

Abstract
In a mouse tumour model for hereditary breast cancer, we previously explored the anti-cancer effects of docetaxel, ritonavir and the combination of both and studied the effect of ritonavir on the intratumoural concentration of docetaxel. The objective of the current study was to apply pharmacokinetic (PK)-pharmacodynamic (PD) modelling on this previous study to further elucidate and quantify the effects of docetaxel when co-administered with ritonavir. PK models of docetaxel and ritonavir in plasma and in tumour were developed. The effect of ritonavir on docetaxel concentration in the systemic circulation of Cyp3a knock-out mice and in the implanted tumour (with inherent Cyp3a expression) was studied, respectively. Subsequently, we designed a tumour growth inhibition model that included the inhibitory effects of both docetaxel and ritonavir. Ritonavir decreased docetaxel systemic clearance with 8% (relative standard error 0.4%) in the co-treated group compared to that in the docetaxel only-treated group. The docetaxel concentration in tumour tissues was significantly increased by ritonavir with mean area under the concentration-time curve 2.5-fold higher when combined with ritonavir. Observed tumour volume profiles in mice could be properly described by the PK/PD model. In the co-treated group, the enhanced anti-tumour effect was mainly due to increased docetaxel tumour concentration; however, we demonstrated a small but significant anti-tumour effect of ritonavir addition (p value <0.001). In conclusion, we showed that the increased anti-tumour effect observed when docetaxel is combined with ritonavir is mainly caused by enhanced docetaxel tumour concentration and to a minor extent by a direct anti-tumour effect of ritonavir.
KEY WORDS: anti-cancer effect, co-administration, docetaxel, pharmacokinetic-pharmacodynamic modelling, ritonavir
INTRODUCTION
Docetaxel is widely used as an anti-cancer agent for several types of cancer, such as lung, breast, gastric and prostate cancer (1). It acts by the inhibition of cell mitosis (1). Oral administration of docetaxel could be a convenient treatment option for patients (2). An oral formulation of docetaxel has been successfully developed in our group (3). One major limitation for oral administration of docetaxel is its low bioavailability due to its transport by P-glycoprotein (Pgp) and presystemic metabolism by Cytochrome P450 (CYP) 3A (2,4). Ritonavir is an HIV protease inhibitor and also a strong CYP3A4 inhibitor (5). It has been reported that co-administration of ritonavir with docetaxel in both mice and humans results in enhanced docetaxel plasma exposure (6,7). Phase I trials conducted with oral docetaxel and ritonavir proved the feasibility of co-administration (8,9).
In addition to its function as exposure booster, ritonavir has been suggested to have anti-cancer effect on its own. It has been reported to cause DNA damage and cell death in human endothelial cells (10). In prostate cancer cells, ritonavir was suggested to inhibit DNA binding activity of nuclear factor κB, which is a contributor to drug resistance (11). It has also been reported that ritonavir contributed to anti-cancer effects in mouse and human lymphoma (12–14), human head and neck carcinoma (15), and human breast cancer (16) models.
In order to explore whether ritonavir co-administration increased anti-cancer efficacy of docetaxel, we performed a study with host mice lacking Cyp3a and orthotopically implanted syngeneic breast tumour tissue with inherent Cyp3a expression (17). The results illustrated that, as expected, administration of docetaxel showed a strong anti-tumour effect. Furthermore, co-administration of ritonavir significantly prolonged survival and substantially reduced average tumour size compared to that of the docetaxel only-treated mice. This might be explained by increased docetaxel concentration in tumour by Cyp3a inhibition, but, ritonavir may also have a direct anti-tumour effect itself. However, ritonavir treatment alone showed no significant effect.
Previously, we have successfully applied pharmacokinetic (PK) modelling in description of oral docetaxel exposure and translation of docetaxel exposure from mice to human (18). Besides, a number of tumour growth inhibition models were successfully applied to explore the characteristics of tumour growth and inhibitory effect of anti-cancer drugs by PK-pharmacodynamics (PD) modelling (19). Accordingly, the aim of this study was to develop a PK-PD model based on the data from a previously performed mouse study to further evaluate and quantify the effects of ritonavir on docetaxel concentration in both plasma and tumour and the anti-tumour effects of docetaxel.
METHODS
Mice Study
Seventy female Cyp3a knockout (Cyp3a−/−) mice, with body weight around 25 g, were implanted with small tumour pieces (1–2 mm) derived from the K14cre;Brca1F/F;p53F/F mouse model (20) for hereditary breast cancer. The implanted tumours had Cyp3a expression. Treatment was started (day 1) when the tumours reached a volume of ~200 mm3 and lasted for 3 weeks unless the mice were sacrificed earlier (when tumours reached a volume of ~1500 mm3). All mice were divided randomly over four groups. Group I was not treated and used as control for tumour growth (control group, n = 20). Group II was treated with ritonavir (ritonavir only-treated group, n = 20), group III was treated with docetaxel (docetaxel only-treated group, n = 30), and group IV was treated with both docetaxel and ritonavir (co-treated group, n = 30).
Docetaxel was administered intravenously at dose of 20 mg/kg once a week, while ritonavir was administered orally at dose of 12.5 mg/kg for five subsequent days per week. Both docetaxel and ritonavir were purchased from Sequoia Research Products (Oxford, UK). As long as tumours were below ~1500 mm3, plasma and tumour samples were collected for PK analysis on days 2, 9 and 16 (n = 5 for each sampling time). All samples were taken at approximately 24 h after docetaxel administration. Tumour volumes were measured daily in all groups. The observed PK and PD data is shown in Fig. 1. Besides, histological analysis and expression of Cyp3a, Mdr1a/b and Bcrp were also monitored in the tumour. For a more detailed description of this study, we refer to (17).
Fig. 1.
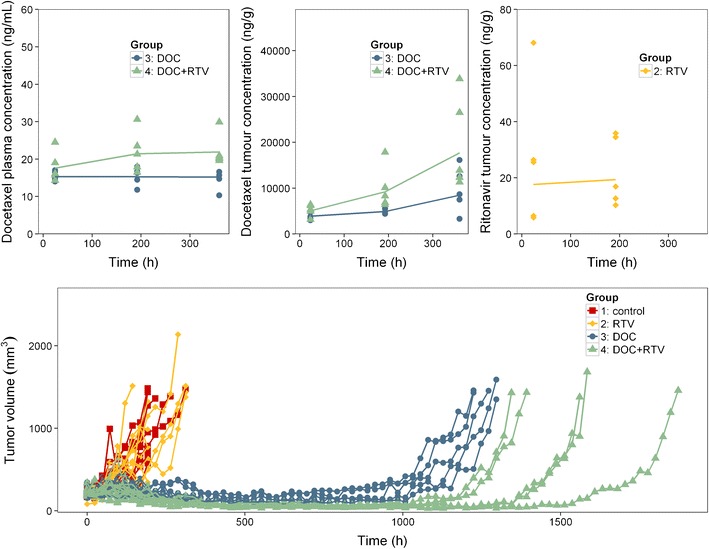
Observed pharmacokinetic and pharmacodynamic data of different study groups in mice. DOC, docetaxel; RTV, ritonavir. The upper panels show the pharmacokinetic data, the symbols represent each observed concentration level, and the line represents the mean concentration levels of each group; the lower panel presents the tumour volumes, the symbols represent each observed tumour volume, and each line represents the tumour volume of one mouse. This figure was originally published in (17). The figure is republished and modified with permission
The available concentration and tumour volume data used for model development is summarized in Table I.
Table I.
Summary of Observed Data Used in Pharmacokinetic-Pharmacodynamic Modelling from Mice Study
| Observation type | Sampling method | Sampled groups | Applied groups | Sampling time | Sample number |
|---|---|---|---|---|---|
| DOC plasma concentration | Naïve pooling | DOC | DOC | 24, 192, 360 h | 15 |
| DOC+RTV | DOC+RTV | 24, 192, 360 h | 15 | ||
| DOC tumour concentration | Population | DOC | DOC | 24, 192, 360 h | 15 |
| DOC+RTV | DOC+RTV | 24, 192, 360 h | 15 | ||
| RTV tumour concentration | Naïve pooling | RTV | RTV | 24, 192 h | 10 |
| DOC+RTV | 24, 192 h | 10 | |||
| Tumour volumes | Population | Control | Control | Every 24 h | 109 |
| RTV | RTV | Every 24 h | 117 | ||
| DOC | DOC | Every 24 h | 403 | ||
| DOC+RTV | DOC+RTV | Every 24 h | 463 |
DOC docetaxel, RTV ritonavir
Structural Model Development
Naïve pooling was applied for docetaxel plasma concentration and ritonavir tumour concentration, while docetaxel tumour concentration and tumour volumes used population method (Table I).
PK model and PK/PD model were performed sequentially using the PPP&D (Population PK Parameters and Data) method (21).
PK Model
A previously established two-compartment model structure was used for docetaxel plasma PK (18). Due to a lack of informative PK sampling in the current study, inter-compartment clearance (QiDOC) and volume of distribution of peripheral compartment (VpDOC) were fixed according to previously established values (18). Docetaxel tumour concentrations were described with a separate compartment with a first-order absorption rate from the central compartment into the tumour compartment (KaTDOC), and with a first-order elimination rate from tumour (KeTDOC). Contribution of the tumour compartment to the total drug exposure was assumed to be negligible.
Ritonavir plasma concentrations were below limit of quantification at the sampling time points in the current study. Therefore, ritonavir systemic PK was fixed with a one-compartment model similar as used in a previous study (18). Measured tumour ritonavir concentrations were modelled analogously to docetaxel. Furthermore, the tumour PK of ritonavir was assumed to be the same in the ritonavir only-treated group and the co-treated group (Table I).
Although host mice were Cyp3a−/−, it was hypothesized that ritonavir might still have an influence on docetaxel plasma PK. This was explored by including a ritonavir effect on docetaxel clearance as described by Eq. 1.
| 1 |
In which CLDOC+RTV represents systemic clearance of docetaxel in the co-treated group; CLDOC represents systemic clearance in the docetaxel only-treated group; and ERTV represents the difference between systemic clearance of docetaxel in the co-treated group and that of the docetaxel only-treated group.
The tumour PK of both docetaxel as ritonavir was only measured at the early phase of treatment (on days 2, 9 and 16). In the co-treated group, the inhibition of docetaxel tumour metabolism by ritonavir was expected to relate to ritonavir tumour concentrations. As tumour PK was considered to be the driver of anti-cancer effect, description of the full tumour PK curve (including the time period after treatment) was necessary. Therefore, assumptions were needed to describe the PK over the full observation period. It was assumed that ritonavir inhibited docetaxel tumour elimination by a concentration-dependent direct effect as described in Eq. 2 (22).
| 2 |
In which KeTDOC+RTV represents the elimination rate constant of docetaxel from the tumour in the co-treated group; KeTDOC represents the elimination rate constant of docetaxel from the tumour in the docetaxel only-treated group; CRTV,tumour represents ritonavir tumour concentration; and IC50RTV represents ritonavir tumour concentration that inhibits half of Cyp3a enzymes. The value of IC50RTV was fixed according to the literature value as 2.5 ng/g (23).
PK/PD Model
Non-perturbed tumour growth in the untreated mice was described by an exponential net tumour growth rate (Kg0) upon the complete tumour size. In the docetaxel-treated mice, it was considered that the tumour growth rate might increase over time, which was described by Eq. 3:
| 3 |
In which Kg represented the net tumour growth rate in the docetaxel-treated groups; Kg0 represents the net tumour growth rate in the untreated groups; and LAMBDA represents the rate of increase in tumour growth over time in the docetaxel-treated groups.
Docetaxel anti-tumour effect was described by a delayed effect through an effect compartment. This assumed that tumour cells were killed related to the docetaxel tumour concentration using a delayed effect described by a transit compartment (Eq. 4):
| 4 |
In which CDOC,tumour represents docetaxel tumour concentration; EC50DOC represents docetaxel tumour concentration with 50% of maximum tumour anti-tumour effect; and KKDOC represents the first-order transition rate of tumour cells to the effect compartment.
These cells in the transit compartment were eventually eliminated by an elimination rate (KKeDOC). Both KKDOC and KKeDOC were first-order rate constants.
In the first step, the aforementioned docetaxel effect parameters were estimated based on data from the docetaxel only-treated and the control group. In the second step, these parameter estimates were used to predict tumour volume profile in the co-treated group taking only the increased docetaxel tumour concentrations as a result of co-administration of ritonavir into account. Subsequently, it was explored whether ritonavir had an additional effect on tumour growth independent from the increased docetaxel tumour concentrations.
Statistical Model Development
Between-subject variability (BSV) was modelled using an exponential distribution according to (Eq. 5).
| 5 |
Where Pi represents the individual parameter estimate for individual i, P represents the typical population parameter estimate and ηi the BSV effect distributed following N (0, ω2). BSV was evaluated for PK model on KaTDOC and for PK/PD model on baseline tumour volume.
Residual errors were described by proportional error model for PK and PK/PD models (Eq. 6).
| 6 |
Where Cobs,ij or Cpred,ij represents, for the ith subject and the jth measurement, the observation or prediction. Proportional error εp,ij was assumed distributed following N (0, σ2). The residual error for tumour concentration of docetaxel was fixed to 15% as reported in bioanalytical study of docetaxel (17,24).
Model Evaluation
Models were required to have successful minimization and covariance steps. Different models were evaluated based on the stability, plausibility and precision of parameter estimates, goodness-of-fit (GOF) plots and drop of objective function value (OFV) with significance level of p < 0.01 (degree of freedom (df) = 1, dOFV >6.63; df = 2, dOFV >9.21) for hierarchical models.
For PK models, GOF plots were conducted as graphical model assessment. For PK/PD models, a visual predictive check (VPC) was performed with upper limit of quantification set as 1500 mm3 (n = 1000). In order to circumvent the bias due to right censoring at 1500 mm3, median of observation in evaluation plotting was presented before the time when the first mouse was sacrificed (tumour volume >1500 mm3) for each study group respectively.
A sensitivity analysis was conducted to test whether the assumed value for IC50RTV has a large influence on final outcome. Therefore, a sensitivity test on fixation of IC50RTV was performed with ±50% difference from the fixed IC50RTV parameter (2.5 ng/g) in the docetaxel PK model.
Software
Model estimation was performed in NONMEM (version 7.3.0) (25) using first-order conditional estimation with interaction as estimation method. Piraña was used as graphical interface (26), and R (version 3.0.3) was used for pre-processing of the data and plotting (27). Besides, the NONMEM toolkit psn (28) and the R-package Xpose (29) were used.
RESULTS
Model Development
The final PK/PD model is schematically shown in Fig. 2. The following sections present the model building stepwise.
Fig. 2.
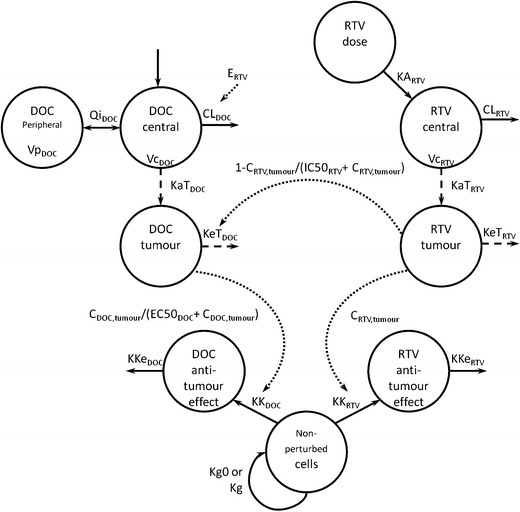
Schematic structure of pharmacokinetic-pharmacodynamic model. C, concentration; CL, clearance; DOC, docetaxel; EC50 DOC, docetaxel tumour concentration with 50% of maximum anti-cancer effect; E RTV, difference between clearance of combined treatment group and single treatment group; IC50 RTV, ritonavir tumour concentration inhibiting half of Cyp3a enzymes in tumour; KA, absorption rate in central compartment; KaT, absorption rate in tumour; KeT, elimination rate in tumour; Kg, net growth rate in treated groups; Kg0, net growth rate in untreated groups; KK, cell transition rate to effect compartment; KKe, cell elimination from effect compartment; Qi, inter-compartment clearance; RTV, ritonavir; Vc, volume of distribution of central compartment; Vp, volume distribution of peripheral compartment
PK Model
Parameter estimates for the PK model of docetaxel and ritonavir are presented in Table II. ERTV was estimated as 0.918 (relative standard error (RSE) 0.4%). This indicated that there was ~8% lowered docetaxel systemic clearance in the Cyp3a−/− host in the co-treated group than that in the docetaxel only-treated group. Based on parameter estimates of the PK model, docetaxel concentration in tumour tissues was increased by ritonavir with a mean area under the concentration-time curve 2.5-fold higher than that of the docetaxel only-treated group.
Table II.
Parameter Estimates for Pharmacokinetic Modelling of Docetaxel and Ritonavir in System and in Tumour
| Parameters | Units | Estimate | RSE (%) | Shrinkage (%) |
|---|---|---|---|---|
| Population parameter—docetaxel | ||||
| Systemic clearance in DOC group (CLDOC) | L/h/kg | 1.39 | 0.4 | – |
| Volume of distribution of central compartment (VcDOC) | L/kg | 1.93 | 0.3 | – |
| Inter-compartment clearance (QiDOC) | L/h/kg | 0.56 FIX | – | – |
| Volume of distribution of peripheral compartment (VpDOC) | L/kg | 2.09 FIX | – | – |
| Absorption rate in tumour (KaTDOC) | /h/kg | 0.32 | 9.6 | – |
| Elimination rate in tumour (KeTDOC) | /h/kg | 0.0044 | 52.4 | – |
| Population parameter—ritonavir | ||||
| Absorption rate in central compartment (KARTV) | /h/kg | 0.87 FIX | – | – |
| Systemic clearance (CLRTV) | L/h/kg | 1.11 FIX | – | – |
| Volume of distribution of central compartment (VcRTV) | L/kg | 1.38 FIX | – | – |
| Absorption rate in tumour (KaTRTV) | /h/kg | 0.0030 | 73.5 | – |
| Elimination rate in tumour (KeTRTV) | /h/kg | 0.031 | 64.3 | – |
| Population parameter—ritonavir effect on docetaxel metabolism | ||||
| Percentage of clearance DOC+RTV versus DOC group (E RTV) | % | 91.8 | 0.4 | – |
| Ritonavir tumour concentration that inhibits half of Cyp3a enzymes (IC50RTV) | ng/g | 2.5 FIX | – | – |
| Between-subject variability | ||||
| Absorption rate in tumour for docetaxel (KaTDOC) | CV% | 35.4 | 15.1 | 28.7 |
| Residual unexplained variability | ||||
| Proportional residual error on central compartment docetaxel | CV% | 20.8 | 17.1 | 0 |
| Proportional residual error on tumour compartment docetaxel | CV% | 15.0 FIX | – | 60.9 |
| Proportional residual error on tumour compartment ritonavir | CV% | 75.6 | 31.3 | 0 |
CV coefficient of variation, DOC docetaxel, RSE relative standard error, RTV ritonavir
PK/PD Model
Introduction of the LAMBDA term to account for an increased tumour growth rate over time resulted in a dramatic model improvement (dOFV = 36, p value <0.001). Parameter estimates for the untreated and the docetaxel only-treated groups are listed in Table IIIA.
Table III.
Parameter Estimates for Pharmacokinetic-Pharmacodynamic Model. A, Parameters Estimated by Control, RTV and DOC Groups; B, Parameter Estimated by Control, RTV, DOC and DOC+RTV Groups
| A | B | ||||||
|---|---|---|---|---|---|---|---|
| Control, RTV, DOC groups | Control, RTV, DOC, DOC+RTV groups | ||||||
| Parameters | Units | Estimate | RSE (%) | Shrinkage (%) | Estimate | RSE (%) | Shrinkage (%) |
| Population parameter | |||||||
| Baseline tumour volume (BASE) | mm3 | 214 | 3.1 | – | 211 | 1.4 | – |
| Net tumour growth rate in untreated groups (Kg0) | /week | 1.18 | 5.7 | – | 1.32 | 3.4 | – |
| Progression appearance factor (LAMBDA) | /week | 0.11 | 16.0 | – | 0.061 | 10.1 | – |
| Cell transition rate by docetaxel to effect compartment (KKDOC) | /week | 1.81 | 4.6 | – | 1.90 | 2.1 | – |
| Cell elimination rate by docetaxel from effect compartment (KKeDOC) | /week | 2.93 | 18.2 | – | 4.62 | 10.1 | – |
| Docetaxel tumour concentration with 50% of maximum anti-cancer effect (EC50DOC) | ng/g | 96.5 | 62.9 | – | 310 | 15.0 | – |
| Cell transition rate by ritonavir to effect compartment (KKRTV) | /week | – | – | – | 0.0054 | 17.4 | – |
| Cell elimination rate by ritonavir from effect compartment (KKeRTV) | /week | – | – | – | 7.45 | 4.6 | – |
| Between-subject variability | |||||||
| Baseline tumour volume (BASE) | CV% | 22.5 | 10.5 | 7.9 | 21.4 | 7.0 | 9.1 |
| Residual unexplained variability | |||||||
| Proportional residual error on tumour volumes | CV% | 23.0 | 10.0 | 3.4 | 24.8 | 8.7 | 2.8 |
CV coefficient of variation, DOC docetaxel, RSE relative standard error, RTV ritonavir
Subsequently, these parameters were used to predict the tumour growth profiles in the co-treated group. As can be observed in Fig. 3a, this resulted in a slight underestimation of the time to tumour re-growth in the co-treated group. Also in early phases of treatment, the anti-tumour effect was underestimated. This indicated that the enhanced anti-cancer effect in the co-treated group, compared to the docetaxel only-treatment, cannot be fully explained by the increased docetaxel tumour concentrations alone.
Fig. 3.
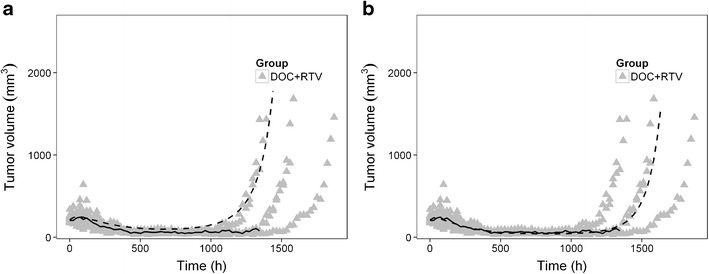
Comparison between observation and population prediction of tumour volume for combined treatment group. a Model prediction with fixation of parameters of docetaxel anti-tumour effect from docetaxel only-treated group with ritonavir effect as exposure booster; b model prediction with estimation of both anti-cancer effect of docetaxel and ritonavir. DOC, docetaxel; RTV, ritonavir. The triangles represent the consecutive observations of individual mouse tumours; the solid line shows the median of observation; and the dashed line represents the population prediction from each model. Observed data was originally published in (17)
Accordingly, a potential ritonavir anti-cancer effect was modelled analogously to that of docetaxel (Fig. 2). Inclusion of this effect of ritonavir resulted in drop of OFV of 59 points (p value <0.001). Bias in the model predictions of the co-treated group disappeared by inclusion of this effect (Fig. 3b).
The parameter estimates of the final model are listed in Table IIIB. Non-perturbed tumour growth was estimated with Kg0 of 1.32 week−1. This rate exponentially increased with a rate as 0.06 week−1 in the docetaxel-treated groups. Docetaxel inhibited tumour growth with EC50DOC of 310 ng/g by tumour PK.
Model Evaluation
The GOF plots showed that the developed final PK model adequately described the observed data (Fig. 4). In Fig. 5, VPC exhibited the simulated model prediction versus the observed tumour volumes for each mice group. This visual check suggested that the final PK/PD model, considering both ritonavir effect on docetaxel metabolism inhibition and anti-cancer effect, sufficiently captured the observed tumour growth profile in all mice groups.
Fig. 4.
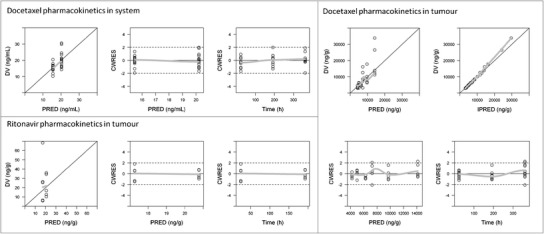
Goodness-of-fit (GOF) plots for docetaxel in system, and for docetaxel and ritonavir in tumour, respectively. CWRES, conditional weighted residuals; DV, observed concentration; IPRED, individual predicted concentration; PRED, population predicted concentration. Observed data was originally published in (17)
Fig. 5.
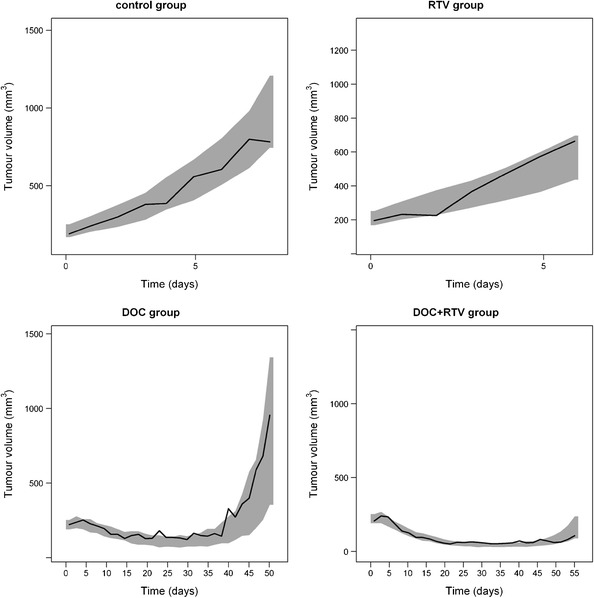
Visual predictive check of model prediction for tumour volumes (n = 1000). DOC, docetaxel; RTV, ritonavir. Solid lines represent the median observed values and grey areas represent simulated 95% confidence intervals. Note the shifts in time and tumour volume scales between the graphs. Observed data was originally published in (17)
Sensitivity test on ±50% of fixation of IC50RTV in docetaxel PK model suggested no influence on the evidence of the anti-cancer effect of ritonavir. There was no obvious change in the prediction of tumour volume profile according to the sensitivity test.
DISCUSSION
In this study, the complex pharmacokinetic interaction between docetaxel and ritonavir and the effects of this interaction on tumour growth were quantified using PK/PD modelling. This study illustrated a useful application of a modelling approach to elaborating on previous observations.
We confirmed and quantified the previous finding that ritonavir inhibits docetaxel metabolism and functions as an exposure booster. Subsequently, a tumour growth inhibition model was developed which showed that the extended tumour growth inhibition observed when combining docetaxel with ritonavir was mainly caused by increased docetaxel concentration in tumour but also by a small yet significant anti-tumour effect of ritonavir itself.
The PK interaction between ritonavir and docetaxel was studied both in the systemic circulation and in the tumour. Although the host mice in this study were Cyp3a−/−, ritonavir showed a small effect on docetaxel systemic clearance. This could be attributed to the fact that in addition to CYP3A, ritonavir also inhibits other metabolizing enzymes or drug transporters which may influence docetaxel systemic exposure (4). However, the effect of ritonavir is based only on samples taken 24 h after administration of docetaxel to mice and might not be clinically relevant.
In the tumour, ritonavir inhibitory effect on docetaxel metabolism was only considered on ritonavir Cyp3a inhibition because Cyp3a is by far the most dominant metabolic enzyme for docetaxel (30) and ritonavir is a very potent CYP3A inhibitor (5). One important assumption that has been made in the current study was the use of a literature value for the inhibition of docetaxel metabolism by ritonavir (IC50RTV). This is because the tumour PK data did not support the estimation of this important parameter. The choice of IC50RTV from the literature was based on two reasons. One was that we observed a continuous increase in docetaxel tumour concentrations during co-treatment (Fig. 1) (17). This phenomenon suggested a potent Cyp3a inhibition by ritonavir in tumour with a relatively low IC50RTV. The other reason was that the use of a relatively high IC50RTV would have resulted in a reduction of estimated docetaxel tumour concentrations in the co-treated group, which may increase the possibility of a type I error in finding a potential ritonavir anti-cancer effect. Accordingly, we have fixed the IC50RTV to the lowest literature data retrieved (23). A sensitivity test with ±50% difference in IC50RTV confirmed the robustness of our findings.
Tumour growth inhibition models for docetaxel were built in order to quantify the relationship between docetaxel tumour concentration and its anti-cancer effect. A delayed anti-cancer effect of docetaxel provided an adequate fit of the observations. This could be explained by the fact that docetaxel inhibits tumour cells during cell division (1) after which the cells go into apoptosis. In addition, model fit of the tumour growth inhibition model enormously improved after the introduction of a term describing an increased tumour growth rate over time. A physiological basis for this could be that cells with less proliferative capacity were relatively prone to be killed by docetaxel, leaving more proliferative tumour cells re-grow with a faster growth rate. Besides, a previous report mentioned that the EC50 of docetaxel in the BRCA2-deficient or BRCA2-proficient mouse mammary tumour cell lines ranged from 0.2 to 1.2 ng/mL (0.23–1.53 nM) (31). Our finding in the orthotopically transplanted syngeneic mammary tumours derived from K14cre;Brca1F/F;p53F/F mice confirmed that docetaxel showed a very potent anti-cancer efficacy in vivo. The observed docetaxel tumour concentration was up to 30,000 ng/g (Fig. 1), compared to which the estimate of EC50DOC from the final model (310 ng/g) was in the lower end of the range of observed docetaxel tumour concentration.
We observed a small anti-cancer effect of ritonavir. The mechanism of this effect was explored previously. As mentioned in the “INTRODUCTION” section, ritonavir may cause DNA damage in human endothelial cells (10). Gaedicke et al. suggested that ritonavir induced significant tumour growth inhibition in mouse lymphoma, which may be associated with perturbation of proteasomal protein degradation (12). Besides, ritonavir was reported to improve the anti-tumour response co-treated with ionizing radiation in mouse HEP-2 head and neck carcinoma model (15). The effect of the co-treatment could be due to a synergistic effect of enhanced radiation-induced apoptosis and anti-angiogenesis. It was also found that ritonavir inhibits breast cancer growth in part by inhibiting heat shock protein 90 substrates (16). With a mathematical approach, the current model also found an anti-cancer effect of ritonavir when co-administered with docetaxel. However, whether this anti-cancer effect is additive or synergistic to the tumour inhibitory effect of docetaxel remains unclear. Our model assumed an additive anti-cancer effect of ritonavir (Fig. 2), which adequately described the observation of both the ritonavir only-treated group and the co-treated group.
The current work not only quantified the tumour growth parameters from the previous study but could be a basis to instruct further study design. For instance, subsequent studies might focus on collecting tumour PK samples after treatment for the estimation of the important parameter—IC50RTV. Besides, the tumour inhibitory effect of ritonavir could be further explored by the studies with treatment started at a lower tumour volume than 200 mm3 or higher ritonavir dose levels than 12.5 mg/kg. PK/PD modelling based on these studies could be a powerful tool to find the best dosing regimen for the combined administration of both compounds. Ultimately, these data could be used to further optimize the application of the combined treatment of docetaxel and ritonavir in humans, which is currently in clinical development (9,32–34).
CONCLUSION
A PK/PD model has been successfully developed describing the complex interaction between docetaxel and ritonavir when co-administered in a mouse model for hereditary breast cancer. We showed that the increased tumour growth inhibition observed in the co-treatment of docetaxel with ritonavir is mainly caused by boosting the docetaxel concentration in tumour and to a minor extent by a direct tumour growth inhibitory effect of ritonavir.
Compliance with Ethical Standards
Conflict of Interest
We appreciate J. G. C van Hasselt for useful discussions. Jan H.M. Schellens and Jos H. Beijnen reported to be patent holder of oral booster of Taxanes. The remaining authors declare no conflict of interest.
REFERENCES
- 1.European Medicines Agency. TAXOTERE 20 mg/0.5 ml concentrate and solvent for solution for infusion. Summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000073/WC500035264.pdf. Accessed 26 Feb 2015.
- 2.Koolen SLW, Beijnen JH, Schellens JHM. Intravenous-to-oral switch in anticancer chemotherapy: a focus on docetaxel and paclitaxel. Clin Pharmacol Ther. 2010;87(1):126–129. doi: 10.1038/clpt.2009.233. [DOI] [PubMed] [Google Scholar]
- 3.Moes JJ, Koolen SLW, Huitema ADR, Schellens JHM, Beijnen JH, Nuijen B. Pharmaceutical development and preliminary clinical testing of an oral solid dispersion formulation of docetaxel (ModraDoc001) Int J Pharm. 2011;420(2):244–250. doi: 10.1016/j.ijpharm.2011.08.041. [DOI] [PubMed] [Google Scholar]
- 4.Schellens JH, Malingré MM, Kruijtzer CM, Bardelmeijer HA, van Tellingen O, Schinkel AH, et al. Modulation of oral bioavailability of anticancer drugs: from mouse to man. Eur J Pharm Sci. 2000;12(2):103–110. doi: 10.1016/S0928-0987(00)00153-6. [DOI] [PubMed] [Google Scholar]
- 5.Eagling VA, Back DJ, Barry MG. Differential inhibition of cytochrome P450 isoforms by the protease inhibitors, ritonavir, saquinavir and indinavir. Br J Clin Pharmacol. 1997;44(2):190–194. doi: 10.1046/j.1365-2125.1997.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bardelmeijer HA, Ouwehand M, Buckle T, Huisman MT, Schellens JHM, Beijnen JH, et al. Low systemic exposure of oral docetaxel in mice resulting from extensive first-pass metabolism is boosted by ritonavir. Cancer Res. 2002;62(21):6158–6164. [PubMed] [Google Scholar]
- 7.Oostendorp RL, Huitema A, Rosing H, Jansen RS, Ter Heine R, Keessen M, et al. Coadministration of ritonavir strongly enhances the apparent oral bioavailability of docetaxel in patients with solid tumors. Clin Cancer Res. 2009;15(12):4228–4233. doi: 10.1158/1078-0432.CCR-08-2944. [DOI] [PubMed] [Google Scholar]
- 8.Koolen SLW, Oostendorp RL, Beijnen JH, Schellens JHM, Huitema ADR. Population pharmacokinetics of intravenously and orally administered docetaxel with or without co-administration of ritonavir in patients with advanced cancer. Br J Clin Pharmacol. 2010;69(5):465–474. doi: 10.1111/j.1365-2125.2010.03621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moes JJ, Stuurman FE, Hendrikx JJMA, Marchetti S, Huitema ADR, Beijnen JH, et al. Pharmacokinetic evaluation of three oral formulations of docetaxel boosted with ritonavir: two single-drug formulations vs. a fixed-dose combination tablet. Drug Deliv Transl Res. 2013;3(3):243–251. doi: 10.1007/s13346-012-0127-6. [DOI] [PubMed] [Google Scholar]
- 10.Zhong D, Lu X, Conklin BS, Lin PH, Lumsden AB, Yao Q, et al. HIV protease inhibitor ritonavir induces cytotoxicity of human endothelial cells. Arterioscler Thromb Vasc Biol. 2002;22(10):1560–1566. doi: 10.1161/01.ATV.0000034707.40046.02. [DOI] [PubMed] [Google Scholar]
- 11.Ikezoe T, Hisatake Y, Takeuchi T, Ohtsuki Y, Yang Y, Said JW, et al. HIV-1 protease inhibitor, ritonavir: a potent inhibitor of CYP3A4, enhanced the anticancer effects of docetaxel in androgen-independent prostate cancer cells in vitro and in vivo. Cancer Res. 2004;64(20):7426–7431. doi: 10.1158/0008-5472.CAN-03-2677. [DOI] [PubMed] [Google Scholar]
- 12.Gaedicke S, Firat-Geier E, Constantiniu O, Lucchiari-Hartz M, Freudenberg M, Galanos C, et al. Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res. 2002;62(23):6901–6908. [PubMed] [Google Scholar]
- 13.Kariya R, Taura M, Suzu S, Kai H, Katano H, Okada S. HIV protease inhibitor Lopinavir induces apoptosis of primary effusion lymphoma cells via suppression of NF-κB pathway. Cancer Lett. 2014;342:52–59. doi: 10.1016/j.canlet.2013.08.045. [DOI] [PubMed] [Google Scholar]
- 14.Dewan MZ, Tomita M, Katano H, Yamamoto N, Yamamoto N, Ahmed S, et al. An HIV protease inhibitor, ritonavir targets the nuclear factor-kappaB and inhibits the tumor growth and infiltration of EBV-positive lymphoblastoid B cells. Int J Cancer. 2009;124:622–629. doi: 10.1002/ijc.23993. [DOI] [PubMed] [Google Scholar]
- 15.Maggiorella L, Wen B, Frascogna V, Opolon P, Bourhis J, Deutsch E. Combined radiation sensitizing and anti-angiogenic effects of ionizing radiation and the protease inhibitor ritonavir in a head and neck carcinoma model. Anticancer Res. 2005;25(6B):4357–4362. [PubMed] [Google Scholar]
- 16.Srirangam A, Mitra R, Wang M, Gorski JC, Badve S, Baldridge L, et al. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin Cancer Res. 2006;12(6):1883–1896. doi: 10.1158/1078-0432.CCR-05-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendrikx JJMA, Lagas JS, Song J-Y, Rosing H, Schellens JHM, Beijnen JH, et al. Ritonavir inhibits intratumoral docetaxel metabolism and enhances docetaxel anti-tumor efficacy in a mouse model for hereditary breast cancer. Int J Cancer. 2015 doi: 10.1002/ijc.29812. [DOI] [PubMed] [Google Scholar]
- 18.Koolen SLW, van Waterschoot RAB, van Tellingen O, Schinkel AH, Beijnen JH, Schellens JHM, et al. From mouse to man: predictions of human pharmacokinetics of orally administered docetaxel from preclinical studies. J Clin Pharmacol. 2012;52(3):370–380. doi: 10.1177/0091270010397051. [DOI] [PubMed] [Google Scholar]
- 19.Ribba B, Holford NH, Magni P, Trocóniz I, Gueorguieva I, Girard P, et al. A review of mixed-effects models of tumor growth and effects of anticancer drug treatment used in population analysis. CPT Pharmacometrics Syst Pharmacol. 2014;3(5):e113. doi: 10.1038/psp.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A. 2007;104(29):12111–12116. doi: 10.1073/pnas.0702969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn. 2003;30(6):387–404. doi: 10.1023/B:JOPA.0000012998.04442.1f. [DOI] [PubMed] [Google Scholar]
- 22.Moltó J, Barbanoj MJ, Miranda C, Blanco A, Santos JR, Negredo E, et al. Simultaneous population pharmacokinetic model for lopinavir and ritonavir in HIV-infected adults. Clin Pharmacokinet. 2008;47(10):681–692. doi: 10.2165/00003088-200847100-00005. [DOI] [PubMed] [Google Scholar]
- 23.Tran TH, Von Moltke LL, Venkatakrishnan K, Granda BW, Gibbs MA, Obach RS, et al. Microsomal protein concentration modifies the apparent inhibitory potency of CYP3A inhibitors. Drug Metab Dispos. 2002;30(12):1441–1445. doi: 10.1124/dmd.30.12.1441. [DOI] [PubMed] [Google Scholar]
- 24.Hendrikx JJMA, Hillebrand MJX, Thijssen B, Rosing H, Schinkel AH, Schellens JHM, et al. A sensitive combined assay for the quantification of paclitaxel, docetaxel and ritonavir in human plasma using liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci. 2011;879(28):2984–2990. doi: 10.1016/j.jchromb.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 25.Beal SL, Sheiner LB. NONMEM user guides. Ellicott City: Icon Development Solutions; 1989. [Google Scholar]
- 26.Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. Elsevier Ireland Ltd; 2011;101(1):72–9. [DOI] [PubMed]
- 27.R Development Core Team . R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2008. [Google Scholar]
- 28.Lindbom L, Pihlgren P, Jonsson EN, Jonsson N. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Prog Biomed. 2005;79(3):241–257. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 29.Jonsson EN, Karlsson MO. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Prog Biomed. 1999;58(1):51–64. doi: 10.1016/S0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 30.Van Herwaarden AE, Wagenaar E, van der Kruijssen CMM, van Waterschoot RAB, Smit JW, Song J-Y, et al. Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. J Clin Invest. 2007;117(11):3583–3592. doi: 10.1172/JCI33435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evers B, Drost R, Schut E, De Bruin M, Der Van Burg E, Derksen PWB, et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res. 2008;14:3916–3925. doi: 10.1158/1078-0432.CCR-07-4953. [DOI] [PubMed] [Google Scholar]
- 32.Marchetti S, Stuurman FE, Koolen SLW, Moes JJ, Hendrikx JJMA, Thijssen B, et al. Phase I study of weekly oral docetaxel (ModraDoc001) plus ritonavir in patients with advanced solid tumors. J Clin Oncol 2012 ASCO Annu Meet Proceedings. 2012;30(15_suppl):2250.
- 33.The Netherlands Cancer Institute. Weekly administration of oral docetaxel in combination with ritonavir for the treatment of a variety of tumour types: an optimal dosing study. 2007. Database: ISRCTN. Identifiers: ISRCTN32770468; N07DOW. http://www.isrctn.com/ISRCTN32770468. Accessed 11 Feb 2015.
- 34.The Netherlands Cancer Institute. Weekly administration of (bi-)daily oral docetaxel in combination with ritonavir. 2010. Database: ClinicalTrials.gov. Identifiers: NCT01173913; N10BOM. https://clinicaltrials.gov/ct2/show/NCT01173913. Accessed 11 Feb 2015.