

Abstract
Veratramine, a major alkaloid from Veratrum nigrum L., has distinct anti-tumor and anti-hypertension effects. Our previous study indicated that veratramine had severe toxicity toward male rats. In order to elucidate the underling mechanism, in vivo pharmacokinetic experiments and in vitro mechanistic studies have been conducted. Veratramine was administrated to male and female rats intravenously via the jugular vein at a dose of 50 μg/kg or orally via gavage at 20 mg/kg. As a result, significant pharmacokinetic differences were observed between male and female rats after oral administration with much lower concentrations of veratramine and 7-hydroxyl-veratramine and higher concentrations of veratramine-3-O-sulfate found in the plasma and urine of female rats. The absolute bioavailability of veratramine was 0.9% in female rats and 22.5% in male rats. Further experiments of veratramine on Caco-2 cell monolayer model and in vitro incubation with GI content or rat intestinal subcellular fractions demonstrated that its efficient passive diffusion mediated absorption with minimal intestinal metabolism, suggesting no gender-related difference during its absorption process. When veratramine was incubated with male or female rat liver microsomes/cytosols, significant male-predominant formation of 7-hydroxyl-veratramine and female-predominant formation of veratramine-3-O-sulfate were observed. In conclusion, the significant gender-dependent hepatic metabolism of veratramine could be the major contributor to its gender-dependent pharmacokinetics.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-016-9870-9) contains supplementary material, which is available to authorized users.
KEY WORDS: 7-hydroxyl-veratramine, enterohepatic recirculation, gender-dependent pharmacokinetics, veratramine, veratramine-3-O-sulfate
INTRODUCTION
Genus Veratrum of Liliaceae, commonly known as American Hellebore or Black False Hellebore and which have a variety of Veratrum alkaloids as active components, has been used for medicinal purposes to treat hypertension, epilepsy, stroke, excessive phlegm, jaundice, scabies, chronic malaria, and wounds in European and Asian countries (1–5). Veratramine (Fig. 1), a C-nor-D-homosteroidal Veratrum alkaloid and the key marker compound of Veratrum (6–8), demonstrated potent anti-tumor (9,10), anti-hypertension (5,11,12), and anti-platelet (13) activities, and also acted as a cardiac (14,15) and 5-HT agonist (16). In addition, the mechanistic properties of its major pharmacological effects have been elucidated. The inhibition of the hedgehog (Hh) signaling pathway, a pathway related to the generation of tumors (17,18), was identified as the anti-tumor mechanism of veratramine against migration and proliferation (19), and the blocking of Na+ channels and reflex suppression of the cardiovascular system were related to its anti-hypertension activity (20). In the clinic, the alternative and complementary medicines, such as “Ji Ming Zi Pills,” primarily containing Veratrum alkaloids, were used for the treatment of breast tumor in China. Veratrum alkaloids have also been used clinically for the treatment of hypertension in Chinese hospitals and used medically by Native Americans and Europeans (3–5,11,21–23). Moreover, the marketed veratramine containing medicine “San Qi Xue Shang Ning Capsule” is listed in the Chinese Pharmacopeia (China, 2010) for the treatment of relieving symptoms of blood congestion. Although veratramine neither induced teratogenicity like most of the other Veratrum alkaloids (24), nor had obvious side effects at the anti-hypertension equivalent dose in Veratrum alkaloid extracts (6), the potential toxicity of genus Veratrum shouldn’t be ignored. So far, it has been reported that some of the alkaloids led to potential gender-dependent toxicities due to their gender-related pharmacokinetics (25–29). Male SD rats were shown to be more susceptible to clivorine (a pyrrolizidine alkaloid) intoxications than female rats due to the gender-associated metabolism (26–28). Therefore, the characterization of veratramine pharmacokinetic profiles of both gender, particularly in gender-related metabolism and elimination, is considered essential for the evaluation of its safety and potential interactions with other drugs or herbs.
Fig. 1.
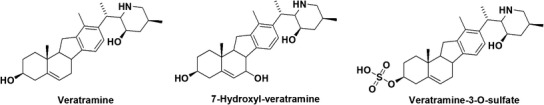
Chemical structures of veratramine, 7-hydroxyl-veratramine and veratramine-3-O-sulfate
Due to their potent biological activities, steroidal alkaloids have been investigated for their pharmacokinetic and pharmacodynamic characteristics (30–33). However, up to now, few studies evaluated the pharmacokinetic profile of veratramine and explored its potential gender-associated pharmacokinetics. It was previously indicated that the structural site of metabolism for veratramine did not occur at the common reactive nitrogen-containing side chain but that reductive and oxidative attacks at rings A and B occurred during the microbial biotransformation (34). Our recent metabolic study had identified seven hydroxylated, two sulfated and two glucuronidated metabolites of veratramine and indicated that CYP2D6-mediating hydroxylation and SULT2A1 and SULT1E1-mediating sulfation were the two major metabolic pathways for the formation of 7-hydroxyl-veratramine (Fig. 1) and veratramine-3-O-sulfate (Fig. 1) (35). During our preliminary pharmacokinetic investigations in rats after oral administration of 30 mg/kg of veratramine, it was surprisingly noticed that male rats easily suffered from toxic reactions, such as severe trembling, with a fatality rate of nearly 50%, whereas female rats were all alive without any toxic response. It is thus hypothesized that gender-dependent pharmacokinetics of veratramine may exist. Therefore, the current study was proposed to provide a comprehensive characterization of the gender-dependent pharmacokinetics of veratramine in rats with further mechanistic illustrations on the potential gender-dependent factors, especially gender-associated metabolism and elimination.
MATERIALS AND METHODS
Materials
Veratramine with purity >99% was purchased from Chembest Bioscience Inc. (Shanghai, China). Male and female rat liver microsomes/cytosols (RLMs/RLCs) and pooled rat intestinal S9 (RIS9) and pooled rat intestinal microsomes (RIMs) of male donor were purchased from XenoTech, LLC (KS, USA). β-Nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate (NADPH), 3′-phosphoadenosine-5′-phosphosulfate (PAPS), and digoxin (internal standard for ultra-performance liquid chromatography tandem mass-spectrometry (UPLC-MS/MS) (I.S.)) were obtained from Sigma-Aldrich (MO, USA). Methanol, acetonitrile, and all other solvents used for high-performance liquid chromatography (HPLC) were HPLC grade. Distilled and deionized water was used for HPLC and preparation of solutions. Other reagents were of at least analytical grade.
For cell experiments, Dulbecco’s modified Eagle’s medium, 0.25% trypsin-EDTA, fetal bovine serum, penicillin-streptomycin, Hank’s balanced salt solution (HBSS), and non-essential amino acids were purchased from Gibco BRL Co. Ltd (Carlsbad, CA, USA) and Life Technologies (Grand Island, NY, USA). Six-well plates with Transwell® inserts (24 mm, ID, 4.71 cm2, polycarbonate filter) were purchase from Corning Costar Corporation (Corning, NY, USA).
Pharmacokinetics of Veratramine in Male and Female Rats
Animal Experiments
Sprague–Dawley rats (male and female, 200–220 g) were obtained from Sino-British SIPPR/BK Lab. Animal Ltd., Co. (Shanghai, China). Rats were housed at 25°C, with a relative humidity of about 60% and a 12-h cycle of light to dark, and food and water were provided ad libitum. Rats were fasted for 12 h with free assess of water prior to drug administration. The right jugular vein of each SD rat was cannulated 1 day before experiments and the bile duct was also cannulated in the hepatobiliary excretion study as previously described (36). The above procedures were under anesthesia by intramuscular injection of ketamine. The rats were allowed free access to food 6 h after oral or intravenous (i.v.) administration of veratramine. All animal procedures were performed in accordance with Animal Ethics Committee guidelines.
For oral administration studies, veratramine, dissolved in PEG400/saline (1:1, v/v) at a final concentration of 5 mg/mL, was administered by gavage to male (n = 5) and female (n = 5) SD rats at a dose of 20 mg/kg. Blood samples (300 μL) were collected into heparinized tubes via a jugular vein catheter at 5, 10, 20, 30, 60, 90, 120, 240, 360, 480, 720, 1440, 2160, and 2880 min after oral administration, with a 300-μL saline given via the indwelling catheter immediately after each blood collection. The plasma was obtained by centrifugation of the collected blood at 4000×g for 5 min. In addition, to characterize the urinary elimination of both veratramine and its major metabolites, urine was collected at different time intervals of 240, 360, 480, 720, 1440, 2160, and 2880 min.
For i.v. administration study, veratramine dissolved in saline containing 5% PEG400 at 50 μg/mL was administrated to male (n = 5) and female (n = 5) rats via the jugular vein catheter at a bolus dose of 50 μg/kg. After the i.v. administration of veratramine, in order to avoid the contamination during the blood collection process, 100 μL of saline was immediately injected via the polyethylene tubing to rinse the drug residue. Such procedure was further verified by the in vitro test. The post-dose time points to gather blood samples were 5, 10, 20, 30, 60, 90, 120, 240, 360, 480, 720, and 1440 min. Urine was also collected at different time intervals of 240, 360, 480, 720, and 1440 min.
To assess the hepatobiliary excretion of veratramine-3-O-sulfate, the right jugular vein and the bile duct of male (n = 5) and female (n = 5) SD rats were cannulated by a polyethylene tube (0.5 mm ID, 1 mm OD, Portex Ltd., England), followed by i.v. (50 μg/kg) or oral (20 mg/kg) administration of veratramine. Bile was collected at different time intervals of 10, 20, 30, 45, 60, 90, 120, 150, 180, 240, 300, and 360 min post-dosing.
Sample Preparation
To prepare the plasma samples collected from oral and i.v. administrations, 400 μL of acetonitrile (ACN)/methanol/0.1% formic acid water (40:40:20, v/v/v) containing 1 μg/mL of I.S. was added into 100 μL of plasma, followed by vortex mixing (Vortex Genie 2; Scientific Industries, Bohemia, NY, USA) for 2 min. The mixture was then centrifuged for 10 min at 16,000×g, and the resulting supernatant was evaporated to dryness under a flow of nitrogen. The residue was then reconstituted in 100 μL of ACN/0.1% formic acid water (60:40, v/v). Aliquots (10 μL) were injected into the UPLC-MS/MS for analysis of veratramine and its major two metabolites.
To prepare the urine and bile samples, aliquots of 100 μL urine and 50 μL bile were mixed with different volumes of ACN/methanol (1:1, v/v) containing I.S. (100 μL for urine and 50 μL for bile), respectively, followed by sonication using Crest Tru-Sweep sonication bath (Crest Ultrasonics corporation, NJ, USA) for 5 min and vortex mixing for 2 min. After centrifugation (16,000×g) of the mixture for 10 min, the resulting supernatant was then transferred to 2 mL glass vials and an aliquot of 10 μL was subjected to UPLC-MS/MS for analysis.
Exploration of Mechanism of Intestinal Absorption of Veratramine
Cell Culture
Caco-2 cells, purchased from American Type Culture Collection, were cultured in Dulbecco’s modified Eagle’s medium at 37°C, supplemented with 10% fetal bovine serum, 1% non-essential amino acids, 100 U/mL of penicillin, and 100 U/mL of streptomycin in an atmosphere of 5% CO2 and 90% relative humidity. Caco-2 cells were subcultured in 0.25% trypsin-EDTA for 5 min, seeded at a density of 2 × 105 cells/cm2 onto Transwell inserts in six-well plates, and incubated for around 21 days. Transepithelial electrical resistance (TEER) values were determined to evaluate the integrity of Caco-2 cell monolayer prior to the final transport study with TEER values above 600 Ω·cm2 being acceptable.
Transport Study in the Caco-2 Monolayer Model
Caco-2 cell monolayers were rinsed twice and equilibrated with HBSS at 37°C for 20 min before the transport studies. For the bi-directional transport study (37), 6 μg/mL veratramine in HBSS (pH 7.4) was loaded into the donor side (1.5 mL for the apical side or 2.6 mL for the basolateral side). Aliquots (0.4 mL) of samples were collected from the receiver side at 10, 30, 60, 90, and 120 min, followed by immediate addition of 0.4 mL blank HBSS into the receiver side. After a 120-min incubation, the polycarbonate components with Caco-2 cell monolayer were carefully rinsed twice with blank HBSS, followed by cutting off these components and lysing the cells with 3 mL ACN. The samples collected from receiver side were prepared by the addition of an equal volume ACN containing I.S. After 2 min vortex mixing and 15 min centrifugation at 16,000×g, all of the sample supernatants were immediately injected to UPLC-MS/MS for analysis.
In Vitro Intestinal Enzymatic Kinetic Study
RIS9 and RIMs Incubation
To investigate the metabolic profiles of veratramine in intestine, veratramine (12.5 μM) was preincubated for 2 min with RIS9 at a final concentration of 1 mg/mL in 100 mM potassium phosphate buffer (pH 7.4) containing 3.3 mM MgCl2 at 37°C. The hydroxylation reaction was initiated by the addition of 1 mM NADPH. The reaction mixture was incubated at 37°C for 60 min. The incubation medium for the sulfation of veratramine consisted of 50 mM potassium phosphate buffer (pH 7.4), 5 mM MgCl2, 200 μM PAPS, and 12.5 μM veratramine with a final RIS9 concentration of 1 mg/mL, and the mixture was incubated at 37°C for 60 min. RIMs were also used to evaluate the hydroxylation metabolism of veratramine in the presence of NADPH using the above procedure. All of the reactions described above were terminated by the addition of an equal volume of ice-cold ACN, followed by vortex mixing (2 min) and centrifugation (16,000×g, 10 min). The resulting supernatants (10 μL) were subjected to UPLC-MS/MS analysis.
To evaluate the potential hydrolysis of veratramine-3-O-sulfate, a range of concentrations of veratramine-3-O-sulfate, collected from the bile of female SD rats after oral administration of veratramine (20 mg/kg), were incubated with RIS9 (2 mg/mL) for 30 min in 50 mM potassium phosphate buffer (pH 7.4) containing 5 mM MgCl2. The reactions were terminated by the addition of equal amount of ice-cold ACN containing I.S. after a 30-min incubation, followed by 2 min of vortex mixing and 10 min of centrifugation at 16,000×g. The corresponding supernatant (10 μL) was injected onto UPLC-MS/MS for analysis of the formation of veratramine due to the hydrolysis of veratramine-3-O-sulfate.
GI Content Incubation
To estimate the stability of veratramine, a GI content incubation study was performed as described elsewhere (38). Briefly, simulated fluids of stomach (pH 1.2), small intestine (pH 6.8), and large intestine (pH 7.4) were prepared as previously reported (39), and the GI content solutions were prepared from the contents taken from stomach, small intestine, and large intestine of SD rats and suspended in 5-fold volumes of the corresponding simulated fluid. The supernatants containing GI contents were kept on ice and used for subsequent experiments after vortex mixing and centrifugation at 8000×g for 5 min. For the stability study, veratramine (6 μg/mL) or veratramine-3-O-sulfate (collected in female bile) was incubated with different simulated fluids and GI content solutions in a shaking water bath (Selecta Unitronic 320 OR; Selecta, Spain) at 37°C. Aliquots (200 μL) of samples were continuously collected at 5, 30, 60, 120, and 240 min. The incubation was terminated by adding 200 μL ice-cold ACN and after standing for 2 min of vortex mixing and 10 min of centrifugation at 16,000×g. Ten-microliter aliquots of the resulting supernatants were subjected to UPLC-MS/MS for analysis. The percentage remaining in each of the different simulated fluids and GI contents, calculated by comparing the concentration at each different time point with that at time zero, was used to evaluate the stability of veratramine and veratramine-3-O-sulfate.
Exploration of Mechanism of Hepatic Metabolism of Veratramine
To evaluate the hepatic enzymatic kinetics for the conversion of veratramine to 7-hydroxyl-veratramine, a series of concentrations of veratramine, ranging from 6.25 to 250 nM, were preincubated for 2 min with pooled male rat liver microsomes (mRLM) or pooled female rat liver microsomes (fRLM) in 100 μL of 100 mM potassium phosphate buffer (pH 7.4) containing 3.3 mM MgCl2 at 37°C for 30 min. The optimized protein concentrations of mRLM and fRLM were both at 0.5 mg/mL. The reaction was initiated by the addition of NADPH with a final concentration of 1 mM, and terminated by the addition of an equal volume of ice-cold ACN-containing I.S. The terminal mixture was vortex mixed and then centrifuged at 16,000×g for 10 min. The resulting supernatant was immediately analyzed by UPLC-MS/MS.
To investigate the hepatic enzymatic kinetics for the formation of veratramine-3-O-sulfate, a range of concentrations of veratramine (1 to 200 nM) were preincubated for 2 min with male rat liver cytosols (mRLC, 0.5 mg/mL) or female rat liver cytosols (fRLC; 0.2 mg/mL) in 100 μL of incubation mixture, containing 50 mM potassium phosphate buffer (pH 7.4) and 5 mM MgCl2 at 37°C. The reactions were performed at 37°C for 30 min after addition of 200 μM PAPS and terminated by the addition of 100 μL of ice-cold ACN-containing I.S., followed by 2 min of vortex mixing and 10 min of centrifugation at 16,000×g. Aliquots (10 μL) of the supernatant were subjected to UPLC-MS/MS for analysis.
UPLC-MS/MS Sample Analyses
For the quantification of veratramine, 7-hydroxyl-veratramine and veratramine-3-O-sulfate, UPLC-MS/MS analysis was developed using a series 1290 UPLC system (Agilent, USA) coupled with an Agilent 6430 triple quadruple mass spectrometer (Agilent, USA). Veratramine, 7-hydroxyl-veratramine, veratramine-3-O-sulfate, and I.S. were separated on a Waters ACQUITY UPLC BEH C18 column (50 × 2.1 mm, 1.7 μm particles). A mixture of 0.1% formic acid water (mobile phase A) and pure ACN (mobile phase B), with a linear gradient elution program at a flow rate of 0.2 mL/min, were used for separation. The gradient program was described as following: 0–6 min (10–50% B), 6–8 min (50–80% B), and 8–10 min (80–10% B), and the injection volume was 10 μL. Electrospray ion source (ESI) was performed in positive ion mode. The multiple reaction monitoring mode, with the transitions of m/z 410 → 295 (collision energy (CE), 27 eV) for veratramine, m/z 426 → 311 (CE: 27 eV) for 7-hydroxyl-veratramine and m/z 490 → 392 (CE: 26 eV) for veratramine-3-O-sulfate, were adopted as the quantification method. The optimized working parameters were: capillary voltage, 4.0 kV; source temperature, 300°C; nebulizer gas, 50 psi; and desolvation gas, 10 L/min. The results of the bioanalytical method validation of veratramine including precision, accuracy, recovery, matrix effect, and stability are shown in Supplemental 1, which indicated that the assay was acceptable for the determination of veratramine in vitro and in vivo. Since the authentic standards of 7-hydroxyl-veratramine and veratramine-3-O-sulfate were unavailable, the signal strength conversion factor was used to adjust the standard curve of veratramine for the quantification of 7-hydroxyl-veratramine and veratramine-3-O-sulfate as previously described (35,40,41). In brief, rat liver cytosolic incubations of veratramine with a series of concentrations were performed in the absence and presence of PAPS. The molar amount of veratramine-3-O-sulfate generated was assumed to be equivalent to the molar amount of veratramine diminished. The signal strength conversion factor could be calculated as the slope of the plot made by peak area/IS of veratramine-3-O-sulfate generated (veratramine-3-O-sulfate/IS) vs. that of veratramine diminished (veratramine/IScontrol − veratramine/ISreaction). A similar method was conducted for 7-hydroxyl-veratramine to calculate the signal strength conversion factor. Due to the lack of the authentic standards of 7-hydroxyl-veratramine and veratramine-3-O-sulfate, the incurred sample reassay (42) was performed to determine the reproducibility with the calculation of ratio limits (RL) and limits of agreement (LA). Finally, the RL and LA were 0.98–1.01 and 0.95–1.04 for 7-hydroxyl-veratramine and 0.96–1.06 and 0.87–1.16 for veratramine-3-O-sulfate. All the RL and LA results fell in the acceptance range with the variability lower than 15%, which indicated the assay for preliminary quantification of 7-hydroxyl-veratramine and veratramine-3-O-sulfate could be used.
Data Analysis
Pharmacokinetic Studies
The pharmacokinetic parameters, including the elimination half-life (t1/2), the area under the concentration-time curve (AUC), and the mean residence time (MRT), were calculated by non-compartmental method using Kinetica™ (Thermo Electron Corporation, PA, USA). Cmax values were obtained from the individual drug plasma concentration-time profile. AUC was evaluated using linear trapezoidal rule. Additionally, to evaluate the gender-related metabolism of veratramine, metabolic ratios (molar concentrations were used for the determination) for the hydroxylation and sulfation were calculated based on Eq. 1:
| 1 |
Absorption Studies Using the Caco-2 Monolayer Model
The permeability coefficients (Papp) (Eq. 2) of veratramine in the Caco-2 cell monolayer model were calculated as described before (43):
| 2 |
Where dC/dt stands for the changing velocity of concentration in receiver chamber. V is the volume of receiver chamber. A stands for the membrane area. C is the drug loading concentration in donor chamber. The corresponding efflux ratio is calculated by Papp (B-A)vs. Papp (A-B).
Metabolic Enzyme Kinetics
The reaction rate, expressed as the amount of metabolite formed per minute per milligram protein, was calculated after incubations using various hepatic or intestinal subcellular fractions. The metabolic enzyme kinetics were obtained by fitting the collected data into the typical form of the Michaelis-Menten equation (Eq. 3), or the atypical equation (Eq. 4) based on the characteristic profile of the Eadie-Hofstee plot (44), using GraphPad Prism 5.00 software (CA, USA).
Typical Michaelis-Menten equation:
| 3 |
Atypical Michaelis-Menten equation (substrate inhibition):
| 4 |
Where V is the velocity, [S] stands for the substrate concentration, Vmax is the maximal velocity, Km stands for the substrate concentration at half of the maximal velocity, and Ki is the inhibition constant. The intrinsic clearance (CLint) was calculated as a ratio of Vmax to Km.
Statistical Analyses
All reported values were presented as mean ± SD. A value of p < 0.05 was considered to be statistically significant, using an unpaired t test by SPSS Statistics 16.0 (SPSS Inc. Chicago, IL, USA).
RESULTS
Gender-Dependent Pharmacokinetics in Rats
Plasma Concentrations vs. Time Profiles
As shown in Fig. 2, veratramine and its major two metabolites (7-hydroxyl-veratramine and veratramine-3-O-sulfate) were formed not only in vitro but also in vivo. Plots of the plasma concentrations vs. time curves of veratramine, 7-hydroxyl-veratramine, and veratramine-3-O-sulfate after oral administration (20 mg/kg veratramine) to both male and female rats are shown in Fig. 3. The plasma concentrations of veratramine reached the first peak at 6 h and rapidly declined to the valley at 8 h, followed by a gradual increasing to a second, smaller peak near 12 h, and subsequently decreased slowly until 48 h after oral administration to male rats. Moreover, double peaks were also noticed in the plasma concentration vs. time profiles of 7-hydroxyl-veratramine and veratramine-3-O-sulfate. As shown in Table I, there were statistically significant differences among the pharmacokinetics parameters for male and female rats after oral dose of veratramine (20 mg/kg). The Cmax and AUC0-∞ of veratramine were 51.1 ng/mL and 53.5 min μg/mL, respectively, in male rats, and 4.33 ng/mL and 2.74 min μg−1 mL−1, respectively, in female rats. The t1/2 and MRT of veratramine in male rats were 2-fold longer than that in female rats, and in comparison with male rats, the CL of veratramine was significantly higher in female rats, suggesting a significant gender-related elimination of veratramine from systemic circulation. The absolute bioavailability of veratramine in male and female rats was 22.5% and 0.9%, respectively, which indicated a potent difference in veratramine pharmacokinetics between male and female rats. From the plasma concentration vs. time profiles of 7-hydroxyl-veratramine and veratramine-3-O-sulfate, the major two metabolites of veratramine in plasma, a significant gender-related difference on the metabolism of veratramine was also revealed. However, for the i.v. administration of veratramine (Fig. 3), the gender-dependent pharmacokinetic differences (Table I) were not obvious.
Fig. 2.
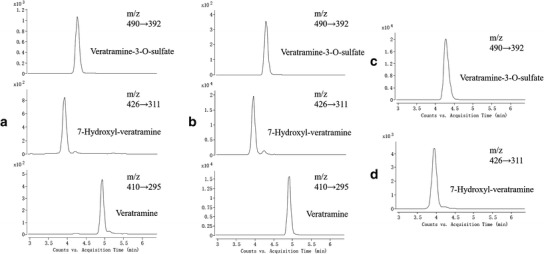
Representative extracted ion chromatographs (MRM) of veratramine, 7-hydroxyl-veratramine and veratramine-3-O-sulfate in a plasma (2 ng/mL of veratramine, 4 ng/mL of 7-hydroxyl-veratramine, and 50 ng/mL of veratramine-3-O-sulfate) and b urine (70 ng/mL of veratramine, 100 ng/mL of 7-hydroxyl-veratramine, and 20 ng/mL of veratramine-3-O-sulfate) after oral administration of 20 mg/kg veratramine; and in incubations of c fRLCs with veratramine in the presence of PAPS (950 ng/mL of veratramine-3-O-sulfate) and d mRLMs with veratramine in the presence of NADPH (20 ng/mL of 7-hydroxyl-veratramine)
Fig. 3.
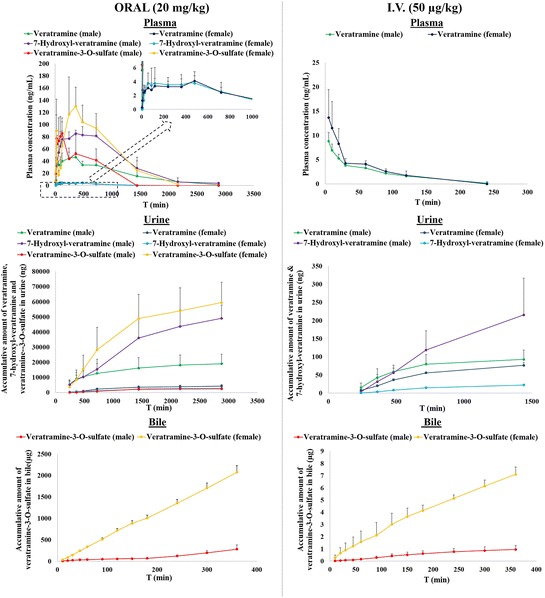
Mean plasma concentrations vs. time profiles and urinary and biliary accumulative amounts of veratramine, 7-hydroxyl-veratramine, and veratramine-3-O-sulfate in male and female rats after oral (20 mg/kg) (left column) or intravenous administration (50 μg/kg) (right column) of veratramine
Table I.
Pharmacokinetic Parameters of Veratramine After Oral (20 mg/kg) and Intravenous (50 μg/kg) Administration to Male and Female Rats
| PK parameters | Oral administration | Intravenous administration | ||
|---|---|---|---|---|
| Male (n = 5) | Female (n = 5) | Male (n = 5) | Female (n = 5) | |
| C max (ng/mL) | 51.1 ± 9.3 | 4.33 ± 1.37** | 8.81 ± 1.81 | 13.7 ± 5.7 |
| AUC0–t (min μg/mL) | 52.6 ± 20.5 | 2.71 ± 0.86** | 0.460 ± 0.080 | 0.581 ± 0.179 |
| AUC0–∞ (min μg/mL) | 53.5 ± 20.9 | 2.74 ± 0.91** | 0.594 ± 0.083 | 0.745 ± 0.339 |
| t 1/2 (min) | 493 ± 130 | 219 ± 148* | 68.6 ± 15.5 | 53.2 ± 27.1 |
| MRT (min) | 833 ± 214 | 494 ± 109** | 92.2 ± 20.4 | 72.8 ± 28.6 |
| CL/F (mL/min) | 95.4 ± 3.0 | 1529 ± 425** | 19.8 ± 3.1 | 17.5 ± 5.8 |
| CLrenal a (mL/min) | 0.343 ± 0.194 | 1.66 ± 0.39** | 0.158 ± 0.040 | 0.126 ± 0.032 |
*p < 0.05, significant difference; **p < 0.01, highly significant difference (vs. the same route of administration for male rats).
aCalculated by the cumulative amount of parent drug in urine (X u) vs. AUC0–∞ of parent drug.
Urine Excretions
After oral administration of veratramine (20 mg/kg), veratramine and its two major metabolites were all detected in urine. The urinary cumulative amounts of these compounds vs. time were plotted in Fig. 3. In male rats, amounts of veratramine, 7-hydroxyl-veratramine, and veratramine-3-O-sulfate during 48 h post-oral dosing (19.1 ± 6.3, 49.1 ± 8.9, and 3.56 ± 2.01 μg, respectively) were all significantly different from those of female rats (4.28 ± 1.04, 2.40 ± 0.72, and 59.5 ± 13.5 μg, respectively). As shown in Fig. 3, the cumulative amount of 7-hydroxyl-veratramine during 24 h in male rats (215 ± 101 ng) after i.v. administration of veratramine (50 μg/mL) was significantly higher than that in female rats (22.3 ± 0.9 ng), also indicating a gender-dependent elimination of 7-hydroxyl-veratramine; however, there was no obvious difference on the cumulative amount of veratramine between male and female rats, which was different from the results after its oral administration.
Bile Excretions
After oral and i.v. administrations of veratramine, only veratramine-3-O-sulfate was observed in bile (Fig. 3). Such more than 7-fold higher secretion of veratramine-3-O-sulfate in female rats (about 2100 μg in females vs. 300 μg in males during the 6-h oral post-dose period, and about 7 μg in females vs. 1 μg in males during the 6-h i.v. post-dose period) provided strong evidence for gender-related metabolism and elimination of veratramine.
Intestinal Absorption of Veratramine Via Passive Diffusion with Minimal Metabolism
Absorption of Veratramine on the Caco-2 Monolayer Model
Solubility and cytotoxicity assays of veratramine were performed prior to the bi-directional permeability study. Consequently, the concentration of veratramine used for absorptive study was 6 μg/mL. The absorptive Papp value (apical-basolateral) in the Caco-2 model was (1.51 ± 0.14) × 10−5 cm/s, suggesting veratramine as a well-absorbed compound with a high permeability (45). In addition, the Papp value (basolateral-apical) of veratramine was (1.74 ± 0.05) × 10−5 cm/s, with an efflux ratio of 1.15, which indicated potential passive diffusion during its absorption. Metabolites of veratramine were not found in either the donor or the receiver side during the bi-directional transport study. The enzymatic protein expressions in the differentiated Caco-2 cells were similar to those in human intestine (46), it is thus suggested that metabolism of veratramine during its absorption across the intestinal epithelium was supposed to be minimal. Such conclusion was further verified by a higher than 99% recoveries calculated based on the mass balance of veratramine in the bi-directional permeability study.
Intestinal Metabolism of Veratramine in Intestinal Subcellular Fractions and GI Contents
It has been reported that the expressions and activities of CYP3A and CYP2D in rat intestine were much lower than those in liver, and SULT2A was deficient in rat intestine (47–52). In our study, male RIS9 and RIMs were used to investigate the stabilities of veratramine in rat intestinal tract. The results revealed that neither 7-hydroxyl-veratramine nor veratramine-3-O-sulfate was found. RIMs were also used to further investigate the potential formation of 7-hydroxyl-veratramine due to the lower P450 enzyme concentrations in RIS9. Veratramine was found to be stable in RIMs, without hydroxylated metabolites generated. After incubation with GI contents, veratramine was also found to be stable with about 100% compound remaining after 4-h incubations in both simulated GI fluids and GI content solutions collected from stomach, small intestine, and large intestine.
Potential Enterohepatic Recirculation of Veratramine
RIS9 and GI content incubations were further performed on veratramine-3-O-sulfate to determine its stability in the gastrointestinal tract. After 4-h incubations, the parent compound veratramine, potentially generated from hydrolysis of veratramine-3-O-sulfate, was not detected in either simulated fluids or in rat gastric/small intestinal/large intestinal content solutions, indicating that veratramine-3-O-sulfate was stable in GI contents. However, when it was incubated with RIS9, the amount of veratramine gradually increased during 2-h incubations, suggesting that veratramine-3-O-sulfate was converted back to veratramine. Further characterization of the hydrolysis kinetics of veratramine-3-O-sulfate provided data best fit by the substrate inhibition equation with Km, Vmax, Ki, and CLint values of 0.141 ± 0.069 μM (1.21 ± 0.39) × 10−3 nmol min−1 mg−1, 0.225 μM and 8.64 μL min−1 mg−1, respectively. These finding suggested that veratramine-3-O-sulfate excreted from bile could be further hydrolyzed and converted back to parent compound during its absorption across rat intestinal epithelium.
Gender-Dependent Liver Hydroxylation and Sulfation of Veratramine
Gender-Dependent Hepatic Metabolism of Veratramine for the Formation of 7-Hydroxyl-Veratramine Using mRLMs and fRLMs
To investigate the potential gender-related metabolism of veratramine for the formation of 7-hydroxyl-veratramine in rat liver, the enzymatic kinetic parameters were compared between mRLMs and fRLMs. The typical Michaelis-Menten equation best fit the data obtained from in vitro incubations using mRLMs and fRLMs, based on the Eadie-Hofstee plots as shown in Fig. 4a. The apparent Km, Vmax, and CLint values were 47.8 ± 3.2 μM, 0.167 ± 0.004 nmol min−1 mg−1, and 3.48 μL min−1 mg−1 in incubations with mRLMs, and 81.2 ± 10.1 μM (3.05 ± 0.15) × 10−2 nmol min−1 mg−1, and 0.375 μL min−1 mg−1 in incubations with fRLMs. There were statistically significant differences in the enzymatic kinetic parameters between the incubations using mRLMs and fRLMs, suggesting that the formation of 7-hydroxyl-veratramine was more active in male rats.
Fig. 4.
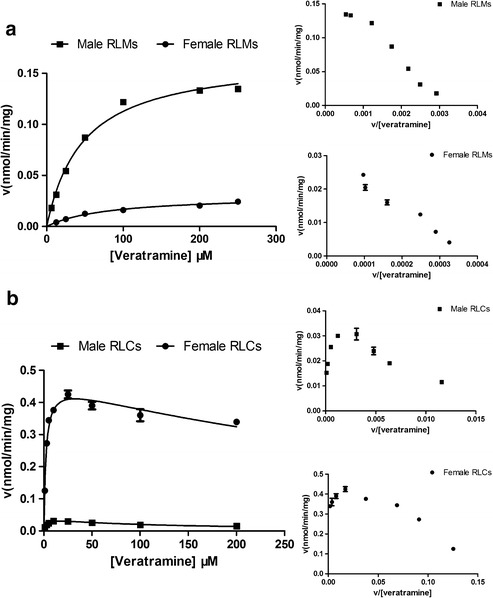
Gender-dependent enzyme kinetic plots of veratramine a for the formation of 7-hydroxyl-veratramine in incubations of male and female RLMs in the presence of NADPH and b for the formation of veratramine-3-O-sulfate in incubations of male and female RLCs in the presence of PAPS. Each data point is expressed as mean ± SD of triplicates. Insert figures, Eadie-Hofstee plots
Gender-Dependent Hepatic Metabolism of Veratramine for the Formation of Veratramine-3-O-sulfate Using mRLCs and fRLCs
Extensive metabolism of veratramine for the exclusive formation of veratramine-3-O-sulfate was observed in both mRLCs and fRLCs. Based on the Eadie-Hofstee plot of the incubations with veratramine using RLCs of both genders (Fig. 4b), substrate inhibition kinetics were clarified with apparent Km, Vmax, Ki, and CLint values of 2.90 ± 0.46 μM (4.04 ± 0.24) × 10−2 nmol min−1 mg−1, 16.2 μM, and 13.9 μL min−1 mg−1 in mRLCs, respectively, and 2.27 ± 0.27 μM, 0.469 ± 0.016 nmol min−1 mg−1, 88.0 μM, and 207 μL min−1 mg−1, respectively, in fRLCs. A significantly higher CLint was noticed in incubations using fRLCs, which indicated gender-dependent sulfation of veratramine for the formation of veratramine-3-O-sulfate in rat liver cytosols.
DISCUSSION
In the first instance, the current study characterized the pharmacokinetics of veratramine in rats. As shown in the plasma concentration vs. time profiles of veratramine and the two major metabolites after oral administration (20 mg/kg veratramine), distinct bimodal phenomena were observed. Although the bimodal and multimodal phenomena have also been observed for other alkaloids, such as berberine, palmatine (53), and some protoberberine-type alkaloids (54), the current work elucidated, for the first time, this phenomenon for Veratrum alkaloids. Re-absorption and enterohepatic circulation are the most prevailing reasons for such phenomena. Data now presented showed that veratramine was a good membrane permeant, underwent rapid passive diffusion, and had a good stability in the gastrointestinal tract during its absorption. After absorption, 7-hydroxyl-veratramine and veratramine-3-O-sulfate were formed in the liver, but only veratramine-3-O-sulfate was secreted into the bile. Neither veratramine nor 7-hydroxyl-veratramine was observed in bile, suggesting that they did not participate in the bimodal enterohepatic circulation of themselves. To further clarify these mechanisms, a study of the stability of veratramine-3-O-sulfate revealed that hydrolysis occurred when it was absorbed across the intestinal epithelium. Therefore, the proposed mechanism of bimodal phenomena of veratramine was described as follows: veratramine was re-absorbed into systemic circulations following the hydrolysis of veratramine-3-O-sulfate. Additionally, in the plasma concentration vs. time curves of veratramine after i.v. administration (50 μg/kg), an extremely slow elimination of veratramine was observed between 30 and 60 min, suggesting that veratramine-3-O-sulfate (secreted into GI tract) was supposed to be converted back into veratramine and re-absorbed into the general circulation. Based on the results, the proposed in vivo biotransformation schematic of veratramine is shown in Fig. 5.
Fig. 5.
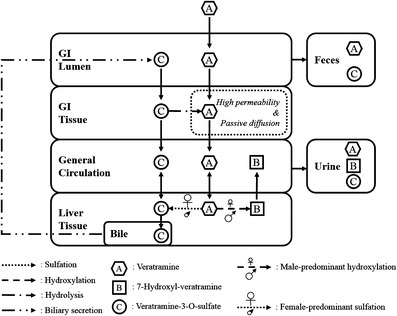
Proposed in vivo biotransformation schematic of veratramine in male and female rats
The systematic investigations of the gender-dependent pharmacokinetic behaviors of steroidal alkaloid veratramine were firstly reported in our study. In the previous published studies, gender-dependent pharmacokinetics leading to a high bioavailability of drug in females were frequently reported. It has been demonstrated that the greater lethality of female rats in comparison with male rats following nicotine administration was mainly due to the gender-dependent pharmacokinetics mediated by the slower nicotine metabolism in females (55). For another example, in comparison with male rats, the much slower elimination and higher bioavailability of schisandrin, schisandrol B, deoxyschisandrin, γ-schisandrin, and schisantherin A in female rats were reported (56). However, in our study, an unusual lower absolute bioavailability of veratramine in female rats than that in male rats was observed. After oral administration of veratramine (20 mg/kg), in comparison with male rats, a significantly lower Cmax (p < 0.01), AUC0–t (p < 0.01), AUC0–∞ (p < 0.01), t1/2 (p < 0.05), and MRT (p < 0.01) and a significantly higher CL (p < 0.01) of veratramine were observed in female rats. Additionally, the AUC0–∞ of 7-hydroxyl-veratramine and veratramine-3-O-sulfate were 10-fold higher and 2-fold lower, respectively, in male rats (Table II). The metabolic ratios of 7-hydroxyl-veratramine and veratramine-3-O-sulfate calculated in plasma were 27.8 ± 4.8% and 33.7 ± 16.7%, respectively, in male rats compared with 3.4 ± 1.2% and 93.8 ± 1.8% in female rats, which indicated distinct gender differences. Mechanisms of gender-dependent pharmacokinetics may include differences in cardiac output, body composition, body weight, hormonal status, and metabolism between genders (57,58). One factor commonly known to contribute to gender-related metabolism, the major reason for gender difference on pharmacokinetic behavior, is the differential expression of drug-processing enzymes including phase I and phase II metabolic enzymes (P450s, sulfotransferases, glutathione transferases, and UDP-glucuronosyltransferases) (59).
Table II.
Pharmacokinetic Parameters of 7-Hydroxyl-Veratramine and Veratramine-3-O-Sulfate After Oral (20 mg/kg) Administration to Male and Female Rats
| PK parameters | 7-Hydroxyl-veratramine | Veratramine-3-O-sulfate | ||
|---|---|---|---|---|
| Male (n = 5) | Female (n = 5) | Male (n = 5) | Female (n = 5) | |
| C max (ng/mL) | 38.8 ± 8.6 | 4.21 ± 0.52** | 98.8 ± 33.2 | 148 ± 40 |
| AUC0–t (min μg/mL) | 39.5 ± 10.9 | 1.83 ± 0.50** | 42.7 ± 11.7 | 106 ± 39* |
| AUC0-∞ (min μg/mL) | 40.1 ± 11.3 | 4.06 ± 2.04** | 68.0 ± 35.2 | 129 ± 28* |
| t 1/2 (min) | 411 ± 159 | 657 ± 466 | 524 ± 453 | 453 ± 111 |
| MRT (min) | 865 ± 234 | 1014 ± 673 | 861 ± 603 | 869 ± 141 |
*p < 0.05, significant difference; **p < 0.01, highly significant difference (vs. the same metabolite for male rats).
Our previous study manifested that CYP2D and CYP3A mainly mediated the formation of 7-hydroxl-veratramine in rats (35). The enzyme activity of CYP2D in male rats was higher than that in female rats, especially for the male dominant CYP2D1 (60–62). The expression and activity of CYP2D2 in SD rats were also slightly greater in males than that in females with CYP2D5 as the male-specific form (63). In addition, male rats manifested the greater expression and activity on CYP3A than female rats (61,64). CYP3A1 and CYP3A2 were the major CYP3A isoforms in rats with CYP3A2 as a distinct male-specific isoform (64,65). Thus, the higher formation of 7-hydroxl-veratramine using mRLMs than that using fRLMs should be explained by the male-specific or male-dominant expressions and activities of CYP2D and CYP3A. Additionally, male-specific CYP2C11, compromising over 40% of the total CYPs in male rat liver, is one of the major gender-dependent CYPs in rats (66). Since the contribution of CYP2C11 for the formation of 7-hydroxyl-veratramine could not be thoroughly ruled out, the male-specific CYP2C11 may also partly mediate the significant gender-dependent formation of 7-hydroxyl-veratramine. SULT2A1 was the major isozyme mediating the sulfation of veratramine in humans (35). Based on the similarity in the sulfation of veratramine in male and female rat liver cytosols with the existence of rat SULT2A (67–69), it could be inferred that SULT2A was responsible for the sulfation of veratramine in rats. The metabolic activity of SULT2A was substantially greater in female rats than that in male rats, with 15-fold higher amounts in female rat liver (69–71). Thus, the significantly high formation of veratramine-3-O-sulfate in female rats was likely due to the high expression and activity of SULT2A. In the meantime, the lack of metabolism of veratramine in rat intestine could be explained by the extraordinarily limited expressions and activities of CYP2D and CYP3A as well as no expression of SULT2A1 in rat intestine (47,52,72,73). Therefore, the 10-fold higher CLint for the formation of 7-hydroxyl-veratramine using mRLMs compared with that using fRLMs and the 14-fold lower CLint for the formation of veratramine-3-O-sulfate using mRLCs compared with that using fRLCs were supposed to be reasonably expected.
After the administration of a rather low i.v. dose of veratramine, a major portion of it was distributed to liver to form veratramine-3-O-sulfate, which was actively transported into bile. It was hypothesized that, due to the low amount formed after i.v. administration, no saturation of biliary excretion of veratramine-3-O-sulfate occurred and the formed veratramine-3-O-sulfate was mainly found in bile rather than blood and urine (Fig. 3). However, due to the much higher oral dose, the uptake of veratramine-3-O-sulfate into bile canaliculi may be slower than the formation of veratramine-3-O-sulfate in the hepatocytes, thus, veratramine-3-O-sulfate was found in plasma and urine in addition to bile. Such explanation for the potential route-dependent disproportionate increase of parent compounds and metabolites across doses due to saturation of biliary excretion was also found in previous studies (74,75). Moreover, although the gender differences on the male-predominant urinary accumulation of 7-hydroxyl-veratramine and female-predominant biliary excretion of veratramine-3-O-sulfate were distinct after both oral and i.v. administration, the gender differences on veratramine after i.v. administration were not obvious. Such discrepancy in gender-dependent pharmacokinetics of veratramine after different routes of administrations is considered to be mainly due to the extensive gender-dependent first-pass metabolism after oral administration. After i.v. administration, all drug directly entered the systemic circulation without significantly different pharmacokinetic profiles observed for male and female rats. One-compartment pharmacokinetic model via Priana estimated the similar pharmacokinetic parameters for male and female rats (ke = 0.677 h−1 and V = 1.81 L for male rats; ke = 1.03 h−1 and V = 0.990 L for female rats). Whereas, after oral administration, due to the fast and extensive first-pass metabolism in liver, it was observed that a large portion of veratramine has been transformed into 7-hydroxyl-veratramine and veratramine-3-O-sulfate with about 35.6% for males and 2.3% for females of the parent drug (estimated from ratio of AUCveratramine/(AUCveratramine + AUCveratramine-3-O-sulfate + AUC7-hydroxyl-veratramine)) entered into the systemic circulation. Such difference during the first-pass metabolism process could be the major cause of the gender-related difference in the pharmacokinetic profiles after oral administration. In the meantime, based on those portion of veratramine entered the systemic circulation after oral administration, one-compartment absorption pharmacokinetic model via Pirana could also be used and similar pharmacokinetic parameters for male and female rats (ka = 2.14 h−1, ke = 0.0434 h−1 and V = 0.366 L for male rats; ka = 2.22 h−1, ke = 0.0519 h−1 and V = 0.264 L for female rats) were observed, which further supported the above hypothesis.
Gender-dependent pharmacokinetics, due to the higher expression and activity of SULT2A in female rats, were rarely reported by others. However, a few studies revealed that female rats had higher clearance than male rats, which was due to the higher expression and activity of sulfotransferase in females, such as anticancer agent PF-02341066 (76). The pharmacokinetic evaluation of PF-02341066 in rats revealed the existence of gender-dependent differences with at least 2-fold higher plasma concentrations of PF-02341066 in male rats than that in female rats when the identical dose was administered, and the more extensive formation of the sulfoconjugated metabolite, mediated by the female-predominant sulfotransferase, was assumed to be the major reason for the lower drug exposure of PF-02341066 in females (76). In addition, the gender-dependent pharmacokinetics in rats mediated by CYP2D has also been reported. Gender-dependent pharmacokinetic study on 3,4-methylenedioxymethamphetamine (MDMA; ecstasy) was preliminarily investigated by Fonsart et al. to explore why male rodents were more sensitive to acute toxicity (62). It was found that MDMA was metabolized to MDA and then to the more toxic HHA mediated by CYP2D via N-demethylation and O-demethylenation (62,77). The much higher AUC and Cmax of MDA in males may lead to more HHA formation and more toxicity in them (62). Gender-dependent pharmacokinetics mediated by rat CYP3A have also been widely studied such as indinavir. The male-predominant expression and activity of CYP3A were verified to be the major reasons for the gender-dependent pharmacokinetics of indinavir in rats (78). Steroidal alkaloids of Liliaceae mainly possess a C27 cholestane carbon skeleton with four or five carbocyclic and one heterocyclic rings (3,79). Among the Liliaceae family, genus Veratrum and Fritillaria, two most important steroidal alkaloids containing herbs, were widely used as folk remedies (3). The pharmacokinetics of peimine, peiminine and verticinone, major steroidal alkaloids from genus Fritillaria of Liliaceae, were significantly influenced by gender in rats, and slower elimination of them was observed in male rat plasma (80,81), which was consistent with the phenomenon found in pharmacokinetic behavior of steroidal alkaloid veratramine. The enterohepatic circulation was also reported after peimine was administrated to rats (80). However, these studies only hypothesized that sulfation was one of the reasons for the gender-related pharmacokinetics of steroidal alkaloids without experimental verification (81). In our study, it was verified that female-predominant sulfation was the major reason for the gender-related pharmacokinetics of steroidal alkaloid veratramine. Therefore, steroidal alkaloids, especial for the hydroxysteroidal alkaloids, may have gender-associated pharmacokinetics due to the gender differences on the metabolism mediated by phase I and phase II enzymes.
Due to the gender differences in drug metabolism being notoriously species dependent, the limitation of the current findings should be noted. Our previous study demonstrated that different enzymes mediated the formation of 7-hydroxyl-veratramine from veratramine in rats (CYP2D and CYP3A) and humans (only CYP2D6) (35). It was previously suggested that CYP2D in rats had gender differences with more predominant expression and activity in males (60,61), whereas no gender difference for CYP2D6 has been identified in humans. Additionally, human solely expressed SULT2A1 for the sulfation of veratramine (35,82), whereas rats expressed four SULT2A isoforms including the female-predominant SULT2A1 and female-specific SULT2A3 and SULT2A4 (67,68). The expression and metabolic activity of SULT2A were substantially greater in female rats than that in male rats (69–71). Although rats exhibited dramatic sexual dimorphism, the gender difference on human SULT2A1 activity was unknown due to few study performed (83,84). These species differences may limit the direct extrapolation of the findings of veratramine in rats to humans. No matter how, current study provided a preliminary evaluation on the potential gender-dependent pharmacokinetics leading to toxicities in rat model. Due to the gender-dependent expression and activity of SULT2A1 in humans being unclear so far, the potential gender-dependent pharmacokinetics in humans should not be overlooked, especially for the narrow therapeutic index drugs metabolized by SULTs, such as steroidal alkaloids.
CONCLUSIONS
In this study, the gender-dependent pharmacokinetic behaviors of veratramine with enterohepatic recirculation were firstly reported. The absolute bioavailability of veratramine in male rats was much higher than that in female rats after oral administration of veratramine, leading to its greater toxicity toward male rats. Further mechanistic studies indicated that male-predominant formation of 7-hydroxyl-veratramine and female-predominant formation of veratramine-3-O-sulfate in liver were the major contributors to such gender-dependent pharmacokinetics of veratramine in rats.
ELECTRONIC SUPPLEMENTARY MATERIAL
(DOCX 16 kb)
(DOCX 16 kb)
ACKNOWLEDGMENTS
This work was financially supported by the Specialized Research Fund for the Doctoral Program of Higher Education (grant no. 20113107110012), the City Board of Education Research and Innovation Project in 2014 (NATURAL SCIENCE), and Science and Technology Commission of Shanghai Research Project (grant no. 13140901800 and 13140901802).
COMPLIANCE WITH ETHICAL STANDARDS
Conflict of Interest
The authors report no commercial interest.
Footnotes
Hai Wei, Ning Zhang and Zhong Zuo contributed equally to this work.
Contributor Information
Hai Wei, Phone: +86 5132 3044, Email: wei_hai@hotmail.com.
Ning Zhang, Phone: +86 5132 2384, Email: ningzh18@126.com.
Zhong Zuo, Phone: +852 3943 6832, Email: joanzuo@cuhk.edu.hk.
REFERENCES
- 1.Li W, Wang L, Zhao D, Liu Y. Steroidal alkaloids from Veratrum nigrum. Chem Nat Compd. 2012;48:919–920. doi: 10.1007/s10600-012-0426-7. [DOI] [Google Scholar]
- 2.Park J, Jeon YD, Kim HL, Lim H, Jung Y, Youn DH, et al. Interaction of Veratrum nigrum with Panax ginseng against obesity: a Sang-ban relationship. Evid Based Complement Alternat Med. 2013;2013. doi: 10.1155/2013/732126 [DOI] [PMC free article] [PubMed]
- 3.Li HJ, Jiang Y, Li P. Chemistry, bioactivity and geographical diversity of steroidal alkaloids from the Liliaceae family. Nat Prod Rep. 2006;23:735–752. doi: 10.1039/b609306j. [DOI] [PubMed] [Google Scholar]
- 4.Coe WS, Best MM, Kinsman JM. Veratrum viride in the treatment of hypertensive vascular disease. JAMA. 1950;143:5–7. doi: 10.1001/jama.1950.02910360007003. [DOI] [PubMed] [Google Scholar]
- 5.Freis ED, Stanton JR. A clinical evaluation of veratrum viride in the treatment of essential hypertension. Am Heart J. 1948;36:723–738. doi: 10.1016/0002-8703(48)90236-1. [DOI] [PubMed] [Google Scholar]
- 6.Wang LS, Li W, Liu YH. Hypotensive effect and toxicology of total alkaloids and veratramine from roots and rhizomes of Veratrum nigrum L. in spontaneously hypertensive rats. Pharmazie. 2008;63:606–610. [PubMed] [Google Scholar]
- 7.Wang LS, Zhao DQ, Tao HM, Liu YH. Determination of vetatramine in Veratrum nigrum by HPLC-ELSD. Zhongguo Zhong Yao Za Zhi. 2008;33:791–792. [PubMed] [Google Scholar]
- 8.Zhang S, Zhou JX, Shou QY, Peng Y, Shen ZW. Determination of jervine and veratramine in Veratrum plants using high performance liquid chromatography coupled with evaporative light scattering detection. Chin J Chromatogr. 2008;26:56–59. doi: 10.1016/S1872-2059(08)60028-0. [DOI] [PubMed] [Google Scholar]
- 9.Tang J, Li HL, Shen YH, Jin HZ, Yan SK, Liu RH, et al. Antitumor activity of extracts and compounds from the rhizomes of Veratrum dahuricum. Phytother Res. 2008;22:1093–1096. doi: 10.1002/ptr.2463. [DOI] [PubMed] [Google Scholar]
- 10.Winkler JD, Isaacs A, Holderbaum L, Tatard V, Dahmane N. Design and synthesis of inhibitors of Hedgehog signaling based on the alkaloid cyclopamine. Org Lett. 2009;11:2824–2827. doi: 10.1021/ol900974u. [DOI] [PubMed] [Google Scholar]
- 11.Wilkins R. Veratrum viride in hypertensive cardiovascular disease. Mod Concepts Cardiovasc Dis. 1951;20:89–90. [PubMed] [Google Scholar]
- 12.Meilman E. The management of hypertensive cardiovascular disease. Circulation. 1956;13:596–607. doi: 10.1161/01.CIR.13.4.596. [DOI] [PubMed] [Google Scholar]
- 13.Tang J, Li HL, Shen YH, Jin HZ, Yan SK, Liu XH, et al. Antitumor and antiplatelet activity of alkaloids from Veratrum dahuricum. Phytother Res. 2010;24:821–826. doi: 10.1002/ptr.3022. [DOI] [PubMed] [Google Scholar]
- 14.Thron C, McCann F. Studies on the bradycardia and periodic rhythm caused by veratramine in the sinoatrial node of the guinea pig. J Electrocardiol. 1998;31:257–268. doi: 10.1016/S0022-0736(98)90141-8. [DOI] [PubMed] [Google Scholar]
- 15.Thron C, McCann F. Pharmacological tests of the mechanism of the periodic rhythm caused by veratramine in the sinoatrial node of the guinea pig. Gen Pharmacol. 1999;32:81–89. doi: 10.1016/S0306-3623(98)00088-3. [DOI] [PubMed] [Google Scholar]
- 16.Nagata R, Izumi K, Iwata SI, Shimizu T, Fukuda T. Mechanisms of veratramine-induced 5-HT syndrome in mice. Jpn J Pharmacol. 1991;55:139–146. doi: 10.1254/jjp.55.139. [DOI] [PubMed] [Google Scholar]
- 17.Mahindroo N, Punchihewa C, Fujii N. Hedgehog-Gli signaling pathway inhibitors as anticancer agents. J Med Chem. 2009;52:3829–3845. doi: 10.1021/jm801420y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shelton CA, Gilmartin AG. Novel receptor antagonists for cancer therapy: hedgehog pathway inhibitors. Drug Discov Today. 2009;6:63–69. [Google Scholar]
- 19.Khanfar MA, El Sayed KA. The Veratrum alkaloids jervine, veratramine, and their analogues as prostate cancer migration and proliferation inhibitors: biological evaluation and pharmacophore modeling. Med Chem Res. 2013;22:4775–4786. doi: 10.1007/s00044-013-0495-6. [DOI] [Google Scholar]
- 20.Honerjäger P. Cardioactive substances that prolong the open state of sodium channels. Rev Physiol Biochem Pharmacol. 1982;92:1–74. doi: 10.1007/BFb0030502. [DOI] [PubMed] [Google Scholar]
- 21.Meilman E, Krayer O. Clinical studies on Veratrum alkaloids I. The action of protoveratrine and veratridine in hypertension. Circulation. 1950;1:204–213. doi: 10.1161/01.CIR.1.2.204. [DOI] [PubMed] [Google Scholar]
- 22.Meilman E, Krayer O. Clinical studies on Veratrum alkaloids II. The dose–response relations of protoveratrine in hypertension. Circulation. 1952;6:212–221. doi: 10.1161/01.CIR.6.2.212. [DOI] [PubMed] [Google Scholar]
- 23.Bolomey A, Lenel R. Clinical experiences with Veratrum alkaloids (veriloid) in the prolonged treatment of essential hypertension. Perm Found Med Bull. 1952;10:57–68. [PubMed] [Google Scholar]
- 24.Gaffield W. The Veratrum alkaloids: natural tools for studying embryonic development. Stud Nat Prod Chem. 2000;23:563–589. doi: 10.1016/S1572-5995(00)80138-6. [DOI] [Google Scholar]
- 25.Reid JM, Kuffel MJ, Ruben SL, Morales JJ, Rinehart KL, Squillace DP, et al. Rat and human liver cytochrome P-450 isoform metabolism of ecteinascidin 743 does not predict gender-dependent toxicity in humans. Clin Cancer Res. 2002;8:2952–2962. [PubMed] [Google Scholar]
- 26.Lin G, Cui YY, Liu XQ. Gender differences in microsomal metabolic activation of hepatotoxic clivorine in rat. Chem Res Toxicol. 2003;16:768–774. doi: 10.1021/tx0340302. [DOI] [PubMed] [Google Scholar]
- 27.Wiedenfeld H, Edgar J. Toxicity of pyrrolizidine alkaloids to humans and ruminants. Phytochem Rev. 2011;10:137–151. doi: 10.1007/s11101-010-9174-0. [DOI] [Google Scholar]
- 28.Lin G, Tang J, Liu XQ, Jiang Y, Zheng J. Deacetylclivorine: a gender-selective metabolite of clivorine formed in female Sprague–Dawley rat liver microsomes. Drug Metab Dispos. 2007;35:607–613. doi: 10.1124/dmd.106.014100. [DOI] [PubMed] [Google Scholar]
- 29.Gurley BJ, Gardner SF, White LM, Wang PL. Ephedrine pharmacokinetics after the ingestion of nutritional supplements containing Ephedra sinica (ma huang) Ther Drug Monit. 1998;20:439–445. doi: 10.1097/00007691-199808000-00015. [DOI] [PubMed] [Google Scholar]
- 30.Heretsch P, Tzagkaroulaki L, Giannis A. Cyclopamine and hedgehog signaling: chemistry, biology, medical perspectives. Angew Chem Int Ed. 2010;49:3418–3427. doi: 10.1002/anie.200906967. [DOI] [PubMed] [Google Scholar]
- 31.Xin GZ, Qi LW, Shi ZQ, Li P, Hao HP, Wang GJ, et al. Strategies for integral metabolism profile of multiple compounds in herbal medicines: pharmacokinetics, metabolites characterization and metabolic interactions. Curr Drug Metab. 2011;12:809–817. doi: 10.2174/138920011797470164. [DOI] [PubMed] [Google Scholar]
- 32.Tremblay MR, Nevalainen M, Nair SJ, Porter JR, Castro AC, Behnke ML, et al. Semisynthetic cyclopamine analogues as potent and orally bioavailable hedgehog pathway antagonists. J Med Chem. 2008;51:6646–6649. doi: 10.1021/jm8008508. [DOI] [PubMed] [Google Scholar]
- 33.Wu X, Chen J, Pan Y. Simultaneous determination of peimine and peiminine in rat plasma by LC‐ESI‐MS employing solid‐phase extraction. Biomed Chromatogr. 2010;24:902–907. doi: 10.1002/bmc.1384. [DOI] [PubMed] [Google Scholar]
- 34.El Sayed KA. Microbial biotransformation of veratramine. J Nat Prod. 1998;61:149–151. doi: 10.1021/np9703611. [DOI] [PubMed] [Google Scholar]
- 35.Lyu C, Zhou W, Zhang Y, Zhang S, Kou F, Wei H, et al. Identification and characterization of in vitro and in vivo metabolites of steroidal alkaloid veratramine. Biopharm Drug Dispos. 2015;36:308–324. doi: 10.1002/bdd.1942. [DOI] [PubMed] [Google Scholar]
- 36.Zhang L, Li C, Lin G, Krajcsi P, Zuo Z. Hepatic metabolism and disposition of baicalein via the coupling of conjugation enzymes and transporters-in vitro and in vivo evidences. AAPS J. 2011;13:378–389. doi: 10.1208/s12248-011-9277-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, Lin G, Kovács B, Jani M, Krajcsi P, Zuo Z. Mechanistic study on the intestinal absorption and disposition of baicalein. Eur J Pharm Sci. 2007;31:221–231. doi: 10.1016/j.ejps.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 38.Kong LT, Wang Q, Xiao BX, Liao YH, He XX, Ye LH, et al. Different pharmacokinetics of the two structurally similar dammarane sapogenins, protopanaxatriol and protopanaxadiol, in rats. Fitoterapia. 2013;86:48–53. doi: 10.1016/j.fitote.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 39.Cheng G, An F, Zou MJ, Sun J, Hao XH, He YX. Time-and pH-dependent colon-specific drug delivery for orally administered diclofenac sodium and 5-aminosalicylic acid. World J Gastroenterol. 2004;10:1769–1774. doi: 10.3748/wjg.v10.i12.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao Q, Zhang Y, Wo S, Zuo Z. Hydrolysis is the dominating in vivo metabolism pathway for arctigenin: identification of novel metabolites of arctigenin by LC/MS/MS after oral administration in rats. Planta Med. 2013;79:471–479. doi: 10.1055/s-0033-1336555. [DOI] [PubMed] [Google Scholar]
- 41.Wong YC, Zhang L, Lin G, Zuo Z. Intestinal first-pass glucuronidation activities of selected dihydroxyflavones. Int J Pharm. 2009;366:14–20. doi: 10.1016/j.ijpharm.2008.08.035. [DOI] [PubMed] [Google Scholar]
- 42.Rocci ML, Devanarayan V, Haughey DB, Jardieu P. Confirmatory reanalysis of incurred bioanalytical samples. AAPS J. 2007;9:336–343. doi: 10.1208/aapsj0903040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun. 1991;175:880–885. doi: 10.1016/0006-291X(91)91647-U. [DOI] [PubMed] [Google Scholar]
- 44.Hutzler JM, Tracy TS. Atypical kinetic profiles in drug metabolism reactions. Drug Metab Dispos. 2002;30:355–362. doi: 10.1124/dmd.30.4.355. [DOI] [PubMed] [Google Scholar]
- 45.Yee S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man-fact or myth. Pharm Res. 1997;14:763–766. doi: 10.1023/A:1012102522787. [DOI] [PubMed] [Google Scholar]
- 46.Meinl W, Ebert B, Glatt H, Lampen A. Sulfotransferase forms expressed in human intestinal Caco-2 and TC7 cells at varying stages of differentiation and role in benzo [a] pyrene metabolism. Drug Metab Dispos. 2008;36:276–283. doi: 10.1124/dmd.107.018036. [DOI] [PubMed] [Google Scholar]
- 47.Mitschke D, Reichel A, Fricker G, Moenning U. Characterization of cytochrome P450 protein expression along the entire length of the intestine of male and female rats. Drug Metab Dispos. 2008;36:1039–1045. doi: 10.1124/dmd.107.019687. [DOI] [PubMed] [Google Scholar]
- 48.Meinl W, Sczesny S, Brigelius-Flohé R, Blaut M, Glatt H. Impact of gut microbiota on intestinal and hepatic levels of phase 2 xenobiotic-metabolizing enzymes in the rat. Drug Metab Dispos. 2009;37:1179–1186. doi: 10.1124/dmd.108.025916. [DOI] [PubMed] [Google Scholar]
- 49.Rao A, Haywood J, Craddock AL, Belinsky MG, Kruh GD, Dawson PA. The organic solute transporter α-β, Ostα-Ostβ, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci. 2008;105:3891–3896. doi: 10.1073/pnas.0712328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou T, Chen Y, Huang C, Chen G. Caffeine induction of sulfotransferases in rat liver and intestine. J Appl Toxicol. 2012;32:804–809. doi: 10.1002/jat.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abrahamson DR, Rodewald R. Evidence for the sorting of endocytic vesicle contents during the receptor-mediated transport of IgG across the newborn rat intestine. J Cell Biol. 1981;91:270–280. doi: 10.1083/jcb.91.1.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishimuta H, Nakagawa T, Nomura N, Yabuki M. Species differences in hepatic and intestinal metabolic activities for 43 human cytochrome P450 substrates between humans and rats or dogs. Xenobiotica. 2013;43:948–955. doi: 10.3109/00498254.2013.787155. [DOI] [PubMed] [Google Scholar]
- 53.Deng Y, Liao Q, Li S, Bi K, Pan B, Xie Z. Simultaneous determination of berberine, palmatine and jatrorrhizine by liquid chromatography-tandem mass spectrometry in rat plasma and its application in a pharmacokinetic study after oral administration of coptis–evodia herb couple. J Chromatogr B. 2008;863:195–205. doi: 10.1016/j.jchromb.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 54.He W, Liu G, Cai H, Sun X, Hou W, Zhang P, et al. Integrated pharmacokinetics of five protoberberine-type alkaloids in normal and insomnic rats after single and multiple oral administration of Jiao-Tai-Wan. J Ethnopharmacol. 2014;154:635–644. doi: 10.1016/j.jep.2014.04.040. [DOI] [PubMed] [Google Scholar]
- 55.Kyerematen GA, Owens GF, Chattopadhyay B, de Bethizy JD, Vesell ES. Sexual dimorphism of nicotine metabolism and distribution in the rat. Studies in vivo and in vitro. Drug Metab Dispos. 1988;16:823–828. [PubMed] [Google Scholar]
- 56.Xu H, Gan J, Liu X, Wu R, Jin Y, Li M, et al. Gender-dependent pharmacokinetics of lignans in rats after single and multiple oral administration of Schisandra chinensis extract. J Ethnopharmacol. 2013;147:224–231. doi: 10.1016/j.jep.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 57.Moody DE, Fang WB, Morrison J, McCance-Katz E. Gender differences in pharmacokinetics of maintenance dosed buprenorphine. Drug Alcohol Depend. 2011;118:479–483. doi: 10.1016/j.drugalcdep.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang Y, Hao H, Xie L, Kang A, Xie T, Zheng X, et al. Development of a systematic approach to identify metabolites for herbal homologs based on liquid chromatography hybrid ion trap time-of-flight mass spectrometry: gender-related difference in metabolism of Schisandra lignans in rats. Drug Metab Dispos. 2010;38:1747–1759. doi: 10.1124/dmd.110.033373. [DOI] [PubMed] [Google Scholar]
- 59.Miao Z, Scott DO, Griffith DA, Day R, Prakash C. Excretion, metabolism, and pharmacokinetics of CP-945,598, a selective cannabinoid receptor antagonist, in rats, mice, and dogs. Drug Metab Dispos. 2011;39:2191–2208. doi: 10.1124/dmd.111.040360. [DOI] [PubMed] [Google Scholar]
- 60.Lewis DFV. Cytochrome P450: structure, function and mechanism. Bristol: Taylor & Francis; 1996. pp. 115–167. [Google Scholar]
- 61.Soucek P, Gut I. Cytochromes P-450 in rats: structures, functions, properties and relevant human forms. Xenobiotica. 1992;22:83. doi: 10.3109/00498259209053106. [DOI] [PubMed] [Google Scholar]
- 62.Fonsart J, Menet MC, Debray M, Hirt D, Noble F, Scherrmann JM, et al. Sprague–Dawley rats display sex-linked differences in the pharmacokinetics of 3,4-methylenedioxymethamphetamine (MDMA) and its metabolite 3,4-methylenedioxyamphetamine (MDA) Toxicol Appl Pharmacol. 2009;241:339–347. doi: 10.1016/j.taap.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 63.Schulz-Utermoehl T, Bennett AJ, Ellis SW, Tucker GT, Boobis AR, Edwards RJ. Polymorphic debrisoquine 4-hydroxylase activity in the rat is due to differences in CYP2D2 expression. Pharmacogenet Genomics. 1999;9:357–366. doi: 10.1097/00008571-199906000-00011. [DOI] [PubMed] [Google Scholar]
- 64.Ghosal A, Sadrieh N, Reik L, Levin W, Thomas PE. Induction of the male-specific cytochrome P450 3A2 in female rats by phenytoin. Arch Biochem Biophys. 1996;332:153–162. doi: 10.1006/abbi.1996.0327. [DOI] [PubMed] [Google Scholar]
- 65.Huang HJ, Tsai ML, Chen YW, Chen SH. Quantitative shot-gun proteomics and MS-based activity assay for revealing gender differences in enzyme contents for rat liver microsome. J Proteome. 2011;74:2734–2744. doi: 10.1016/j.jprot.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 66.Morgan ET, MacGeoch C, Gustafsson JA. Hormonal and developmental regulation of expression of the hepatic microsomal steroid 16 alpha-hydroxylase cytochrome P-450 apoprotein in the rat. J Biol Chem. 1985;260:11895–11898. [PubMed] [Google Scholar]
- 67.Klaassen CD, Liu L, Dunn RT., II Regulation of sulfotransferase mRNA expression in male and female rats of various ages. Chem Biol Interact. 1998;109:299–313. doi: 10.1016/S0009-2797(97)00141-5. [DOI] [PubMed] [Google Scholar]
- 68.Blanchard RL, Freimuth RR, Buck J, Weinshilboum RM, Coughtrie MW. A proposed nomenclature system for the cytosolic sulfotransferase (SULT) superfamily. Pharmacogenet Genomics. 2004;14:199–211. doi: 10.1097/00008571-200403000-00009. [DOI] [PubMed] [Google Scholar]
- 69.Rajkowski KM, Robel P, Baulieu EE. Hydroxysteroid sulfotransferase activity in the rat brain and liver as a function of age and sex. Steroids. 1997;62:427–436. doi: 10.1016/S0039-128X(97)00013-5. [DOI] [PubMed] [Google Scholar]
- 70.Runge-Morris M, Wilusz J. Age and gender-related gene expression of hydroxysteroid sulfotransferase—a in rat liver. Biochem Biophys Res Commun. 1991;175:1051–1056. doi: 10.1016/0006-291X(91)91671-X. [DOI] [PubMed] [Google Scholar]
- 71.Davis W, Hewer A, Rajkowski KM, Meinl W, Glatt H, Phillips DH. Sex differences in the activation of tamoxifen to DNA binding species in rat liver in vivo and in rat hepatocytes in vitro: role of sulfotransferase induction. Cancer Res. 2000;60:2887–2891. [PubMed] [Google Scholar]
- 72.Martignoni M, Groothuis GMM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2:875–894. doi: 10.1517/17425255.2.6.875. [DOI] [PubMed] [Google Scholar]
- 73.Dunn RT, Klaassen CD. Tissue-specific expression of rat sulfotransferase messenger RNAs. Drug Metab Dispos. 1998;26:598–604. [PubMed] [Google Scholar]
- 74.Pottenger LH, Domoradzki JY, Markham DA, Hansen SC, Cagen SZ, Waechter JM. The relative bioavailability and metabolism of bisphenol A in rats is dependent upon the route of administration. Toxicol Sci. 2000;54:3–18. doi: 10.1093/toxsci/54.1.3. [DOI] [PubMed] [Google Scholar]
- 75.Gao Q, Zhang Y, Wo S, Zuo Z. Elucidation of arctigenin pharmacokinetics after intravenous and oral administrations in rats: integration of in vitro and in vivo findings via semi-mechanistic pharmacokinetic modeling. AAPS J. 2014;16:1321–1333. doi: 10.1208/s12248-014-9664-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhong WZ, Zhan J, Kang P, Yamazaki S. Gender specific drug metabolism of PF-02341066 in rats-role of sulfoconjugation. Curr Drug Metab. 2010;11:296–306. doi: 10.2174/138920010791514207. [DOI] [PubMed] [Google Scholar]
- 77.Carvalho M, Milhazes N, Remiao F, Borges F, Fernandes E, Amado F, et al. Hepatotoxicity of 3,4-methylenedioxyamphetamine and alphamethyldopamine in isolated rat hepatocytes: formation of glutathione conjugates. Arch Toxicol. 2004;78:16–24. doi: 10.1007/s00204-003-0510-7. [DOI] [PubMed] [Google Scholar]
- 78.Lin JH, Chiba M, Chen IW, Nishime JA, Vastag KJ. Sex-dependent pharmacokinetics of indinavir: in vivo and in vitro evidence. Drug Metab Dispos. 1996;24:1298–1306. [PubMed] [Google Scholar]
- 79.Gaffield W, Benson M, Lundin RE, Keeler RF. Carbon-13 and proton nuclear magnetic resonance spectra of Veratrum alkaloids. J Nat Prod. 1986;49:286–292. doi: 10.1021/np50044a014. [DOI] [Google Scholar]
- 80.Chen LH, Zhang HM, Guan ZY, Zhu WF, Yi WJ, Guan YM, et al. Sex dependent pharmacokinetics, tissue distribution and excretion of peimine and peiminine in rats assessed by liquid chromatography-tandem mass spectrometry. J Ethnopharmacol. 2013;145:77–84. doi: 10.1016/j.jep.2012.09.054. [DOI] [PubMed] [Google Scholar]
- 81.Wu X, Sun JG, Peng Y, Liang Y, Wang GJ, Chen H, et al. Pharmacokinetics, tissue distribution and excretion of verticinone from F. hupehensis in rats. Molecules. 2014;19:20613–20626. doi: 10.3390/molecules191220613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Comer KA, Falany JL, Falany CN. Cloning and expression of human liver dehydroepiandrosterone sulphotransferase. Biochem J. 1993;289:233–240. doi: 10.1042/bj2890233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu L, Klaassen CD. Regulation of hepatic sulfotransferases by steroidal chemicals in rats. Drug Metab Dispos. 1996;24:854–858. [PubMed] [Google Scholar]
- 84.Gamage N, Barnett A, Hempel N, Duggleby RG, Windmill KF, Martin JL, et al. Human sulfotransferases and their role in chemical metabolism. Toxicol Sci. 2006;90:5–22. doi: 10.1093/toxsci/kfj061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 16 kb)
(DOCX 16 kb)