

Abstract
Immunogenicity assessment is important for biological products due to potential impacts of immunogenicity on safety and efficacy. We reviewed the prescribing information and the FDA’s clinical pharmacology review of 121 approved biological products for evaluating and reporting of immunogenicity data. Of the 121 products, 89% (n = 108) reported the incidence of immunogenicity and 49% (n = 59) reported immunogenicity impact on efficacy. However, only 26% (n = 31) reported whether the immunogenicity affected pharmacokinetics. A subset of 16 products reported effects of anti-drug antibodies (ADA) on both systemic clearance and efficacy; 8 of 16 products had increased systemic clearance coinciding with reduced efficacy, and 6 of 16 products had no changes in either clearance or efficacy. Factors contributing to infrequent reporting of the ADA effect on exposure and methods for determining the effect of ADA on exposure are summarized. Measuring ADA and drug concentrations concurrently over time enables the evaluation of ADA impact on pharmacokinetics. Within-subject comparison of concentration data (before vs. after ADA formation) is a useful alternative to between-subject (ADA+ vs. ADA−) comparison when sample size is limited or when the majority of subjects developed ADA. The biological complexity of immune responses presents challenges to quantifying the ADA impact on pharmacokinetics using model-based methods. Our findings support that pharmacokinetic exposure is more sensitive than efficacy endpoints for evaluating ADA effects. A decrease in drug concentration due to formation of ADA during treatment can serve as an early indicator for potential reduced efficacy occurring at a later time.
Key words: clinical pharmacology assessment, immunogenicity data for approved biological products, impact on clinical pharmacokinetics and efficacy, incidence of anti-drug antibodies and neutralizing antibodies
INTRODUCTION
A growing number of biological products have been approved by regulatory agencies and some of them have been in clinical use for decades. Cumulative clinical experience with approved biological products highlighted the importance of evaluating immunogenicity given its effect on clinical outcomes (1,2). When an individual develops immunogenicity to a biological treatment, the body’s immune system produces anti-drug antibodies (ADA) which bind to the biological product in circulation at various epitopes of the molecule and can potentially lead to variable effects on the pharmacokinetics and/or efficacy. By binding to the biological products, some ADA can influence the clearance of the biological products (3), either shortening the elimination half-life (drug-clearing ADA) or extending the elimination half-life (drug-sustaining ADA). Some ADA may have neutralizing activity (neutralizing ADA, or NAb) leading to a reduction of the efficacy while others may be non-neutralizing. Many factors can influence the probability of ADA formation including product-related and patient-related factors (4–6).
Numerous literature reports have stated that immunogenicity negatively impacted the therapeutic outcomes of biological treatments, such as immunogenicity-associated secondary treatment failure (5,7,8) for monoclonal antibodies and recombinant proteins. A reduction in the systemic exposure can be associated with the observed immunogenicity and a potential explanation for the reduced efficacy to biologics. It is worth noting that clinical studies reported in the literature and conducted by individual institutions often relied on in-house assay methods for measuring not only the ADA but also the drug concentrations. A cross-study comparison of immunogenicity data in the literature may not be as informative as within-study comparisons because the immunogenicity results are highly dependent on the assay methodology used (3,9). As such, almost all prescribing information of biological products cautioned that comparison of incidence of ADA across products may be misleading. However, multiple independent studies showing consistency of an association between the ADA formation and the reduction of efficacy for a given product, in spite of the methodological differences, would be rather informative.
Bartelds et al. (10) reported the time course of pre-dose (i.e., trough) adalimumab concentration and the proportion of dropout (discontinued participation due to treatment failure) over 156 weeks in adalimumab-treatment naïve rheumatoid arthritis patients who were treated with adalimumab 40 mg every other week. At each time point, the data were stratified into three groups: (i) patients who developed high-titer ADA, (ii) patients who developed low-titer ADA, and (iii) patients who did not develop ADA (i.e., ADA- group). At week 4, the median pre-dose adalimumab concentrations were relatively similar (in the range of 4–7 mcg/mL) among the three groups; however, the median pre-dose concentration of adalimumab was different among three groups at week 16. Compared to the week 4 data in each group, the median concentration at week 16 was higher (12 vs. 7 mcg/mL) in the ADA- group and the concentration sustained as the steady state level, slightly higher (6 vs. 5 mcg/mL) in the low-titer ADA+ group and maintaining the steady state concentration in the range of 4–8 mcg/mL, and lower (<2 vs. 4 mcg/mL) in the high-titer ADA+ group and maintained below 2 mcg/mL through week 156. In contrast, the proportion of dropout due to treatment failure was relatively similar between ADA+ and ADA− groups at week 16 and only at the next scheduled assessment (week 28) the dropout rate in ADA+ group exceeded that of the ADA− group. Because the formation of ADA is first associated with a reduction of drug concentration followed by a treatment failure, one could infer that the drug concentration was a more sensitive parameter than the clinical consequence of the ADA formation. Such inference has scientific support because the achievement of therapeutically effective drug concentrations is a prerequisite for efficacy, for many products if not all. Therefore, the observed reduction in drug concentration is likely an early marker of the influence of ADA on efficacy; especially when the systemic exposure correlates with the efficacy.
The negative impact of immunogenicity on treatment response to biologics reported in clinical publications motivated our research on the reporting and impact of immunogenicity as described in the product labeling for approved biological products. We reviewed primarily the prescribing information and the clinical pharmacology review documents of biological products published on the Drugs@FDA website, which was sometimes supplemented by unpublished documents in the internal databases at the FDA. Our main focus was the clinical pharmacology reporting of immunogenicity impact, specifically whether immunogenicity affected the systemic exposure and/or efficacy. When the effect of ADA on systemic exposure was not reported, we investigated into what, if any, factors could have affected the ability to make such a determination of the effect. In this paper, we will report our findings and share our perspectives of lessons learned.
Immunogenicity Data Reporting in the Prescribing Information
For approved biological products, the extent of immunogenicity assessment during clinical development is reflected in the prescribing information under Section 6 Adverse Reactions and Section 12.3 Pharmacokinetics. Broadly speaking, the immunogenicity information includes five elements: two on immunogenicity incidence including those for the binding antibodies and neutralizing antibodies, and three on the impact of immunogenicity on clinical outcomes including pharmacokinetics, efficacy, and safety. Among these five elements, the frequency of reporting was the highest for immunogenicity incidence (specifically binding antibodies) and the lowest for immunogenicity impact on pharmacokinetics. For the assessment of immunogenicity impact, our evaluations are mainly based on the data for binding anti-drug antibodies because of the data availability.
We reviewed the prescribing information of 121 products approved before February 2015 (majority approved over the past 2 decades), which included 43 monoclonal antibodies, 26 enzyme products, 11 cytokines, 12 growth factors and hormones, and peptides, proteins, and toxins for the remaining 29 products (Fig. 1). As illustrated in Fig. 2, most (n = 108, 89%) of these 121 products reported the ADA incidence, i.e., incidence of binding anti-drug antibodies. Fewer products reported the incidence of neutralizing antibodies (n = 73, 60%) and the immunogenicity impact on safety (n = 73, 60%), followed by the impact on efficacy (n = 59, 49%) and pharmacokinetics (n = 31, 26%). When the prescribing information included text about the immunogenicity impact on efficacy, a substantial proportion (16 of 59) mentioned that the clinical significance of ADA was unclear/unknown or the limited available data precluded a determination of the effect of ADA. In other words, about 27% of products reported an inconclusive result despite the attempt to evaluate the immunogenicity impact on efficacy. Whereas only 2 of 31 products (<7% of products) stated the impact of ADA on pharmacokinetics was inconclusive. The smaller proportion of inconclusive results for the pharmacokinetic endpoints suggests that pharmacokinetics is a more sensitive metric for evaluating the impact of immunogenicity, although the number of products that reported the impact of ADA on pharmacokinetics was about half of that reported the ADA impact on efficacy (n = 31 vs. 59).
Fig. 1.

Summary of the database of 121 biological products: a product categories and b number of products approved by 5-year intervals
Fig. 2.
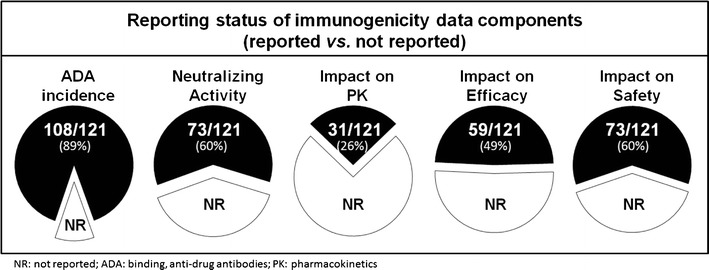
Summary of immunogenicity impact reporting in the prescribing information
Among the 31 products that included immunogenicity impact on pharmacokinetics in the prescribing information, the observed effect was clearly stated for 29 products, whereas 2 products had inconclusive ADA status and no information on the ADA effect on pharmacokinetics (Table I). Of the 29 products, drug-clearing ADA was observed in 13 products, which was associated with an increase in systemic clearance and a reduction in systemic exposure. Eight of 13 products with drug-clearing ADA, including 5 monoclonal antibodies and 3 enzyme products, also had a reduction in efficacy, whereas no information was available on the remaining 5 products (2 monoclonal antibodies, 1 enzyme product, and 2 protein/peptide products). Overall, there was a striking concordance between an increase in systemic clearance of products and a reduction of efficacy associated with ADA. Six of 29 products had drug-sustaining ADA, which resulted in a reduced clearance possibly due to the formation of complex (drug molecules bound to ADA) with prolonged elimination half-life. The effect of drug-sustaining ADA on efficacy varied—one product (a protein/peptide) had a reduced efficacy, one product (an enzyme) had no change in efficacy, and the remaining four products (three enzyme products and one protein/peptide product) did not report the effect of ADA on efficacy. Ten products reported that the formation of ADA did not affect the systemic exposure of the product. Six of these ten products, including four monoclonal antibodies, one enzyme, and one peptide/protein product, reported that the efficacy was not affected, whereas no information was available for the remaining four products (three monoclonal antibodies and one protein/peptide product). Overall, 15 of the 31 products did not report ADA effect on efficacy (i.e., all NR combined in Table I), either by stating that a conclusion could not be drawn based on limited available data in the product labeling or remaining conspicuously silent in the labeling. We noted that no monoclonal antibodies developed drug-sustaining ADA. Apart from that, there appeared no clear association of the product modality (monoclonal antibody, enzyme, or protein/peptide) with the type of ADA formed. For instance, five of nine monoclonal antibody products had drug-clearing ADA, but the ADA did not affect pharmacokinetics for the remaining four monoclonal products.
Table I.
Summary of the Effect of Immunogenicity on Pharmacokinetics and Efficacy for 31 Products; Data Grouped by the Effect on Pharmacokinetics (↑ = increase, ↔ = no change, ↓ = decrease)
| ADA type | CL | Number of products | Percent of total number of drugs | Efficacy | Number of products |
|---|---|---|---|---|---|
| Clearing | ↑ | 13 | 42.0 | ↓ | 8 |
| NRa | 5 | ||||
| No effect | ↔ | 10 | 32.2 | ↔ | 6 |
| NR | 4 | ||||
| Sustaining | ↓ | 6 | 19.4 | ↓ | 1 |
| ↔ | 1 | ||||
| NR | 4 | ||||
| Inconclusive | Unknown | 2 | 6.4 | NR | 2 |
| Total number of drugs | 31 | 100 | 31 | ||
a NR not reported, including about equal number of products with no mention in the labeling or labeling mentioned a conclusion could not be drawn based on limited available data
Factors Influencing the Ability to Report the Immunogenicity Impact on Pharmacokinetics
Because majority of the products (74%) did not report the effect of immunogenicity on pharmacokinetics, we further looked into potential factors related to such an outcome. We identified some limiting factors and summarized them in three categories:
Study design limitations—Examples included a lack of pharmacokinetic sampling and not measuring drug concentrations at the time of immunogenicity sampling. Additionally, inadequate sampling schedule for either immunogenicity samples or pharmacokinetic samples would fall into this category.
Data limitations—Examples included a small sample size in clinical trials, a small number of ADA+ subjects, and a small number of ADA− subjects.
Assay limitations—When the drug concentration was at a level that interfered with the detection of ADA in study samples, immunogenicity incidence reporting and impact assessments were affected.
We recognize that the identified limitations relevant to the database may not be comprehensive from the perspectives of today’s best practices (3,4,11). For instance, historically, researchers used the general category of ADA, whereas today’s researchers are increasingly providing a greater granularity for ADA data, e.g., transient versus persistent ADA, ADA with high titers versus low titers, among others. Given the current database, we are unable to determine if the data granularity with respect to ADA characteristics is a limiting factor.
Methods of Data Analysis May Influence the Reporting of Immunogenicity Impact on Pharmacokinetics
We observed that methods used to analyze the data could influence the ability to draw a conclusion and sometimes the reliability of the conclusion. In this section, we will describe three data analysis methods: two conventional methods, namely between-subject comparison and within-subject comparison of drug concentration data, and a model-based method using covariate analysis in population pharmacokinetics (PopPK) modeling.
Conventional Methods—Between-Subject or Within-Subject Comparison of Drug Concentration Data
The most common method used to evaluate the effect of ADA on pharmacokinetics was by comparing the systemic exposures in ADA+ subgroup and ADA− subgroup, i.e., between-subject comparison (panel A in Fig. 3). The comparative evaluation could be based on data at one time-point or at multiple time-points depending on the schedule of assessments in the protocol design. Most studies had a measurement of ADA at or near the time of primary efficacy assessment, which was often the time-point used for evaluating the between-group (ADA+ vs. ADA−) differences in pharmacokinetics, pharmacodynamics, and efficacy. Comparing data at multiple time-points was advantageous in that it can provide information on the time course of the effect of ADA, including the onset of effect, the magnitude of effect over time, and possibly the decline of ADA influence or reversal of ADA status from positive to negative over time, as shown in literature examples (3,10,12).
Fig. 3.
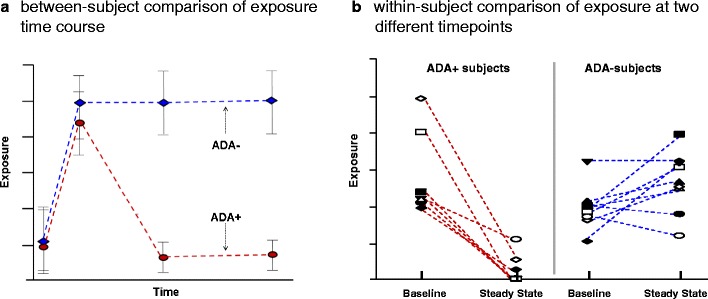
Schematics for two separate case examples showing the effect of ADA on exposure by between-subject comparison (a), i.e., group average of ADA− subjects vs. group average of ADA+ subjects, at a single time-point or multiple time-points, and within-subject comparison (b) of exposures observed at baseline and at steady state stratified by the subject ADA status at steady state
Another common method used is to compare the exposure data before ADA formation to those after ADA formation in individuals who developed ADA during treatment (ADA+ subjects of panel B in Fig. 3). The within-subject comparison approach was a necessary alternative when an overwhelming majority of subjects developed ADA and/or when the sample size was extremely small as often seen in clinical trials for products intended to treat patients with rare diseases. This approach would also be useful when only a few subjects developed ADA. A lower systemic exposure after repeated dosing compared to that obtained after the first dose could reflect the effect of immunogenicity in ADA+ subjects. Panel B of Fig. 3 showed a graphical illustration of within-subject comparison using an example product for which the pharmacokinetic exposure data were available at baseline and at steady state. Subjects who were ADA+ at the time of steady state pharmacokinetic measurement had an apparent decrease in exposure compared to the baseline exposure; in contrast, the exposures in ADA− subjects were similar at baseline and at steady state. In addition, when ADA titer data were available, a semi-quantitative evaluation of the effect of ADA titer may be feasible. As an example, the alglucosidase alfa prescribing information used within-subject comparison and further indicated that an increase in systemic clearance after repeated dosing was observed only in a subgroup of subjects with sustained high ADA titers (13).
By visual inspection of steady state data in panel B of Fig. 3, the group of ADA+ subjects had about two units lower median exposure than the group of ADA− subjects at steady state, whereas at the individual subject level, the ADA+ subjects had a drop in exposure by up to four units from baseline to steady state due to ADA formation. This observation suggests that within-subject comparison may be more sensitive than between-subject comparison for detecting the effect of ADA. If it is feasible to conduct both within-subject and between-subject comparisons, a concordance from both analyses could further strengthen the conclusion when the sample size is small. As an example, the idursulfase data in 27 children younger than 7.5 years old were analyzed using both approaches to support the conclusion that formation of ADA was associated with a reduction in systemic exposure (14).
Model-Based Method—Covariate Analysis in PopPK Modeling
Applications of covariate analysis in PopPK modeling have been successful in integrating data from multiple trials, including small molecule drugs and biologics. In particular, it has been proven useful to elucidate the influence of various intrinsic factors and extrinsic factors on the pharmacokinetic properties at the population level. In some cases, the results of covariate analyses supported dose adjustments in specific populations. A logical extension is to apply the same approach to quantify the effect of ADA as an intrinsic factor. Of the 121 products we reviewed, the clinical pharmacology program of at least 8 products included PopPK analysis and incorporated the ADA status as a covariate for the clearance parameter in the model. The estimated covariate effect showed that the clearance value for ADA+ group increase by <80% compared to the ADA- group which was the designated reference group with a typical clearance value, except for one product with a >2-fold increase in clearance value and one product with a 50% lower clearance value in ADA+ group when compared to the ADA− group. However, the results of PopPK modeling analysis were mainly supportive of the traditional comparative analysis described above.
Using a monoclonal antibody product as the case example, Fig. 4 conceptually illustrated the challenges for assessing ADA effect based on covariate analysis in PopPK modeling. Per study design, pharmacokinetic samples were collected more frequently than immunogenicity samples. Specifically, Fig. 4 showed an example subject who developed ADA with the first ADA+ sample occurring at T2. For PopPK modeling analysis, the subject’s ADA profile was implemented in three different ways according to the principles described below. Scenario 1 treated ADA as a time-invariant variable and assigned a positive value for all time-points starting from the baseline. Scenarios 2 and 3 treated ADA as a time-varying variable. Scenario 3 assigned ADA+ value to pharmacokinetic observations from T2 through T3, whereas scenario 2 assumed that pharmacokinetic observations between T1 and T2 were associated with ADA+ status given the lack of experimentally determined value, in addition to conservatively assigning the sample at T1 as ADA+. In scenario 1, the estimated CL value for ADA+ subjects was 1.25-fold of the clearance value in a typical ADA− subject. In scenarios 2 and 3, at the time when subjects were ADA+, their clearance values were, on average, 2.25-fold and 3.5-fold of the typical clearance value in ADA− subjects. In this case example, the model-based analysis method was able to show that ADA formation was associated with a higher clearance, similar to the conclusion from the conventional between-subject comparison method. However, the model-based approach concluded smaller numeric differences between ADA+ subjects and ADA− subjects for various scenarios compared to an estimated greater than tenfold difference in pharmacokinetic exposure between ADA− subjects and ADA+ subjects based on the observed drug concentrations which were largely not detectable in ADA+ subjects. As such, opportunities remain for further improvements in the model-based approach to better describe the observed data.
Fig. 4.
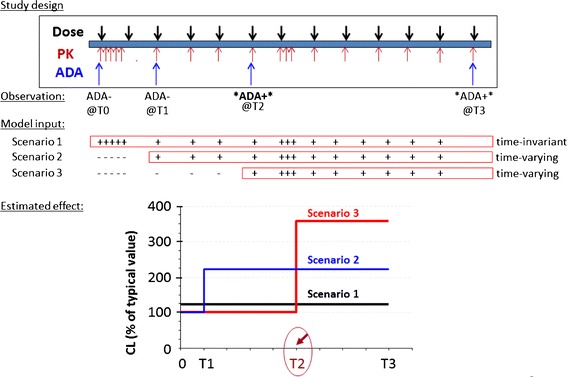
A conceptual illustration of the use of PopPK modeling method to evaluate the immunogenicity effect on systemic clearance with ADA implemented as time-invariant covariate or time-varying covariate. (PK pharmacokinetic(s), ADA anti-drug antibodies; T0, T1, T2, T3 four immunogenicity sampling time-points with T0 representing the baseline; plus sign: ADA+ for a PK sample; minus sign: ADA− for a PK sample; CL clearance; typical value: the estimated value for a typical subject without ADA formation)
DISCUSSIONS
Biological products can be targets of human adaptive immune response as the immune system is well-equipped to react to the invasion of foreign proteins and/or peptides and can produce antibodies against exogenously administered biological products. Structurally complex biological products may present multiple immunogenic epitopes and can induce the formation of a heterogeneous mixture of ADA with varying degrees of affinity to various epitopes. Within an individual subject, the composition of ADA mixture may vary over time. ADA heterogeneity can also exist among study subjects administered the same product because the development of immunogenicity may depend on patient-related factors in addition to product-related factors.
The current approach for immunogenicity assessment consists of two steps: first identifying samples with the presence of ADA using mainly ligand binding assays and subsequently evaluating the ADA+ samples for the capability of neutralizing the biological function of the product using in vitro systems (3,11). In addition to reporting the presence or absence of binding antibodies (ADA+ vs. ADA−) or neutralizing antibodies (NAb+ vs. NAb−) for each study sample, immunogenicity assay results may be reported as ADA titer, a semi-quantitative determination of the magnitude of ADA response. These ADA data could be used to explore the relationship with clinical outcomes in order to assess the clinical impact of immunogenicity (3,10). Inherently, the impact of immunogenicity on clinical outcomes reflects the effects of a mixture of ADA with varying compositions over time within an individual and across individuals. The composite nature and variability of ADA may have contributed in part to a high frequency of inconclusive results with respect to the impact of immunogenicity on efficacy endpoints. We suspect that the heterogeneity and variability of ADA produced after treatment may also have partially contributed to the difficulties in quantifying the extent to which ADA affect the systemic clearance, and presented unique challenges for the PopPK modeling approach as illustrated in Fig. 4.
Findings from our current research support an assertion that the drug concentration (a pharmacokinetic exposure endpoint) is more sensitive than efficacy endpoints for the determination of whether or not ADA have a negative influence on clinical treatment response. The supporting evidence includes (i) among the prescribing information of 121 products, the impact of ADA is less often reported with uncertainty for pharmacokinetic endpoints than for efficacy endpoints (<7% [2/31] vs. 27% [16/59]), and (ii) a strong concordance exists between the ADA influence on pharmacokinetic exposure and clinical efficacy as shown in Table I, which is consistent with the general principle that a lower exposure leads to a reduced efficacy. A subset of 16 products reported effects of ADA on both systemic clearance and efficacy; 8 of 16 products had increased systemic clearance coinciding with reduced efficacy, and 6 of 16 products had no changes in either clearance or efficacy. The effect of ADA within one product modality appears to vary. For example, among nine monoclonal antibodies, five products had clearing ADA versus ADA of four products had no effect on pharmacokinetics.
These findings are striking despite the fact that the database is small, i.e., only 16 out of 121 products reported the impact of immunogenicity on both pharmacokinetics and efficacy. Scientifically, the concordance is logical because sustaining drug concentrations within a therapeutic range is essential for maintaining treatment effects. A reduction in pharmacokinetic exposure observed in clinical trials can provide insight into the potential for ADA to negatively influence efficacy outcomes at a later time when the product is used more extensively for a longer duration after approval. However, two products in our dataset had drug-sustaining ADA resulting in higher exposure without an increase in efficacy; specifically, one reported a reduction of efficacy and the other reported no change in efficacy. Interpreting the relationship of effects of drug-sustaining ADA on pharmacokinetics and efficacy is more challenging as it is often unclear whether the drug molecules bound to ADA retain the pharmacological activity. Because the pharmacokinetic exposure is a more sensitive endpoint compared to efficacy, monitoring systemic exposure in late-stage clinical trials would provide valuable return on the investment.
As well-recognized, the effect of immunogenicity on efficacy needs to be determined based on the efficacy endpoints during clinical trials. A simultaneous assessment of immunogenicity effect on pharmacokinetic endpoints and on efficacy endpoints can provide added values. At the minimum, a negative effect of immunogenicity on the systemic exposure without accompanying changes in efficacy over the duration of clinical trials could raise the awareness to carefully monitor treatment effects for extended treatment duration. Whether or not the early effect of immunogenicity on pharmacokinetics ultimately manifests as a clinically meaningful impact on the efficacy may vary among products and may depend on the characteristics of the anti-drug antibodies formed. For instance, the antibody levels may wane over time in some cases, whereas they may continue to rise over time in some other cases; the resulting impact on long-term efficacy would differ. In cases where antibody levels maintain at a very low level and have a negative, but limited, impact on systemic exposure, the effect on efficacy endpoints may not be clinically meaningful. For some products, a reduction of exposure may not have associated reduction in the clinical efficacy. As an example, some products do not show a clear dose-response (D-R) or exposure-response (E-R) relationship; increasing plasma concentrations may not result in increasing response to treatment for these products. The reasons for a lack of E-R relationship are often unclear and likely multifaceted, which may include but not limited to the sensitivity of the endpoints used for E-R analysis, dose/exposure range evaluated, the site and mechanism of action, and the pharmacokinetic characteristics of the product, among others. A potential explanation for the lack of E-R could be a lack of correlation between the concentration in systemic circulation and the concentration at the target site, because the plasma or serum drug concentrations are typically used in the E-R analyses. In such a scenario, the impact of ADA on the target site concentration may not be apparent despite a change in systemic exposure due to ADA formation.
During clinical development, a well-designed schedule of assessment for both pharmacokinetics and immunogenicity is warranted to enhance the ability to evaluate the impact of immunogenicity on pharmacokinetics. For example, determining drug concentration and ADA at the same time-points is desirable for the purpose of evaluating whether the ADA can alter the drug levels; on the other hand, immunogenicity assessment solely for the purpose of safety evaluation is not as desirable. Furthermore, studies designed to establish the time course of ADA and drug concentration allow a more robust evaluation of immunogenicity impact by capturing not only the formation but also the rise and fall of ADA titer over time. For instance, a clinical study with exenatide (12) showed that ADA against exenatide first rose to peak at week 6 and subsequently declined over 30 weeks. The study of Barteld et al. showed that the proportion of subjects became ADA+ increased over a 52-week study duration; when subjects were divided into three groups (ADA−, low-titer ADA, and high-titer ADA), the effect of ADA on systemic adalimumab exposure correlated with the ADA titer, i.e., the high ADA titer group had the lowest adalimumab concentration which persisted over time.
The quest to numerically quantify the effect of ADA formation on systemic exposure is likely the motivation for using covariate analysis in PopPK modeling. The implementation of ADA status as a covariate on clearance in the PopPK model has evolved from being a time-invariant covariate (either ADA+ or ADA− throughout the entire study duration) to being a time-varying covariate (ADA status changing over time), and it continues to evolve. Recently, we observed one variation of time-varying covariate implementation where two clearance values were estimated for each ADA+ subject with the assumption that ADA+ occurred after 3–4 weeks of treatment regardless of the observed ADA status for the study samples. Thereby, the clearance value estimated based on data from the initial 3–4 weeks (assumed to be ADA−) was compared to the estimated clearance value based on data collected afterwards (assumed to be ADA+) to assess the effect of ADA on systemic clearance, which is akin to within-subject comparison. This model-based within-subject comparison appears to be more sensitive than the model-based comparison of population average (a form of between-subject comparison); similar to the observations with conventional methods. With the continued exploration of additional variations of modeling implementation, someday, it may be feasible to integrate the temporal profile of ADA titer into the PopPK model provided that adequate data are available to inform the model-building process. One additional technical aspect of PopPK modeling to be considered is the handling of concentration data that are below the limit of quantification. Exclusion of such data can introduce bias, but this practice has been consistently observed thus far.
Above all, the complex nature of ADA in blood samples, and their variability over time and across subjects may prove to be the greatest challenges to achieving precision in describing the effect of ADA on systemic drug concentration by mathematical modeling. Fundamentally, ADA differ from conventional covariates of PopPK model, such as body weight and sex, in that ADA consist of a heterogeneous mixture of differing amounts, binding epitopes, and/or potency of antibodies, and its composition can differ over time in a given subject as well as among subjects. Although covariate analysis approach has been influential to dosing recommendation based on conventional covariates, its application in quantifying ADA effect on systemic exposure has not been translated into an influence on dosing recommendation yet. Nonetheless, proposals for dose adjustments upon ADA formation have been empirically derived and described in literature reports of several monoclonal antibodies indicated for the treatment of rheumatoid arthritis, Crohn’s disease, and ulcerative colitis to cope with secondary treatment failure (15,16). Generally, the proposed dose adjustment approaches rely on multiple laboratory tests for decision making, including the systemic drug concentration, the ADA titer or ADA status, and other clinical parameters in conjunction with the physicians’ assessment of clinical symptoms.
The availability of an immunogenicity assay and a drug concentration assay is critical for evaluating immunogenicity impact on pharmacokinetics. The quality of these assays undoubtedly has profound impact on the reliability of the outcome of assessment (17). Drug interference has been identified as a major factor influencing the interpretation of immunogenicity data. As described in an earlier publication (9), our database contains many products that suffered from drug interference to the immunogenicity assay, which in part contributed to the infrequent reporting of ADA impact on pharmacokinetics. When immunogenicity assays are interfered by the drug concentration present in ADA samples, the assays are only able to detect high-titer ADA. Very often the high-titer ADA negatively impacted the systemic exposure and the efficacy outcome. Whether the low-titer ADA presumably masked by the drug interference could have a similar effect on the systemic exposure and the efficacy outcome remains to be elucidated. The development of newer immunogenicity assay technologies less prone to drug interferences (18–20) will likely address this knowledge gap soon and possibly can further facilitate quantitative evaluations of immunogenicity impact.
Similarly, the assay for drug concentration plays an important role in drug development, including the evaluation of immunogenicity effect on systemic exposure (3,21–24). When the assay measures the active moiety of drug that is relevant to clinical efficacy, the exposure data are more likely to correlate with the clinical response and show an E-R relationship. In the event that the formation of ADA is associated with an observed increase in clearance (i.e., a decrease in exposure) based on an assay that detects the active moiety, a reduction in efficacy is more likely to follow. On the other hand, if the drug concentration assay can detect the drug molecule bound to ADA even when the ADA have neutralized the pharmacological activity of the drug molecule, the measured drug concentration may not correlate with the clinical efficacy. With the currently available methodologies, it is well-known that the determined concentrations from pharmacokinetic assays are highly dependent on the capture reagents and detection reagents which are specifically selected for each drug molecule of interest. As such, special attention to what moiety(ies) are measured by the assay for drug concentrations and whether the detected moiety is relevant to the clinical response would be highly important for both pharmacokinetic samples not containing ADA and pharmacokinetic samples containing ADA. Overall, a good understanding of the performance of bioanalytical assays and immunogenicity assays is critical for data interpretation (21,22,24).
CONCLUSION
In summary, evaluating the effect of ADA on systemic exposure during clinical development would be highly informative and can supplement the traditional evaluation of ADA impact on the efficacy outcomes because a reduction in the systemic exposure, in principle, would precede a reduction in efficacy. Measuring drug concentrations and ADA levels in the same schedule and obtaining both measurements at multiple time-points throughout the study are among study design features that would facilitate the evaluation of ADA effects on systemic exposure. With respect to data evaluation, we found that multiple approaches have been applied. Between-subject comparison (ADA+ subgroup vs. ADA− subgroup) has been the traditional approach, and within-subject comparison of exposure data (before vs. after ADA formation) serves as an alternative or complementary approach for the determination of pharmacokinetic alteration due to ADA formation. Model-based covariate analysis within the framework of PopPK modeling is an up-and-coming new methodology which is still evolving. Although not being evaluated in this research project, considerations of the assay methods for drug concentrations and ADA measurement are necessary when interpreting the observed ADA impact, or lack thereof, on systemic exposure.
Acknowledgments
The authors wish to acknowledge David E. Matthews for his contribution to the preliminary data collection as a student intern from The Ohio State University. The authors would like to thank Dr. E. Dennis Bashaw for his critical review of the manuscript.
Compliance with Ethical Standards
Disclaimer
The opinions expressed in this article are those of the authors who are affiliated with the United States Food and Drug Administration (FDA) and do not necessarily reflect the official views or policies of the FDA.
References
- 1.Mckoy JM, Stonecash RE, Cournoyer D, Rossert J, Nissenson AR, Raisch D, et al. Epoetin-associated pure red cell aplasia: past, present, and future considerations. Transfusion. 2008;48(8):1754–1762. doi: 10.1111/j.1537-2995.2008.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nanda KS, Cheifetz AS, Moss AC. Impact of antibodies to Infliximab on clinical outcomes and serum Infliximab levels in patients with inflammatory bowel disease (IBD): a meta-analysis. Am J Gastroenterol. 2013;108(1):40–47. doi: 10.1038/ajg.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shankar G, Arkin S, Cocea L, Devanarayan V, Kirshner S, Kromminga A, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J. 2014;16(4):658–673. doi: 10.1208/s12248-014-9599-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.FDA Guidance for Industry: Immunogenicity Assessment for Therapeutic Protein Products (2014).
- 5.Moss AC, Brinks V, Carpenter JF. Review article: immunogenicity of anti-TNF biologics in IBD—the role of patient, product and prescriber factors. Aliment Pharmacol Ther. 2013;38(10):1188–1197. doi: 10.1111/apt.12507. [DOI] [PubMed] [Google Scholar]
- 6.Shankar G, Shores E, Wagner C, Mire-Sluis A. Scientific and regulatory considerations on the immunogenicity of biologics. Trends Biotechnol. 2006;24(6):274–280. doi: 10.1016/j.tibtech.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Garces S, Demengeot J, Benito-Garcia E. The immunogenicity of anti-TNF therapy in immune-mediated inflammatory diseases: a systematic review of the literature with a meta-analysis. Ann Rheum Dis. 2013;72(12):1947–1955. doi: 10.1136/annrheumdis-2012-202220. [DOI] [PubMed] [Google Scholar]
- 8.Keiserman M, Codreanu C, Handa R, Xibille-Friedmann D, Mysler E, Briceno F, et al. The effect of antidrug antibodies on the sustainable efficacy of biologic therapies in rheumatoid arthritis: practical consequences. Expert Rev Clin Immunol. 2014;10(8):1049–1057. doi: 10.1586/1744666X.2014.926219. [DOI] [PubMed] [Google Scholar]
- 9.Wang YM, Fang L, Zhou L, Wang J, Ahn HY. A survey of applications of biological products for drug interference of immunogenicity assays. Pharm Res. 2012;29(12):3384–3392. doi: 10.1007/s11095-012-0833-2. [DOI] [PubMed] [Google Scholar]
- 10.Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–1468. doi: 10.1001/jama.2011.406. [DOI] [PubMed] [Google Scholar]
- 11.FDA Guidance for Industry: Assay Development for Immunogenicity Testing of Therapeutic Proteins (2009).
- 12.Drucker DJ, Buse JB, Taylor K, Kendall DM, Trautmann M, Zhuang D, et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study. Lancet. 2008;372(9645):1240–1250. doi: 10.1016/S0140-6736(08)61206-4. [DOI] [PubMed] [Google Scholar]
- 13.US Prescribing information of Myozyme (alglucosidase alfa). May 20, 2014.
- 14.US Prescribing Information of Elaprase (idursulfase). June 24, 2013.
- 15.Eser A, Primas C, Reinisch W. Drug monitoring of biologics in inflammatory bowel disease. Curr Opin Gastroenterol. 2013;29(4):391–396. doi: 10.1097/MOG.0b013e328361f7f6. [DOI] [PubMed] [Google Scholar]
- 16.Khanna R, Sattin BD, Afif W, Benchimol EI, Bernard EJ, Bitton A, et al. Review article: a clinician’s guide for therapeutic drug monitoring of Infliximab in inflammatory bowel disease. Aliment Pharmacol Ther. 2013;38(5):447–459. doi: 10.1111/apt.12407. [DOI] [PubMed] [Google Scholar]
- 17.Bendtzen K. Critical review: assessment of interferon-beta immunogenicity in multiple sclerosis. J Interferon Cytokine Res. 2010;30(10):759–766. doi: 10.1089/jir.2010.0091. [DOI] [PubMed] [Google Scholar]
- 18.Patton A, Mullenix MC, Swanson SJ, Koren E. An acid dissociation bridging ELISA for detection of antibodies directed against therapeutic proteins in the presence of antigen. J Immunol Methods. 2005;304(1–2):189–195. doi: 10.1016/j.jim.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 19.Wang SL, Ohrmund L, Hauenstein S, Salbato J, Reddy R, Monk P, et al. Development and validation of a homogeneous mobility shift assay for the measurement of Infliximab and antibodies-to-Infliximab levels in patient serum. J Immunol Methods. 2012;382(1–2):177–188. doi: 10.1016/j.jim.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Schantz A, Schwegler M, Shankar G. Detection of low-affinity anti-drug antibodies and improved drug tolerance in immunogenicity testing by Octet((R)) biolayer interferometry. J Pharm Biomed Anal. 2011;54(2):286–294. doi: 10.1016/j.jpba.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 21.Swann PG, Shapiro MA. Regulatory considerations for development of bioanalytical assays for biotechnology products. Bioanalysis. 2011;3(6):597–603. doi: 10.4155/bio.11.27. [DOI] [PubMed] [Google Scholar]
- 22.Lee JW, Kelley M, King LE, Yang J, Salimi-Moosavi H, Tang MT, et al. Bioanalytical approaches to quantify “total” and “free” therapeutic antibodies and their targets: technical challenges and PK/PD applications over the course of drug development. AAPS J. 2011;13(1):99–110. doi: 10.1208/s12248-011-9251-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bashaw ED, DeSilva B, Rose MJ, Wang YM, Shukla C. Bioanalytical method validation: concepts, expectations and challenges in small molecule and macromolecule—a report of PITTCON 2013 symposium. AAPS J. 2014;16(3):586–591. doi: 10.1208/s12248-014-9597-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang YM, Jawa V, Ma M. Immunogenicity and PK/PD evaluation in biotherapeutic drug development: scientific considerations for bioanalytical methods and data analysis. Bioanalysis. 2014;6(1):79–87. doi: 10.4155/bio.13.302. [DOI] [PubMed] [Google Scholar]