

Abstract
Background
Astragalus membranaceus Bge. var. mongolicus (Bge.) Hsiao (A. mongolicus) is an important traditional Chinese herb that is cultivated on a large scale in northwestern China. Understanding plant responses to drought has important effects on ecological environment recovery and local economic development. Here, we combined transcriptomics (Illumina Hiseq 2000) and metabolomics (1H-NMR) to investigate how the roots of two-year-old A. mongolicus responded to 14 days of progressive drought stress.
Results
The dried soil reduced the relative water content (RWC) of the leaves and biomass, induced the differential expression of a large fraction of the transcriptome and significantly altered the metabolic processes. PCA analysis demonstrated that the sucrose, proline, and malate metabolites contributed greatly to the separation. Strikingly, proline was increased by almost 60-fold under severe stress compared to the control. Some backbone pathways, including glycolysis, tricarboxylic acid (TCA) cycle, glutamate-mediated proline biosynthesis, aspartate family metabolism and starch and sucrose metabolism, were significantly affected by drought. An integrated analysis of the interaction between key genes and the altered metabolites involved in these pathways was performed.
Conclusions
Our findings demonstrated that the expression of drought-responsive genes showed a strong stress-dose dependency. Analysis of backbone pathways of the transcriptome and metabolome revealed specific genotypic responses to different levels of drought. The variation in molecular strategies to the drought may play an important role in how A. mongolicus and other legume crops adapt to drought stress.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2554-0) contains supplementary material, which is available to authorized users.
Keywords: Drought, Metabolism, Gene expression, RNA-seq, A. mongolicus
Background
Plants colonised the land over 400 million years ago. Thereafter, they successfully evolved to cope with water deficits. For instance, some plants have an excellent tolerance to water scarcity during drought [1, 2]. The physiology, biochemistry, and molecular biology of plants are affected by some responses [3]. When faced with mild soil water stress, plants maintain their turgor pressure for a short time to ensure steady growth [4]. However, under mild to severe drought stress, plants show decreased photosynthesis and employ several measures to minimise water loss [5–7], including stomata closure and leaf rolling [8, 9], biomass desiccation [10], cell turgor maintenance, and osmotic adjustment [11]. Ultimately, the physiological responses to water stress are underpinned by reprogramming of the metabolism and gene expression [12–14]. Therefore, we need to elucidate these protective mechanisms to better understand how plants adapt to drought conditions.
Drought resistance is a complex process that is regulated by interactions among multiple genes [15]. Therefore, manipulation of a single gene or a subset of genes is not sufficient to study drought resistance. Instead, the complex responses of plants to drought stress need to be considered on a systems biology scale. Over the past decade, technological innovations have made it possible to overview the changes that occur at the transcriptomic level under environmental stress conditions. Many published studies have reported the use of microarrays and RNA sequencing techniques to identify changes in genes induced by drought treatments in a variety of plant species [16–21]. These results primarily identified genes that were up-regulated and/or down-regulated under drought stress. However, the plant response to drought is affected not only by the drought-responsive transcriptome but also by interactions among genes, metabolites and proteins. The relationship between the transcriptome and metabolome under water stress conditions has been characterised in different species [22–24].
The response of plants to soil water deficits may be governed by changes in small molecules to reach a new state. A number of previous reports have described metabolomics responses of plants to water and/or salt stress [25–30]. Several compounds synthesised by plants, such as sugars, ferulic acid, amino acids, and ABA have shown tolerance to drought stress [31, 32]. Osmotic adjustment via the accumulation of these solutes is one of the principal mechanisms by which plants adapt to water stress [33, 34]. With the reduction in osmotic potential, the plants can maintain cell turgor pressure and growth by changing the turgor-raising potential of the existing cellular water [35]. Some compatible solutes, such as mannitol, raffinose, polyols, trigonelline and proline, can reduce the osmotic potential [36, 37]. The trigonelline content increased when plants were subjected to stress, such as salt stress or drought stress [38, 39]. In many higher plants, proline accumulation was correlated with a water deficit [22, 40, 41]. These compounds also protect membranes and enzymes [37] during the drought process. Hence, we can better understand the mechanisms underlying plant resistance to drought stress by combining transcriptomics and metabolomics approaches.
The shortage of the water resource is a common phenomenon in Northwestern China and seriously hampers development and production. Therefore, we need to search for drought-adapted plants and study plant responses to drought stress to effectively tackle the problem. Astragalus membranaceus Bge. var. mongolicus (Bge.) Hsiao (A. mongolicus) is a traditional Chinese herb that has successfully evolved in arid and semi-arid environments. At present, it is primarily cultivated on a large scale by farmers in the northern and northwestern regions of China due to the shortage of wild resources. Understanding the herb resistance to drought stress has contributed to guiding planting and optimising the yield.
The purpose of this study is to present a comprehensive overview of the changes in the A. mongolicus transcriptome and metabolome under progressive drought stress and to identify important pathways through which this herb adapts to and tolerates drought stress. Moreover, we provide candidate genes that may provide useful information for translational approaches to investigate A. mongolicus or other legume crops in future studies.
Results
Physiological changes during drought acclimation
We grew the plants for 14 days while withholding water to make the drought stress treatments as realistic as possible. First, we tested the soil field capacity under drought stress. The soil relative water content (SWC) rapidly decreased from 70.0 % to 53.1 % on the 6th day of the experiment. Then, the SWC gradually declined from 53.1 % to 32.3 % on the 14th day (Fig. 1a). The relative water content (RWC) of leaves started to decline on the 4th day and then it gradually declined to 73.5 % on the 10th day. Following water withholding, the RWC of the leaves substantially decreased to 35.5 % on the 14th day of the experiment (Fig. 1b).
Fig. 1.
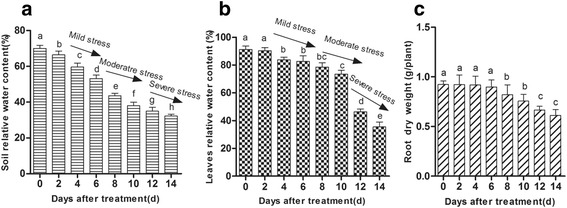
Physiological changes of A. mongolicus during progressive drought stress. a Effects of drought stress on soil water content (SWC). b Effects of drought stress on relative water content (RWC) of leaves. c Effects of drought stress on root dry weight (n = 8)
We defined three phases of progressing drought stress according to the literature [41], the RWC, and phenology. During the mild stress stage (days 4–6), the RWC of the leaves declined by 8 %–10 % (Fig. 1b) without affecting the visible signs of plant growth relative to the control. During moderate stress (days 8–10), the soil water content decreased to 38.0 % (Fig. 1a). Although the RWC of the leaves remained higher than 50 % (Fig. 1b), the leaves were withered. The effects of severe stress (days 12–14) were quite intense. On the 14th day, most plants exhibited yellowing and rolling of the leaves with a very low RWC (35.5 %). Furthermore, drought stress caused a significant reduction in the root dry weight (Fig. 1c). Taken together, these results indicated that drought stress had a significant impact on A. mongolicus. We performed transcriptomic and metabonomic analyses to elucidate the effects of drought stress on A. mongolicus. Based on the physiological changes under drought stress, we chose the following key time points for the study: 0d (A), 6d (B), 10d (C), and 14d (D).
Transcriptomic analysis of A. mongolicus under progressive drought stress
We used the Illumina Hiseq 2000 technology to generate a reference transcriptome of A. mongolicus under drought stress. We generated 18,885,163,680 bp and detected 88,662 unigenes. Table 1 displayed a total of 55,424,432, 56,050,320, 55,691,302, and 55,972,712 raw reads that were obtained from the control and water stress treatments. After removing low quality sequences, 52,178,802, 52,783,838, 52,231,504 and 52,641,008 clean reads were retained and used for assembly, respectively. The Q20 values (sequencing error rate < 1 %) were greater than 97.82 %, and the GC percentages were 42.13, 42.25, 42.29, and 42.08 %, respectively (Table 1). These results demonstrated that the sequencing data were of sufficient quantity and quality to ensure accurate sequence assembly and adequate transcriptome coverage. Figure 2a showed 60,707 unigenes annotated in the nr database according to the Basic Local Alignment Search Tool (BLASTx) (E-value < 1e−5) (NCBI, Bethesda, MD, USA) and 39,322 unigenes annotated according to the Swiss-Prot protein database (Swissprot, The European Bioinformatics Institute, Cambridge, UK). Additionally, 36,048 unigenes were annotated according to the Kyoto Encyclopaedia of Genes and Genomes (KEGG, Kanehisa Laboratories, Kyoto, Japan) and 23,222 unigenes were annotated using the Cluster of Orthologous Groups of Protein (COG) database (NCBI, Bethesda, MD, USA). A total of 13,993 unigenes were annotated only in the nr database, 74 unigenes were assigned only in Swissprot, and 3 unigenes and 146 unigenes were annotated only by COG and KEGG, respectively. Approximately 29 % (18,190/60,850) of the unigenes were assigned to a homolog in all four databases (Fig. 2a).
Table 1.
Summary statistics of sequencing results
| Samples | Total raw reads | Total clean reads | Total clean nucleotides(nt) | Q20 percentage | N percentage | GC percentage |
|---|---|---|---|---|---|---|
| A | 55,424,432 | 52,178,802 | 4,696,092,180 | 97.91 % | 0.01 % | 42.13 % |
| B | 56,050,320 | 52,783,838 | 4,750,545,420 | 97.99 % | 0.01 % | 42.25 % |
| C | 55,691,302 | 52,231,504 | 4,700,835,360 | 97.82 % | 0.01 % | 42.29 % |
| D | 55,972,712 | 52,641,008 | 4,737,690,720 | 97.96 % | 0.01 % | 42.08 % |
A control, B water-stress for 6 days, C water-stress for 10 days, D water-stress for 14 days, respectively
Fig. 2.
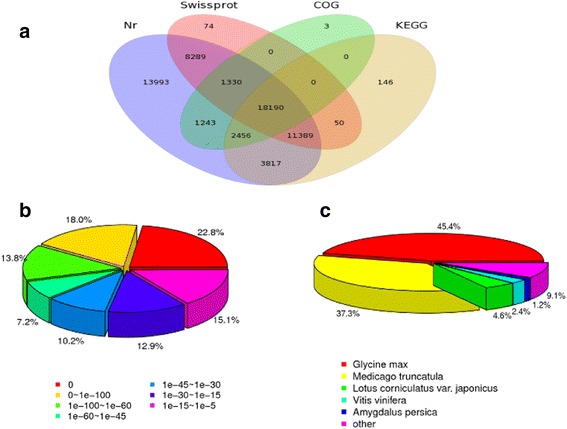
Distribution of the homology search of unigenes of against the database. a Venn diagram of a number of annotated unigenes by Blastx against protein databases. b E-value distribution of Blastx hits for each unigene against the NR database. c Species distribution of unigenes matching the top species using Blastx in the NR database
The E-values and species distributions were analysed by evaluating the matched unigenes from the returned Blastx results of the nr protein database. Based on the E-value distribution of the top hits in the nr databases, 61.8 % of the aligned sequences showed strong homology (<1e-45), moreover, 58.29 % of the homologous sequences ranged from 1e-45 to 1e-5 (Fig. 2b). The species distribution of the top Blast hits for the A. mongolicus transcriptome showed that these unigenes had the greatest numbers of matches with the genes of Glycine max (45.4), followed by Medicago truncatula (37.3), Lotus corniculatus var. japonicas (4.6), Vitis vinifera (2.4) and Amygdalus persica (1.2 %). The other 9.1 % of the unigenes had hits with other species (Fig. 2c). This result indicates that the transcriptome of A. mongolicus is more closely related to Glycine max than to other plant genomes in the current public databases.
Functional analysis of DE genes
Functional analysis provides information and clarifies how A. mongolicus regulates its biological functions in response to drought stress at the molecular level. As shown in Fig. 3a, 18,292, 15,085, and 20,986 genes were differentially expressed on days 6, 10, and 14, respectively, compared to the control (0 day). However, only 6881 DE genes were co-expressed at all three levels of drought stress (2,127 DE genes were only significantly up-regulated and 4599 DE genes were only down-regulated) (Fig. 3b, c). We performed hierarchical clustering to obtain a complete transcriptional profile of the co-expressed transcripts under different stages of drought stress (Fig. 4). The clustered profiles indicated that drought stress significantly affected the transcriptional profiles of the co-expressed transcripts. The number of down-regulated genes was more than the number of up-regulated genes in these co-expressed transcripts compared to the control. Severe stress resulted in more down-regulated genes than the early stages of drought stress.
Fig. 3.
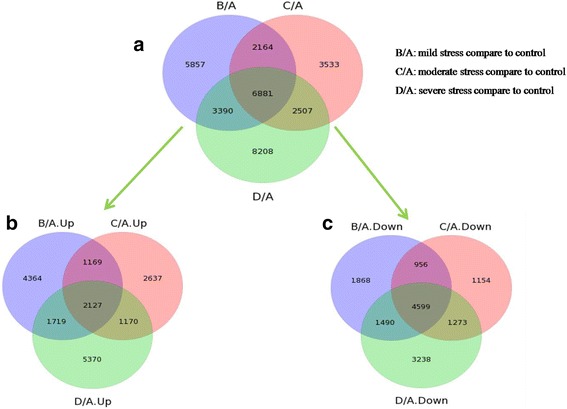
Transcriptomic analysis of A. mongolicus by high-throughput sequencing. a Venn diagram showing the number of differentially expressed (DE) genes, which were found at different treatment points. b Venn diagram showing the number of DE genes, which were only up-regulated at different treatment points. c Venn diagram showing the number of DE genes, which were only down-regulated at different treatment points (A: control, B: water stress for 6 days, C: water stress for 10 days, D: water stress for 14 days)
Fig. 4.
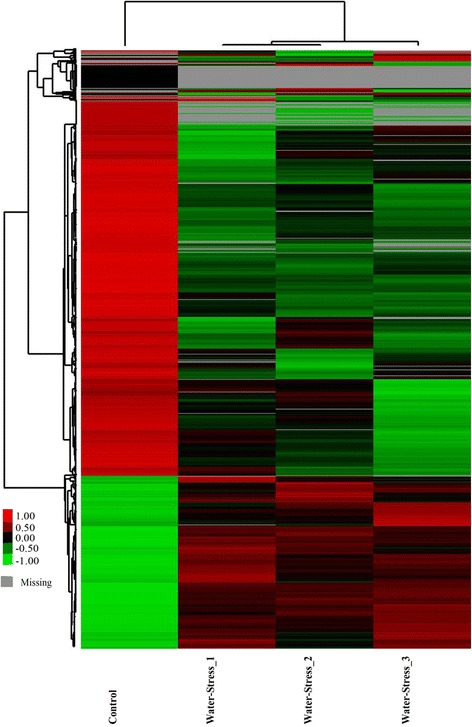
The hierarchical analyses of co-expression transcripts at different stages of drought stress. Green indicates the down-regulated and red indicates the up-regulated expression of genes (Control: 0 day, Water-stress-1: water stress for 6 days, Water-stress-2: water stress for 10 days, Water-stress-3: water stress for 14 days)
The DE genes were mapped to 20 gene ontology (GO) terms from the biological processing category (Fig. 5a, b, c, d). The most abundant DE genes were classified in the metabolism category, followed by cellular process and single-organism process. Interestingly, almost all of the related GO terms were enriched by the co-expressed down-regulated DE genes, such as genes related to growth, immunity and stimulus processes (Fig. 5a). The metabolism category contained more up-regulated DE genes than down-regulated DE genes under mild and moderate stress conditions (Fig. 5b, c). However, 1392 down-regulated genes were enriched under severe drought stress, resulting in a greater than 2-fold increase compared to mild stress (Fig. 5d). Additionally, the regulation of biological processes, signalling, and growth were more enriched in the down-regulated DE genes than the up-regulated genes (Fig. 5d). This finding indicates that severe stress can seriously affect the growth and regulation of biological processes in plants. Nine sub-categories were found under the metabolism category (Fig. 5e, f, g). In the metabolism category, the most genes were related to carbohydrate metabolism. A total of 1053 unigenes that mapped to carbohydrate metabolism were affected under mild stress conditions (Fig. 5e), 953 unigenes were affected by moderate stress (Fig. 5f), and 1,194 unigenes were affected by severe stress (Fig. 5 g). These changes were followed by genes related to lipid metabolism, secondary metabolism and amino acid metabolism, which were also affected by all stages of drought stress. The unigenes annotated in the metabolism category exercised their biological functions to catalyse metabolic processes or generate energy for primary and secondary metabolite production during progressive drought stress.
Fig. 5.
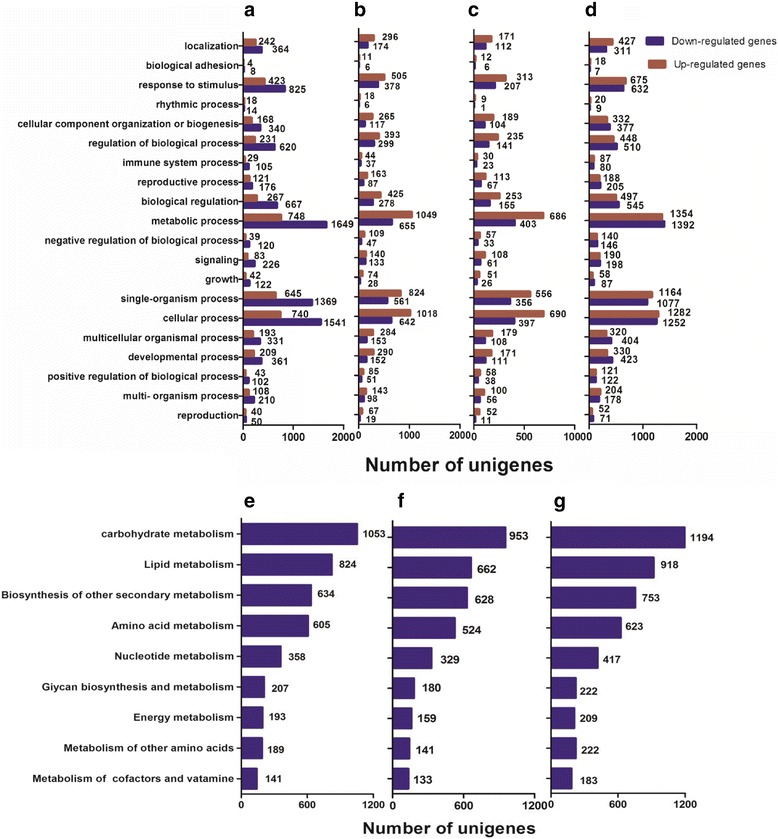
Plots of gene ontology (GO) terms from the biological process category of genes: a DE genes in total. b DE genes in mild stress (compare to the control). c DE genes in moderate stress (compare to the control). d DE genes in severe stress (compare to the control). e GO categories of a major metabolic process under mild stress (compare to the control). f GO categories of a major metabolic process under moderate stress (compare to control). g GO categories of a major metabolic process under severe stress (compare to control)
Gene expression response to drought
Based on the expression ratio between mild stress relative to the control, we selected the 100 most highly expressed DE genes (Additional file 1: Table S1). Three of these highly elevated genes encoded P5CS, which was involved in the metabolism of the compatible solute proline. Two of the top 100 DE genes encoded a late embryogenesis abundant (LEA) protein. Interestingly, two genes were related to defensins, which influenced biotic stress [42]. Several genes encoded proteins of unknown function. Some of the top 100 highly expressed genes under severe stress (Additional file 2: Table S2) were related to ATP, three of these genes were auxin-regulated genes, whereas the functions of several genes were unknown.
We found that 35 DE genes were only up-regulated or only down-regulated during all three stages of drought stress. Among these genes, 25 genes were down-regulated DE genes (Additional file 3: Table S3). Furthermore, galactose metabolism was governed by two genes (the polygalacturonase gene and the fasciclin-like arabinogalactan gene). We also found one MADS-box transcription factor, two phosphoenolpyruvate carboxykinase (PEPC) genes, and one gene encoding cytochrome P450. However, 7 genes lacked functional annotation. In total, there were five enzymatic genes in this list, including a gene encoding a peroxidase enzyme, one probable gene encoding the carboxyl esterase 15-like enzyme, one gene encoding a methylmalonate-semialdehyde dehydrogenase and two genes encoding pectinesterase. Additionally, one gene was annotated as the pectate lyase enzyme. Other genes were also identified, including one auxin-induced gene, one formin-like gene, and two genes encoding laccase. Correspondingly, one gene might have encoded the toleration protein DRT100, which was probably associated with the repair of damaged DNA. Among these DE genes, the 10 up-regulated DE genes included one gene that probably encoded the U3 small nucleolar RNA, and one gene related to ATP. However, six genes had no functional annotation.
Assignment of A. mongolicus metabolic changes during drought acclimation
To monitor the metabolic acclimation processes of A. mongolicus under drought stress, we performed nuclear magnetic resonance (NMR) in this study. Based on the 1H-NMR spectra of the methanol extracts from the A. mongolicus roots (Additional file 4: Figure S1), we identified sixteen amino acids and their derivatives, four carbohydrates, nine organic acids/amines, one nucleic acid component, three alcohols, and four other compounds (Additional file 5: Table S4). Sucrose was the highest out of the metabolites, and had a weight of > 30 mg per gram of freeze-dried plant at all time points. Other compounds, including asparagine, fructose, glucose and malate, had weights of >1 mg/g in the freeze-dried plant samples at all time points (Additional file 6: Table S5).
During progressive drought stress, we found significantly changed metabolites based on the ratios of metabolite concentration changes to the control (Fig. 6a, b). Compared with the control, the most obviously changed metabolites included proline and trigonelline. The proline content was almost 60-fold higher during severe stress than in the control (Fig. 6a). Some other metabolites, such as galactose, ethanolamine, fumarate and acetate, also exhibited a strong stress-dose dependency. However, the levels of the other metabolites changed in a non-linear manner under water stress (Fig. 6b). For example, the concentration of most amino acids, such as valine, threonine, isoleucine, glycine, arginine, alanine, asparagines, leucine and tyrosine, the sugars fructose and glucose, and the organic acid malate, increased in response to the water deficit within 6 to 10 days reached an approximate plateau and then decreased to the control level on the 14th day. Some metabolites, such as trimethylamine and pyruvate did not exhibt any significant changes in their concentrations (Additional file 6: Table S5). Based on the heat map analysis (Fig. 6c), we deduced that the abundant primary metabolites were significantly affected under drought stress. Most of the amino acids were significantly enriched under moderate stress, but dramatically decreased under severe stress. In contrast, some sugars, such as galactose, fumarate and sucrose, were increased under severe stress compared to the control.
Fig. 6.
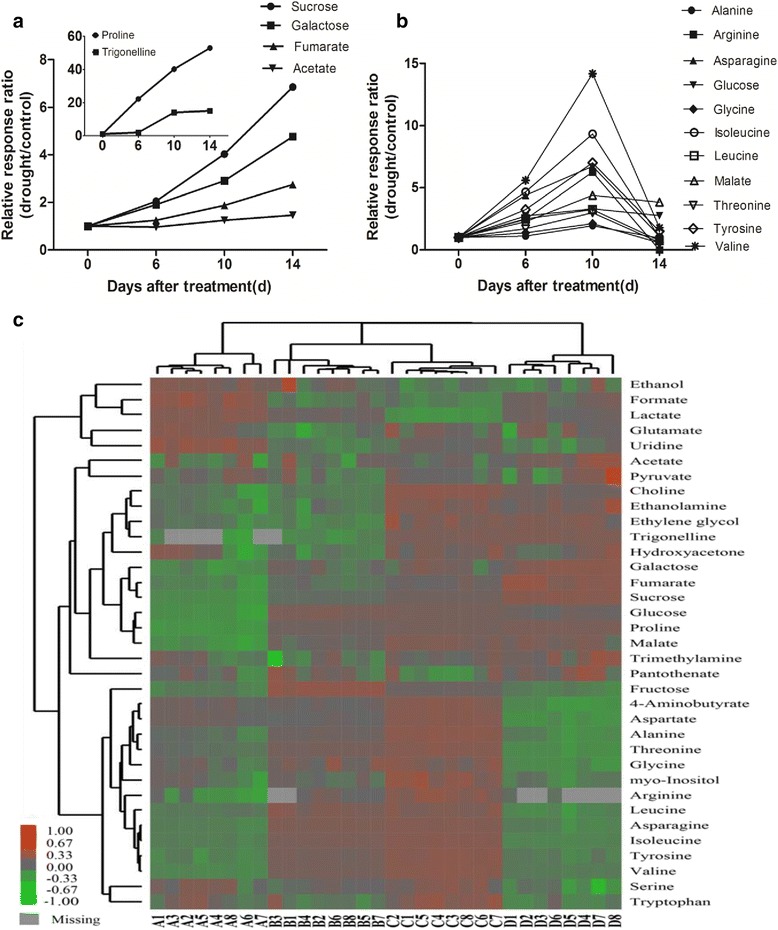
An overview of metabolic changes in A. mongolicus, which was exposed to drought stress. a, b Drought–induced metabolites concentration had relatively changed as compared to the control (0 day). c Differences in primary metabolite profiles during drought, heatmap color indicate the abundance of each metabolite in different stages of drought stress (A: control; B: water stress for 6 days; C: water stress for 10 days; D: water stress for 14 days)
We performed a multivariate analysis of the metabonomic responses. Principal component analysis (PCA) revealed the component data without prior metabolite identity information. The two component plot accounted for 93 % of the total variability. The first principal component accounted for 68 % of the variability, which was primarity reflected during mild and moderate drought stress, whereas the second principle component accounted for 24.8 % of the variability reflected control and severe drought stress (Fig. 7a). A stress-induced trend of dynamic changes in metabolites was clearly associated with the dosage of the drought stress. The PCA visualised sample classifications and implied good reproducibility of the 1H-NMR–based metabonomic method based on the trend of clustered samples. Moreover, from the loading plot (Fig. 7b), we inferred that sucrose, proline, malate, asparagine, fructose, and glucose greatly contributed to the separation of the different treatments.
Fig. 7.
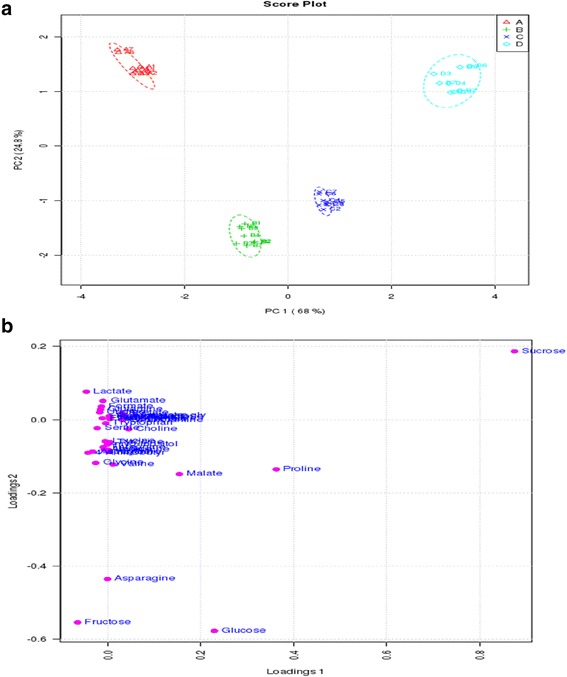
Metabolic analysis using a non-supervised principal component analysis (PCA). a Score plot of PCA. b Loading plot of PCA
Integrative analysis of gene expression and metabolite levels
We identified a relationship between gene expression and the abundance of metabolites. The list of metabolites, correlated genes, and primary metabolic pathways is provided in Table 2. We found that these pathways were significantly affected by drought stress. The major known pathways glycolysis, the tricarboxylic acid (TCA) cycle, glutamate-mediated proline biosynthesis and aspartate family metabolism were shown in the metabolism overview (Fig. 8).
Table 2.
Annotation of differentially expressed genes associated with metabolites of A. mongolicus in response to drought
| Pathway:Glycolysis | ||
|---|---|---|
| Metabolites: Sucrose, Fructose, Glucose | ||
| Gene ID | Gene annotation detail | P-value |
| MTR8g104540 | Phosphoglucomutase | 3.97E-10 |
| MTR1g102010 | Phosphoglucomutase | 0 |
| MTR5g009000 | Hexokinase | 3.29E-06 |
| MTR5g065880 | Glucose-6-phosphate isomerase | 6.38E-19 |
| MTR6g090130 | 6-phosphofructokinase | 1.62E-26 |
| MTR091s0008 | 6-phosphofructokinase | 0 |
| MTR1g090300 | 6-phosphofructokinase | 3.13E-11 |
| MTR5g069050 | Fructose-bisphosphate aldolase | 0 |
| MTR5g096670 | Fructose-bisphosphate aldolase | 0.000103 |
| LOC100778356 | Phosphoglycerate kinase, chloroplastic-like | 3.04E-07 |
| MTR7g074570 | 2,3-bisphosphoglycerate-independent phosphoglycerate mutase | 0 |
| MTR4g055260 | 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase | 1.37E-09 |
| MTR145s0008 | 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase-like | 1.47E-16 |
| MTR3g087540 | Pollen-specific protein C13 | 1.59E-14 |
| MTR5g096970 | Enolase | 2.05E-06 |
| LOC101509496 | Heat shock factor binding protein | 4.08E-07 |
| MTR7g100760 | Pyruvate kinase, cytosolic isozyme-like | 6.14E-37 |
| MTR1g061630 | Pyruvate kinase | 8.60E-14 |
| MTR5g014140 | Pyruvate kinase | 6.07E-46 |
| MTR3g111120 | Phosphoenolpyruvate carboxykinase | 2.78E-86 |
| MTR5g030620 | Phosphoenolpyruvate carboxykinase | 2.08E-248 |
| Pathway: Tricarboxylic acid (TCA)cycle | ||
| Metabolites: Citrate, Fumarate, Malate | ||
| MTR5g091930 | Citrate synthase | 8.17E-101 |
| MTR4g048190 | Aconitate hydratase | 5.04E-19 |
| MTR1g075520 | Aconitate hydratase | 3.58E-19 |
| MTR5g022940 | Aconitate hydratase | 1.82E-45 |
| LOC101493754 | Isocitrate dehydrogenase [NAD] catalytic subunit 5 | 1.28E-09 |
| MTR5g020050 | Succinate dehydrogenase | 0 |
| LOC101492705 | Succinate dehydrogenase | 4.91E-51 |
| LOC101512712 | Fumarate hydratase 1, mitochondrial-like | 1.24E-14 |
| MTR8g005980 | Malate dehydrogenase | 3.78E-38 |
| MTR5g014710 | Malate dehydrogenase, glyoxysomal | 1.96E-08 |
| MTR5g033920 | ATP-citrate synthase | 4.24E-08 |
| MTR2g062920 | ATP-citrate synthase | 6.30E-12 |
| Pathway: Glutamate-mediated amino acids biosynthesis | ||
| Metabolites: Glutamate, Glutamine, GABA, Proline | ||
| LOC101509615 | NADH-dependent glutamate synthase | 3.91E-57 |
| MTR3g064740 | Glutamate decarboxylase | 1.71E-79 |
| MTR3g064740 | Glutamate decarboxylase | 1.70E-09 |
| MTR3g065250 | Glutamine synthetase | 2.38E-05 |
| MTR5g077950 | Glutamine synthetase | 2.69E-10 |
| LOC100780873 | Glutamine synthetase | 6.70E-05 |
| MTR4g107940 | Delta-1-pyrroline-5-carboxylate dehydrogenase 1 protein | 2.89E-115 |
| MTR7g020820 | Proline dehydrogenase | 2.09E-13 |
| Pathway: Aspartate family metabolisms | ||
| Metabolites: Aspartate, Alanine, Threonine | ||
| MTR4g080140 | Aspartate aminotransferase | 1.88E-14 |
| LOC101495687 | Peptide transporter PTR2-like | 9.89E-18 |
Unigene sequences are aligned with @blastdb using blastx (evalue < 1e−5)
Fig. 8.
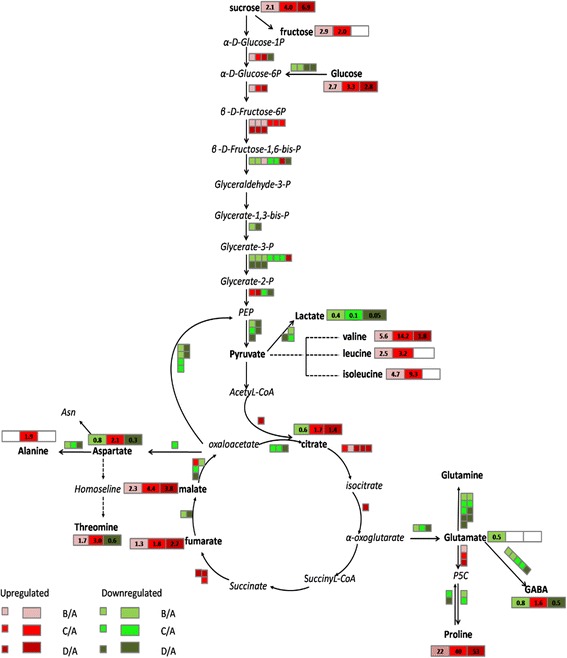
Pathway analysis related to A. mongolicus under progressive drought stress. The proposed metabolic pathways were based on literature and a web-based database of metabolic pathways. (A: control; B: water stress for 6 days; C: water stress for 10 days; D: water stress for 14 days). Metabolites, which were written in bold, were detected in this study. Metabolites in italic were not detected. Some metabolites have been omitted from this graph, because they were unaffected by drought stress. The small icons represent the genes, while the big icons represent the metabolites. Green indicates the down-regulated expression, while red indicates the up-regulated expression
As shown in Fig. 8, genes encoding glycolytic enzymes, such as phosphoglucomutase, glucose-6-phosphate isomerase and phosphofructokinase, were upregulated during the three different phases of drought stress compared to the control. Regulation by phosphofructokinase is the most important step in glycolysis. During the metabolism of pyruvate and phosphoenolpyruvate (PEP), genes encoding pyruvate orthophosphatedikinase (PPDK) and phosphoenolpyruvate carboxylase are downregulated in the energy payoff phase of the glycolytic pathway. Genes encoding L-lactate dehydrogenase were decreased after drought stress relative to the control, which prevented the conversion of pyruvate into lactate. The regulation of glycolysis indicates that the changes in the abundance of adenosine triphosphate (ATP) may occur during different phases of drought stress. Each glucose molecule oxidised to pyruvate, which produce two ATP molecules. These ATP molecules provide energy for other physiological functions [43].
In the TCA cycle pathway, genes encoding aconitate hydratase were up-regulated during the three phases of drought stress. Genes related to succinate dehydrogenase were up-regulated under moderate and severe drought stress, whereas the genes encoding fumarate hydratase were down-regulated. These changes in gene expression may have increased the fumarate content by 2.7-fold compared to the control. Malate plays an important role in stress and usually responds to changes in stomatal conductance and osmotic potential [44, 45]. Using metabolomics analysis, we found that the malic acid content was increased under drought stress, whereas the expression of malate dehydrogenase (MDH) was down-regulated. The expression of genes involved in malate synthase was not consistent with the changes in the malic acid content (Fig. 8).
The changes in the genes involved in glycolysis affected other key primary metabolic pathways, such as starch and sucrose metabolism. Most genes associated with starch synthesis were up-regulated under drought stress (Fig. 9), such as the genes encoding the starch branching enzyme. However, genes related to glucose-1-phosphate adenylyl transferase were down-regulated compared to the control. Sucrose metabolism was also affected. The expression of the sucrose-phosphate synthase (SPS) gene was up-regulated under drought stress and the invertase genes were down-regulated, which might be responsible for the progressive increase in sucrose in metabolomics results (Fig. 6a). These changes in key genes could alter the source–sink relationship in plants.
Fig. 9.
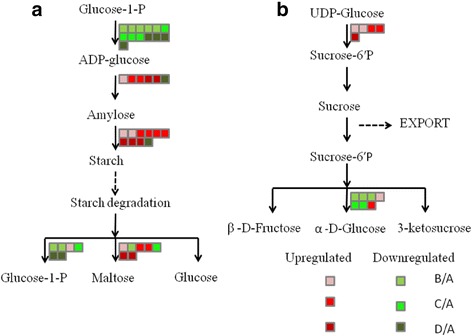
Changes in the expression of genes in starch and sucrose pathways of A. mongolicus under progressive drought stress. Altered expression of genes involved in starch metabolism (a) and sucrose metabolism (b). (A: control, B: water-stress for 6 days, C: water stress for 10 days, D: water stress for 14 days). Green indicates the down-regulated gene expression, while red indicates the up-regulated gene expression
Discussion
Our experiments were designed to study the response of A. mongolicus to drought stress. With a gradual water deficit, the plants showed significantly adaptive responses to stress. We determined the RWC of the soil and leaves during a progressive 14 day drought treatment. Based on the physiological and biochemical responses of A. mongolicus to drought stress, we imposed mild (6d), moderate (10d), and severe (14d) drought stress as references to reflect the stress intensity. Then, we gathered precise information from transcriptomics and metabolomics based on the drought intensity of the plants. We discussed the metabolic pathways and the correlated genes that were affected throughout the progressive drought stress.
The total number of drought-regulated genes was specifically correlated to the intensity of the imposed drought stress. However, we must note that drought tolerance-related genes may be activated earlier. For instance, most early response of genes may have participated in the stimulus response and growth process. We also found that most of the down-regulated genes participated in the signalling process. These genes were induced or repressed during the early stage of drought stress to enable the plant to adapt to the progressive water deficit. For instance, a gene encoding zeaxanthin epoxidase (ZEP) was detected under the mild stress condition and an up-stream enzyme of abscisic acid (ABA) was identified in the biosynthesis pathway. ABA is a major regulator of drought stress and can clearly trigger a wide range of avoidable mechanisms and physiological responses, including stomatal closure and osmoregulation [46, 47].
One of the consequences of stomatal closure due to progressive drought stress is the overproduction of reactive oxygen species (ROS), which cause oxidative stress. Transpirational cooling also leads to thermal stress. ROS can damage cells and the photosynthetic apparatus by oxidising amino acids and proteins [46, 48]. Our results indicated that the PSII (photosystem II) genes were down-regulated and several heat shock protein genes were induced after drought stress. These findings were consistent with previous studies on drought stress [49, 50] and demonstrated the plant responses to oxidative damage and thermal stress.
Glycine max and A. mongolicus are both legume crops but are not in the same genus. Our results showed that the A. mongolicus transcriptome had the largest number of matches with the genes of Glycine max (Fig. 2c). However, Glycine max and A. mongolicus showed different responses to water deficits. For instance, the growth and development of Glycine max are extremely sensitive to water deficits [51] with the exception of a few genotypes, whereas our results revealed that A. mongolicus exhibited good tolerance and adaption to drought stress. Chen et al. (2013) compared drought-tolerant and drought-sensitive Glycine max genotypes under dehydration and rehydration conditions and found that the genes encoding the auxin protein and zinc finger protein were up-regulated in the roots of the drought-tolerant genotype compared to the control. Zinc finger proteins play a central role in drought tolerance [52]. Our results also showed that genes in A. mongolicus related to these proteins were up-regulated under drought stress. Furthermore, cytochrome P450s can eliminate ROS, participate in photosynthesis, and serve as precursors to ABA synthesis [53]. They are more likely to determine drought stress tolerance [1]. In both drought-tolerant Glycine max and A. mongolicus, the expression of the genes encoding cytochrome P450s were suppressed after drought stress. These results indicated that A. mongolicus adapted to and tolerated to the drought.
Metabolite profiling can provide a view of the metabolic phenotypes. Thus, we can discover novel metabolic phenotypes not found by traditional targeted analyses and the identification of chemical signatures of specific phenotypes [54, 55]. A less biased NMR-based metabolomics approach was employed to determine a larger number of metabolites affected by drought stress. Our results identified the glycolysis, TCA cycle, glutamate-mediated proline biosynthesis, shikimate-mediated metabolism, and aspartate family metabolism pathways. The increase in the concentration of amino acids probably occurred due to the inhibition of protein biosynthesis or the increase in protein degradation induced by moderate drought stress. In the severe phase of drought stress, many amino acids decreased to the control level, probably due to secondary metabolism. Aromatic amino acids, such as tyrosine and phenylalanine are precursors for the biosynthesis of polyphenols and flavonoids. These compounds are natural secondary metabolites that act as endogenous antioxidants in the defence mechanisms of plants.
By comparing the metabolomics data with the transcriptomic data, we gathered more information about the metabolic pathways. Trigonelline and proline had striking regulatory profiles among the metabolites.
Trigonelline (TG) is an alkaloid that is found in the legume crop Trigonella foenum graecum fabaceae (fenugreek). TG also exists in other edible plants, such as peas, corn, and coffee. Because TG is an osmotic agent, its accumulation decreases the cellular osmotic potential, therefore TG plays an important role in protecting the stability of cell membranes when plants are subjected to stress [39]. Our results indicated that the TG content increased under mild stress and accumulated to a level that represented a 15-fold under severe stress compared to the control.
Proline has been widely considered to be a key drought-inducible metabolite because it plays an osmoprotective role in plants. As an osmotic agent, proline protects cell membranes by decreasing the cellular osmotic potential. Additionally, it has also been recognised as a regulator of the cellular redox status and a ROS scavenger [56–58]. In our study, we found that proline was one of the most significantly changed metabolites under different phases of drought stress. Proline increased from the 6th day and reached its peak on the 14th day of the experiment. Proline is synthesised from glutamate by the key enzyme 1-pyrroline-5-carboxylate synthetase (P5CS). Our study also revealed that A. mongolicus tightly regulated proline accumulation during progressive water stress by up-regulating the genes encoding P5CS. Two of the related genes (Unigene42840_All and CL3765.Contig3_All) exhibited very slight changes under drought stress compared to the control, whereas the expression of the third gene (CL8276.Contig2_All) was increased by four to five-fold under severe drought stress. Moreover, some of the identified genes encoded proline degrading enzymes, such as proline dehydrogenase (PDH). The down-regulation of the PDH gene occurred on the 6th day of the experiment. Interestingly, proline was increased by 53-fold compared to the control under severe drought stress. However, the increase in gene expression was not as high as the proline content. This discrepancy suggests that proline synthesis may not occur first in the roots. Instead, proline may be first synthesised in the above-ground parts and then transported to the roots. Zhang [22] has that the proline content in the roots of the model legume forage Medicago truncatula was several fold higher than in the shoots, although the P5CS gene showed a higher expression level in the shoots under drought stress. Our results were also in agreement with the findings of Sharma’s [56] report.
In addition to these metabolites, soluble sugars often accumulate and perform functions such as signalling and osmotic adjustment [59]. In our study, we found that monosaccharide content (glucose and fructose) was increased and the genes involved in degradative metabolism were down-regulated under drought stress. Sugars can indirectly affect gene expression as an energy source. Additionally, sugars are direct precursors of metabolic reactions, such as osmotic regulators or signalling molecules [60]. The expression of invertase and its genes, which are involved in the conversion of amylose–starch and starch–maltose, were significantly affected by drought stress. These changes may significantly alter the communication of the source–sink.
In this study, metabolite profiling revealed an abundant accumulation of metabolites under drought stress. Moreover, we discovered pathways related to the tolerance and adaptation of A. mongolicus under drought stress conditions. We also elucidated the transcriptomic variation induced by different stages of drought stress. These findings provided an insight into the tolerance mechanisms of A. mongolicus and other legume crops subjected to drought stress.
Conclusions
The findings of this study elucidate the mechanisms by which A. mongolicus responds to different stages of progressive drought stress. The data demonstrated the differential expression of several key genes, such as P5CS, PDH, MDH, zinc finger proteins genes, heat shock protein genes and cytochrome P450s genes, which are more likely to determine tolerance to drought stress, and identified the altered metabolites involved in the important glycolysis, TCA cycle, sucrose and starch metabolism, and glutamate-mediated proline biosynthesis pathways. These data may be used to develop methods to detect genes and metabolites of the host that respond to various stages of drought stress. Our findings help clarify the mechanism by which A. mongolicus and other legume crops tolerate and adapt to drought stress. Furthermore, by using various bioengineering approaches we can improve crop resistance to drought and optimise the yield. The findings of this study have a positive effect on solving the drought problem in arid and semi-arid areas.
Methods
Plant material and experimental design
The study was performed at Inner Mongolia University (39°40′ N, 110°52′ E, elevation 1040 m), Huhhot City, Inner Mongolia Province, Northwest China. Astragalus membranaceus Bge. var. mongholicus (Bge.) Hsiao seeds were collected from Wuchuan City, Inner Mongolia Province, China. The seeds were treated with concentrated sulfuric acid for 8 min and sterilised in 30 % bleach and 0.1 % Tween-20 for 10 min. Thereafter, the seeds were pre-germinated at 4 °C for 3d on a wet filter paper. On 30 March 2013, the germinated seeds were selected and sown in plastic pots (17 cm diameter × 25 cm height) filled with soil collected from the surface layer (0–30 cm) of a vegetable garden field located near the university. The soil pH was maintained at 7.5, and the soil contained 20.7 g/kg of organic matter, 114.7 mg/kg of hydrolysed nitrogen, 22.8 mg/kg of available phosphorus, and 207 mg/kg of available potassium. The plants were regularly watered and kept outside under natural conditions for one year. Each pot contained five seedlings. One month before the drought period, we chose healthy seedlings with an average height of 15.0 ± 0.4 cm. We moved these seedlings into a waterproof plastic shed. We weighed the pots and watered them every two days at 8:00 A.M.to maintain the field capacity conditions.
On 12 June 2014, the pots of seedlings were randomly divided into two groups: one group was watered well (control group) and the second group was not watered to simulate 14d of drought stress. During the experimental period, the average temperature was approximately 12 °C-18 °C at night and 22 °C-30 °C during the day. Light and relative humidity were maintained at constant levels. On days 0, 2, 4, 6, 8, 10, 12, and 14, six pots were randomly selected from each group and the seedlings were harvested at 8:00 A.M.. The harvested plants were kept in an ice box and immediately transported to the laboratory within five minutes. Three whole root samples were quickly cleaned frozen in liquid nitrogen and stored at −80 °C prior to RNA isolation. Furthermore, eight root samples were frozen in liquid nitrogen and stored at −20 °C prior to metabolite analysis. Other samples were used to measure the physiological indices. The RWC of the leaves was calculated using the formula:
Here, FW represents the fresh weight of the leaf samples, SW represents the weight of leaves that were submerged in distilled water for 48 h, and DW represents the weight of leaves dried in an oven at 80 °C for 48 h. Eight root samples were washed with tap water and rinsed twice with distilled water. The samples were gently dried on a paper towel and then in a 60 °C oven for 48 h. Finally, we determined the dry weight of these leaves.
RNA isolation and analysis
The A. mongolicus roots were ground into a fine powder for the total RNA isolation. From these powdered samples, we purified 200 ng of total RNA using oligo-dT beads. The poly (A)-containing mRNA was fragmented into small pieces using a one–cycle cDNA synthesis protocol. The purified and fragmented cDNA was combined with the End Repair Mix. Then, the end-repaired DNA was purified with Ampure XP Beads (AGENCOURT). Next, we added the A-Tailing Mix and mixed the samples well using a pipette. The adenylated 3′ ends of the DNA were combined with the RNA Index Adapter and Ligation Mix, and the reagents were mixed well by pipetting. Then, the end-repaired DNA was purified with Ampure XP Beads (AGENCOURT). We performed several rounds of PCR amplification using the PCR Primer Cocktail and PCR Master Mix to enrich the cDNA fragments. Then, the PCR products were purified using Ampure XP Beads (AGENCOURT).
The average length of the molecule was determined using the Agilent 2100 bioanalyser instrument (Agilent DNA 1000 Reagents). To quantify the library, we performed real-time quantitative PCR (QPCR) (TaqMan Probe). The qualified libraries were subjected to amplification using the cBot system, which generated the cluster on the flowcell (TruSeq PE Cluster Kit V3–cBot–HS, Illumina). The amplified flowcell was used for paired-end sequencing on a HiSeq 2000 System (TruSeq SBS KIT-HS V3, Illumina). Then, we determined that the average read length was 90 bp.
To analyse the correlations between gene functions, we performed a cluster analysis of the gene expression patterns using Cluster 3.0 and the JavaTreeview software. The Venn diagram and expression analysis of different transcripts were performed using R packages, GO term enrichment was calculated by comparing the number of annotations within the list of query transcripts to all of the annotated transcripts on the Poplar Affymetrix Genome Array. We characterised the important pathways associated with the drought transcriptome/metabolome using the Kyoto Encyclopaedia of Genes and Genomes (KEGG).
Metabolomics profiling by 1H-NMR spectrometry
We prepared eight A. mongolicus root samples for the metabolomics investigation. The samples were separately ground into freeze-dried powder. Approximately 50 mg was extracted from each of these samples using 500 μL of water and 500 μL of methanol by vortexing for 1 min. Thereafter, the extracts were subjected to four seconds of ultrasound at three second intervals in one cycle for a total of eight cycles. Finally, we centrifuged the samples for 15 min at 13,000 rpm and 4 °C. The supernatants of all samples (700 μL) were transferred into a centrifuge tube and dried completely under nitrogen gas. Then, the residues were ground into a freeze-dried powder. Thereafter, 450 μL of water was added by vortexing for 30s, and 50 μL of a standard ACDSS was added to the sample by vortexing for 10s. This step was followed by centrifugation of the sample for 2 min at 13,000 rpm and 4 °C. The supernatants of all samples (480 μL) were transferred to standard NMR tubes for the 1H-NMR analysis.
All 1H-NMR spectra were collected at 25 °C on a 600 MHz Bruker spectrometer (AV III) equipped with an inverse cryoprobe. The 1H-NMR spectra were acquired using the first transient of the Bruker “noesygppr1d.comp” sequence. The spectra were collected using 32 transients at a recycle delay of 1 s. All free induction decay (FID) signals were input into the Chenomx NMR Suite Professional software (version 7.7, Chenomx, Edmonton, Canada). Then, the automatically adjusted phase was corrected to the baseline. The DSS-d6 groups were used as internal standard references while determining the chemical shift (set to 0 ppm) in all spectra; the convolution was reversed and adjusted based on the chemical shift index (CSI) and peak shape in the spectrum. According to the related information in the 1H-NMR spectra (chemical shift, peak shape, half band width, and coupling split classification), we used the DSS-d6 concentration and the peak area as standards. Finally, the Chenomx NMR suite software and an internal database (a standard Chenomx metabolite database) were used to perform qualitative and quantitative analyses of the NMR spectra.
Statistical analysis
The multivariate statistical analysis and PCA were performed using the Chenomx NMR Suite 7.7 software. PCA was used to present the variance in the data matrix. Statistical significance was calculated using Fisher’s exact test in R. We adjusted the false recovery rate (FDR) in the transcriptome analysis by applying the Benjamini-Hochberg correction. Both t tests and ANOVA were performed using the SPSS software (version 19, IBM, Armonk, NY, USA). The tests were used to analyse the effects of water stress on the metabolite concentrations. Significant differences were declared at the level of p < 0.05.
Ethics
All researches were carried out in accordance with institutional and local regulations. No special ethical consent or approval was required.
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (grants 81460578), the Key Scientific and Technological Foundation of Inner Mongolia, China (grants 20120414), the Natural Science Foundation of Inner Mongolia, China (grants 2014MS0308).
Abbreviations
- 1H-NMR
1H nuclear magnetic resonance
- DE genes
Differentially expressed (DE) genes
- DW
Dry weight
- FDR
False discovery rate
- FW
Fresh weight
- GO
Gene ontology
- P5CS
1-pyrroline-5-carboxylatesynthetase
- PCA
Principal component analysis
- PEP
Phosphoenolpyruvate
- PPDK
Pyruvate orthophosphatedikinase
- PSII
Photosystem II
- ROS
Reactive oxygen species
- RWC
Relative water content
- SW
Saturated weight
- SWC
Soil water content
- TCA
Citrate cycle
Additional files
Table S1. List of the top 100 highly expressed genes in the early phase of drought stress (day 6). (DOCX 19 kb)
Table S2. List of the top 100 highly expressed genes in the severe stage of drought stress (day 14). (DOCX 18 kb)
Table S3.. The genes that were only up-regulated or only down-regulated during the three stages of drought stress. (DOCX 17 kb)
Figure S1. A typical 600 MHz 1H NMR spectra of methanol extracts obtained from the A. mongolicus roots. (DOCX 256 kb)
Table S4. Raw metabolomics data under different stages of drought stress (n = 8). (CSV 13 kb)
Table S5. The metabolite content in the A. mongolicus roots under different stages of progressive drought stress. (DOCX 21 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The project was conceived by XJ, CSS, YCZ, GYL, GBL, LYR and GLC. GLC and XJ designed the experiment. XJ, CSS and YCZ conducted transcriptional profiling and bioinformatics analyses. XJ performed the metabomic analyses. GYL and GBL performed the physiological work. All authors interpreted biological relevance of results. XJ, LYR and GLC prepared the manuscript. All the authors have studied and approved the final manuscript.
Contributor Information
Xin Jia, Email: jiaxin198761231@163.com.
Chuangshu Sun, Email: 215210919@qq.com.
Yongchun Zuo, Email: yongchunzuo@163.com.
Guangyue Li, Email: 498084982@qq.com.
Guobin Li, Email: 1198516491@qq.com.
Liangyu Ren, Email: ndrly@imu.edu.cn.
Guilin Chen, Email: guilinchen61@163.com.
References
- 1.Lichtenthaler HK. Vegetation stress: an introduction to the stress concept in plants. J Plant Physiol. 1996;148:4–14. doi: 10.1016/S0176-1617(96)80287-2. [DOI] [Google Scholar]
- 2.Wilson RS, Franklin CE. Testing the beneficial acclimation hypothesis. Trends Ecol Evol. 2002;17:66–70. doi: 10.1016/S0169-5347(01)02384-9. [DOI] [Google Scholar]
- 3.Shao HB, Chu LY, Jaleel CA, Manivannan P, Panneerselvam R, Shao MA. Understanding water deficit stress-induced changes in the basic metabolism of higher plants-biotechnologically and sustainably improving agriculture and the ecoenvironment in arid regions of the globe. Crit Rev Biotechnol. 2009;29:131–51. doi: 10.1080/07388550902869792. [DOI] [PubMed] [Google Scholar]
- 4.Boyer JS. Leaf enlargement and metabolic rates in corn, soybean, and sunflower at various leaf water potentials. Plant Physiol. 1970;46:233–5. doi: 10.1104/pp.46.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawlor DW, Cornic G. Photosynthetic carbon assimilation and associated metabolism in relation to water deficits in higher plants. Plant Cell Environ. 2002;25:275–94. doi: 10.1046/j.0016-8025.2001.00814.x. [DOI] [PubMed] [Google Scholar]
- 6.Forster BP, Ellis RP, Moir J, Talame V, Sanguineti MC, Tuberosa R, et al. Genotype and phenotype associations with drought tolerance in barley tested in North Africa. Ann Appl Biol. 2004;144:157–68. doi: 10.1111/j.1744-7348.2004.tb00329.x. [DOI] [Google Scholar]
- 7.Morison JIL, Baker NR, Mullineaux PM, Davies WJ. Improving water use in crop production. Philos Trans R Soc B Biol Sci. 2008;363:639–58. doi: 10.1098/rstb.2007.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckley TN. The control of stomata by water balance. New Phytol. 2005;168:275–92. doi: 10.1111/j.1469-8137.2005.01543.x. [DOI] [PubMed] [Google Scholar]
- 9.Macfarlane C, White DA, Adams MA. The apparent feed-forward response to vapour pressure deficit of stomata in droughted, field-grown Eucalyptus globulus Labill. Plant Cell Environ. 2004;27:1268–80. doi: 10.1111/j.1365-3040.2004.01234.x. [DOI] [Google Scholar]
- 10.Rawat JS, Banerjee SP. The influence of salinity on growth, biomass production and photosynthesis of Eucalyptus camaldulensis Dehnh. and Dalbergia sissoo Roxb. seedlings. Plant Soil. 1998;205:163–9. doi: 10.1023/A:1004381021039. [DOI] [Google Scholar]
- 11.Zhang JX, Nguyen HT, Blum A. Genetic analysis of osmotic adjustment in crop plants. J Exp Bot. 1999;50:291–302. doi: 10.1093/jxb/50.332.291. [DOI] [Google Scholar]
- 12.Talame V, Ozturk NZ, Bohnert HJ, Tuberosa R. Barley transcript profiles under dehydration shock and drought stress treatments: a comparative analysis. J Exp Bot. 2007;58:229–40. doi: 10.1093/jxb/erl163. [DOI] [PubMed] [Google Scholar]
- 13.Oono Y, Seki M, Nanjo T, Narusaka M, Fujita M, Satoh R, et al. Monitoring expression profiles of Arabidopsis gene expression during rehydration process after dehydration using ca. 7000 full-length cDNA microarray. Plant J. 2003;34:868–87. doi: 10.1046/j.1365-313X.2003.01774.x. [DOI] [PubMed] [Google Scholar]
- 14.Boominathan P, Shukla R, Kumar A, Manna D, Negi D, Verma PK, et al. Long term transcript accumulation during the development of dehydration adaptation in Cicer arietinum. Plant Physiol. 2004;135:1608–20. doi: 10.1104/pp.104.043141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tardieu F, Tuberosa R. Dissection and modelling of abiotic stress tolerance in plants. Curr Opin Plant Biol. 2010;13:206–12. doi: 10.1016/j.pbi.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 16.Hayano KC, Calderon VC, Ibarra LE, Herrera EL, Simpson J. Analysis of gene expression and physiological responses in three Mexican maize landraces under drought stress and recovery irrigation. Plos One. 2009;4:7531. doi: 10.1371/journal.pone.0007531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang D, Wu W, Abrams SR, Cutler AJ. The relationship of drought-related gene expression in Arabidopsis thaliana to hormonal and environmental factors. J Exp Bot. 2008;59:2991–3007. doi: 10.1093/jxb/ern155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Juenger TE, Sen S, Bray E, Stahl E, Wayne T, Mckay J, et al. Exploring genetic and expression differences between physiologically extreme ecotypes: comparative genomic hybridization and gene expression studies of Kas-1 and Tsu-1 accessions of Arabidopsis thaliana. Plant Cell Environ. 2010;33:1268–84. doi: 10.1111/j.1365-3040.2010.02146.x. [DOI] [PubMed] [Google Scholar]
- 19.Kreps JA, Wu YJ, Chang HS, Zhu T, Wang X, Harper JF. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002;130:2129–41. doi: 10.1104/pp.008532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shinozaki K, Yamaguchi SK. Gene networks involved in drought stress response and tolerance. J Exp Bot. 2007;58:221–7. doi: 10.1093/jxb/erl164. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell DS, Laramy SE, Teresa JDR, Frederick PB, Blair DS, Tiffany MHM. Transcriptional response of soybean to thiamethoxam seed treatment in the presence and absence of drought stress. BMC Genomics. 2014;15:1055. doi: 10.1186/1471-2164-15-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang JY, Carvalho MHC, Jerez IT, Kang Y, Allen S, Huhman DV, et al. Global reprogramming of transcription and metabolism in Medicago truncatula during progressive drought and after rewatering. Plant Cell Environ. 2014;37:2553–76. doi: 10.1111/pce.12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamanishi ET, Barchet GLH, Dauwe R, Mansfield SD, Campbell MM. Poplar trees reconfigure the transcriptome and metabolome in response to drought in a genotype- and time-of-day-dependent manner. BMC Genomics. 2015;16:329. doi: 10.1186/s12864-015-1535-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyer E, Aspinwall M, Lowy DB, Palacio-Mejía JD, Logan TL, Fay PA, et al. Integrating transcriptional, metabolomic, and physiological responses to drought stress and recovery in switchgrass (Panicum virgatum L.) BMC Genomics. 2014;15:527. doi: 10.1186/1471-2164-15-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charles RW, Ismael A, Cano FJ. Metabolomics demonstrates divergent responses of two Eucalyptus species to water stress. Metabolomics. 2012;8:186–200. doi: 10.1007/s11306-011-0299-y. [DOI] [Google Scholar]
- 26.Sanchez DH, Schwabe F, Erban A, Udvardi MK, Kopka J. Comparative metabolomics of drought acclimation in model and forage legumes. Plant Cell Environ. 2012;35:136–49. doi: 10.1111/j.1365-3040.2011.02423.x. [DOI] [PubMed] [Google Scholar]
- 27.Canam T, Li X, Holowachuk J, Yu M, Xia JG, Mandal R, et al. Differential metabolite profiles and salinity tolerance between two genetically related brown-seeded and yellow-seeded Brassica carinata lines. Plant Sci. 2013;198:17–26. doi: 10.1016/j.plantsci.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 28.Charlton AJ, Donarski JA, Harrison M, Jones SA, Godward J, Oehlschlager S, et al. Responses of the pea (Pisum sativum L.) leaf metabolome to drought stress assessed by nuclear magnetic resonance spectroscopy. Metabolomics. 2008;4:312–27. doi: 10.1007/s11306-008-0128-0. [DOI] [Google Scholar]
- 29.Foito A, Byrne SL, Shepherd T, Stewart D, Barth S. Transcriptional and metabolic profiles of Lolium perenne L. genotypes in response to a PEG-induced water stress. Plant Biotechnol J. 2009;7:719–32. doi: 10.1111/j.1467-7652.2009.00437.x. [DOI] [PubMed] [Google Scholar]
- 30.Sanchez DH, Szymanski J, Erban A, Udvardi MK, Kopka J. Mining for robust transcriptional and metabolic responses to long-term salt stress: a case study on the model legume Lotus japonicus. Plant Cell Environ. 2010;33:468–80. doi: 10.1111/j.1365-3040.2009.02047.x. [DOI] [PubMed] [Google Scholar]
- 31.Pelleschi S, Leonardi A, Rocher JP, et al. Analysis of the relationships between growth, photosynthesis and carbohydrate metabolism using quantitative trait loci (QTLs) in young maize plants subjected to water deprivation. Mol Breeding. 2006;17:21–39. doi: 10.1007/s11032-005-1031-2. [DOI] [Google Scholar]
- 32.Hura T, Grzesiak S, Hura K, Thiemt E, Tokarz K, Wedzony M. Physiological and biochemical tools useful in drought-tolerance detection in genotypes of winter triticale: accumulation of ferulic acid correlates with drought tolerance. Ann Bot-London. 2007;100:767–75. doi: 10.1093/aob/mcm162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan JM. Osmoregulation and water-stress in higher plants. Annu Rev Plant Physiol Plant Mol Biol. 1984;35:299–319. doi: 10.1146/annurev.pp.35.060184.001503. [DOI] [Google Scholar]
- 34.Sanguinetti MC, Tuberosa R, Landi P, Salvi M, Maccaferri E, Casarini E, et al. QTL analysis of drought-related traits and grain yield in relation to genetic variation for leaf abscisic acid concentration in field-grown maize. J Exp Bot. 1999;50:1289–97. doi: 10.1093/jxb/50.337.1289. [DOI] [Google Scholar]
- 35.Chaves MM, Maroco J, Pereira JS. Understanding plant responses to drought—from genes to the whole plant. Funct Plant Biol. 2003;30:239–64. doi: 10.1071/FP02076. [DOI] [PubMed] [Google Scholar]
- 36.Verslues PE, Kim YS, Zhu JK. Altered ABA, proline and hydrogen peroxide in an Arabidopsis glutamate: glyoxylate amino transferase mutant. Plant Mol Biol. 2007;64:205–17. doi: 10.1007/s11103-007-9145-z. [DOI] [PubMed] [Google Scholar]
- 37.Shen B, Jensen RG, Bohnert HJ. Increased resistance to oxidative stress in transgenic plants by targeting mannitol biosynthesis to chloroplasts. Plant Physiol. 1997;113:1177–83. doi: 10.1104/pp.113.4.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho Y, Njiti VN, Chen X, Lightfoot DA, Wood AJ. Trigonelline concentration in field-grown soybean in response to irrigation. Biol Plantarum. 2003;46:405–10. doi: 10.1023/A:1024390522259. [DOI] [Google Scholar]
- 39.Tramontano WA, Jouve D. Trigolline accumulation in salt-stress legumes and the roll of other osmergulators as cell cycle control agents. Phytochemistry. 1997;44:1037–40. doi: 10.1016/S0031-9422(96)00715-7. [DOI] [Google Scholar]
- 40.Barnett NM, Naylor AW. Amino acid and protein synthesis in Bermuda grass during water stress. Plant Physiol. 1966;41:1222–1230. doi: 10.1104/pp.41.7.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsiao TC. Plant responses to water stress. Annu Rev Plant Physiol. 1973;24:519–70. doi: 10.1146/annurev.pp.24.060173.002511. [DOI] [Google Scholar]
- 42.Stotz HU, Thomson JG, Wang Y. Plant defensins: defense, development and application. Plant Signal Behav. 2009;4:1010–2. doi: 10.4161/psb.4.11.9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernie ARF, Carrari F, Sweetlove L. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol. 2004;7:254–61. doi: 10.1016/j.pbi.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 44.Evans RD, Black RA, Loescher WH, Fellows RJ. Osmotic relations of the drought tolerant shrub Artemisia tridentate in response to water stress. Plant Cell Environ. 1992;15:49–59. doi: 10.1111/j.1365-3040.1992.tb01457.x. [DOI] [Google Scholar]
- 45.Lee M, Choi Y, Burla B, Kim YY, Jeon B, Maeshima M, et al. The ABC transporter AtABCB14 is a malate importer and modulates stomatal response to CO2. Nat Cell Biol. 2008;10:1217–23. doi: 10.1038/ncb1782. [DOI] [PubMed] [Google Scholar]
- 46.Reddy AR, Chaitanya V, Vivekanandan M. Drought-induced responses of photosynthesis and antioxidant metabolism in higher plants. J Plant Physiol. 2004;161:1189–1202. doi: 10.1016/j.jplph.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 47.Hubbard KE, Nishimura N, Kenichi H, Getzoff ED, Schroeder JI. Early abscisic acid signal transduction mechanisms: newly discovered components and newly emerging questions. Gene Dev. 2010;24:1695–1708. doi: 10.1101/gad.1953910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawlor DW, Tezara W. Causes of decreased photosynthetic rate and metabolic capacity in water-deficient leaf cells: a critical evaluation of mechanisms and integration of processes. Ann Bot-London. 2009;103:561–79. doi: 10.1093/aob/mcn244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bogeat-Triboulot MB, Brosché M, Renaut J, Laurent J, Thiec DL, Fayyaz P, et al. Gradual soil water depletion results in reversible changes of gene expression, protein profiles, ecophysiology, and growth performance in Populus euphratica, a poplar growing in arid regions. Plant Physiol. 2007;143:876–92. doi: 10.1104/pp.106.088708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swarbreck SM, Lindquist EA, Ackerly DD, Andersen GL. Analysis of leaf and root transcriptomes of soil-grown Avena barbata plants. Plant Cell Physiol. 2011;52:317–32. doi: 10.1093/pcp/pcq188. [DOI] [PubMed] [Google Scholar]
- 51.Chen LM, Zhou XA, Li WB, Chang W, Zhou R, Wang C, et al. Genome-wide transcriptional analysis of two soybean genotypes under dehydration and rehydration conditions. BMC Genomics. 2013;14:687. doi: 10.1186/1471-2164-14-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sugano S, Kaminaka H, Rybka Z, Catala R, Salinas J, Matsui K, et al. Stress-responsive zinc finger gene ZPT2-3 plays a role in drought tolerance in petunia. Plant J. 2003;36:830–41. doi: 10.1046/j.1365-313X.2003.01924.x. [DOI] [PubMed] [Google Scholar]
- 53.Narusaka Y, Narusaka M, Seki M, Umezawa T, Ishida J, Nakajima M, et al. Crosstalk in the responses to abiotic and biotic stresses in Arabidopsis: analysis of gene expression in cytochrome P450 gene superfamily by cDNA microarray. Plant Mol Biol. 2004;55:327–42. doi: 10.1007/s11103-004-0685-1. [DOI] [PubMed] [Google Scholar]
- 54.Cramer GR, Ergül A, Grimplet J, Tillett RL, Tattersall EAR, Bohlman MC, et al. Water and salinity stress in grapevines: early and late changes in transcript and metabolite profiles. Funct Intrgr Genomic. 2007;7:111–34. doi: 10.1007/s10142-006-0039-y. [DOI] [PubMed] [Google Scholar]
- 55.Fiehn O. Metabolomics-the link between genotypes and phenotypes. Plant Mol Biol. 2002;48:155–71. doi: 10.1023/A:1013713905833. [DOI] [PubMed] [Google Scholar]
- 56.Sharma S, Villamor JG, Verslues PE. Essential role of tissue-specific proline synthesis and catabolism in growth and redox balance at low water potential. Plant Physiol. 2011;157:292–304. doi: 10.1104/pp.111.183210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smirnoff N, Cumbes QJ. Hydroxyl radical scavenging activity of compatible solutes. Phytochemistry. 1989;28:1057–60. doi: 10.1016/0031-9422(89)80182-7. [DOI] [Google Scholar]
- 58.Szabados L, Savouré A. Proline:a multifunctional amino acid. Trends Plant Sci. 2010;15:89–97. doi: 10.1016/j.tplants.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 59.Chaves MM, Flexas J, Pinheiro C. Photosynthesis under drought and salt stress: regulation mechanisms from whole plant to cell. Ann Bot-London. 2009;103:551–60. doi: 10.1093/aob/mcn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jang JC, Sheen J. Sugar sensing in higher plants. Plant Cell. 1994;6:1665–79. doi: 10.1105/tpc.6.11.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]