone pYFP-NLS-L10, which harbours a YFP–L10 fusion with a nuclear localization signal (NLS) under the control of the 35S promoter, was constructed by first inserting the amplified coding region of RPL10A into the SacI site of the pGR vector. The resulting clone, pGR-L10, which contains the RPL10A coding region fused to a glucocorticoid receptor (GR) domain with an NLS at the 5′ end, was then used as the template for the amplification of the NLS-containing L10 fusion with specific primers. The resulting product was inserted by recombination into pDONR207 and transferred to 35S-YFP-casseteA-Nos-pCAMBIA1300, generating pYFP-NLS-L10.
For the induction of T474D expression, the mutant cDNA T474D was transferred by recombination from the pDON201-T474D entry vector to the destination vector pBAV150 (ref. 17). The resulting clone pBAV150-NIK1 T474D–GFP harbours the mutant T474D open reading frame (ORF) under the control of a dexamethasone-inducible promoter and fused to a C-terminal GFP tag.
Arabidopsis growth conditions and transformation
The Columbia (Col-0) ecotype of A. thaliana was used as the wild-type control, and the nik1 knockout line10 was used for plant transformation. The nik1 lines10 were transformed with pK7F-NIK1T474D or pBAV150- NIK1 T474D-GFP using the floral dip method. Two independently transformed lines expressing the T474D transgene (T474D-4 and T474D-6) were selected for the infection assays. The transgenic lines ectopically expressing NIK1–GFP (NIK1-5 and NIK1-8) or an inactive kinase, the double mutant of NIK1, and the G473V/T474A–GFP dead kinase (G473V/T474A-10 and G473V/T474A-8) have been previously described11,12 (Supplementary Table 1).
RT–PCR and real-time RT–PCR analyses
Total RNA was extracted from Arabidopsis leaves using TRIzol (Invitrogen). RT–PCR assays were performed using gene-specific primers, as previously described18. For gene expression analysis by qRT–PCR, gene-specific primers were designed using Primer Express 3.0 (Life Technologies). Real-time RT–PCR assays were performed on an ABI7500 instrument (Life Technologies) using the SYBR Green PCR Master Mix (Life Technologies). The amplification reactions were performed as follows: 2 min at 50 °C, 10 min at 95 °C and 40 cycles of 94 °C for 15 s and 60 °C for 1 min. The variation in gene expression was quantified using the comparative Ct method (2−ΔΔCt), and absolute gene expression was quantified using the 2−ΔCt method. The values were normalized to endogenous actin and ubiquitin as control genes11.
RNA sequencing and data analysis
The transgenic and wild-type lines were infected at the seven-leaf stage with CaLCuV, as described later. After 10 dpi or after 21 dpi, total RNA from systemically infected leaves, as diagnosed by PCR, and mock-inoculated leaves from wild-type, 35S∷NIK1-5 and 35S∷T474D-4 lines was isolated using TRIzol (Invitrogen). For the RNA-sequencing experiments, we used two biological replicates of a pool of ten plants at 10 dpi, when we detected high levels of viral DNA in systemic leaves but symptoms had not yet developed, or at 21 dpi, when symptoms were visible. The RNA-sequencing data were obtained using an Illumina Hi-seq 2000. The paired-end 100-bp protocol was used with the following quality filter parameters: 5 bases trimmed at the 3′ and 5′ ends of the reads, a minimum average quality of 30 phred score. The data were stored in a comma-separated value spreadsheet file, and differential gene expression (DGE) analysis was performed using the R/Bioconductor package edger19. The raw data were normalized using the TMM normalization factor20. The dispersion was estimated by the tagwise edger parameter. Differential expression was determined using the cut-off P value of 0.1 adjusted by the false discovery rate (FDR). The read mapping process was executed using the Bowtie program21 with the cDNA data set retrieved from The Arabidopsis Information Resource (TAIR) database (http://www.arabidopsis.org), tenth release. Gene ontology classification was performed using the R/Bioconductor packages GSEABase and GOstats. Clustering analysis was performed using the R package pvclust (Hierarchical Clustering with P-Values via Multiscale Bootstrap Resampling) using Ward’s method22, and heatmaps were generated using gplots. The results were stored in a relational database created in PostgreSQL, and a web interface was created using PHP to allow the database to be accessed and navigated (http://arabidopsisnik.inctipp.ufv.br).
In vivo labelling of leaf proteins
Arabidopsis seedlings (300 mg) were incubated with 1 ml of nutrient solution containing 30 μg ml−1 dexamathosone, 10 μg ml−1 cycloheximide, 10 μg ml−1 puromycin or 25 μg ml−1 chloramphenicol for 1 h, 4 h or 8 h. After the incubation period, 20 μCi of [35S]Met (EasyTag Protein Labelling Mix, [35S]-, 2 mCi (74 MBq), Perkin Elmer) was added for 1 h at room temperature. To quantitate the incorporation of [35S]Met into protein, aliquots of protein extracts were placed in 10% (w/v) TCA and incubated on ice for 30 min. The samples were filtered onto glass microfibre filters, and the filters were washed three times with 5 ml cold 5% (w/v) TCA and two times with 5 ml 95% ethanol. After drying, the filters were counted with a scintillation counter.
PS fractionation
PSs were fractionated from 500 mg of 15-day-old Arabidopsis seedlings over sucrose gradients as described23. Fractions were collected manually from the top, and total RNA was extracted with phenol/chloroform/isoamyl alcohol, precipitated with isopropanol, and treated with DNase I. The specific transcripts were examined by northern blot analysis. For the infection assays, PSs from wild type, NIK1-4-overexpressing and T474D-6-overexpressing lines were isolated 10 dpi with infectious CaLCuV clones.
PS gradients of dexamethasone-inducible T474D seedlings were prepared from 8-day-old seedlings, treated or not with 30 μg ml−1 dexamathosone. After 8 h of dexamethasone treatment, the seedlings were dried, frozen and ground in liquid nitrogen. Cytoplasmic extracts were prepared from 350 mg of powder that were re-suspended in the ice-cold extraction buffer. Cell debris was removed by centrifugation and 0.6 ml of the supernadant were loaded onto a 4.5 ml 15–55% sucrose density gradient and separated by ultracentrifugation. Fractions (16) of 310 μl were collected from the top. Fractions 2–7 correspond to density regions of complexes ≤80S (NPSs) and fractions 8–15 correspond to density regions containing small and large polysomes (PSs). Quantitation of NPSs and PSs from the absorbance profile was performed as described previously24. The quantitation of total RNA from the combined NPS and PS fractions was performed as described previously25. To quantify mRNA loading into PSs, total RNA isolated from combined NPS (2–7) and PS (9–15) fractions was extracted using Trizol (Invitrogen), precipitated with isopropanol and quantified by qRT–PCR, as described earlier. qRT–PCR on each fraction from the PS gradient of dexamethasone-inducible T474D seedlings was also performed for quantitation of the distribution of AtWWP1 and S39 transcripts after induction of T474D expression. Each sample was amplified with 1 cycle at 50 °C for 2 min, 95 °C for 10 min and 40 cycles at 95 °C for 15 s and 60 °C for 1 min. Values were normalized to actin.
CaLCuV inoculation and viral DNA accumulation
A. thaliana plants at the seven-leaf stage were inoculated with plasmids containing partial tandem repeats of CaLCuV DNA-A and DNA-B by biolistic delivery26, and the course of infection was monitored as described previously10. We used an attenuated form of the virus in which the coat protein ORF in the CaLCuV DNA-A was interrupted by the introduction of a stop codon at amino acid position 47. The inoculated plants were transferred to a growth chamber and examined for symptom development (leaf necrosis, chlorosis, leaf epinasty, leaf curly, young leaf death and stunted growth). Total nucleic acids were extracted from systemically infected leaves, and viral DNA was detected by PCR with DNA-B CaLCuV-specific primers (566CLCVBFBR1v, 5′-GGCGTGGGGTATCTTACTC-3′, and 1253CLCVBRBR1c, 5′-GACATAGCATCGGACATCC-3′) as well as the actin-specific primers as an endogenous control. In each experiment, 20 plants of each line (Col-0, nik1 and nik1-expressing NIK1 mutant proteins) were inoculated with 2 μg of tandemly repeated DNA-A plus DNA-B per plant. The course of infection was examined using data from three independent experiments.
Viral DNA accumulation was measured by qPCR. The reactions were prepared in a final volume of 10 μl using the Fast SYBR Green Master Mix (Life Technologies) according to the manufacturer’s instructions and analysed on a 7500 Real Time PCR System. Virus-specific primers were designed using Primer Express 3.0 (Life Technologies) and tested by conventional PCR using plasmids containing the complete DNA-B of each virus (106 copies per reaction). The genomic copies of CaLCuV were normalized against an internal control (18S rRNA) to consider template input variation between tubes. CaLCuV DNA was amplified with primers B-Fwd (5′-GGGCCTGGGCCTGTTAGT-3′) and B-Rvs (5′-ACGGAAGATGGGAGAGGAAGA-3′). In this case, the genomic unit refers to one copy of the DNA-B of CaLCuV. PCR reactions were run in parallel with primers 18S-Fwd (5′-TAATTTGCGCGCCTGCTGCC-3′) and 18S-Rvs (5′-TGTGCTGGCGACGCATCATT-3′) for the reference plant gene 18SRNA. Standard curves were obtained by regression analysis of the Ct values of each of the three replicates of a given dilution as a function of the log of the amount of DNA in each dilution. Standard curves containing viral DNA and host DNA served as references. Each sample was analysed in triplicate from at least two biological replicates.
Two-hybrid screening
The yeast reporter strain MaV203 (MATαleu2-3,112 trp1-901 his3200 ade2-101 gal4 gal80 SPAL10∷URA3 GAL1∷lacZ HIS3UAS GAL1∷HIS3-LYS2 can1R cyh2R; Trp- Leu- Ura-) was transformed sequentially with pBD-L10 and 25 μg of the pEXAD502 cDNA library previously prepared27 and approximately 5 × 106 transformants were screened for histidine protoprophy an β-galactosidase activity, as described27,28.
LIMYB-based plasmid constructs
The At5g05800 cDNA (LIMYB), which was isolated based on its ability to bind to rpL10A in yeast, was amplified by PCR from the cDNA library vector using AT5G05800-specific primers, re-amplified with the primers AttB1-Fwd and AttB1-Rvs, and cloned by recombination into the entry vectors pDONR201 and pDONR207. The resulting products were then transferred to different destination vectors for expression in yeast (pDEST32 and pDEST 22) and plants (pK7FWG2, 35S-YFP-casseteA-Nos-pCAMBIA1300, pSPYNEGW and pSPYCEGW). The resulting clones, pBD-At5g05800ST (pUFV1903) and pAD-At5g05800ST (pUFV1480), enabled the expression of AT5G05800 (LIMYB) in yeast as GAL4 binding domain (BD) or activation domain (AD) fusions, respectively. The clone pAt5g05800-GFP (pUFV1395) harboured the AT5G05800 (LIMYB) coding region fused to the GFP N terminus under the control of the 35S promoter, whereas in pYFP-At5g05800 (pUFV1886), the At5g05800 coding region was fused to the YFP C terminus. In the resulting clones, pSPYNE-At5g05800 (pUFV1658) and pSPYCE-At5g05800 (pUFV1657), the At5g05800 cDNA was linked to the YFP NE and the YFP CE, respectively, under the control of the 35S promoter.
The HA epitope was fused to the At5g05800 C terminus using the triple Gateway system, which consisted of the vector p5′, in which the 35S (2×) promoter had been previously cloned, the clone pUFV1378 (At5g05800 cDNA without a translational stop codon in pDONR201) and the vector p3′, in which a HA (6×) epitope had been previously inserted. The At5g05800 cDNA was transferred by recombination into the destination vector pK7M34GW along with 2×35S promoter and the HA tail, yielding the clone p2×35S-At5g05800-6HA (pUFV1984), which enabled the expression of At5g05800 fused in frame to HA under the control of the 35S promoter in plants. The same triple Gateway system was used to place the LIMYB fused in frame with Cherry or GFP under the control of the LIMYB promoter.
Transient expression in N. benthamiana leaves
To determine the subcellular localization of the proteins, N. benthamiana leaves were agro-infiltrated with pAt5g05800-GFP, pYFP-At5g05800 and pYFP-NAC81 (ref. 29), while for the co-immunoprecipitation assays, they were agro-infiltrated with pL10-GFP and p2×35S-At5g05800-6HA. Agrobacterium tumefaciens strain GV3101 was used for agroinfiltration. Agrobacterium infiltration was performed in the leaves of 3-week-old N. benthamiana, as previously described12.
Confocal microscopy
Approximately 72 h after agro-infiltration, 1-cm2 leaf ex-plants were excised, and the GFP and YFP fluorescence patterns were examined in epidermal cells with a ×40 or ×60 oil immersion objective and a Zeiss inverted LSM510 META laser scanning microscope equipped with an argon laser and a helium laser as excitation sources, as described27.
Co-immunoprecipitation assays
The co-immunoprecipitation assay was performed using the μMACSTM Epitope Tag Protein Isolation Kit (MACS/Miltenyi Biotec) according to the manufacturer’s instructions. Total protein extracts were prepared from N. benthamiana leaves that had been agro-infiltrated with pL10-GFP and/or p2×35S-At5g05800-6HA. At 48 h after infiltration, 200 mg of leaves were homogenized with 1 ml of lysis buffer (50 mM Tris-HCl pH 8.0, 1% (v/v) Nonidet P-40) and incubated for 2 h with anti-GFP magneticbeads (MACS/Miltenyi Biotec) at 4 °C undergentle agitation. After extensive washing of the magnetic beads, the bound proteins were eluted using 50 ml of elution buffer pre-warmed to 95 °C. The immunoprecipitated proteins were separated by 10% (w/v) SDS–PAGE and immunoblotted with anti-HA (Miltenyi Biotec, catalogue number 130-091-972) or anti-GFP (Miltenyi Biotec, catalogue number 130-091-833) monoclonal antibodies. The reacting antibodies were detected using Signal West Pico Chemiluminescent Substrate (Thermo Scientific) according to the manufacturer’s instructions.
BiFC
N. benthamiana leaves were agro-infiltrated as previously described for the transient expression experiments using the following combinations of recombinant plasmids: pSPYNE-At5g05800 + pSPYCE-rpL10A; pSPYCE-At5g05800 + pSPYNE-rpL10A; pSPYNE-At5g05800 + pSPYCE empty vector; pSPYNE-rpL10A + pSPYCE empty vector; pSPYCE-At5g05800 + pSPYNE empty vector; pSPYCE-rpL10A + pSPYNE empty vector. After incubation for 72 h, the leaf sectors were examined by confocal microscopy. YFP was excited at 514 nm using an argon laser, and YFP emission was detected using a 560–615 nm filter. The stability of the CE and NE YFP regions was monitored by immunoblotting of transfected leaf protein extracts with a polyclonal anti-YFP serum.
LIMYB- orRPL10A-expressing lines and T474D-expressinglimyb lines
Arabidopsis Col-0 was transformed with pAt5g05800-GFP, generating the transgenic line LIMYB-1; with pYFP-At5g05800, generating the transgenic lines LIMYB-2 and LiMYB-3; and with pYFP-NLS-L10, generating the transgenic lines RPL10-1, RPL10-2 and RPL10-3. Homozygous seeds of the T-DNA insertion limyb mutants (Salk_032054C and Salk_082995C) were obtained from the Arabidopsis Biological Resource Center. The limyb-32 mutant was transformed with the gain-of-function mutant T474D, generating the transgenic lines limyb/T474-1, limyb/T474D-2 and limyb/T474D-3. Primary transformants were selected using the appropriate antibiotic (50 μg ml−1 kanamycinor 30 μg ml−1 hygromycin), and the stable incorporation of the transgene in the plant genome was evaluated by PCR with gene-specific primers. The expression of the transgenes was monitored by qRT–PCR.
LIMYB promoter construct and GUS histochemical assay
Approximately 1 kb of the 5′ flanking sequences of NIK1, RPL10A and At5g05800 (LIMYB) were amplified from Arabidopsis genomic DNA using Taq Platinum and specific oligonucleotides and inserted into the cloning vector pCR8/GW/TOPO (Invitrogen). The promoter sequences were then transferred by recombination into the destination vector pMDC162. The resulting clones, pNIK1-MDC162, pL10A-MDC162 and pAt5g05800-MDC162 (pUFV 1892), harboured the respective promoter sequences of the three genes fused to the β-glucuronidase (GUS) reporter gene and were used to transform Arabidopsis Col-0 plants. GUS expression was assayed histochemically.
Luciferase reporter gene assay
The At1g29970 (RPL18A) promoter was isolated from Arabidopsis DNA by PCR and cloned into pDONR-P4-P1R, generating pUFV2155. The clone LUCF-term-pDON221 (pUFV 2132) harbours the firefly luciferase cDNA in the entry vector pDON221, whereas the clone 2×35S-RLUCF-pDONR-P2R-P3 (pUFV 2131) contains Renilla luciferase cDNA under the control of a 2× 35S promoter. The RPL18A promoter was transferred from pUFV2155 to the destination vector pK7m34GW (pUFV 1918), along with 2×35S-RLUCF-pDONR-P2R-P3 (pUFV 2131) and LUCF-term-pDON221 (pUFV 2132), by triple recombination. The resulting clone, prAt1g29970-Lucif-term-2X35S RLucif pH7M34GW (pUVF2231), harboured the firefly luciferase cDNA under the control of the rpL18A promoter, as well as the Renilla luciferase cDNA under the control of a 2× 35S promoter. The same procedure was used to clone the firefly luciferase cDNA under the control of the RPL28e, RPS13A and ubiquitin promoters for luciferase transactivation assays.
N. benthamiana leaves were agro-infiltrated with A. tumefaciens GV3101 strains carrying the following combinations of DNA constructs: At1g29970-Lucif-term-2×35S RLucif pH7M34GW; At1g29970-Lucif-term-2X35S RLucif pH7M34GW + AT5G05800NS-pK7FWG2; At1g29970-Lucif-term-2X35S RLucif pH7M34GW + AT5G05800NS-pK7FWG2 + rpL10ANS-pK7FWG2; and At1g29970-Lucif-term-2×35S RLucif pH7M34GW + rpL10ANS-pK7FWG2. Forty-eight hours after infiltration, 200 mg of leaf tissue was harvested for total protein extraction. Luciferase activity was assayed with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions.
Protoplast preparation fromA. thaliana leaves and transient expression assays
Protoplasts were prepared from 15-day-old Arabidopsis seedlings as previously described30, except that digestion was initiated for 30 s under vacuum and then prolonged for 6 h with agitation at 80 r.p.m. The transient expression assays were performed by electroporating (250 V, 250 μF) 10 mg of the expression cassette DNA and 30 μg of the sheared salmon sperm DNA into 2 × 105–5 × 106 protoplasts in a final volume of 0.8 ml. The protoplasts were diluted into 8 ml of MS medium supplemented with 0.2 mg ml−1 2,4-dichlorophenoxyacetic acid and 0.8 M mannitol at pH 5.7. After 36 h of incubation in the dark, the protoplasts were washed with 0.8 M mannitol plus 20 mM MES at pH 5.7 and frozen in liquid N2 until further use.
Statistical analyses
All statistical analyses (including gene expression clustering, qRT–PCR and protein synthesis data) were performed in R.
Cluster analysis
Cluster analysis of the RNA-sequencing data was used to classify the treatments (mock-inoculated and infected wild type, T474D and NIK1) according to similarities in the profiles of genome-wide expression at 10 and 21 dpi. The uncertainty of clustering results caused by sampling variations was verified by the probability-based cluster analysis, which was implemented using the pvclust package of R software. In this context, the bootstrap (BP) and approximately unbiased (AU) probability are used to validate the reported cluster. The BP and AU values are the percentage that a given cluster appears in the bootstrap andmultiscale bootstrap replicates, respectively.
RNA-sequencing differential gene expression analysis
The edgeR package of R/bioconductor software was used to carry out the gene expression analysis. This package assumes negative binomial distribution for the read counts, and the used normalization is given by the TMM (trimmed mean of M value) method. The significance of differential gene expression was reported in terms of q values (FDR-adjusted P values).
Enrichment analysis
Gene set enrichment analysis (GSEA) was used to uncover biological processes associated with sets of differentially expressed genes instead of focusing on individual genes. This analysis was implemented in the GOstats package of R/bioconductor software by using the function GSEAGOHyperGParams, which uses the relationships among the gene ontology terms as extra information in the statistical inference of groups.
Percentage of protein synthesis analysis
Analysis of protein synthesis was based on the one-proportion one-sided (less) test using chi-squared distribution. The tested hypothesis was the 100% synthesized labelled protein for each treatment; thus, if a given treatment is significant, it indicates that the proportion (or percentage) is different from 100%.
Confidence interval analysis
Nonparametric bootstrap confidence intervals were used on our graphics to increase the accuracy of the confidence limits by an overlapping analysis31. This method was introduced as a nonparametric device for estimating standard errors and biases. It is an automatic algorithm for producing highly accurate confidence limits from a bootstrap distribution implemented with the ‘boot’ package in R software.
Phylogenetic analysis
MYB family sequences were retrieved from the Agris database (http://arabidopsis.med.ohio-state.edu). The alignment was performed by Maft aligner software32, and the tree was built by Fasttree33 software.
Extended Data
Extended Data Figure 1. Characterization of the Arabidopsis transgenic lines.
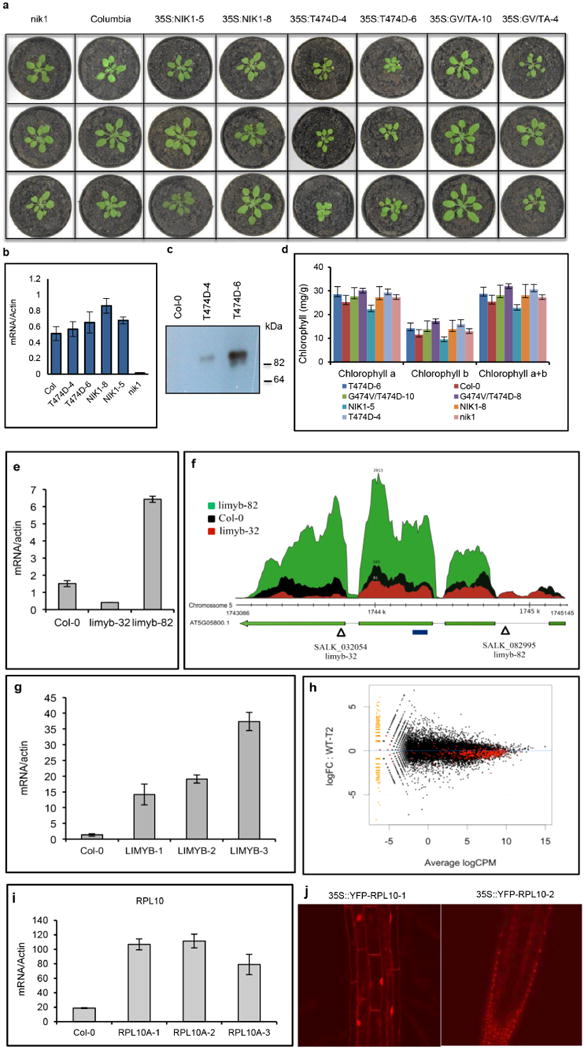
a, Phenotypes of wild-type (Col-0) and nik1 plants transformed with NIK1 (NIK1-5 and NIK1-8), T474D (T474D-4 and T474D-6) or the double-mutant G4743V/T474A (inactive kinase, GV/TA-10 and GV/TA-4). Transgenic plants (R2 generation, n = 15) were grown in soil at 22 °C under short day conditions and photographed 2 weeks after planting. b, T474D transcript accumulation in transgenic lines (R2 generation). The expression of T474D or NIK1 in the leaves of independent transgenic lines was monitored by quantitative RT–PCR. Mean ± 95% confidence intervals (n = 3) based on bootstrap resampling replicates of three independent experiments. c, Accumulation of T474D–GFP in transgenic lines. Total protein was extracted from the leaves of independent transgenic lines (as indicated), immunoprecipitated and immunoblotted with an anti-GFP antiserum. d, Chlorophyll content of transgenic lines. Total chlorophyll, chlorophyll a and chlorophyll b were determined in leaf sectors of the indicated transgenic lines. Error bars, 95% confidence intervals (n = 3) based on bootstrap resampling replicates of four independent experiments. e, Transcript accumulation of LIMYB in T-DNA insertion mutant lines. LIMYB expression was monitored by qRT–PCR of RNA prepared from Col-0, limyb-32 (SALk_032054) and limyb-82 (SALK_082995) plants. Gene expression was calculated using the 2−ΔCt method, and actin was used as an endogenous control. Error bars, 95% confidence intervals (n = 3) based on bootstrap resampling replicates of three independent experiments. f, Schematic representation of the At5g05800 (LIMYB) locus in the chromosome 5 and RNA sequencing data. The At5g05800 gene harbours three introns and four exons. Triangles show the positions of the T-DNA insertion in the limyb-32 and limyb-82 mutants, and the blue line indicates the position of the amplicon from e. The relative abundance of the mapped RNA hits in the At5g05800 locus is shown in red in limyb-32, black in Col-0 and green in limyb-82. The accumulation of LIMYB transcripts was much lower in limyb-32 and higher in limyb-82 than in Col-0. Sequencing of the limyb-32 and A-82 transcripts revealed unprocessed intron sequences and premature stop codons that would have prevented the translation of a functional protein in these mutant lines. Therefore, limyb-32 and limyb-82 were confirmed as loss-of-function limyb mutant lines. g, LIMYB transcript accumulation in LIMYB-overexpressing lines (R2 generation). LIMYB expression in the leaves of independent transgenic lines was monitored by quantitative RT–PCR. Error bars, 95% confidence intervals (n = 3) based on bootstrap resampling replicates of three independent experiments. h, General downregulation of translational machinery-related genes in LIMYB-1 seedlings. The ‘MA’ plots show the log of the ratio of the expression levels against log concentration, and each dot represents a gene. This plot visualizes the contrast of LIMYB-1 and Col-0 seedlings. The smear of points on the left side indicates those genes that were observed in only one group of replicated samples, and the red points denote ribosomal and protein synthesis-related genes. CPM, counts per million; FC, fold change; WT, wild type. i, RPL10 transcript accumulation in RPL10-overexpressing lines (R2 generation). The expression of an NLS-containing RPL10 transgene in the leaves of independent transgenic lines was monitored by quantitative RT–PCR. Error bars, 95% confidence intervals (n = 3) based on bootstrap resampling replicates of three independent experiments. j, Nuclear localization of the NLS-containing YFP–RPL10 fusion in transgenic lines. Root tips from transgenic seedlings expressing the NLS-containing YFP–RPL10 fusion were directly examined under a laser confocal microscope. The figure shows representative confocal images from five independent biological replicates.
Extended Data Figure 2. Expression of immune-system-related genes in T474D-overexpressing lines and in LIMYB-overexpressing lines.
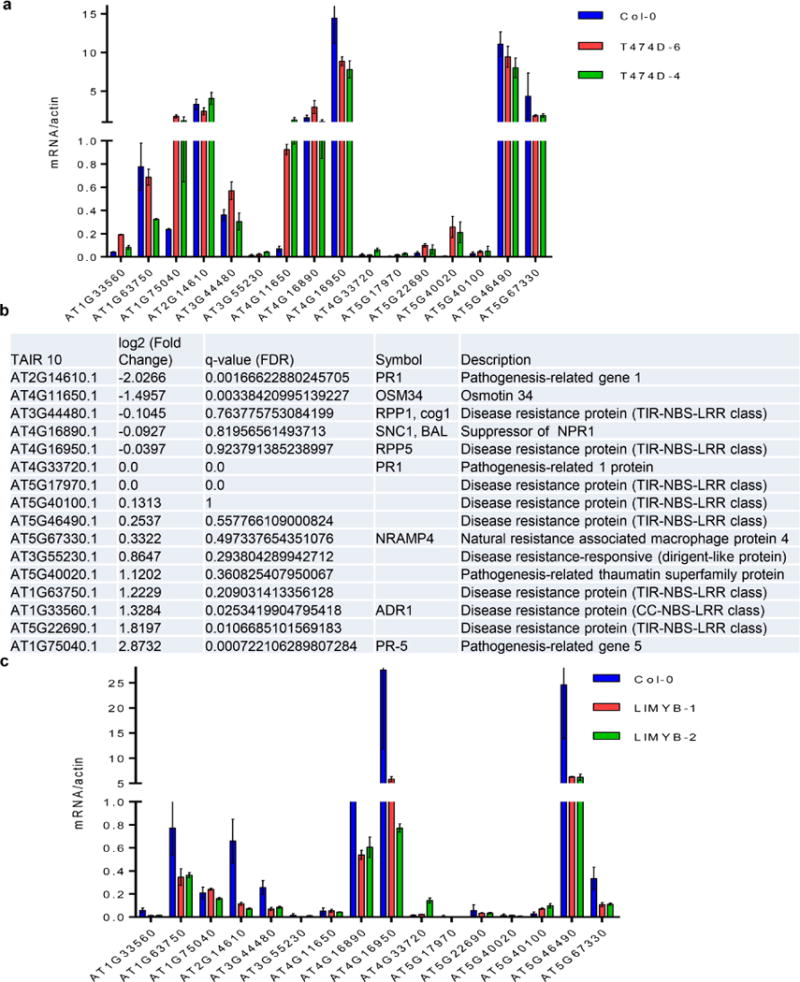
a–c, On the basis of our global comparison of EST sequences (Fig. 1a) and the role of NIK as an antiviral receptor, we asked whether the constitutive activation of NIK would elicit a defence response similar to that induced by geminivirus infection via the salicylic acid pathway or typical defence responses to virus. No significant gene enrichment was detected in the virus-induced gene silencing (GO:0009616) and viral defence response (GO:0051607) categories using the gene set enrichment analysis (GSEA) method (Supplementary Table 2). For the immune system category, gene enrichment was found in both up- and downregulated changes using the GSEA method. However, the typical markers of salicylic acid signalling, such as PR1 and SNC1, were either non-differentially expressed or downregulated, and the expression of T474D did not enhance the salicylic acid level in the transgenic lines. Collectively, these results indicate that ectopic expression of T474D did not activate typical viral defences, such as salicylic acid signalling or gene silencing. a, b, Transcript accumulation of selected immune-system-related gene markers by RT–PCR (a) or RNA-sequencing in T474D-overexpressing lines (b). qRT–PCR of a representative sample confirmed an 80% match with the RNA-sequencing results. c, Transcript accumulation of the immune-system-related genes in LIMYB-overexpressing lines. The expression of the indicated genes in the leaves of independent transgenic lines was monitored by qRT–PCR. a, c, Mean ± 95% confidence intervals (n = 3) are shown, based on bootstrap resampling replicates of three independent experiments.
Extended Data Figure 3. Ectopic expression of T474D-D downregulates translational-machinery-related genes and suppresses de novo protein synthesis.
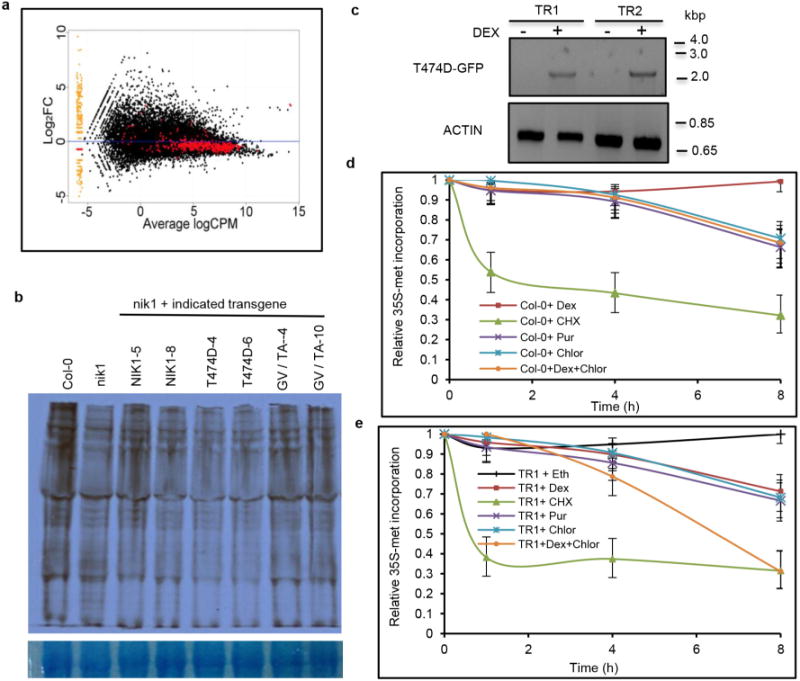
a, Representation of the translational-machinery-related genes differentially expressed in the T474D lines. The ‘MA’ plots show the log of the ratio of expression levels versus the log concentration, and each dot represents a gene. This plot represents the contrast between the T474D mock-inoculated lines and the Col-0 mock-inoculated lines. The red points denote ribosomal and protein synthesis-related genes (as shown in Supplementary Table 3). b, Downregulation of global translation by ectopic expression of T474D in Arabidopsis. The in vivo labelling of leaf proteins with [35S]Met was performed in 20-day-old Col-0 plants and T474D transgenic lines. The total protein extracts were fractionated by SDS–polyacrylamide gel electrophoresis (SDS– PAGE), and the radioactive bands were quantified by densitometric analysis of the images obtained by autoradiography. The labelling percentage was normalized to the leaf chlorophyll content, and the protein loading on the gel was adjusted to the Coomassie-stained band of the large subunit of rubisco. c, Induction of T474D expression by dexamethasone (DEX) in Arabidopsis transgenic seedlings. The constitutive expression of T474D was associated with stunted growth in the transgenic lines (Extended Data Fig. 1a) and repression of global protein synthesis (Fig. 1b). These phenotypes precluded the use of an appropriate normalization method for protein loading in comparative gels of contrasting genotypes to estimate precisely the T474D-mediated protein synthesis inhibition in our assay. To overcome this limitation, we used a dexamethasone-inducible promoter to control the T474D expression in the transgenic lines. Arabidopsis seedlings independently transformed with a T474D–GFP fusion under the control of a dexamethasone-inducible promoter (TR1 and TR2) were treated with 30 mM dexamethasone for 8 h, and the induction of T474D–GFP expression was monitored by semi-quantitative RT– PCR. The dexamethasone-induced expression of T474D for 8 h led to a higher inhibition of de novo protein synthesis in the transgenic lines, as measured by TCA-precipitable radioactivity, which could be normalized to total protein (TR1 and TR2; Fig. 1c). d, Inhibition of de novo protein synthesis by protein synthesis inhibitors in Col-0, untransformed lines. We also compared the T474D-mediated suppression of translation with known global translation inhibitors, such as the cytosolic protein synthesis inhibitors cycloheximide and puromycin and the chloroplast translation suppressor chloramphenicol. Arabidopsis seedlings (10 days old) were treated with 10 μM cycloheximide (Cyclo), 10 μM puromycin (Pur), 25 μM chloramphenicol (Chlor) or 30 μM dexamethasone (Dex) for the indicated periods of time, and then they were pulse labelled with L-[35S]Met for 60 min. Lysates of treated cells were measured by liquid scintillation counting and normalized to total protein. The relative [35S]Met incorporation was normalized to wild-type (WT = 1) control without treatment. Means ± 95% confidence intervals (n = 3) based on bootstrap resampling replicates of three independent experiments are shown. e, Inhibition of de novo protein synthesis by inducible expression of T474D. Seedlings (10 days old) from the TR1 transgenic line were treated with dexamethasone and the protein synthesis inhibitors for the times as indicated in the figure, and then they were pulse labelled with L-[35S]Met for 60 min. Lysates were processed as described in d. Means 6 95% confidence interval (n = 3) based on bootstrap resampling replicates of three independent experiments are shown. Cycloheximide was the most effective inhibitor of translation in both the wild-type and T474D-expressing lines. T474D expression inhibited global translation to the same extent as puromycin and caused a further inhibition in the level of chloramphenicol translational inhibition in a combined treatment. The increase in translational inhibition by combining T474D expression and chloramphenicol treatment may indicate that T474D inhibits cytosolic protein synthesis, which is consistent with the T474D-mediated downregulation of components of the cytosolic translational machinery (Supplementary Table 3).
Extended Data Figure 4. Isolation of PS fractions from Arabidopsis seedlings and LIMYB-mediated inhibition of protein synthesis.
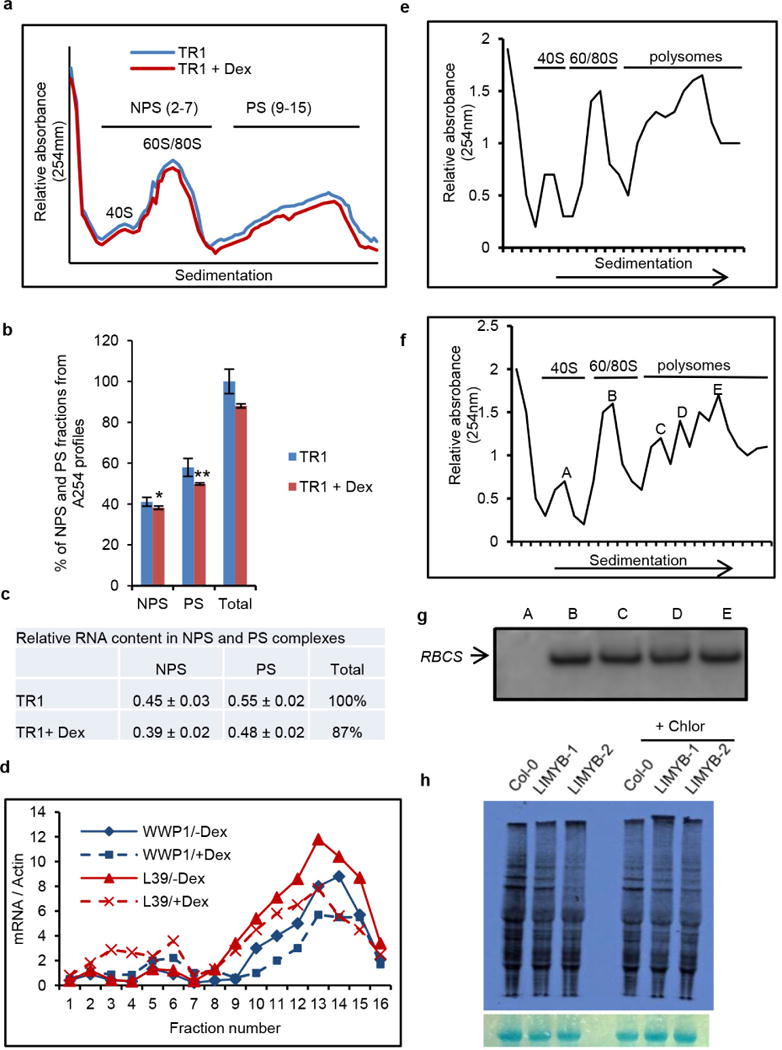
a, Ultraviolet absorbance profiles of the sucrose gradient used for RNA fractionation of dexamethasone (Dex)-inducible T474D transgenic lines. Sixteen fractions of 310 ml were collected. NPS RNA includes complexes ≤ 80S that fractionated in the top half of the gradient (fractions 2–7) and PS (polysome) represents complexes fractionated in the bottom of the gradient (fractions 9–15). b, Distribution (%) of NPS and PS fractions on polysome density gradients. The percentage of NPS or PS was calculated by integrating the areas under the corresponding peaks in the A254nm profile delimited by a gradient baseline absorbance (buffer density). Values are the average ± standard deviation (s.d.) of three biological replicates. NPS and PS fractions from T474D + Dex samples were significantly different from the corresponding fractions of the T474D samples by the t-test (greater). NPS, *P value = 0.03725; PS, **P value = 0.009137 (t-test; greater). c, Relative RNA content in NPS and PS complexes. RNA was precipitated from NPS and PS density regions of sucrose gradients (as in a) and quantified. Relative NPS RNA and PS RNA contents from T474D and T474D + Dex samples were calculated in relation to the total NPS + PS content from T474D. Values for the relative NPS and PS RNA content are the average ± s.d. of three biological replicates and they were significantly different between the samples (P < 0.01, t-test). d, Distribution of specific mRNAs in the PS gradient fractions from extracts prepared from T474D seedlings treated (or not) with dexamethasone. The RNA on each fraction was reverse transcribed and aliquots amplified with specific primers for the indicated genes by qPCR. e, Ultraviolet absorbance profiles of the sucrose gradient used for the RNA fractionation of Col-0 seedlings. PSs from 15-day-old Col-0 seedlings were fractionated on a sucrose gradient, and the fractions were manually collected. f, Ultraviolet absorbance profiles of the sucrose gradient used for RNA fractionation from T474D seedlings. PSs from 15-day-old T474D-overexpressing seedlings were fractionated on a sucrose gradient, and the fractions were manually collected. g, Levels of the small subunit of rubisco (RBCS) mRNA per fraction. The levels of mRNA of RBCS were examined by northern blotting. This control was used to ensure the quality and distribution of a specific mRNA. h, Overexpression of LIMYB suppresses cytosolic translation. In vivo labelling of leaf proteins with [35S]Met was performed in Col-0 and LIMYB-1 transgenic seedlings in the presence and absence of chloramphenicol treatment. The total protein extracts were fractionated by SDS–PAGE, and the radioactive bands were quantified by densitometric analysis of the images obtained by autoradiography. The labelling percentage was normalized to the leaf chlorophyll content, and protein loading is shown by Coomassie staining of the radioactive gel.
Extended Data Figure 5. Ectopic expression of T474D and LIMYB confers tolerance to begomovirus infection.
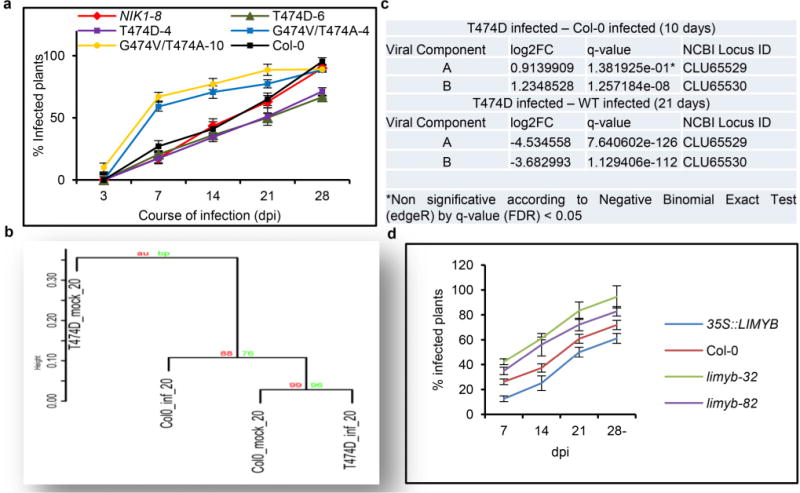
a, Delayed onset of infection in the T474D-4- and T474D-6-overexpressing lines. Ecotype Col-0 plants, as well as the T474D-4- and T474D-6-overexpressing lines, and the NIK1-8 and G473V/T474A-overexpressing lines were infected with CaLCuV DNA by the biolistic method. The progression of the infection was monitored by PCR detection of viral DNA in the systemic leaves of the inoculated plants. The values represent the percentages of systemically infected plants at different dpi. Error bars, 95% confidence intervals (n = 3) based on bootstrap resampling replicates of four independent experiments. b, Upon symptom development, the T474D-induced transcriptome diverges from the infected Col-0 transcriptome. The mock-inoculated T474D-overexpressing lines exhibited a constitutively infected wild-type transcriptome at 10 dpi (Fig. 1a). Nevertheless, these T474D transgenic lines did not phenocopy the infected wild-type plants because they did not develop symptoms of viral infection. In fact, the wild-type plants displayed typical symptoms of CaLCuV infection at 21 dpi, such as leaf distortion, stunting with epinasty and chlorosis (Fig. 1e). The symptoms in the T474D-expressing lines at 21 dpi, however, were greatly attenuated, with no visible leaf distortion or chlorosis. To examine these phenotypes, we performed a Ward hierarchical clustering of the gene expression data (normalized by the trimmed mean of M-values (TMM) normalization method) from the Arabidopsis infection experiments at 21 dpi. The TMM normalization method assumes that the majority of genes are not differentially expressed, and it adjusts genes with larger read counts and lower variance on the logarithmic scale. The dendrogram provides two types of P values: AU (black) and BP (grey). The AU P value comes from multiscale bootstrap resampling, while the BP value represents normal bootstrap resampling. These P values were calculated by multiscale bootstrap resampling using the R-cran package pvclust with a cut-off of 0.05. These P values show the significance of the proximity of each gene expression experiment profile. The cluster analysis at 21 dpi indicated that when symptoms had developed in the infected Col-0 leaves, the T474D-induced transcriptome diverged from the infected Col-0 transcriptome. The mock T474D transcriptome formed a unique clade, while the infected T474D transcriptome was more closely related to the mock-inoculated Col-0 transcriptome. c, Reduced viral transcript accumulation in T474D-overexpressing lines at 21 dpi. RNA-sequencing data of viral gene transcripts in the systemic leaves of infected wild-type and T474D-overexpressing plants at 10 dpi and 21 dpi. d, The onset of infection is delayed in LIMYB-overexpressing lines. Ecotype Col-0 plants and LIMYB-overexpressing and limyb mutant lines were infected with CaLCuV DNA using the biolistic method. The progression of the infection was monitored by the PCR detection of viral DNA in the systemic leaves of the inoculated plants. The values represent the percentages of systemically infected plants at different dpi. Error bars, 95% confidence intervals (n = 3) based on bootstrap resampling replicates of four independent experiments.
Extended Data Figure 6. LIMYB, which belongs to the MYB domain-containing superfamily, interacts with RPL10 in the yeast two-hybrid system.
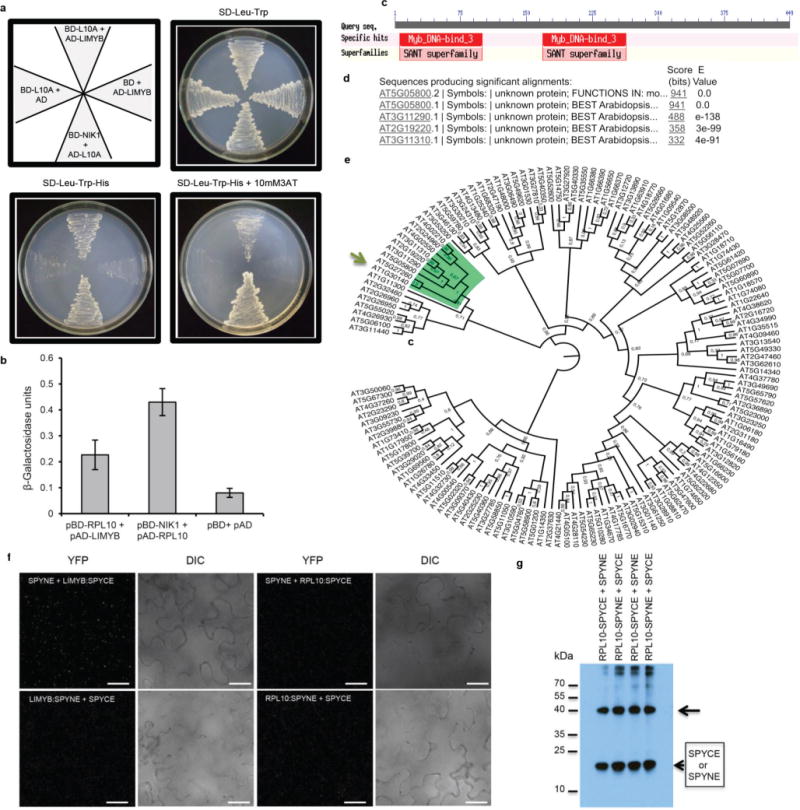
a, LIMYB and RPL10 interact in yeast. LIMYB was expressed in yeast as a GAL4 activation domain (AD) fusion (pAD–LIMYB), and RPL10 was expressed as a GAL4 binding domain (BD) fusion (pBD–RPL10). The interactions between the tested proteins were examined by monitoring His prototrophy. b, The interactions were further confirmed by measuring the expression activity of the β-galactosidase reporter enzyme for the second reporter gene, β-galactosidase. The interaction between pAD–RPL10 and pBD–NIK1 was monitored as a positive control. Means ± 95% confidence intervals (n = 3) based on bootstrap resampling replicates of three technical replicates are shown. c, LIMYB harbours two MYB domains. The position of these MYB domains is indicated in the schematic representation of the LIMYB primary structure. d, Sequence identity of the closest related LIMYB (At5g05800) homologues. e, Dendrogram of MYB domain-containing proteins from Arabidopsis. The MYB family sequences were retrieved from the Agris database (http://arabidopsis.med.ohio-state.edu). The alignment was performed by Maft aligner software using full-length sequences, and the tree was built by Fasttree software (the bootstrap values are indicated close to the branch divisions). The arrow indicates LIMYB (At5g05800). f, g, The negative controls used in the BiFC analysis. f, Confocal fluorescent image of SPYNE + LIMYB:SPYCE, LIMYB:SPYNE + SPYCE, SPYNE + RPL10:SPYCE and RPL10:SPYNE + SPYCE, as indicated in the figure. We used a BiFC assay to determine whether RPL10 and LIMYB interact in the nuclei of plant cells. The formation of a RPL10–LIMYB complex occurred in the nuclei of transfected cells independent of the orientation of the LIMYB or RPL10 fusions (amino terminus or carboxy terminus of YFP; Fig. 2a), and the reconstituted fluorescent signal was much higher than that of the background (control panels with combinations of the protein fusions with empty vectors). The figure displays representative samples from three independent biological repeats. Scale bars, 20 μm. g, The C-terminal (SPYCE) and N-terminal region (SPYNE) of YFP accumulates detectably in co-transfected leaves. Total protein extracts from leaves co-transfected with the indicated constructs were immunoblotted with anti-YFP serum. The arrow indicates the position of RPL10–SPYCE and RPL10–SPYNE fusions, and arrowheads indicate the positions of the C-terminal (SYYCE) and N-terminal (SPYNE) regions of YFP.
Extended Data Figure 7. Nuclear localization of LIMYB.
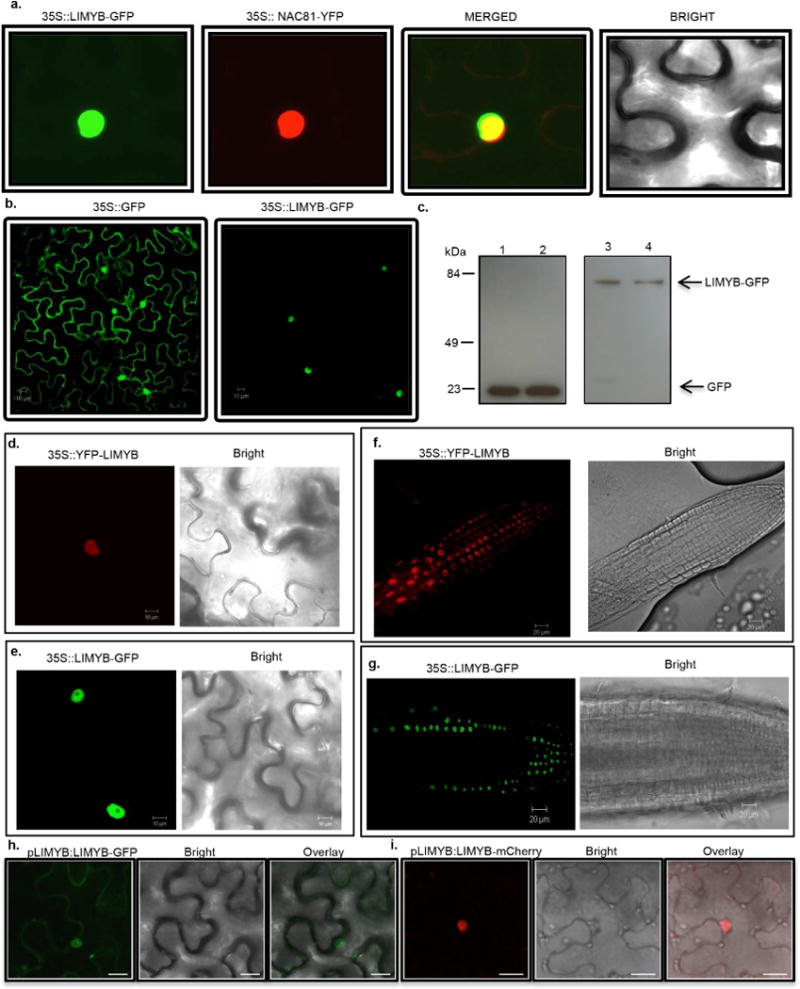
a, Colocalization of LIMYB with the nuclear marker gene Glycine max (Gm)NAC81. N. benthamiana leaves were co-infiltrated with A. tumefaciens carrying a 35S∷LIMYB–GFP construct and a 35S∷YFP–GmNAC81 construct. Forty-eight hours after infiltration, the subcellular localizations of the fluorescent fusion proteins were examined by confocal microscopy. The figure shows representative confocal images from two independent experiments. b, Confocal fluorescence image of transiently expressed GFP (left) or LIMYB–GFP (right) in epidermal cells of tobacco leaves. Scale bars, 10 μm. The figure shows representative confocal images from two independent experiments. c, Immunoblotting of transiently expressed LIMYB–GFP in epidermal cells of tobacco leaves. Total protein was extracted from agro-infiltrated N. benthamiana leaves containing the 35S∷GFP (left lanes) or 35S∷LIMYB–GFP (right lanes) constructs and immunoblotted with an anti-GFP monoclonal antibody to examine the stability of the fusion protein. The positions of molecular mass are shown in kDa. d, Confocal fluorescence image of transiently expressed GFP–LIMYB in epidermal cells of tobacco leaves. Scale bars, 10 μm. The figure shows representative confocal images from four independent experiments. e, Confocal fluorescence image of transiently expressed LIMYB– GFP in epidermal cells of tobacco leaves. Scale bars, 10 μm. The figure shows representative confocal images from four independent experiments. f, g, Confocal fluorescence image of root cells stably transformed with YFP– LIMYB or LIMYB–GFP. Root tips from transgenic seedlings expressing YFP–LIMYB (f) or LIMYB–GFP (g) were directly examined under a laser confocal microscope. Scale bars, 20 μm. The figures show representative confocal images from three biological replicas. Neither the fusion of YFP to the LIMYB N terminus nor GFP to its C terminus altered the nuclear localization of LIMYB in either agro-inoculated N. tabacum leaves or stably transformed Arabidopsis roots. h, i, Confocal fluorescent image of LIMYB fused to GFP or mCherry under the control of its own promoter. The figures show representative confocal images from two independent experiments. The fluorescence was also concentrated in the nucleus of transfected cells by expression of LIMYB–GFP or LIMYB–mCherry fusions under the control of the LIMYB endogenous promoter. Scale bars, 20 μm. Collectively, these results indicate that LIMYB was localized in the nucleus.
Extended Data Figure 8. LIMYB, RPL10 and NIK1 display overlapping expression profiles.
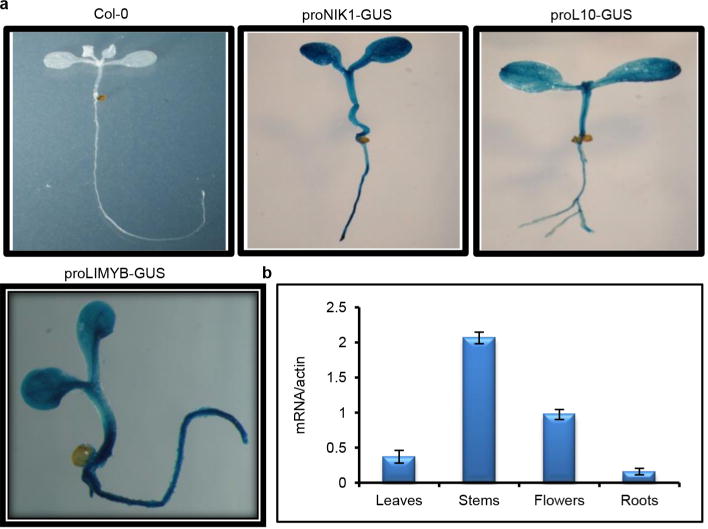
a, pLIMYB∷GUS, pRPL10∷GUS and pNIK1∷GUS are ubiquitously expressed in seedling tissues. GUS reporter gene expression was histochemically monitored in 2-week-old seedling leaves and roots from transgenic lines harbouring a β-glucuronidase (GUS) reporter gene expressed from the LIMYB, RPL10 and NIK1 promoters. The figure shows representative GUS staining images of three seedlings per genotype. All three genes were ubiquitously expressed in all seedling tissues. b, Expression analysis of LIMYB in various plant organs. LIMYB expression was monitored by qRT–PCR of RNA prepared from leaves, roots, stems or flowers of Col-0 plants. Gene expression was calculated using the 2−ΔCt method, and actin was used as an endogenous control. Error bars, 95% confidence intervals (n = 3) based on bootstrap resampling replicates of three independent experiments. LIMYB was also expressed in the leaves, roots, stems and flowers, indicating that LIMYB, RPL10 and NIK1 are co-expressed in several organs.
Extended Data Figure 9. Controls for the regulation of RP gene expression experiments.
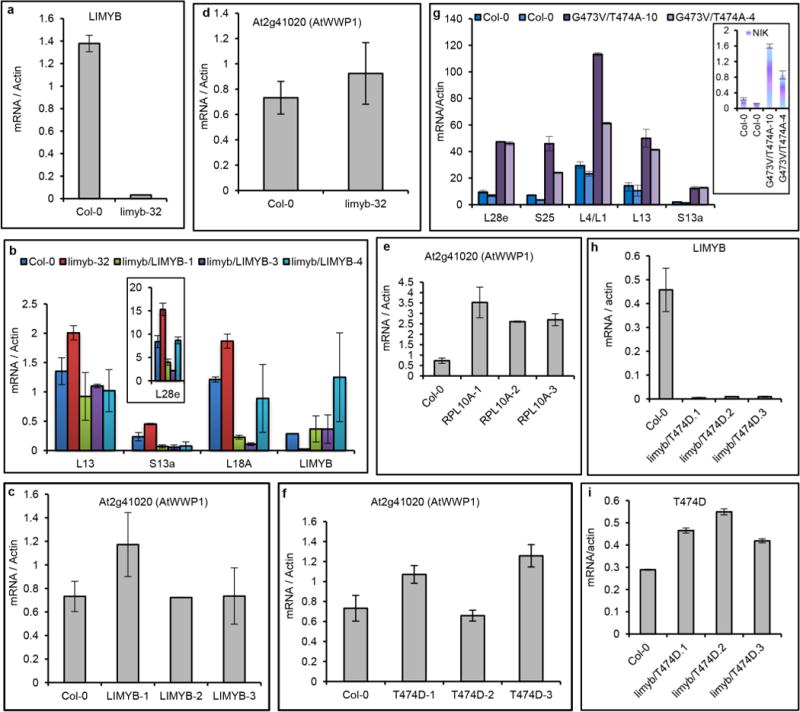
a–i, Expression of the indicated genes in the leaves of independent transgenic lines was monitored by qRT–PCR. a, LIMYB expression in the limyb-32 mutant was examined. b, LIMYB expression in limyb-32 mutant restores wild-type expression of the RP genes. Expression of the S13a, L18A and L28e genes was monitored in three independently transformed limyb-32 knockout plants with the LIMYB gene. c–f, Expression of the unrelated gene AtWWP1 was monitored as a negative control in three independently transformed LIMYB-, RPL10- and T474D-overexpressing lines in addition to the limyb-32 mutant. g, The double-mutant inactive kinase, G4743V/T474A, does not downregulate the RP genes. The transcript accumulation of the indicated RP genes was quantified by qRT–PCR in two independently transformed nik1 knockout lines expressing the G4743V/T474A double mutant. h, i, Expression of LIMYB (h) and the transgene T474D (i) was monitored in the limyb-32 lines, which were transformed with T474D. a–i, Means ± 95% confidence intervals (n = 3) based on bootstrap resampling replicates of three independent experiments are shown.
Extended Data Figure 10. T474D requires LIMYB function to mediate the downregulation of translational machinery-related genes.
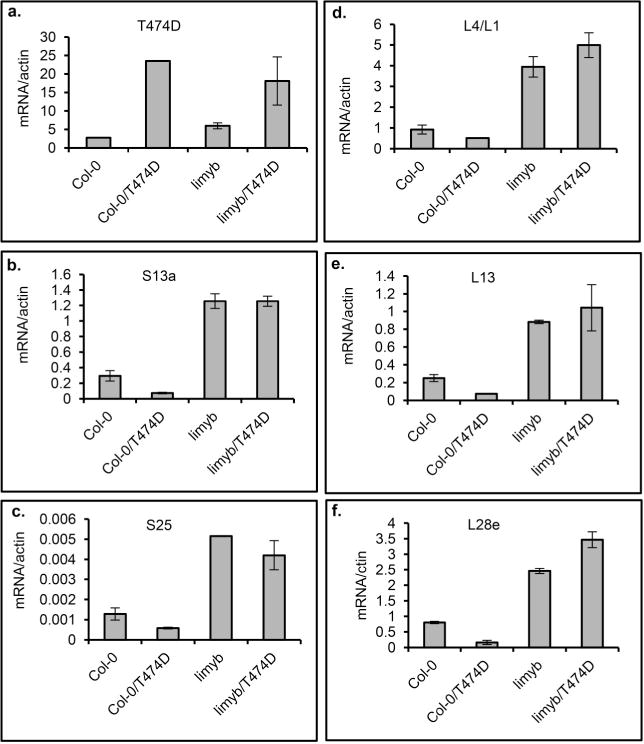
a–f, A 35S∷T474D–GFP construct was electroporated into protoplasts prepared from Col-0 and limyb-32 seedlings, and the expression of the indicated RP genes was monitored by qRT–PCR of RNA prepared from untransfected and transfected protoplasts. Gene expression was calculated using the 2−ΔCt method, and actin and ubiquitin were used as an endogenous control. Means ± 95% confidence intervals (n = 3) based on bootstrap resampling replicates of three independent experiments are shown.
Supplementary Material
Acknowledgments
This research was financially supported by the following grants from Brazilian government agencies: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) grants 573600/2008-2 and 470287/2011-0 (to E.P.B.F.) and FAPEMIG grant CBB-APQ-00070-09 (to E.P.B.F.); and by the US National Institutes of Health grant 5R01-GM94428 (to J.C.). O.J.B.B. was supported by a Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) graduate fellowship; C.Z., K.V.G.L., J.P.B.M., I.P.C., B.C.G. and P.A.B.R. were supported by CNPq graduate fellowships; K.J.T.N., W.A.P. and M.D. were supported by postdoctoral fellowships from CNPq. A.A.S. was supported by postdoctoral fellowships from CAPES; and M.A.C.S. was the recipient of an undergraduate scholarship from CNPq. J.C. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
Author Contributions C.Z. and J.P.B.M. co-wrote the manuscript and performed most of the experiments related to LIMYB isolation and characterization. K.V.G.L. performed the T474D-related experiments. K.J.T.N. performed qPCR for viral DNA. W.A.P. generated the RPL10 constructs and conducted plant transformation. O.J.B.B. and F.F.S. performed the bioinformatic analysis of the RNA-sequencing data and the statistical analysis of the data. M.D. performed the infectivity assays. B.C.G. conducted qRT–PCR. P.A.B.R. and I.P.C. performed the protein synthesis assays. V.A.P.L. performed complementation assays. G.S.-M. performed the bimolecular fluorescence complementation experiments. M.A.C.S. performed the tissue expression experiments. A.A.S. constructed the mutant proteins and designed the infectivity assays. J.C. conceived the experiments and edited the final draft. E.P.F.B. co-wrote the manuscript, designed the experiments and directed the project.
RNA-sequencing data have been deposited in the Gene Expression Omnibus under accession number GSE56922
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Pumplin N, Voinnet O. RNA silencing suppression by plant pathogens: defence, counter-defence and counter-counter-defence. Nature Rev Microbiol. 2013;11:745–760. doi: 10.1038/nrmicro3120. [DOI] [PubMed] [Google Scholar]
- 2.Mandadi KK, Scholthof KB. Plant immune responses against viruses: how does a virus cause disease? Plant Cell. 2013;25:1489–1505. doi: 10.1105/tpc.113.111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones JD, Dangl JL. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 4.Hanley-Bowdoin L, Bejarano ER, Robertson D, Mansoor S. Geminiviruses: masters at redirecting and reprogramming plant processes. Nature Rev Microbiol. 2013;11:777–788. doi: 10.1038/nrmicro3117. [DOI] [PubMed] [Google Scholar]
- 5.Santos AA, Lopes KVG, Apfta JAC, Fontes EPB. NSP-interacting kinase, NIK: a transducer of plant defence signalling. J Exp Bot. 2010;61:3839–3845. doi: 10.1093/jxb/erq219. [DOI] [PubMed] [Google Scholar]
- 6.Mariano AC, et al. Identification of a novel receptor- like protein kinase that interacts with a geminivirus nuclear shuttle protein. Virology. 2004;318:24–31. doi: 10.1016/j.virol.2003.09.038. [DOI] [PubMed] [Google Scholar]
- 7.Sakamoto T, et al. The tomato RLK superfamily: phylogeny and functional predictions about the role of the LRRII-RLK subfamily in antiviral defense. BMC Plant Biol. 2012;12:229. doi: 10.1186/1471-2229-12-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shiu SH, Bleecker AB. Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc Natl Acad Sci USA. 2001;98:10763–10768. doi: 10.1073/pnas.181141598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chinchilla D, Shan L, He P, de Vries S, Kemmerling B. One for all: the receptor-associated kinase BAK1. Trends Plant Sci. 2009;14:535–541. doi: 10.1016/j.tplants.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fontes EPB, Santos AA, Luz DF, Waclawovsky AJ, Chory J. The geminivirus NSP acts as virulence factor to suppress an innate transmembrane receptor kinase-mediated defense signaling. Genes Dev. 2004;18:2545–2556. doi: 10.1101/gad.1245904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santos AA, Carvalho CM, Florentino LH, Ramos JJO, Fontes EPB. Conserved threonine residues within the A-loop of the receptor NIK differentially regulate the kinase function required for antiviral signaling. PLoS ONE. 2009;4:e5781. doi: 10.1371/journal.pone.0005781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carvalho CM, et al. Regulated nuclear trafficking of rpL10A mediated by NIK1 represents a defense strategy of plant cells against virus. PLoS Pathog. 2008;4:e1000247. doi: 10.1371/journal.ppat.1000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rocha CS, Santos AA, Machado JPB, Fontes EPB. The ribosomal protein L10/QM-like protein is a component of the NIK-mediated antiviral signaling. Virology. 2008;380:165–169. doi: 10.1016/j.virol.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Oh HS, Kwon H, Sun SK, Yang CH. QM, a putative tumor suppressor, regulates proto-oncogene c-Yes. J Biol Chem. 2002;277:36489–36498. doi: 10.1074/jbc.M201859200. [DOI] [PubMed] [Google Scholar]
- 15.Imafuku I, et al. Presenilin 1 suppresses the function of c-Jun homodimers via interaction with QM/Jif-1. J Cell Biol. 1999;147:121–134. doi: 10.1083/jcb.147.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monteclaro FS, Vogt PK. A Jun-binding protein related to a putative tumor suppressor. Proc Natl Acad Sci USA. 1993;90:6726–6730. doi: 10.1073/pnas.90.14.6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vinatzer BA, et al. The type III effector repertoire of Pseudomona syringae pv. syringae B728a and its role in survival and disease on host and non-host plants. Mol Microbiol. 2006;62:26–44. doi: 10.1111/j.1365-2958.2006.05350.x. [DOI] [PubMed] [Google Scholar]
- 18.Delú-Filho N, et al. A sucrose binding protein homologue from soybean affects sucrose uptake in transgenic tobacco suspension-cultured cells. Plant Physiol Biochem. 2000;38:353–361. [Google Scholar]
- 19.Robinson MD, McCarthy DJ, Smyth GK. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ward JH., Jr Hierarchical grouping to optimize an objective function. J Am Stat Assoc. 1963;58:236–244. [Google Scholar]
- 23.Kim TH, Kim BH, Yahalom A, Chamovitz DA, von Arnim AG. Translational regulation via 59 mRNA leader sequences revealed by mutational analysis of the Arabidopsis translation initiation factor subunit eIF3h. Plant Cell. 2004;16:3341–3356. doi: 10.1105/tpc.104.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawaguchi R, William AJ, Bray EA, Bailey-Serres J. Water deficit-induced translational control in Nicotiana tabacum. Plant Cell Environ. 2003;26:221–229. [Google Scholar]
- 25.Kawaguchi R, Girke T, Bray EA, Bailey-Serres J. Differential mRNA translation contributes to gene regulation under non-stress and dehydration stress conditions in Arabidopsis thaliana. Plant J. 2004;38:823–839. doi: 10.1111/j.1365-313X.2004.02090.x. [DOI] [PubMed] [Google Scholar]
- 26.Santos AA, Florentino LH, Pires ABL, Fontes EPB. Geminivirus: biolistic inoculation and molecular diagnosis. Methods Mol Biol. 2008;451:563–579. doi: 10.1007/978-1-59745-102-4_39. [DOI] [PubMed] [Google Scholar]
- 27.Florentino LH, et al. A PERK-like receptor kinase interacts with the geminivirus nuclear shuttle protein and potentiates viral infection. J Virol. 2006;80:6648–6656. doi: 10.1128/JVI.00173-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carvalho CM, et al. A novel nucleocytoplasmic traffic GTPase identified as a functional target of the bipartite geminivirus nuclear shuttle protein. Plant J. 2008;55:869–880. doi: 10.1111/j.1365-313X.2008.03556.x. [DOI] [PubMed] [Google Scholar]
- 29.Pinheiro GL, et al. Complete inventory of soybean NAC transcription factors: sequence conservation and expression analysis uncover their distinct roles in stress response. Gene. 2009;444:10–23. doi: 10.1016/j.gene.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 30.Costa MDL, et al. A new branch of endoplasmic reticulum-stress signaling and the osmotic signal converge on plant specific asparagine-rich proteins to promote cell death. J Biol Chem. 2008;283:20209–20219. doi: 10.1074/jbc.M802654200. [DOI] [PubMed] [Google Scholar]
- 31.DiCiccio TJ, Efron B. Bootstrap confidence intervals. Stat Sci. 1996;11:189–228. [Google Scholar]
- 32.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.