

SUMMARY
Oncocytomas are predominantly benign neoplasms possessing pathogenic mitochondrial mutations and accumulation of respiration-defective mitochondria, characteristics of unknown significance. Using exome and transcriptome sequencing, we identified two main subtypes of renal oncocytoma. Type 1 is diploid with CCND1 rearrangements, whereas type 2 is aneuploid with recurrent loss of chromosome 1, X or Y, and/or 14 and 21, which may proceed to more aggressive eosinophilic chromophobe renal cell carcinoma (ChRCC). Oncocytomas activate 5′ adenosine monophosphate-activated protein kinase (AMPK) and Tp53 (p53) and display disruption of Golgi and autophagy/lysosome trafficking, events attributed to defective mitochondrial function. This suggests that the genetic defects in mitochondria activate a metabolic checkpoint, producing autophagy impairment and mitochondrial accumulation that limit tumor progression, revealing a novel tumor-suppressive mechanism for mitochondrial inhibition with metformin. Alleviation of this metabolic checkpoint in type 2 by p53 mutations may allow progression to eosinophilic ChRCC, indicating that they represent higher risk.
Graphical abstract
INTRODUCTION
Altered metabolism in cancer that promotes the fermentation of glucose was proposed more than 50 years ago by Otto Warburg to originate from respiratory system damage (Warburg, 1956). Only recently with the discovery of cancer driver mutations in nuclear genes encoding mitochondrial tricarboxylic acid (TCA) cycle enzymes have we begun to unravel the underlying oncogenic functions linked to altered metabolism (Ward and Thompson, 2012). Moreover, it is now clear that many oncogenic events common in cancer indirectly regulate cellular metabolism (Vander Heiden et al., 2009). These metabolic alterations may provide substrates and energy necessary for cell growth, promote antioxidant defense, and produce signals that alter gene expression and drive pro-oncogenic signaling pathways. These discoveries are revealing new approaches to cancer therapy that target metabolic functions important for tumorigenesis. One underexplored area within the sphere of metabolic alterations in cancer is represented by the tumor type oncocytoma.
Oncocytomas are rare, predominantly benign neoplasms mostly of epithelia that have inactivating mutations in mitochondrial genome-encoded enzymes or control regions that cause respiration defects. Oncocytomas are also characterized by the dramatic accumulation of these defective mitochondria (Gasparre et al., 2011). Whether the mitochondrial impairments are functionally neutral, promote tumor growth as Warburg envisioned, or to the contrary are a liability is unknown. It is also unclear why defective mitochondria accumulate and if this plays any role in disease. Oncocytomas may represent an evolutionary dead end or an intermediate that progresses to more aggressive cancer. Identifying mechanisms that restrict some tumors such as these to benign disease can inform novel approaches to cancer therapy.
Mitochondria are essential for eukaryotic cell function. Cells contain hundreds of mitochondria, with their numbers controlled by both a transcription program of biogenesis (Wallace, 2012) and removal of defective mitochondria through mitophagy, a selective form of autophagy (Youle and Narendra, 2011). Thus, the presence of tumor cells with the accumulation of defective mitochondria suggests increased biogenesis, possibly as a compensatory mechanism (Simonnet et al., 2003), or an underlying defect in removal by mitophagy (Guo et al., 2013b). As autophagy deficiency in genetically engineered mouse models for KrasG12D- and BRAFV600E-driven lung cancers converts carcinomas to more benign oncocytomas, defects in mitochondrial removal may underlie the oncocytoma phenotype and produce benign instead of malignant disease (Guo et al., 2013a; Karsli-Uzunbas et al., 2014; Strohecker et al., 2013). This also suggests that autophagy and mitochondrial quality control are important for promoting tumorigenesis (Guo et al., 2013b).
To address the cause and role of oncocytomas in the spectrum of cancer, we sequenced the exome and transcriptome of the most common oncocytoma, those that arise in the kidney (Linehan and Ricketts, 2013). We identified two major subtypes, one without (type 1) and the other with (type 2) recurrent whole-chromosome losses that contain few other mutations. Both subtypes had recurrent inactivating mutations in nuclear- and mitochondria-encoded proteins critical for respiration and displayed activation of AMPK and p53. Recurrent CCND1 chromosomal rearrangement and overexpression was a likely cancer driver mutation in type 1. Both subtypes had evidence of defective autophagy attributed to metabolic-deficiency-induced Golgi disassembly and lysosome dysfunction that blocked LAMP-2 trafficking and lysosomal protease activation. Pharmacologic inhibition of mitochondrial complex I with metformin to replicate mitochondrial dysfunction in oncocytomas caused AMPK activation and Golgi disassembly and blocked LAMP-2 trafficking and autophagy. Thus, mitochondrial respiration defects that arise early in renal tumorigenesis trigger a metabolic checkpoint, trafficking and lysosome defects, and failure of mitochondrial quality control, activating p53 and AMPK as a barrier to tumor progression. Moreover, the similarities in the mutational landscape and transcriptome suggest that type 2 oncocytoma and eosinophilic subtype of chromophobe renal cell carcinoma (ChRCC) are closely related, with ChRCC having acquired additional driver mutations in p53 and PTEN and further genetic instability. Thus, treatment of type 2 oncocytomas requires more consideration.
Genomic Analysis Indicates Two Main Subtypes of Renal Oncocytoma
Whole-exome sequencing of 12 renal oncocytomas and matched adjacent normal tissue and RNA sequencing of 9 of these pairs were performed. Copy-number variation (CNV) analysis from exome sequencing indicates that tumors are clonal, with two distinct subtypes based on the pattern of aneuploidy. One tumor was an outlier that was excluded from further analysis. Type 1 is diploid and type 2 is hypodiploid with loss of chromosome 1, X or Y, and/or 14 and 21 (Figures 1A and S1A; Table S1A). Type 1 shows no sex bias, but type 2 has male predominance (five male/one female), suggesting strong selection for loss of the Y chromosome. The one type 2 tumor arising in a female has lost one X chromosome (Figure 1A). Strong selection for loss of Y in males or X in females suggests possible involvement of gene loss on the pseudoautosomal region (PAR).
Figure 1. Identification of Two Main Subtypes of Renal Oncocytoma.
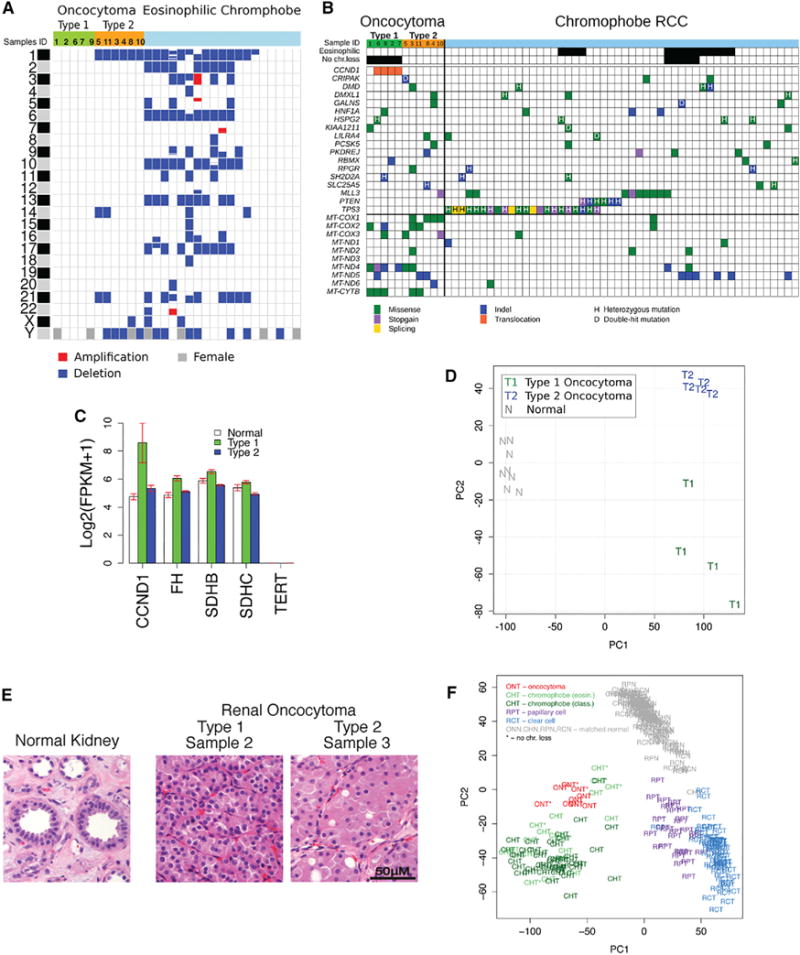
(A) Copy-number variations in renal oncocytomas in comparison to eosinophilic ChRCC (eosinophilic ChRCC sequencing data were downloaded from the TCGA CGHub repository [https://cghub.ucsc.edu/]) (Davis et al., 2014). Blue corresponds to loss of one copy, red corresponds to a gain, and gray on chromosome Y indicates a female.
(B) Somatic mutations in renal oncocytomas in comparison to ChRCC (ChRCC sequencing data were downloaded from the TCGA CGHub repository [https://cghub.ucsc.edu/]) (Davis et al., 2014). Only SNVs common between oncocytoma and ChRCC are shown; the complete list of detected somatic SNVs and indels is reported in Tables S2 and S3.
(C) Expression levels of key genes from the transcriptome analysis. Each bar represents log2-transformed transcript abundance. Error bars show SEs.
(D) Principal component analysis (PCA) of gene expression identifies two subtypes of oncocytoma. The first principal component (PC1) separates tumor and normal samples; the second principal component (PC2) separates oncocytomas into two subtypes.
(E) Type 1 and type 2 oncocytomas have distinct histology, with type 2 having a larger volume of eosinophilic cytoplasm relative to the nucleus. Error bar, 50 μm.
(F) Gene expression similarities between oncocytoma and ChRCC. PCA of gene expression for oncocytoma, ChRCC, clear cell RCC, and papillary cell RCC identify the three RCC subtypes as separate clusters with the corresponding matched normal samples forming one cluster. Oncocytomas cluster with ChRCC demonstrating similarities in gene expression patterns.
Few and no recurrent single-nucleotide variants (SNVs) (162 nonsynonymous SNVs in 11 tumors; Tables S1B and S2) or insertions/deletions were detected (Table S3). There were no mutations in common cancer-causing genes (p53, PTEN) with the exception of one tumor with an MLL3 mutation (Figure 1B). Three tumors have mutations in nuclear-encoded mitochondrial genes NDUFV1, UQCRC2 (type 1), and SLC25A5 (ANT2) (type 2). ANT2 is located on the X chromosome, and this ANT2 mutant tumor likely has a loss-of-function mutation as it has a frameshift mutation with X chromosome loss (Figure 1B; Table S3). Mutations in FLCN, VHL, FH, SDHB, SDHC, or SDHD, known drivers in other renal cell carcinomas (RCCs), were not detected. Despite loss of chromosome 1 in type 2, expression levels of FH, SDHB, and SDHC located on chromosome 1 were not affected (Figure 1C).
The nuclear genome mutation spectrum observed in oncocytomas was C > A (132), C > G (98), C > T (329), T > A (64), T > C (147), and T > G (97). The analysis showed transition to transversion ratio (Ts/Tv) of 1.22 in somatic mutations in contrast to 2.33 for germline mutations (found in dbSNP) consistent with expected values (Ebersberger et al., 2002). The comparison displayed strong enrichment for transversion mutations among somatic mutations, suggesting reactive oxygen species (ROS) exposure at some point in oncocytoma evolution.
Oncocytomas have mitochondrial genome mutations responsible for respiration defects (Gasparre et al., 2007; Mayr et al., 2008; Simonnet et al., 2003), with all oncocytomas examined here having mutations, and mutations in MT-COX1, MT-COX2, MT-COX3, MT-ND4, MT-ND5, and MT-CYTB found in three or more tumors (Figures 1B and S1B; Table S4). Thus, the distinguishing feature of both subtypes of renal oncocytoma is recurrent inactivating mutations in mitochondrial genes encoded by both the nuclear or mitochondrial genomes.
The mitochondrial point mutation spectrum for oncocytomas was C > T (46) and A > G (10) on the heavy strand and C > T (10) and A > G (3) on the light strand. The only other point mutations detected were C > A (7) on the light strand. The dominance and strand bias of C > T and A > G mutations on the heavy strand are consistent with findings from other human cancers and postulated to result from mitochondrial DNA replication errors (Ju et al., 2014). In contrast, the C > A mutations are consistent with damage by ROS (Thilly, 2003), which were elevated (9%) compared to other cancers (4%), (Ju et al., 2014). While the majority of oncocytoma mitochondrial mutations showed the signature of DNA replication error indicating that DNA replication errors are the dominant source of mutations, most of the pathogenic mutations with high allelic frequencies (>75%) are indels and occure in the complex I subunits. In summary, 7 out of 11 oncocytomas have a pathogenic mitochondrial DNA mutation frequency greater than 75% (Table S4).
The gene expression pattern also represents type 1 and 2 oncocytomas as distinct groups and indicates that the matched normal adjacent tissue (NAT) from sample 5 is likely contaminated with tumor (Figures S1C and S1D, A). Principle component analysis (PCA) shows type 1 and 2 grouping separately with type 2 forming a tighter cluster showing greater similarity (Figure 1D). The separate groups persist using only gene expression from diploid chromosomes from types 1 and 2 (Figure S1D, B and C). Both types are distinct from normal controls (Figures 1D and S1C). Type 1 and 2 are distinguished histologically, with type 2 having a more expansive eosinophilic cytoplasm (Figures 1E and S2), suggesting greater accumulation of defective mitochondria, thought to be a main contributing factor to the histological appearance of these tumors.
Type 2 Oncocytoma May Be the Precursor of Eosinophilic ChRCC
The relationship between oncocytomas and other renal tumors is not currently known. Thus, we compared the genomic and transcriptomic signature of oncocytomas with RCC subtypes. The pattern of gene expression in oncocytoma was clearly distinct from clear cell and papillary cell RCC (Figure 1F), consistent with the vastly different mutational landscape of these tumors (Gao et al., 2013; Varela et al., 2011). In contrast, ChRCC has some histologic similarities and common biomarkers with oncocytoma, and distinguishing these tumor types is important, as ChRCC is a more aggressive disease with higher risk of metastasis (Ng et al., 2014).
There are two types of ChRCC, classic and eosinophilic, with the eosinophilic subtype possessing some similarities with oncocytoma (Davis et al., 2014). The global gene expression pattern of both oncocytoma and ChRCC are similar (Figure 1F), and both have a gene expression signature of distal tubule, suggesting that they originate from a common cell type (Figure S1E). Eosinophilic ChRCC has two subgroups: one is chromosomally diploid like type 1 but without rearrangement or overexpression of CCND1, and the other has a pattern of chromosome loss similar to type 2 with loss of chromosomes 1, X, or Y, but with further losses including 2, 6, 10, 13, and 17 (Figures 1A and 1B) (Davis et al., 2014). Like type 2, eosinophilic ChRCC also has a sex bias (15 male/4 female) whereas classic ChRCC has no gender bias (24 male/23 female) (Figure 1A; Table S5). The main nuclear genomic difference between oncocytomas and ChRCC are mutations in p53 and PTEN in ChRCC (Figure 1B). TERT promoter mutations and upregulation of telomerase reverse transcriptase (TERT) expression occur in 10% of ChRCC (Davis et al., 2014), and both oncocytoma subtypes lacked TERT expression (Figure 1C), indicating the absence of this driver mutation. The similarities in gene expression, aneuploidy pattern, and histology suggest type 2 may be closely related to eosinophilic ChRCC. One possibility is that ChRCC may have acquired additional mutations in TP53, PTEN, or TERT, leading to further genomic instability and a less indolent natural history. Alternatively, there could be a single cell type precursor for both type 2 oncocytoma and ChRCC, where chromosome 1 is lost followed by either pathogenic mitochondrial genome or p53 mutations, directing fate to either oncocytoma or ChRCC. This, however, does not account for the common mitochondrial mutations in type 1 and 2 oncocytoma without chromosome 1 loss in type 1.
In contrast to the increased nuclear genomic mutation burden in ChRCC compared to oncocytoma, there was less mutational burden of the mitochondrial genome in ChRCC compared to oncocytoma (p < 0.0001) (Figure 1B) (Davis et al., 2014). Heteroplasmy may be a prerequisite to selection for a less defective pool of mitochondria during progression of benign type 2 to a more malignant ChRCC.
Recurrent CCND1 Rearrangement and Amplification in Type 1 Oncocytomas
Four of the five type 1 oncocytomas had high expression of CCND1 in comparison to type 2 and NAT (Figure 1C). CCND1 encodes the cell-cycle regulator and oncoprotein CyclinD1. All three tumors exhibit monoallelic expression of CCND1 consistent with chromosomal rearrangement that led to aberrant high expression of one CCND1 allele (Figure 2A) consistent with previous observations (Sukov et al., 2009). Western blot shows high CyclinD1 protein expression in some tumors with high mRNA expression (Figure 2B). CyclinD1 expression is also elevated in some of the type 2 tumors, reflecting non-transcriptional mechanisms for CyclinD1 activation (Figure 2B).
Figure 2. Chromosome Rearrangements and Overexpression of CCND1 in Type 1 and p53 Activation in Type 1 and 2 Oncocytomas.
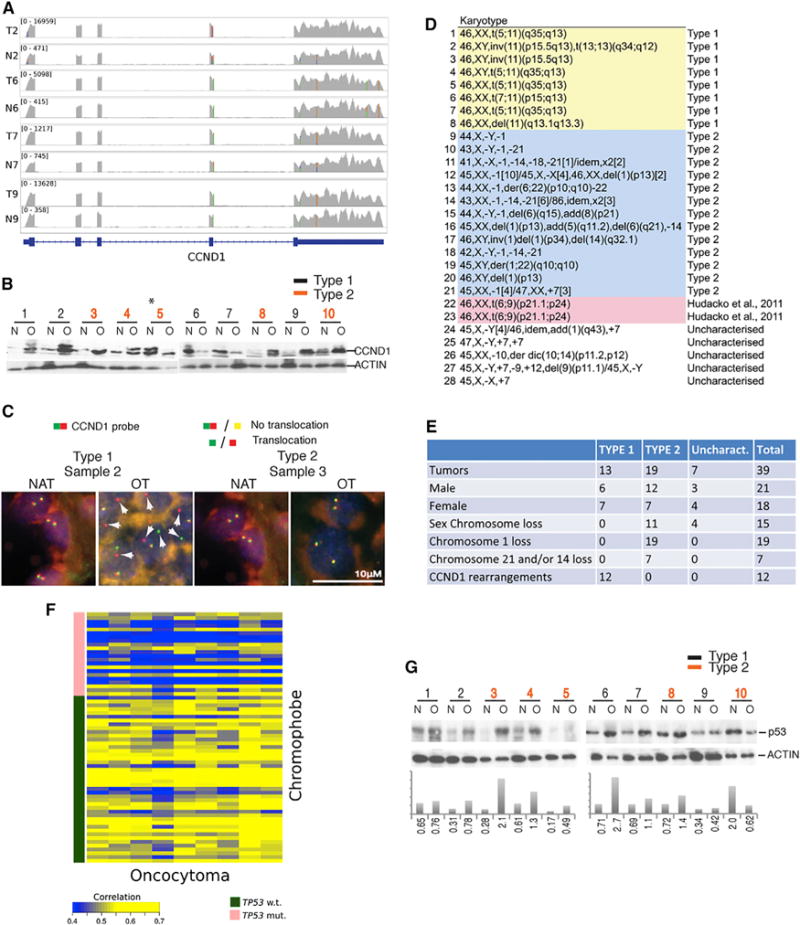
(A) Overexpression of a single allele of CCND1. The RNA-sequencing coverage tracks for four tumors (T2, T6, T7, and T9) and matched normal samples (N2, N6, N7, and N9) are shown in gray. The numbers in brackets display the coverage for this region and reflect the level of the gene expression, showing that CCND1 is over 10-fold higher in tumors. SNPs present in the exons of CCND1 are shown as colored bars. Some of the SNPs are heterozygous and are indicated by the two-color bars showing the relative expression of the two alleles. The appearance of homozygous SNPs in tumor where the same SNPs appear heterozygous in the matched normal sample, as in T2 versus N2, is a result of the overexpression of a single allele of CCND1.
(B) Western blot showing elevated CCND1 protein expression in tumors. The asterisks marks sample 5, which has normal (N) tissue mixed with oncocytoma (O) tissue. Multiple bands represent different phosphorylated forms of CCND1.
(C) Representative fluorescence images of CCND1 FISH show rearrangement in type 1 (sample 2), but not type 2 (sample 3), tumors. White arrowheads show segregation of CCND1 probe.
(D) Karyotype of renal oncocytomas shows chromosomal rearrangement including translocations, inversions, and deletions near CCND1 in type 1 and loss of chromosome 1 and Y in type 2.
(E) Summary of genetic alterations in all 39 oncocytoma samples.
(F) Oncocytomas have a gene expression signature similar to ChRCC without p53 mutation as shown by correlation heat map between oncocytoma and ChRCC gene expression values. The correlation is calculated for the set of genes (Table S6), which are differentially expressed between ChRCCs with and without a mutation in p53.
(G) Western blot for p53 showing induction in oncocytomas with quantitation relative to ACTIN.
Fluorescence in situ hybridization (FISH) analysis of three each of type 1 and 2 oncocytomas confirmed translocation of 11q13 at the CCND1 locus in type 1 and not in type 2 (Figure 2C). Cytogenetic analysis of 28 additional renal oncocytomas showed eight tumors with translocations, inversions, or deletions near CCND1 (11q13) but an otherwise chromosomally intact genome (Figure 2D). These findings are consistent with the presence of two mutually exclusive categories of oncocytoma: type 1 with CCND1 rearrangement and no other gross chromosomal losses, and type 2 with chromosome 1 loss. Of the 28 oncocytomas, two were chromosomally stable, harboring a translocation between chromosomes 6 and 9 of unknown significance (Figure 2D) (Hudacko et al., 2011). Five others had unique chromosome rearrangements and CNVs, suggesting low-level diversity of this tumor type (Figures 2D and 2E). The expanded characterization of 39 oncocytomas confirmed the association of type 1 with CCND1 rearrangements (12/13) and type 2 with chromosome 1 loss (19/19), sex chromosome loss (11/19), and male bias (12/19 male, 7/19 female) (Figure 2E). No aberrant high expression of CCND1 was detected in chromosomally stable ChRCC (six tumors) so there is no evidence that type 1 progresses to a subset of ChRCC that also lacks chromosomal losses. Since translocation or rearrangement of CCND1 and overexpression of CyclinD1 is a well-known oncogenic driver, it is likely a driver mutation in type 1 oncocytomas.
p53 Is a Barrier to Oncocytoma Progression
p53 is mutated in 22 out of 66 (33%) ChRCC tumors (Davis et al., 2014) and not in oncocytoma (Figure 1B). We found a set of genes differentially expressed between ChRCC with and without p53 mutation (Table S6) and oncocytomas have the gene signature more similar to ChRCC without p53 mutation (Figure 2F). This finding is consistent with changes in gene expression linked to p53 mutational status in both ChRCC and oncocytomas.
p53 protein is stabilized as a barrier to tumorigenesis by stress, oncogene activation including CCND1 amplification, and genome damage. To test if p53 was activated in benign oncocytomas explaining its mutation upon progression to ChRCC in type 2, we examined p53 levels. In comparison to NAT, wild-type p53 protein was induced in most oncocytomas independent of subtype, suggesting that p53 activation in oncocytomas may contribute to their slow growth and benign characteristics (Figure 2G).
Type 1 and 2 Oncocytomas Display Mitochondrial Defects and Metabolic Stress
Since metabolic stress, triggered by mitochondrial defects, is a potential cause of p53 activation in oncocytomas, we assessed mitochondrial genome copy number, gene expression signature, mitochondrial complex protein expression, and metabolic signaling. Both type 1 and 2 have elevated mitochondrial genome copy number relative to NAT, with type 2 being greater, suggesting increased mitochondrial accumulation (Figure 3A; Table S7).
Figure 3. Oncocytomas Show a Gene Expression Signature of Oxidative Metabolism and Mitochondrial and Energetic Defects.

(A) Oncocytomas have increased mitochondrial DNA copy number. p value was calculated for a one-tailed t test.
(B–D) Canonical pathway analysis. Each graph represents the top 15 canonical pathways enriched for the corresponding gene set (http://www.ingenuity.com/). Numbers on top of the bars indicate the number of genes in the pathway. Differential expression analysis of type 2 versus type 1 identifies 50 genes with FDR-corrected p value < 0.05.
(B) Pathway analysis of a set of 1,042 genes differentially expressed between tumor and normal samples. The set of genes was selected based on the FDR-corrected p value < 0.05. Red corresponds to genes that are upregulated in oncocytomas, and green corresponds to genes downregulated in oncocytomas.
(C) Pathway analysis of a set of 457 genes that are upregulated in oncocytomas compared to normal samples. The set of genes was selected based on the FDR-corrected p value < 0.05. Red arrowhead marks AMPK signaling.
(D) Pathway analysis of a set of 500 genes differentially expressed between type 1 and type 2 oncocytoma. The set of genes was selected based on the lowest p values. Red corresponds to genes upregulated in type 2. Red arrowheads mark AMPK and p53 signaling.
(E) Oncocytomas show upregulation of a gene expression signature for mitochondrial respiration. Red and blue correspond to up- or downregulation of nuclear-encoded genes as for ChRCC (Davis et al., 2014) for ease of comparison. Yellow and gray correspond to mitochondrial genes with and without somatic mutations. Star indicates somatic mutations in nuclear-encoded genes.
(F) Deregulation of mitochondrial protein expression in oncocytomas determined by western blots. The asterisk marks sample 5 with mixed normal and tumor.
(G) Oncocytomas have loss of mTOR as indicated by reduction of P-S6 (Ser 235/236) and induction of AMPK signaling as indicated by induction of P-AMPK (α-subunit, Thr 172) in western blots.
Pathway analysis of the transcriptome indicated that in comparison to NAT, oncocytomas have a predominance of downregulated genes that are represented in metabolic and degradative pathways (Figure 3B; Tables S8 and S9). Mitochondrial accumulation in oncocytomas may be due to compensatory increased biogenesis or decreased clearance by autophagy, concepts that are not mutually exclusive. Indeed, PPARGC1a expression is induced several fold in oncocytomas compared with NAT (Table S9), indicating activation of mitochondrial biogenesis. Pathway analysis of genes upregulated in oncocytomas identified AMPK, creatine-phosphate biosynthesis, and ketolysis pathways suggestive of energy crisis (Figure 3C; Table S9). Oncocytomas have high expression of genes encoding the TCA cycle and other mitochondria-expressed gene products involved in respiration (Figure 3C; Table S10). Comparison of differential gene expression between types 1 and 2 identified metabolic and cancer pathways such as AMPK and p53 (Figure 3D; Tables S11 and S12). Both type 1 and type 2 oncocytomas have AMPK pathway upregulated compared to NAT, with upregulation more pronounced in type 1. Type 1 has 3.4-, 6.7-, 2.4-, and 4-fold higher levels of PRKAB2, PRKAG2, ACACA (ACC1), and ACACB (ACC2), whereas type 2 has 8-, 2.2-, and 3.3-fold higher levels of PRKAG2, ACACA, and ACACB, respectively. The mitochondrial accumulation and genome mutations, oxidative metabolism gene expression signature, and patterns of chromosome loss suggest similarities between oncocytomas and eosinophilic ChRCC (Figure 3E) (Davis et al., 2014). This may reflect an increased number of mitochondria and not necessarily increased mitochondrial metabolism.
Increased mitochondrial genome copy number was reflected in high cytochrome c levels in oncocytomas relative to NAT in all tumors with the exception of sample 5, where normal tissue was contaminated with tumor (Figure 3F). Mitochondrial genome mutations resulting in defects in complex I of the electron transport chain (ETC) are common in oncocytomas (Figures 1B and S1B) (Gasparre et al., 2007; Mayr et al., 2008; Simonnet et al., 2003). Low or undetectable levels of the subunit NDUFS1 (complex I), UQCRC2 (complex III), and MT-COX2 (complex IV) were observed in a subset of oncocytomas indicative of impairment in mitochondrial respiration at multiple points in the ETC (Figure 3F). Compared to NAT, ANT2, and SLC25A6 (ANT3), protein expression levels were low or undetectable in type 2 in contrast to the increased mitochondrial mass (Figure 3F). ANT3 is on the PAR and had a reduced mRNA expression (log2 fold change = −0.9, uncorrected p = 0.04) associated with loss of the Y or X chromosome and correspondingly reduced protein expression (Figure 3F). Tumor 8 had no detectable ANT2, likely due to a frameshift mutation and X chromosome loss containing the other allele (Figures 1B and 3F). Interestingly, mutations in ANT2 also occur in ChRCC (Figure 1B). ANT2 and ANT3 modulate the mitochondrial permeability transition pore, and their deficiency impairs mitochondrial function (Kokoszka et al., 2004). These findings are consistent with unifying defects in mitochondrial function in oncocytomas.
To examine the potential manifestation of mitochondrial metabolic defects on oncocytoma, we assessed the activity of the nutrient sensor and master regulator of cell growth, the mammalian target of rapamycin (mTOR), and the metabolic stress response regulator AMPK. Both subtypes had reduced phospho(P)-S6, indicating loss of mTOR activity, and increased phospho(P)-AMPK (Thr 172) (Figures 3G and S1F), indicating activation of AMPK signaling, consistent with upregulation of the gene expression signature (Figure 3C, arrow). These findings reflect metabolic and growth impairment consistent with defective mitochondrial function in oncocytomas.
Oncocytomas Manifest Phenotypes of Defective Autophagy
To test if autophagy defects underlie the accumulation of defective mitochondria in oncocytomas, we examined the levels of autophagy substrates that accumulate with autophagy inactivation. Oncocytomas show accumulation of mitochondria that are normally degraded by autophagy as determined by immunohistochemistry (IHC) and western blotting for the mitochondrial proteins TOM20 and cytochrome c (Figures 4A and 4B). Levels of the essential autophagy regulator BECLIN1 were reduced (Figures 4B and S2), and puncta of LC3 indicative of autophagosomes were present in NAT and absent in oncocytomas (Figure 4A). Consistent with the absence of LC3 puncta and autophagy, oncocytomas had higher levels of the unprocessed form of LC3 (LC3-I) and lower levels of the processed, autophagosome-associated form of LC3 (LC3-II) than NAT as indicated by western blotting and LC3I/II ratios (Figure 4B). The absence of LC3-II and autophagosomes and the accumulation of LC3-I is a characteristic of genetic ablation of an essential autophagy gene in mice (Karsli-Uzunbas et al., 2014). Oncocytomas also displayed high levels of the autophagy substrate p62 that accumulates in autophagy-defective cells and tissues (Figures 4A and 4B).
Figure 4. Oncocytomas Lack Autophagosomes, Accumulate Autophagy Substrates and Dysfunctional Mitochondria, and Are Primed for Mitophagy.
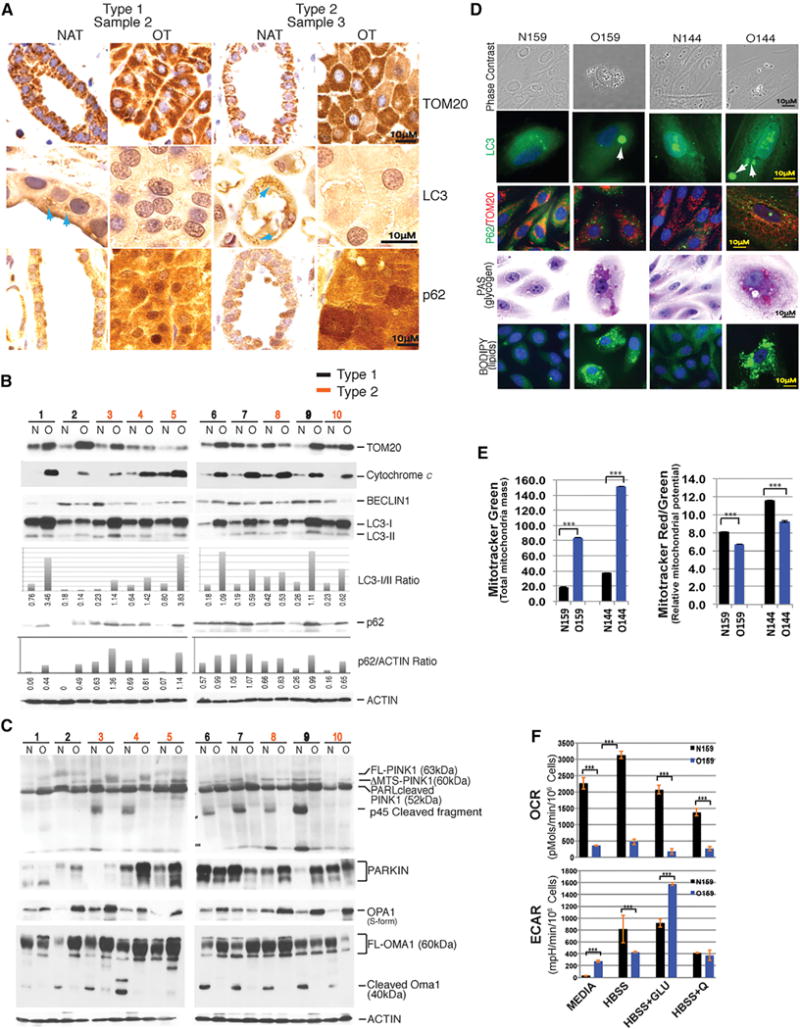
(A) Human oncocytomas accumulate mitochondria (TOM20) and p62 (autophagy substrate) and lack LC3 puncta (autophagosomes) indicative of autophagy inactivation. Representative IHC for each of the two subtypes with all in Figure S2.
(B) Western blots of indicated mitochondrial and autophagy proteins in normal tissues and oncocytomas. Quantitation of LC3-I/II and p62 levels is shown below.
(C) Oncocytomas have evidence for activation of the fission and the PINK1/PARKIN machinery. Western blots of normal and oncocytoma tissues for indicated proteins. FL, full length; ΔMTS, without mitochondrial translocation sequence.
(D) Primary cultures of oncocytoma cells display autophagy substrate accumulation. In contrast to primary cells derived from NAT (NK159/N144), primary oncocytoma cells (O159/O144) accumulate LC3 and p62 aggregates, mitochondria, glycogen, and lipids, characteristics of autophagy impairment.
(E) Primary cultures of oncocytoma cells (0159/O144) show accumulation of dysfunctional mitochondria with several fold more mitochondrial mass but significantly lower relative membrane potential than normal cell controls NK159 and N144.
(F) Primary oncocytoma cells (O159) have reduced OCR and increased ECAR when provided a fermentable carbon source in comparison to normal control cells (N159).
Mitophagy Is Primed, but Not Executed, in Oncocytomas
Since oncocytomas accumulate defective mitochondria, we examined oncocytomas for evidence of mitophagy dysfunction. Mitochondria undergo fission in response to stress to allow their segregation and elimination by mitophagy. Fission is induced when the protease OMA1 cleaves the dynamin-related GTPase OPA1 (McBride and Soubannier, 2010). Mitophagy is subsequently activated by loss of mitochondrial membrane potential that prevents cleavage and inactivation of the kinase PTEN-induced putative kinase 1 (PINK1), which recruits the E3 ligase PARKIN to mitochondria (Youle and Narendra, 2011). PARKIN then adds ubiquitin chains to mitochondrial outer membrane proteins to signal recruitment of autophagy adaptors such as p62, which interact with LC3-II in autophagosome membranes, allowing their engulfment within autophagosomes.
In normal conditions, OMA1 is cleaved and OPA1 is not, allowing fusion and preventing fission and mitophagy. This was the case in NAT, where cleaved OMA1 (40 kDa) and less cleaved OPA1 (S-form) were apparent (Figure 4C). In contrast, oncocytomas have uncleaved OMA1 (60 kDa) and accumulation of cleaved OPA1 (S-form) consistent with activation of fission. Since LC3-II-positive autophagosomes were not detected in oncocytomas (Figures 4A and 4B), this suggests that the initial steps of mitochondrial fission are initiated but the process of autophagosome sequestration is not. Oncocytomas have uncleaved PINK1 (FL-PINK1) and accumulation of PARKIN, consistent with priming of mitophagy without autophagosome formation (Figure 4C). PINK1 is cleaved by PARL and further cleaved to the p45kDa fragment to inactivate fission and mitophagy when mitochondria are healthy. Importantly, normal tissue but not oncocytomas have the p45kDa cleaved PINK1, consistent with inactivation of PINK1 and priming of mitophagy (Figure 4C). Interestingly, cells with pathogenic mitochondrial DNA mutations are defective for mitochondrial membrane fusion (Mishra et al., 2014), consistent with activation of the fission machinery and the PINK1/PARKIN system shown here in oncocytomas (Figure 4C). Thus, upstream signaling of mitophagy at mitochondria occurs in oncocytomas, but sequestration is absent, suggesting a block in autophagy.
Impaired Mitochondrial Respiration in Primary Oncocytoma Cells
Attempts to generate cell lines from renal oncocytomas by others and us have been unsuccessful. To address mitochondrial function, two pairs of normal adjacent kidney (NK159/N144) and oncocytoma-derived (O159/O144) primary cell cultures were examined. Primary oncocytoma cells remain viable in short-term culture (<1 week) without undergoing cell division, permitting limited analysis. In contrast to NK159 and N144, O159 and O144 primary cultures were composed of large cells with LC3 and p62 aggregates and increased mitochondria, glycogen, and lipids indicative of defective autophagy (Figure 4D). Even though primary oncocytoma cells had severalfold greater mitochondrial mass, relative mitochondrial membrane potential was significantly lower when compared to normal cells, consistent with ETC defects (Figure 4E). One of the primary cultures (O159) provided sufficient material for determination of the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). Compared to normal cells, O159 cells were defective for respiration and more glycolytic, consistent with pathogenic mitochondrial genome mutations (Figures 4F and S1B).
Oncocytomas Display LAMP-2 Trafficking, Golgi, and Lysosome Defects
Autophagy delivers cargo to lysosomes for degradation, and interference with lysosome function and cargo degradation feeds back to inhibit autophagy (Tanaka et al., 2000). Moreover, mutations in the major and essential lysosome transmembrane component lysosome-associated membrane protein-2 (LAMP-2) cause lysosomal storage disease and interfere with autophagy (Eskelinen, 2006). To test if lysosome defects in oncocytomas interfere with autophagy, lysosome numbers and localization were examined by monitoring LAMP-2. LAMP-2-positive vesicle distribution in types 1 and 2 was distinct from NAT (Figure 5A). In contrast, and suggestive of lysosome abnormality, localization of LAMP-2-positive vesicles was perinuclear in type 1 and peripheral in type 2 (Figure 5A).
Figure 5. Golgi Disassembly, Altered LAMP-2, and Defective Cathepsin Activation in Oncocytomas.
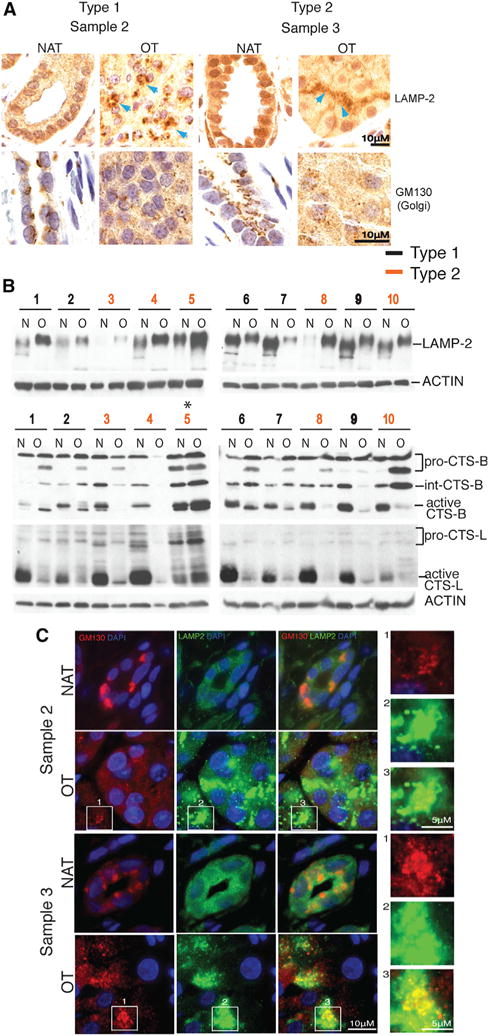
(A) Golgi disassembly and altered LAMP-2 localization in human oncocytomas. Representative IHC for each subtype with all shown in Figure S2.
(B) Western blots showing enhanced LAMP-2 glycosylation (increased molecular weight) in human oncocytomas. Oncocytomas have defective CTS-B and -L activation. Low molecular weight, cleaved, activated forms of cathepsins predominate in normal tissue. Pro, proenzyme; Int, intermediate.
(C) Golgi disassembly and LAMP-2 co-localization in human oncocytomas. Representative fluorescence images of each subtype stained for Golgi (GM130), LAMP-2, and nuclei (DAPI). The fluorescence images of all are shown in Figure S2.
LAMP-2 is synthesized and heavily polylactosamine glycosylated in the Golgi apparatus prior to transit to lysosomes, which serves to protect the protein from proteolysis in the lysosome lumen where most of the polypeptide resides. Oncocytomas have enhanced glycosylation of LAMP-2 compared to NAT as indicated by its higher molecular weight (Figure 5B). Increased glycosylation of LAMP-2 occurs when it is retained in the Golgi, where this post-translational modification takes place (Nabi and Rodriguez-Boulan, 1993). In contrast to perinuclear stacks of Golgi in NAT, oncocytomas displayed disrupted Golgi organization with LAMP-2 co-localization (Figures 5A and 5C), consistent with Golgi disassembly and LAMP-2 retention and enhanced glycosylation (Figures 5B and 5C).
The major lysosomal proteases, cathepsins (CTSs), are also processed in the Golgi and cleaved and activated to mature forms upon reaching the acidic environment of lysosomes (Ishidoh and Kominami, 2002). In contrast to NAT, most oncocytomas had predominantly inactive CTS-B and -L and lacked the cleaved, active forms, indicative of defective trafficking and lysosome function (Figure 5B). CTS-B and -L are cysteine proteases involved in lysosomal protein turnover, and failure to activate them is consistent with the accumulation of autophagic substrates in oncocytomas. These findings suggested that mitochondrial defects and energy crisis in oncocytomas produce a general disruption of Golgi-lysosome protein trafficking that impairs autophagy.
The Golgi is a highly structurally organized organelle essential for protein sorting, trafficking, and post-translational modification. The Golgi undergoes disassembly at mitosis for proper partitioning in daughter cells and in response to metabolic stress to conserve energy (Mao et al., 2013). To test if metabolic stress triggers Golgi disassembly and enhances LAMP-2 glycosylation that is alleviated by autophagy, isogenic tumor cells wild-type or null for the essential autophagy gene ATG7 (Guo et al., 2013a) were starved and Golgi and LAMP-2 localization and glycosylation were examined. Wild-type cells displayed no changes in LAMP-2 glycosylation but had increased P-AMPK, inhibition of P-S6, perinuclear Golgi with distinct LAMP-2 localization, and only modest Golgi disruption with prolonged starvation (Figures 6A and 6B). Autophagy-deficient ATG7 null cells, however, showed Golgi disruption and enhanced LAMP-2 glycosylation even under normal growth conditions, which was increased by starvation (Figures 6A and 6B). LAMP-2 co-localized with the Golgi in the starved ATG7-deficient cells, consistent with Golgi retention (Figure 6B). Thus, metabolic crisis induced by either starvation or autophagy deficiency disrupts the Golgi and alters LAMP-2 glycosylation.
Figure 6. Sustained Metabolic Stress Induces AMPK Signaling, Golgi Disassembly, Golgi-LAMP-2 Co-localization, and LAMP-2 Hyper-glycosylation and Inhibits Autophagy Flux.
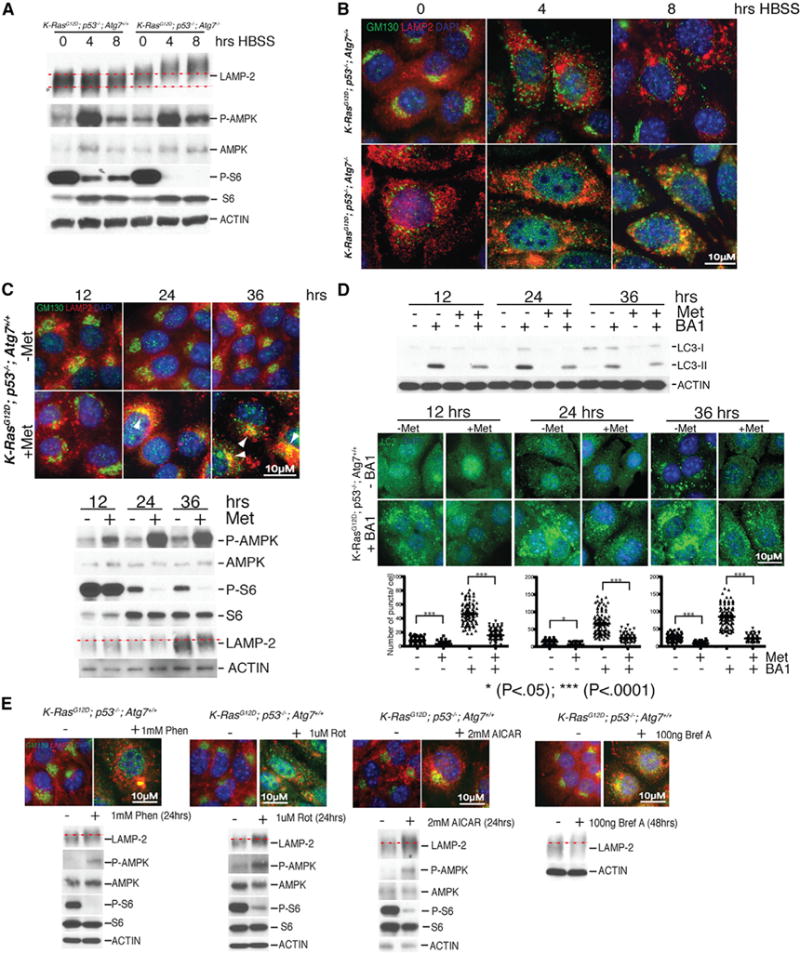
(A) Autophagy deficiency and starvation increase LAMP-2 glycosylation. Western blots of autophagy-competent and autophagy-deficient TDCLs following 0, 4, and 8 hr of starvation (HBSS) for the indicated proteins. Starvation induces AMPK signaling (increased P-AMPK) and reduces mTOR activity (decreased P-S6).
(B) Autophagy deficiency and starvation cause Golgi disassembly and LAMP-2 co-localization. Representative fluorescence images of autophagy-competent and autophagy-deficient TDCLs stained for Golgi (GM130), LAMP-2, and DNA (nuclear DAPI).
(C) Metformin (Met) disrupts Golgi and causes LAMP-2 co-localization. Autophagy-competent TDCLs stained for Golgi (GM130), LAMP-2, and nuclei (DAPI) following metformin treatment for the indicated times. White arrowheads show Golgi-LAMP2 co-localization. Bottom panel: western blots from samples.
(D) Metformin disrupts autophagic flux. Western blots of metformin-treated wild-type TDCLs for LC3 showing less LC3-II in BA1. Metformin disrupts autophagic flux indicated by reduced puncta of endogenous LC3 with BA1 with quantitation.
(E) Phenformin (Phen), Rotenone (Rot), 5-aminoimidazole 4-carboxamide ribonucleotide (AICAR), and brefeldin A (Bref A) disrupt Golgi and cause Golgi-LAMP-2 co-localization and LAMP-2 hyper-glycosylation. Autophagy-competent TDCLs stained for Golgi (GM130) and LAMP-2 following Phen, Rot, AICAR or Bref A treatment for indicated times. Nuclei (DAPI). Bottom panel: corresponding western blots.
Complex I Inhibition and Golgi Disassembly Block Autophagy
Metabolic stress and AMPK activation triggers Golgi disassembly by phosphorylation of Golgi-specific brefeldin A resistance factor 1 (GBF1), a guanine nucleotide exchange factor (GEF) for Arf-GTPases. Phosphorylation of GBF1 disassociates GBF1 from the Golgi membrane, producing Golgi fragmentation (Miyamoto et al., 2008). To test the hypothesis that mitochondrial mutations impairing complex I in oncocytomas produce metabolic crisis that disrupts Golgi function, thereby inhibiting autophagy, we examined the cellular response to the complex I inhibitor metformin (Bridges et al., 2014). Metformin potently induced AMPK activation (increased P-AMPK), inhibited mTOR (reduced P-S6), disrupted the Golgi, produced Golgi-LAMP-2 co-localization, and enhanced LAMP-2 glycosylation (Figure 6C). Importantly, metformin reduced autophagic flux as measured by inhibition of LC3-I to -II processing and LC3 puncta in the presence of BA1 (Figure 6D). The other complex I inhibitors phenformin and rotenone acted similarly (Figure 6E). Treatment with the AMPK activator AICAR confirmed that these changes are AMPK mediated (Figure 6E). Pharmacological disruption of the Golgi with brefeldin A induced Golgi-LAMP2 co-localization and LAMP-2 hyper-glycosylation (Figure 6E). Thus, mutational inactivation of the mitochondrial ETC in oncocytomas is likely the cause of AMPK activation and disruption of Golgi, autophagy, and lysosome function.
DISCUSSION
Molecular characterization of benign renal oncocytomas revealed two main subtypes that share recurrent inactivating mutations in mitochondrial genes encoded by both nuclear and mitochondrial genomes. Clonal expansion of somatic mitochondrial mutations in oncocytomas suggests that they are an early event (Gasparre et al., 2008). This might indicate a causal role for mitochondrial defects in tumor promotion as previously suggested (Gasparre et al., 2011). The genomic analysis of renal oncocytomas performed here was not able to resolve this issue; however, alterations of the nuclear genome consistent with oncogene activation and tumor suppressor gene inactivation were found in almost all cases.
Several issues we were able to address were the relationship between oncocytomas and other renal tumors, the cause of the metabolic defects, and the reasons for mitochondrial accumulation. Diploid type 1 has chromosomal rearrangements and upregulation of CCND1, a known oncogene. While type 2 lacked a clear driver mutation, it was hypodiploid, which could be the underlying basis for tumor formation, with the consistent loss of chromosome 1 being the prime candidate for location of a tumor suppressor gene. Loss of X or Y also suggests the presence of a tumor suppressor on the PAR that escapes X inactivation. Despite differences at the genomic level, the two subtypes were qualitatively similar in phenotype.
Our data suggest that oncocytomas have activated p53, and type 2 may progress to more malignant eosinophilic ChRCC upon acquiring p53, PTEN, and other mutations. Recent sequencing findings also support similarities between renal oncocytomas and ChRCC and the absence of p53 mutations in oncocytomas (Durinck et al., 2015). Autophagy deficiency in genetically engineered mouse models causes energy crisis, activates p53, and suppresses tumor growth, effects lessened by p53 deficiency (Guo et al., 2013b). Analogously, oncocytomas may represent benign disease whose growth is limited by metabolic crisis and activation of p53. Subsequent mutation in p53 in type 2 may restore growth capacity and progression to eosinophilic ChRCC (Guo et al., 2013a; Strohecker et al., 2013). It is also worth noting that ChRCCs have significantly fewer mitochondrial mutations than oncocytomas, suggesting that progression from oncocytoma might be aided by heteroplasmy and the ability to partly recover the functional pool of mitochondria (Ju et al., 2014). This might be achieved by restoration of autophagy and lysosomal biogenesis by TFE3 or TFEB activation (Linehan and Ricketts, 2013). Alternatively, tumor cells depleted of mitochondrial DNA recover respiration, ETC function, and tumor growth by horizontal transfer of normal mitochondria from non-tumor to tumor tissue (Tan et al., 2015). These findings point to the need for tumors to preserve mitochondrial ETC function. In contrast, type 1 may represent an evolutionary dead end, as these tumors with CCND1 rearrangements are less susceptible to recurrence (Sukov et al., 2009).
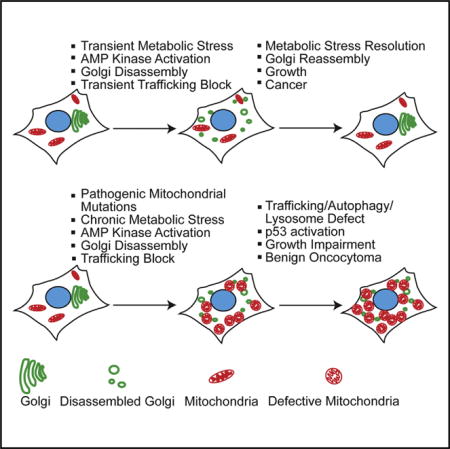
Recurrent pathogenic mitochondrial gene mutations encoded within the mitochondrial and nuclear genomes are the likely cause of the respiration defects in oncocytomas, consistent with and expanding on previous work. Inactivating mutations are found in complex I, III, IV, and control regions, in nuclear encoded NDUFA13, and now also in complex V, ANT2, NDUFV1, and UQCRC2. Alterations in key mitochondrial components are associated with the accumulation of defective mitochondria, suggesting a link between chronic metabolic impairment and autophagy suppression. Indeed, depletion of mitochondrial DNA and the resulting respiration defect inactivates autophagy in budding yeast and mammalian cells (Graef and Nunnari, 2011; Tan et al., 2015). In agreement with these findings, the pathogenic mitochondrial mutations and respiration defects in the oncocytomas reported here similarly suppress autophagy with the resulting metabolic insufficiency attributed to disruption of Golgi and lysosome function (Figure 7).
Figure 7. Model for Oncocytoma Genesis.
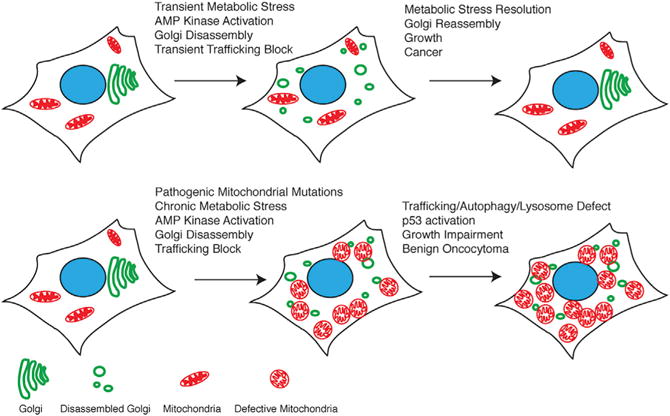
Chronic mitochondrial metabolic defect causes mitochondrial accumulation.
A major consequence of compromised mitochondrial respiration in oncocytomas is chronic metabolic deficiency exemplified by AMPK activation and loss of mTOR signaling. This is also seen in genetically engineered mouse models for cancer with autophagy inactivation (Guo et al., 2013b; Karsli-Uzunbas et al., 2014) and in response to pharmacologic inhibition of complex I (e.g., metformin) in cancer cells in vitro. Normally, transient energy crisis and AMPK activation cause Golgi disassembly to block trafficking and secretion that is thought to conserve energy. In contrast, the dysfunctional mitochondria in oncocytomas causes a chronic metabolic defect, sustained energy crisis, AMPK activation, and disruption of Golgi and lysosome function essential for autophagic cargo degradation (Figure 7). Importantly, inhibition of complex I is sufficient to disrupt the Golgi, trafficking, and autophagic flux. Thus, cancer cells need to sustain mitochondrial respiration to prevent metabolic crisis that inhibits trafficking. It is worth noting that PTEN and p53 mutations, which alleviate AMPK activation, occur in ChRCC and may facilitate progression of type 2 tumors by restoring Golgi, autophagy, and lysosome function.
Oncocytic features are common to many different subsets of renal tumors such as those with SDHB, SDHC, SDHD, and FLCN mutations and in hybrid ChRCC tumors (Linehan and Ricketts, 2013). This may be due to the occurrence of numerous means to impair mitochondrial respiration, producing chronic metabolic and trafficking defects, and the resulting mitochondrial accumulation as described here in oncocytomas. Metformin and other means to inhibit mitochondrial function have anti-cancer activity (Pollak, 2013; Viale et al., 2014; Weinberg et al., 2010). Our findings suggest that this may be due in part to chronic metabolic stress-induced trafficking defects, necessitating the preservation of ETC function to overcome a metabolic barrier to tumorigenesis.
EXPERIMENTAL PROCEDURES
Tumor Samples
Tumor and adjacent normal kidney tissues from ten sporadic oncocytomas at MSKCC were collected. All patients had previously consented to a tissue protocol. This study was preapproved by the institutional review board (IRB), and all patients signed informed consent. All samples were reviewed by a dedicated uropathologist. Paired fresh-frozen normal and primary tumor tissue blocks were identified, marked, and macrodissected for maximal tumor density. The two tumor specimens with normal adjacent tissue from CINJ were obtained from a de-identified tissue bank maintained at the Biospecimen Repository Service under the auspices of an IRB-approved protocol. All specimens were pre-existing and de-identified. Tissues for cytogenetics were obtained by reviewing records to identify all cases of renal oncocytomas and ChRCC for which cytogenetics was performed at Robert Wood Johnson University Hospital in the past 10 years.
Whole-Exome and RNA-Sequencing Data Analysis
The Agilent SureSelect Human All Exon V5 Enrichment protocol was used for exome capture. The exome was sequenced on Illumina HiSeq 2500, 2 × 100 bp paired-end reads to a minimum depth of 100 million reads per sample. The reads were preprocessed with Picard to remove PCR duplicates and aligned to the human reference genome (hg19) with Bowtie2. Somatic point mutations and small indels were called with MuTect and GATK Somatic Indel Detector algorithms. CNVs were detected with exomeCNV. Library preparation and RNA sequencing were carried out using the Illumina TruSeq RNA Sample Prep Kit v2, according to the manufacturer’s protocol (Illumina), and samples were sequenced on the Illumina HiSeq 2500 (2 × 100 bp paired-end reads) to a minimum depth of 50 million reads per sample. Sequence reads were mapped using TopHat2/Bowtie2 algorithm. Analysis of gene expression was performed using Cufflinks and DESeq. The complete description of the sequencing and genomic analysis and all other methods can be found in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Type 2 renal oncocytoma may precede eosinophilic ChRCC
Mitochondrial defects disrupt Golgi organization and trafficking
Mitochondrial accumulation in oncocytomas is due to defective elimination
Impaired mitochondrial function is a barrier to tumorigenesis
Acknowledgments
E.W. acknowledges support from the NIH (grants R01 CA163591, R01 CA130893, and CA188096), the Val Skinner Foundation, and CINJ (grant P30 CA072720). C.S.C. and D.T. acknowledge support from CINJ. S.G. acknowledges support from the Triple Negative Breast Cancer Foundation, the Hugs for Brady Foundation, and the NIH (grants R01 CA169182 and R01 CA130893). S.J. received support from a postdoctoral fellowship from the New Jersey Commission for Cancer Research (DFHS13PPCO24). This work used the Functional Genomics/Rutgers University Cell and DNA Repository, Biospecimen Repository Service, and Flow Cytometry/Cell Sorting CINJ core facilities supported by grant P30 CA72720. We thank R. Muthuramam for technical assistance. E.W. is on the SAB of Forma Therapeutics. J.H. is on the EAB of Chugai Pharmaceuticals, is a consultant for Novartis, and receives research support from Pfizer and Novartis.
Footnotes
ACCESSION NUMBERS
The accession number for the sequencing data reported in this paper is SRA: SRP051509.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, two figures, and twelve tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.10.059.
AUTHOR CONTRIBUTIONS
S.J. designed and performed the histology and biological experiments. D.T. performed sequencing data analysis. H.A. performed karyotype analysis. A.A.H. and J.J.H. guided the renal tumor biology and tumor procurement. M.Y. helped perform FISH analysis. S.G. helped conceive and direct the project and human tissue and chromosome analysis. C.S.C. directed project and bioinformatics analysis. E.W. conceived and directed the project.
References
- Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J. 2014;462:475–487. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Stawiski EW, Pavia-Jimenez A, Modrusan Z, Kapur P, Jaiswal BS, Zhang N, Toffessi-Tcheuyap V, Nguyen TT, Pahuja KB, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet. 2015;47:13–21. doi: 10.1038/ng.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersberger I, Metzler D, Schwarz C, Pääbo S. Genomewide comparison of DNA sequences between humans and chimpanzees. Am J Hum Genet. 2002;70:1490–1497. doi: 10.1086/340787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27:495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparre G, Porcelli AM, Bonora E, Pennisi LF, Toller M, Iommarini L, Ghelli A, Moretti M, Betts CM, Martinelli GN, et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc Natl Acad Sci USA. 2007;104:9001–9006. doi: 10.1073/pnas.0703056104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparre G, Hervouet E, de Laplanche E, Demont J, Pennisi LF, Colombel M, Mège-Lechevallier F, Scoazec JY, Bonora E, Smeets R, et al. Clonal expansion of mutated mitochondrial DNA is associated with tumor formation and complex I deficiency in the benign renal oncocytoma. Hum Mol Genet. 2008;17:986–995. doi: 10.1093/hmg/ddm371. [DOI] [PubMed] [Google Scholar]
- Gasparre G, Romeo G, Rugolo M, Porcelli AM. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim Biophys Acta. 2011;1807:633–642. doi: 10.1016/j.bbabio.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Graef M, Nunnari J. Mitochondria regulate autophagy by conserved signalling pathways. EMBO J. 2011;30:2101–2114. doi: 10.1038/emboj.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013a;27:1447–1461. doi: 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013b;155:1216–1219. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudacko R, May M, Aviv H. A new translocation between chromosomes 6 and 9 helps to establish diagnosis of renal oncocytoma. Ann Diagn Pathol. 2011;15:278–281. doi: 10.1016/j.anndiagpath.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Ishidoh K, Kominami E. Processing and activation of lysosomal proteinases. Biol Chem. 2002;383:1827–1831. doi: 10.1515/BC.2002.206. [DOI] [PubMed] [Google Scholar]
- Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik-Zainal S, Ramakrishna M, Davies HR, Papaemmanuil E, Gundem G, Shlien A, et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife. 2014;3:3. doi: 10.7554/eLife.02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, White E. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914–927. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan WM, Ricketts CJ. The metabolic basis of kidney cancer. Semin Cancer Biol. 2013;23:46–55. doi: 10.1016/j.semcancer.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Li N, Guo Y, Xu X, Gao L, Xu Y, Zhou L, Liu W. AMPK phosphorylates GBF1 for mitotic Golgi disassembly. J Cell Sci. 2013;126:1498–1505. doi: 10.1242/jcs.121954. [DOI] [PubMed] [Google Scholar]
- Mayr JA, Meierhofer D, Zimmermann F, Feichtinger R, Kogler C, Ratschek M, Schmeller N, Sperl W, Kofler B. Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin Cancer Res. 2008;14:2270–2275. doi: 10.1158/1078-0432.CCR-07-4131. [DOI] [PubMed] [Google Scholar]
- McBride H, Soubannier V. Mitochondrial function: OMA1 and OPA1, the grandmasters of mitochondrial health. Curr Biol. 2010;20:R274–R276. doi: 10.1016/j.cub.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19:630–641. doi: 10.1016/j.cmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Oshiro N, Yoshino K, Nakashima A, Eguchi S, Takahashi M, Ono Y, Kikkawa U, Yonezawa K. AMP-activated protein kinase phosphorylates Golgi-specific brefeldin A resistance factor 1 at Thr1337 to induce disassembly of Golgi apparatus. J Biol Chem. 2008;283:4430–4438. doi: 10.1074/jbc.M708296200. [DOI] [PubMed] [Google Scholar]
- Nabi IR, Rodriguez-Boulan E. Increased LAMP-2 polylactosamine glycosylation is associated with its slower Golgi transit during establishment of a polarized MDCK epithelial monolayer. Mol Biol Cell. 1993;4:627–635. doi: 10.1091/mbc.4.6.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KL, Rajandram R, Morais C, Yap NY, Samaratunga H, Gobe GC, Wood ST. Differentiation of oncocytoma from chromophobe renal cell carcinoma (RCC): can novel molecular biomarkers help solve an old problem? J Clin Pathol. 2014;67:97–104. doi: 10.1136/jclinpath-2013-201895. [DOI] [PubMed] [Google Scholar]
- Pollak M. Potential applications for biguanides in oncology. J Clin Invest. 2013;123:3693–3700. doi: 10.1172/JCI67232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonnet H, Demont J, Pfeiffer K, Guenaneche L, Bouvier R, Brandt U, Schagger H, Godinot C. Mitochondrial complex I is deficient in renal oncocytomas. Carcinogenesis. 2003;24:1461–1466. doi: 10.1093/carcin/bgg109. [DOI] [PubMed] [Google Scholar]
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, White E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3:1272–1285. doi: 10.1158/2159-8290.CD-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukov WR, Ketterling RP, Lager DJ, Carlson AW, Sinnwell JP, Chow GK, Jenkins RB, Cheville JC. CCND1 rearrangements and cyclin D1 overexpression in renal oncocytomas: frequency, clinicopathologic features, and utility in differentiation from chromophobe renal cell carcinoma. Hum Pathol. 2009;40:1296–1303. doi: 10.1016/j.humpath.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21:81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- Thilly WG. Have environmental mutagens caused oncomutations in people? Nat Genet. 2003;34:255–259. doi: 10.1038/ng1205. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.