

Abstract
Background
Soil-borne fungi of the Fusarium oxysporum species complex cause devastating wilt disease on many crops including legumes that supply human dietary protein needs across many parts of the globe. We present and compare draft genome assemblies for three legume-infecting formae speciales (ff. spp.): F. oxysporum f. sp. ciceris (Foc-38-1) and f. sp. pisi (Fop-37622), significant pathogens of chickpea and pea respectively, the world’s second and third most important grain legumes, and lastly f. sp. medicaginis (Fom-5190a) for which we developed a model legume pathosystem utilising Medicago truncatula.
Results
Focusing on the identification of pathogenicity gene content, we leveraged the reference genomes of Fusarium pathogens F. oxysporum f. sp. lycopersici (tomato-infecting) and F. solani (pea-infecting) and their well-characterised core and dispensable chromosomes to predict genomic organisation in the newly sequenced legume-infecting isolates. Dispensable chromosomes are not essential for growth and in Fusarium species are known to be enriched in host-specificity and pathogenicity-associated genes. Comparative genomics of the publicly available Fusarium species revealed differential patterns of sequence conservation across F. oxysporum formae speciales, with legume-pathogenic formae speciales not exhibiting greater sequence conservation between them relative to non-legume-infecting formae speciales, possibly indicating the lack of a common ancestral source for legume pathogenicity. Combining predicted dispensable gene content with in planta expression in the model legume-infecting isolate, we identified small conserved regions and candidate effectors, four of which shared greatest similarity to proteins from another legume-infecting ff. spp.
Conclusions
We demonstrate that distinction of core and potential dispensable genomic regions of novel F. oxysporum genomes is an effective tool to facilitate effector discovery and the identification of gene content possibly linked to host specificity. While the legume-infecting isolates didn’t share large genomic regions of pathogenicity-related content, smaller regions and candidate effector proteins were highly conserved, suggesting that they may play specific roles in inducing disease on legume hosts.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2486-8) contains supplementary material, which is available to authorized users.
Keywords: Fusarium, Conditionally dispensable chromosomes (CDC), Effectors, Pathogenicity, Legume, Pulse, Fungal pathogen
Background
Fusarium wilt and root rot caused by members of the Fusarium oxysporum species complex (FOSC) are major constraints to the production of horticultural, cotton, and legume crops worldwide. F. oxysporum is a globally ubiquitous soil-borne fungus [1, 2] and is one of the most important plant-pathogens of the Fusarium genus, having been ranked 5th in a list of the top 10 plant pathogens of scientific/economic importance [3]. While some F. oxysporum isolates are non-pathogenic saprophytes and may even have symbiotic or bio-control properties [4] this species notably contains more than 150 host-specific plant-pathogenic sub-species [5], known as formae speciales (ff. spp. singular forma specialis, abrv. f. sp.). Each of which cause disease on a narrow range of host plant species and which may be further divided into races or pathotypes and additionally vegetative compatibility groups [6].
Many fungi have evolved the ability to attack living plants rather than obtain nutrients saprophytically and the invasion is often facilitated by effector molecules that interact with the host plant’s immune system (reviewed in [7, 8]). In some fungal genera, including Fusarium, genes encoding the production of these molecules have evolved on chromosomal regions that are not required for saprophytic growth and are thus known as ‘conditionally dispensable chromosomes’ (CDCs, also known as supernumerary, accessory, lineage-specific, B-chromosomes or mini-chromosomes) in contrast to ‘core’ chromosomes whose gene content is essential and conserved across generations [9, 16]. Dispensable genomic regions encoding genes that play a role in pathogenicity and host-specificity, including effector genes, have been identified in Fusarium isolates infecting a range of plant hosts [9–13]. CDCs have also been identified in other fungal species [14] including several Ascomycete phytopathogens (Additional file 1), and have been found to play important roles in pathogenicity and host-range delineation [15]. The first Fusarium CDC identified was from F. solani (syn. Nectria haematococca) and contained a cluster of six pea-pathogenicity (PEP) genes involved in detoxifying the plant defence compound pisatin produced by the garden pea, Pisum sativum [11–13]. For some Fusarium species including the tomato-infecting F. oxysporum f. sp lycopersici (Fol), the genes residing on CDCs define its host range and these chromosomes when transferred to non-pathogenic species can confer pathogenicity on a new host. Dispensable regions of the genome are presumed to be maintained while they convey an evolutionary advantage, either to allow for adaptation of novel genes separately from regions containing core conserved genes, or to allow for the transfer of genetic material e.g. conferring pathogenicity on a new host. The clustering of genes important for pathogenicity on a small CDC chromosome (e.g. as has been demonstrated for CDC 14 of Fol) that could be transferred would provide a highly selective advantage for a ‘one step’ horizontal transfer event that could enable an isolate to become pathogenic on a new host [9, 16]. Presumably genes that do not confer pathogenicity on the new host would be more susceptible to shuffling and subsequent loss, as has been observed for Fol and F. solani CDCs relative to core chromosomes [11].
Genomic mapping and sequencing of Fusarium species has revealed chromosome numbers to be highly plastic, ranging from 4–17 [9, 11, 17]. The common ancestor species has been proposed to have only 11 chromosomes, with the increase in chromosome number due in part to the presence of CDCs which are thought to have originated in F. oxysporum via horizontal transmission from other Fusarium species [9]. Gene content in CDCs is often relatively sparse but enriched in transposable elements. For example, less than 1/8 of Fol pathogenicity CDC 14 is predicted to encode protein coding genes and these are predominately proteins of unknown function. In comparison to core chromosomes, CDCs are enriched for pathogenicity-associated proteins, secreted proteins and proteins involved in secondary metabolism [18]. Some Fol CDC genes important for pathogenicity encode the SECRETED-IN-XYLEM (SIX) effector proteins [9], and are often associated with distinct repeat types [10, 18]. SIX proteins, first identified in the xylem sap of Fol-infected tomato plants, are small, secreted and often cysteine-rich [18, 19]. So far 14 families of SIX proteins have been identified, sharing little similarity with each other or other known fungal proteins (except in Colletotrichum sp.- also a member of the class Sordariomycetes). Several have characterised roles in virulence and/or avirulence with their cognate host R-genes identified [9, 18–24], although for the most part their biological function within the host remain unknown.
Members of the Fusarium genus are major constraints to global grain and forage legume production. Fusarium wilts and root rots caused by species such as F. oxysporum, F. solani, F. udum, and F. virguliforme are a major problem for a number of important legume crops including chickpea, pea, soybean, lentil, lupin, alfalfa, common bean and pigeon pea causing losses upwards of 10 % annually, but in many cases complete loss [25, 26]. These crops provide a high protein food source to a large proportion of the world’s population as well as serving as a source of livestock feed [27]. In addition, they improve the soil through nitrogen fixation and are often used in rotational cropping systems to provide disease breaks.
In this study we generate, inspect and compare the assembled genome sequences and functional annotation of three legume-infecting formae speciales of the FOSC, adding to the increasing list of available F. oxysporum ff. spp. genome assemblies, with none that infect legumes, the third-largest family of higher plants, previously published. These isolates, F. oxysporum f. sp. medicaginis (Fom, Fom-5190a), F. oxysporum f. sp. ciceris (Foc, Foc-38-1) and F. oxysporum f. sp. pisi (Fop, Fop-37622) are causal agents of Fusarium wilt on Medicago species (including Medicago sativa (alfalfa/lucerne) and the model legume Medicago truncatula), chickpea (Cicer arietinum) and pea (Pisum sativum) respectively. The legume-infecting ff. spp. discussed herein exhibit a similar infection cycle to Fol, favoured by warm soil temperatures [28–32]. The hyphae of germinated conidia colonize and penetrate the root epidermis, move inter-cellularly through the root cortex and into the xylem. As shown for the model legume M. truncatula in Additional file 2a and for C. arietinum in 2b, extensive colonisation of the vascular system leads to water stress, wilting and bleaching of stems and leaves, followed by necrosis and eventually plant death.
Chickpea and pea are the second and third most important legume crops worldwide, with chickpea the most important in India, due to its high protein content (FAO: www.fao.org). Foc is a major pathogen of chickpea typically accounting for 10-15 % of yield losses worldwide [33, 34] and can be transmitted via seed [35] but can also survive in the soil for long periods. Foc has two known pathotypes, that cause either yellowing or wilt [36], and eight pathogenic races (Races 0, 1, 1B/C, 2, 3, 4, 5 and 6) although it is proposed to be one of the few F. oxysporum ff. spp. that is monophyletic [37]. The isolate sequenced in this study Foc-38-1, represents the most virulent race of this forma specialis (race 1) which shows wide geographic distribution throughout India, the largest producer of chickpeas [38, 39] and is capable of causing complete loss of grain yield [30, 36, 40, 41]. In Fop four races 1, 2, 5 and 6 have been described [42] and the isolate sequenced in this study belongs to race 5. Medicago spp. are pasture crops typically used for rotation and fodder [43], although alfalfa (M. sativa) is also grown for human consumption, and M. truncatula is a notable model plant species [44]. The corresponding pathogenic f. sp. (Fom) of M. truncatula is therefore of relevance as a model for the study of plant-pathogen interactions in legumes while also having bearing on alfalfa/lucerne, the world’s major temperate forage crop [45]. The race of the isolate sequenced in this study Fom-5190a is not known, as in this f. sp. races are yet to be defined.
In this work, we focus our analysis on identifying regions of the new legume-infecting F. oxysporum genomes that may be relevant to plant pathogenicity in part by predicting potential CD regions and coupling this knowledge with in planta expression during F. oxysporum- infection of the model legume M. truncatula. This process led to the identification of several effector candidates and conserved pathogenicity factors across the legume infecting ff. spp., that we speculate play a role in inducing disease on legume hosts.
Results and discussion
Genome features and organisation
The Fom-5190a and Foc-38-1 genomes were assembled using a combination of paired-end, mate-pair and long-jumping distance Illumina libraries with additional 454 sequencing used for Fom-5190a as described in the Methods and Additional file 3. The Fom-5190a genome was sequenced at ~170x coverage (trimmed Illumina and 454 data) and assembled into 4034 scaffolds with a total length of ~51.1 Mb and encoding 16,858 proteins. The Foc-38-1 genome was sequenced at ~577x coverage (trimmed Illumina data) and assembled into 1482 scaffolds, totalling ~54.8 Mb encoding for 16,124 proteins. The whole-genome sequence of Fop NRRL strain 37622 had a final 260× physical coverage generated from two libraries using Illumina sequencing technology. The final assembly encompassed 472 scaffolds with a total length of 55.1 Mb encoding 19,623 genes. Approximately 98 % of highly conserved protein-coding genes were estimated by CEGMA [46] to be represented in all three assemblies, highlighting their comprehensiveness in core regions, and as compared to other Fusarium reference assemblies which had similarly high percentages (Table 1). The majority of proteins from all three genomes were functionally annotated based on comparisons to publicly available databases Pfam, InterPro and KEGG (summarised in Additional file 4).
Table 1.
Genome assembly characteristics and comparisons across Fusarium species
| F. oxysporum f. sp. medicaginis | F. oxysporum f. sp. ciceris | F. oxysporum f. sp. pisi | F. oxysporum f. sp .lycopersici [9]b | F. oxysporum brassica [57] | F. solani (syn. Nectria haematococca) [11]b | F. verticilliodes (syn. Gibberella fujikuroi) [9] | F. graminearum (syn. Gibberell zeae) [17] | |
|---|---|---|---|---|---|---|---|---|
| Genome Abbreviation | Fom | Foc | Fop | Fol | Fob | Fs | Fv | Fg |
| Isolate | 5190a | 38-1 | NRRL37622 | 4287 | 5176 | MPVI 77-13-4 | 7600 | PH-1 |
| Chromosome number | Unknown | Unknown | 16 | 15 | Unknown | 17 | 11 | 4 |
| Host | Medicago spp. | Chickpea | Pisum | Tomato | Brassica spp. including Arabidopsis thaliana | Pea | Maize and cereals | Wheat and barley |
| Total assembly length (bp) | 51,139,932 | 54,813,009 | 55,188,000 | 57,720,560 | 54,767,602 | 47,328,059 | 41,104,290 | 36,346,967 |
| Average length (bp) | 12,674 | 36,961 | 116,923 | 3,607,535 | 6969 | 2,629,337 | 3,425,358 | 7,269,393 |
| Maximum length (bp) | 4,225,797 | 2,918,844 | 5,895,957 | 6,854,980 | 415,898 | 6,369,736 | 6,219,215 | 11,694,295 |
| Minimum length (bp) | 200 | 1000 | 2000 | 1,646,460 | 200 | 215,166 | 2,040,847 | 7,711,129 |
| L50 (bp) | 1,632,076 | 182,371 | 2,840,000 | 4,589,937 | 60,340 | 3,621,839 | 4,236,349 | 8,911,601 |
| N50 | 10 | 58 | 54 | 6 | 222 | 6 | 5 | 2 |
| Total sequences | 4034 | 1482 | 472 | 15 | 7858 | 17 | 11 | 4 |
| Total unknown bases (N) | 1,929,171 | 2,262,803 | 12,221 | 1,238,430 | 33 | 1915 | 87,573 | 212,843 |
| GC%c | 48.2 | 48.1 | 47.6 | 48.5 | 47.8 | 51.5 | 48.7 | 48.4 |
| Percentage of de novo predicted repetitive sequencesc | 1.5 | 5.4 | 8.2 | 21.9 | 9.4 | 3.1 | 1.3 | 1.5 |
| Percentage of predicted repetitive sequences based on comparison to known fungal repeats in Repbase | 1.0 | 2.4 | 2.8 | 4.4 | 2.4 | 1.4 | 0.8 | 0.9 |
| CEGMA Analysisa | ||||||||
| CEGMA partial % | 98 | 98 | 98 | 97 | 98 | 98 | 96 | 97 |
| CEGMA partial | 243 | 244 | 242 | 241 | 243 | 243 | 239 | 241 |
| CEGMA complete | 239 | 240 | 237 | 237 | 238 | 238 | 235 | 234 |
aNumber of complete Conserved Eukaryotic Genes found in the assembly of the core set of 248 defined by Parra et al. [46]
bReconstructed chromosome mapped regions only
cGC content and de novo repeat percentage based on RepeatMasker analysis of scaffolds versus de novo repeats
The most in-depth studied genome of an F. oxysporum ff. spp. to date belongs to Fol, which was assembled into near-complete chromosome sequences via an optical map [9]. We therefore used Fol as our primary point of reference for subsequent comparative genomics. In some analyses we have also made additional comparisons to the similarly high-quality chromosome assembly of F. solani [11] which is a more distantly related species but shares some legume hosts with the novel isolates presented here. The main protein features of Fom-5190a, Fop-37622 and Foc-38-1 are compared in Table 2 with those of other Fusarium species which shows that Fom-5190a and Foc-38-1 had similar gene numbers despite their differing assembly sizes (Fom-5190a 51.1 Mb versus Foc-38-1 54.8 Mb), which appear to be influenced mostly by repetitive DNA content (Table 1). The number of small secreted proteins (SSPs), indicative of putative roles as effectors, across the legume-infecting ff. spp. was comparable to those predicted in the two other F. oxysporum ff. spp. genomes analysed (Fol and Fob-5176), using the criteria of protein length ≤ 300 amino acid, predicted to be secreted and containing ≤ one transmembrane domain in the N-terminal region.
Table 2.
Protein set comparisons across Fusarium species
| Foc | Fom | Fop | Fob | Fol | Fsol | Fg | Fv | |
|---|---|---|---|---|---|---|---|---|
| Total number of proteins | 16,124 | 16,858 | 19,623 | 17,817 | 17,701 | 15,707 | 13,321 | 20,553 |
| SSPsa | 537 | 580 | 620 | 588 | 597 | 426 | 483 | 621 |
| Number of species specific proteins (including paralogs) | 1090 | 863 | 832 | 1606 | 1825 | 2083 | 1632 | 1852 |
| Number of unique species specific proteinsb | 1058 | 808 | 785 | 1540 | 1645 | 1919 | 1622 | 1687 |
aSmall secreted proteins
bNo orthologs or paralogs in any of the other 43 spp. tested including all publicly available Fusarium spp.
Isolate details are as detailed in Additional file 5
Foc F. oxysporum f. sp. ciceris, Fom F. oxysporum f. sp. medicaginis, Fop F. oxysporum f. sp. pisi, Fob F. oxysporum 5176, Fol F. oxysporum f. sp. lycopersici, Fsol F. solani, Fg F. graminearum, Fv F. verticilliodes
To expand our analysis and aid identification of proteins common to the legume-infecting ff. spp., we next conducted protein orthology comparisons across 44 diverse fungal species (Additional file 5) both closely and distantly related but sharing similar hosts (Additional file 6). This analysis identified 1090 paralog groups unique to Foc-38-1, 823 in Fop-37622 and 863 unique to Fom-5190a (containing 1–5 paralogs per gene) (Table 2). Those genes that do not have orthologs in another ff. spp. are presumed likely to encode proteins that play a role in host specificity, and were used in subsequent analyses predicting effectors. However, host specificity may also be governed by small differences in orthologous proteins that may affect their interaction with host protein. There were 10,602 ortholog groups shared by the legume-infecting ff. spp. (Fom, Foc and Fop) of which 8118 were also shared with the legume pathogen F. solani. A similar number of F. solani sequences (over 9000) was observed to be conserved within three distinct species of the genus Fusarium (Fol, F. graminearum and F. verticilliodes [9]) suggesting that this corresponds roughly to the number of core genes conserved amongst Fusarium species.
We next examined G:C content in the new genomes as G:C variation in other fungal phytopathogens has highlighted key pathogenicity regions. For example, Leptosphaeria maculans (blackleg disease, stem canker of Brassicas) has AT-rich isochores throughout its core genome [47], while Z. tritici (septoria leaf blotch of wheat) possesses AT-rich CDCs [48]. However we found that G:C content across the legume-infecting ff. spp. scaffolds, as well across chromosomes of the Fol reference, were relatively constant at ~46–48 % with, in general, no large regions of atypical G:C % (isochores) observed, even within Fol CDCs. There were however small local variations on CDCs around transposons and other repeated sequences, resulting in a marginally lower chromosome average G:C% for core versus dispensable scaffolds of Fol and F. solani which was also observed for predicted dispensable versus core scaffolds from the legume-infecting ff. spp. (Additional file 7).
Next a comparison of repetitive DNA content across Fusarium spp. was conducted as it is an important feature in many fungal genomes, driving evolution through repeat induced point mutation, gene duplication or altering gene expression through insertion into or near other genes [49–51]. Prediction of repetitive DNA in assemblies that primarily use short-read next-generation sequencing data, such as the recently assembled Fusarium spp. genomes, is generally underestimated compared to their Sanger-based counterparts (e.g. Fol-4287) due to the presence of repeats in unassembled sequences. Consequently the de novo predicted repeat content of the novel Foc-38-1, Fop-37622 and Fom-5190a genomes was considerably lower than that of Fol (Table 1) although, the repeat content of other F. oxysporum ff. spp. sequenced and assembled using similar methods and analysed via this method were found to be within similar ranges (3.9–9.4 %, Additional file 8a).
In Fol CDCs, DNA transposons were over-represented and Ma et al. (2010) speculate that their expansion in Fol may correlate with the formation of the Fol lineage-specific regions as well as segmental duplications of some regions of the genome [9]. Additionally, the predicted effector genes of Fol, and the F. solani and F. oxysporum f. sp. betae pea pathogenicity (PEP) clusters have been observed to reside within genomic subregions enriched with DNA transposons [9, 18, 52, 53]. To aid localisation of pathogenicity-associated gene content, we therefore scanned the legume-infecting F. oxysporum ff. spp. genomes for transposable elements (TEs). The Foc-38-1 and Fop-37622 assemblies contained a larger number of DNA transposons and retrotransposons than Fom-5190a (Additional file 8b), with the majority of predicted TEs in all three belonging to the Tc1-mariner superfamily of DNA transposons which includes the impala family [54]. Remnants of impala and Fot5 transposons have been observed to occur in the vicinity of several Fol SIX effector genes [18, 22] and have been used to predict new effector-like genes in Fol and F. oxysporum f. sp. melonis [10, 18].
Known conditionally dispensable chromosomes in Fusarium spp. exhibit varying sequence conservation across legume-infecting isolates and other Fusarium spp.
CDCs of Fol are thought in part to define host range, are enriched for effectors and can be transferred to non-pathogenic species to confer pathogenicity [1]. We therefore aimed to isolate lineage specific gene content in the legume-infecting isolates by identifying their potential CDC sequences. To do this we aligned their scaffold sequences to the non-repetitive regions of chromosomes of Fol and F. solani in which CDCs are well defined [9, 11], as well as those of other publicly available Fusarium spp. to compare the levels of conservation. The chromosome sequences of Fol [9] and F. solani [11] were masked based on the presence of de novo-predicted repetitive DNA sequences and then translated and aligned to the repeat-masked genomes of other Fusarium spp. using MUMmer [55]. A distinct pattern of variation in levels of sequence conservation between core and dispensable chromosomes was observed across F. oxysporum ff. spp. and other Fusarium spp. (Fig. 1). Similar patterns were also observed for F. solani chromosomes (Additional file 9). The percentage length of each Fol and F. solani chromosome (excluding masked repetitive sequences) that was covered by one or more matches is summarised in Additional file 10a and b.
Fig. 1.
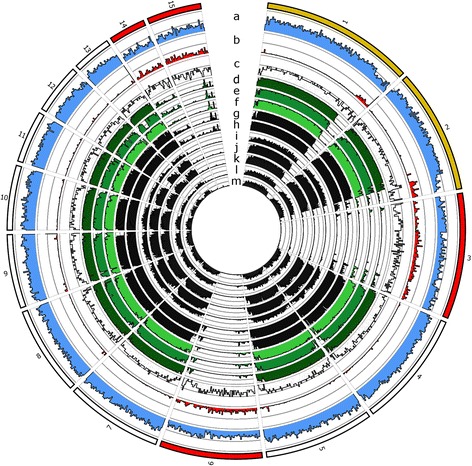
F. oxysporum f. sp. lycopersici chromosomes highlighting sequence conservation and other features of CDCs in comparison to core chromosomes. The circos plot illustrates the gene-sparse, repeat-rich nature of Fol CDCs and their lower sequence conservation across related species in comparison to core chromosomes. Outer ring-Fol chromosomes highlighting CDCs (red) and chromosomes that are partially dispensable (yellow). Inner rings: (a) gene density in 100 kb windows, (b) repeat density in 100 kb windows, (c) GC content in 50 k bp windows, range 45–55 %, (d) Region of Fol chromosomes overlapped by Fom-5190a sequences, (e) Foc-38-1, (f) Fop-37622, (g) F. oxysporum f. sp. brassica Fo5176, (h) F. oxysporum f. sp. melonis, (i) F. solani, (j) F. fujikuori, (k) F. verticilliodes, (l) F. virguliforme, and (m) F. graminearum
Presence-absence variation relative to chromosomes of Fol indicated a distinctive pattern of widespread absence of sequences homeologous to Fol CDCs 3, 6, 14 and 15 across Fusarium spp. as previously described [9]. These Fol CDCs are distinct from core chromosomes in having markedly higher repetitive DNA content (as determined by the total length of sequences masked as de novo-predicted repeats). For the legume-infecting isolates an average of ~93 % sequence conservation to the masked Fol core scaffolds was observed but only ~25 % for the CDCs (Additional file 10a). Partial conservation of the non-repetitive sequences of Fol pathogenicity CDC 14 (42–51 %) was observed in Foc-38-1 and Fom-5190a, as well as the Arabidopsis- and melon-infecting isolates Fo5176 and Fom-26406 respectively, however similar levels of conservation were not observed in Fop-37622 or across other publicly-available F. oxysporum ff. spp. genomes (BROAD MIT, Additional file 10a). This observation is interesting as most CDCs were initially thought to lack homology or synteny to related species [9, 11, 56], although consistent with the finding that Fo5176 shared an average of 34.5 % of the total sequence of the Fol CDCs (described in supplementary data presented in [9]) and that SIX genes originally thought to be unique to Fol have been identified in several other ff. spp. [57–62]. The fact that Foc-38-1 and Fom-5190a show greater sequence conservation of Fol CDC 14 (51 % and 42 % respectively) than Fop-37622 which shares only 20 % non-repetitive sequence indicates that legume infecting ff. spp. may not derive their pathogenicity from common sequences conserved with Fol. The length of conserved sequence with Fol pathogenicity CDC 14 was also low in several other F. oxysporum ff. spp. including unsurprisingly the non-pathogenic biocontrol species (Fo47, 8 %) as well as pathogens of other plant species (less than 20 % for pathogens of tomato and banana, ff. spp. radicis lycopersici and cubense). Although conservation as high as 88 % was observed in another tomato-infecting isolate and 45–50 % in F. oxysporum f. sp. conglutinans and F. oxysporum f. sp. raphani. Similar patterns of variation in conservation across ff. spp. were also observed for other Fol CDCs in isolates included in this study (Additional file 10).
We postulated that pathogenicity on legumes may be due to conserved CD sequences within the legume-infecting ff. spp. and possibly shared with the legume pathogen F. solani. A presence-absence variation analysis relative to F. solani chromosomes found distinct absences across Fusarium spp. for known CDCs 14, 15 and 17 - all of which have distinctively high repetitive content relative to core chromosomes (Additional files 9 and 10b). Genes important for F. solani pathogenicity on legumes are known to be encoded on CDC 14 which is proposed to have been acquired via horizontal transfer [11]. This includes the PEP cluster identified in F. solani mating population IV [63]. Several genes from this cluster are thought to have been transferred to F. oxysporum f. sp. pisi [64], with orthologs of four genes demonstrated to contribute to virulence on pea identified in this f. sp. (PDA1, PEP1, PEP2 and PEP5). Yet the non-repetitive sequence of F. solani CDC 14 shared only 17 % sequence conservation with Fop-37622, 16 % with Fom-5190a, and 21 % with Foc-38-1, with similar values also observed in the non-legume pathogens Fo5176 (brassicas) and Fom-26406 (melon). Therefore F. solani appears not to be the common origin for legume-infecting ff. spp. pathogenicity content, although we did identify orthologs of some PEP genes with known roles in virulence as discussed in later sections.
Phylogenetic and orthology analysis indicates independent origins of legume host- specificity
One possible explanation for the evolution of legume-host specificity in F. oxysporum isolates is a common ancestor shared by only legume-infecting ff. spp., however this was not supported by a phylogenetic analysis of 100 randomly selected orthologous genes across Fusarium spp. (Additional file 11). Thus legume-pathogenicity appears to have arisen more than once within this species. Another possibility is that the legume-infecting ff. spp. may share a set of similar proteins governing legume-host pathogenicity, arrived at via either lateral gene transfer or convergent evolution. However, based on the orthology analysis, only one protein was common to just the four legume-infecting Fusarium species and the encoding gene was not detected as expressed under the in planta conditions examined in the following sections (Additional file 12).
Comparative analysis of predicted core and dispensable sequences in legume-infecting isolates and their gene content with other F. oxysporum ff. spp.
Drawing upon sequence comparisons to core chromosomes and experimentally demonstrated CDCs of Fol and F. solani [9, 11], scaffolds from Fom-5190a, Fop-37622 and Foc-38-1 were predicted as either “core” or putatively “dispensable” (Additional file 13a, b and c). Scaffolds with unique matches across more than 30 % of their length to core chromosomes in Fol or F. solani were designated “core” scaffolds, whilst those with no match, or that matched to a CDC in either genome, were designated as potentially “dispensable”. In general we observed that the newly predicted conditionally dispensable sequences of the legume-infecting ff. spp. shared known characteristics of Fusarium CDCs. They had increased repetitive content, reduced gene density, smaller average size of predicted proteins relative to those encoded on core scaffolds and a slightly lower average G:C% (Table 3). The predicted dispensable scaffolds were also more numerous and shorter in average length than those scaffolds predicted to form part of core chromosomes (Additional file 14), presumably due the influence of repetitive sequence on the assembly of those genomic regions.
Table 3.
Properties of scaffolds predicted to form part of either core or dispensable chromosomes in legume-infecting formae speciales
| Fom-5190a core | Fom-5190a dispensable | Foc-38-1 core | Foc-38-1 dispensable | Fop-37622 core | Fop-37622 dispensable | |
|---|---|---|---|---|---|---|
| Number of scaffold sequences | 446 | 3529 | 413 | 1069 | 116 | 356 |
| Total length (bp) | 42,167,838 | 8,766,042 | 41,518,444 | 13,294,565 | 43,587,736 | 11,600,480 |
| Average length (bp) | 94,547 | 2484 | 100,529 | 12,436 | 375,756 | 64,988 |
| Length gene coding (bp) | 23,149,588 | 2,802,952 | 21,935,481 | 4,117,674 | 23,633,062 | 4,042,338 |
| Length repetitive (bp) | 277,400 | 234,489 | 707,976 | 580,264 | 1,354,046 | 3,172,535 |
| % Gene coding | 54.90 % | 31.98 % | 52.83 % | 30.97 % | 54.2 % | 34.8 % |
| % Repetitive | 0.66 % | 2.67 % | 1.71 % | 4.36 % | 3.1 % | 27.3 % |
| Number of predicted genes | 14,427 | 2424 | 12,985 | 3139 | 16,405 | 3218 |
| Percentage of genes with Pfam annotaion | 71 % | 48 % | 72 % | 47 % | 66 % | 51 % |
| Gene density (per 10 Kb) | 3.4 | 2.8 | 3.1 | 2.4 | 3.7 | 2.7 |
| Average protein length (aa) | 479 | 330 | 486 | 361 | 443 | 374 |
In order to facilitate the search for genomic regions with roles in plant pathogenicity we compared the sequences of predicted dispensable scaffolds from the legume-infecting ff. spp. with CDCs of Fol and F. solani or other F. oxysporum ff. spp. to identify those with high conservation levels. A comparison between the predicted CD sequences of Fom-5190a and Foc-38-1 revealed that although the total size difference between the predicted dispensable regions was 4.5 Mb (Table 4), the length of conserved non-repetitive sequence between these isolates was very similar (~3.1 Mb). We speculate that increased repetitive content in the predicted Foc-38-1 dispensome is reflective of and potentially accounts for its overall increased assembly length. Interestingly, in contrast to the phylogenetic studies based on genes encoded on “core” scaffolds (Additional file 11), after masking repetitive and low complexity sequences Foc-38-1, Fop-37622 and Fom-5190a predicted dispensable scaffolds shared highest sequence conservation with the Brassica-infecting isolate Fo5176 (34.7, 38.3 and 44.8 % respectively), closely followed by the melon-infecting F. oxysporum f. sp. melonis (NRRL 26406) and the other legume-infecting isolates. The masked predicted dispensable scaffolds of Foc-38-1 and Fom-5190a share a greater length of conserved sequence with the pea-infecting isolate Fop-37622 than with each other (Table 4). No long runs of consecutive conserved genes between the ff. spp. were observed in predicted dispensable sequences although this may be due to the fragmented assembly of these repeat-rich genomic regions. These data suggest that the legume-infecting ff. spp. may not have acquired and retained whole chromosome sized segments of CD content specific to pathogenicity on legume hosts. They may however, share smaller conserved segments.
Table 4.
Summary of sequence conservation between Fom-5190a and Foc-38-1 predicted dispensable scaffolds and other Fusarium species
| Foc-38-1 | (Masked)a | Fom-5190a | (Masked)a | Fop-37622 | (Masked)a | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total length of predicted dispensable scaffolds (bp) | 13,294,565 | (12,714,301) | 8,766,042 | (8,531,553) | 11,600,480 | (8,427,945) | ||||||
| Foc length conserved | Foc sequence conserved | Foc length conserved maskeda | Foc sequence conserved masked | Fom length conserved | Fom sequence conserved | Fom length conserved masked | Fom sequence conserved masked | Fop length conserved | Fop sequence conserved | Fop length conserved masked | Fop sequence conserved masked | |
| (bp) | (%) | (bp) | (%) | (bp) | (%) | (bp) | (%) | (bp) | (%) | (bp) | (%) | |
| Foc-38-1 | - | - | - | - | 3,504,836 | 40 | 3,051,034 | 34.8 | 5,820,869 | 50.2 | 3,747,704 | 32.3 |
| Fom-5190a | 3,618,892 | 27.2 | 3,134,089 | 23.6 | - | - | - | - | 5,200,782 | 44.8 | 3,852,673 | 33.2 |
| Fop-37622 | 5,645,201 | 42.5 | 4,105,088 | 30.9 | 4,413,435 | 50.3 | 3,484,062 | 39.7 | - | - | - | - |
| F. solani | 1,417,711 | 10.7 | 1,099,653 | 8.3 | 1,215,395 | 13.9 | 1,037,655 | 11.8 | 1,458,181 | 12.6 | 954,748 | 8.2 |
| F. oxysporum f. sp. lycopersici | 4,580,988 | 34.5 | 2,041,608 | 15.4 | 3,174,649 | 36.2 | 1,543,921 | 17.6 | 5,107,489 | 44.0 | 1,636,670 | 14.1 |
| F. oxysporum f. sp. melonis | 5,635,252 | 42.4 | 4,242,674 | 31.9 | 4,684,280 | 53.4 | 3,907,121 | 44.6 | 6,686,394 | 57.6 | 3,999,585 | 34.5 |
| Fo5176 | 6,429,341 | 48.4 | 4,618,139 | 34.7 | 5,013,128 | 57.2 | 3,930,676 | 44.8 | 7,604,002 | 65.5 | 4,446,346 | 38.3 |
aMasked sequences exclude repetitive regions
As CDCs from Fusarium spp. are known to be enriched for pathogenicity-associated genes (e.g. including those encoding cell-wall degrading enzymes, SIX effectors and other effector-like proteins, transcription factors, and proteins involved in signal transduction and lipid metabolism), but lack ‘housekeeping’ genes [9], we next compared the gene content and assigned functions of genes encoded on the predicted dispensable scaffolds with that of predicted core scaffolds. A larger proportion of the proteins encoded on scaffolds assigned as dispensable had no known function based on Pfam annotation (Table 3). Over half of the manually-curated non-TE ORFs from Fol CDC 14 also have no known function [18], highlighting one of the main obstacles in assigning biological roles to potential pathogenicity genes due to the lack of conserved domains identified in most fungal effectors, which thus require experimental evaluation. For genes assigned functional annotation based on comparisons to the Pfam database [65], most functional groups enriched on predicted dispensable scaffolds were similar to those observed on Fol CDCs [18] (details in Additional files 15, 16 and 17). These included: Major Facilitator Superfamily (MFS) transporters, transcriptional regulators, sugar transporters, methyl transferases, chitin-binding domains (LysM), p450s, HET domains (with possible roles in vegetative compatibility that may influence potential sequence transfer), NACHT domains (may be associated with proteins involved in heterokaryon incompatibility-HET-E in P. anserina, or apoptosis [66]), and Carbohydrate-Active Enzymes (CAZymes: (GH3 and GH43) [67, 68]) as well as several peptidases. Additionally in Foc-38-1 and Fop-37622 an enrichment of TE related ORFs were observed relative to predicted core scaffolds, including those with domains related to reverse transcription, transposition, DNA binding and dimerisation (Additional files 16 and 17), which is consistent with their elevated repeat content relative to Fom-5190a.
Expression of predicted-CD sequence-encoded genes in Fom-5190a with conservation across legume-infecting F. oxysporum ff. spp.
In order to further narrow in on predicted CD scaffold gene content with potential roles in pathogenicity that may be conserved within the legume-infecting ff. spp., we used our model Fom-5190a legume pathosystem to identify genes expressed during an early stage of infection. From RNA sequencing of three pooled biological replicates of infected M. truncatula roots at 2 days post inoculation (dpi) we found that for each replicate 0.1, 0.08 and 0.07 % of the reads mapped to the Fom-5190a assembly respectively, giving combined support for the expression of 6448 genes (out of 16,858 predicted). Due to the early time-point and the low fungal biomass at this stage of infection, only 201 genes had 100 % coverage of the predicted gene model with RNA-seq reads, with 1181 genes having 50 % or more coverage, and the remaining overlapped by one or more reads. Of the 6448 genes with expression data, 367 genes were encoded on predicted Fom-5190a dispensable chromosomes.
Comparison of predicted Fom-5190a dispensable scaffolds that shared greater than 40 % sequence identity with the pea pathogen F. solani identified 87 scaffolds encoding 102 genes, 16 of which were expressed by Fom-5190a at 2 dpi. These included genes with potential roles in pathogenicity such as cytochrome p450s, glycoside hydrolases (GH28), peptidases, a sodium/hydrogen-exchanger family protein, and fungal transcription factors.
The same comparison between Fom-5190a dispensable scaffolds and Foc-38-1 identified 176 genes expressed at 2 dpi. This included several small clusters of 3–4 genes expressed in planta, on scaffold 29 (19 genes), scaffold 24 (29 genes) and a cluster of ‘restless-like’ transposons on scaffold 124. TEs have been previously observed to be active in several F. oxysporum genomes [9, 69], and many in Fom-5190a were also found to be transcribed during infection. This indicates they are still active and may be involved in the rearrangement of the genome. The genes encoded on scaffold 29 and the smaller gene clusters predominantly encoded proteins of unknown function but also included fungal transcription factors, proteases, peptidases, and MFS and ABC transporters. Fom-5190a scaffold 24 is ~380 kb in length, encodes 142 genes and many of those detected as expressed in planta (29) have possible associations with pathogenicity, including pectate lyases, MFS transporters, peptidases, cytochrome P-450 s, and components of the F. solani PEP cluster (proteins with similarity to the pisatin demethylase PDA1, two other PDAs and PEP5) [52]. However, this scaffold shares only 23 % sequence conservation with F. solani, indicating that there has not been a large scale transfer of the CDC, or that if there was, this region of the Fom-5190a genome has since undergone significant reshuffling. Fom-5190a scaffold 24 does however share 71.8 % sequence similarity with Foc-38-1 and 83.5 % with Fop-37622, suggesting that these sequences may share a similar source. This scaffold also shares 87 % sequence conservation with Fo5176 and F. oxysporum f. sp. melonis, but only 12.7 % with Fol. The source and route of transfer amongst F. oxysporum ff. spp. of the CDC that this scaffold is thought to represent, may become apparent with further comparative studies enabled by the growing number of available Fusarium genomes.
Finally, a comparison between Fom-5190a predicted dispensable scaffolds and Fop-37622 identified two scaffolds with high sequence similarity. Firstly, Fom-5190a scaffold 31 shared more than 183 kb (88.4 %) with Fop-37622. This scaffold encodes 82 genes including several MFS transporters, a cytochrome p450 and several fungal transcription factors. Twelve of these genes were expressed at 2 dpi, with three predicted to be secreted, and four consecutively encoded (FOXM-5190a_14251-14254) including those with similarity to a FAD dependent oxidoreductase, a protein-arginine deiminase and an MFS transporter as well as a nearby encoded isochorismatate hydrolase. This scaffold also shares over 86 % homology with F. oxysporum f. sp. melonis and Fo5176 but not Foc-38-1, F. solani or Fol, suggesting it hasn’t been derived from a common legume-infecting isolate. Another Fom-5190a scaffold, scaffold 113, shares high sequence conservation with only Fop-37622 (80 % versus ~30 % in the other ff. spp. compared) and encodes 7 genes (FOXM_5190a_15729-15735) all of which were expressed in planta at 2 dpi including a fungal transcription factor, an ABC transporter, a monoxygenase (FAD_binding_3) and a glutathione S-transferase. Four of these genes (FOXM_5190a_15729-15732) are also co-linear with Fop-37622 genes (FOVG_17777-17780). The properties of these genes: location on predicted CD sequences, expression early in infection and conserved synteny, collectively suggests important roles in the infection process and thus these genes will be prioritised for follow up in future functional studies.
Proteins with potential roles in legume phytoalexin detoxification
As orthologs or possible components of the F. solani PEP cluster were identified several times in the above analysis, we investigated this cluster in further detail. Legumes are known to produce low molecular weight antimicrobial compounds, known as phytoalexins including maackiain in chickpea, pisatin in pea, and medicarpin in Medicago sp. [70, 71]. These pterocarpan molecules are structurally similar and are toxic to several genera of fungi and legume pathogens with the ability to detoxify or export these compounds are more virulent [72–75]. In the pea pathogens Fop and F. solani the phytoalexin pisatin is demethylated by a cytochrome P450 known as pisatin demethylase (PDA) shown to be important for virulence on this host [64, 71, 76] and demonstrated to be able to detoxify pisatin in isolation [77, 78]. In F. solani the PEP cluster of genes containing PDA1 is found in the reference mating population on CDC 14 and exhibits altered codon usage compared to core genes [52]. The cluster contains six genes in F. solani, four of which have demonstrated roles in virulence (PEP1, 2, 5 and PDA1) [52], all of which are induced in response to pisatin and during infection of pea [79] and function independently as virulence factors [52, 79]. Apart from PDA1 only two have proposed biological functions PEP5 is a potential MFS and PEP2 possibly has a role in RNA binding [52]. Our analysis showed that none of the legume infecting F. oxysporum ff. spp. contained the PEP cluster in its entirety. Fop-37622 has two copies of PEP2 (FOVG_17451T0, FOVG_16839T0), whilst Foc-38-1 has one (FOC38_09209) and Fom-5190a has none. Genes similar to PEP5 and containing MFS domains [Pfam:PF07690.11] were found in Fop (FOVG_16838T0), Foc-38-1 (FOC38_09210), and Fom-5190a (FOXM_5190a_13563, FOXM_5190a_15270) where FOXM_5190a_13563 was next to a F. solani PDA1 ortholog. We identified two orthologs of PDA1 in Foc-38-1 and four in Fom-5190a, three of which were detected as expressed in planta (Additional file 18). Several of these orthologs had greater homology to F. solani PDAs, while the others were closer to PDA genes from Fop. Previous analyses of F. oxysporum f. sp. pisi isolates showed that while homologs of genes from the F. solani PEP cluster are often present amongst isolates of the different Fop races, their location can differ across races and they are rarely identified as a cluster [64], supporting the idea of multiple origins for Fop races unrelated to PDA gene content [64]. These studies also show that orthologs of PDA1 are present within a group of related ff. spp. that are pathogenic on dicots (f. sp. lini, pisi, dianthi) although the encoded proteins are not always functional due to small but important amino acid changes [64]. A functional homolog, demonstrated to be more closely related to Fop PDA1 than F. solani PDA1, was identified in f. sp. phaesoli (cause of wilt on common beans, Phaseolus vulgaris) which was also virulent on pea and that the authors suggest may have arisen via HGT [64]. Another possible explanation proposed in a recent study [80] is that PDA1 is vertically inherited within the FOSC, rather than via HGT from F. solani as previously proposed [53, 81].
The F. solani PEP cluster also contains Nht1 transposons [82], however none were found in the genome assemblies of Foc-38-1, Fom-5190a or Fop-37622, another possible indicator of the separate evolution of these genes in these ff. spp. from a common ancestor rather than transfer of a whole region. It is possible that some of the genes characterised as PDAs are detoxifying pterocarpans other than pisatin, such as medicarpin or sativan which are produced by both alfalfa and M. truncatula. However further investigation including biochemical examination of the breakdown products of these fungal enzymes will be required to determine this.
Prediction of effector genes in Fom-5190a– pathogen of the model legume Medicago truncatula
Like many other plant pathogens, Fusarium spp. are known to produce small secreted proteins and secondary metabolites to manipulate and evade their host plant’s defences [83, 84]. In addition to identification of proteins with known roles in plant pathogenicity in other fungal species (Additional file 18) a combination of multiple sources of evidence was used to predict putative legume host-specific effectors. This incorporated predictions of secretion and protein size, orthology across Fusarium spp. and orthology-based lineage-specificity, functional annotations, predictions of dispensable sequences, proximity to pathogenicity gene-associated repetitive DNA, and RNA-seq data derived from our model legume pathosystem.
We identified 580 SSPs in Fom-5190a, and 537 and 620 respectively in Foc-38-1 and Fop-37622 (Table 2, Additional files 19, 20 and 21). This number is comparable to that predicted in other Fusarium oxysporum ff. spp. and Fusarium spp. (Table 2). In Fom-5190a, 75 SSPs were predicted to occur on potentially dispensable scaffolds, with 94 and 98 respectively found in Foc-38-1 and Fop-37622. RNA-seq data from infected M. truncatula roots showed that 19 of these SSPs were expressed at 2 dpi. This included four homologs of the Fol SECRETED IN XYLEM (SIX) genes, with proposed roles in virulence/avirulence on tomato (SIX1, SIX8, SIX9, SIX13). Of these 19 proteins, only five had characterised Pfam domains including a GH16, CFEM and LysM, a peroxidase and a DUF3129 domain (Table 5, Additional file 19).
Table 5.
Properties of Fom-5190a effector candidates
| Gene | FOXM-5190a_15788 | FOXM-5190a_16235 | FOXM-5190a_16257 | FOXM-5190a_16301 | FOXM-5190a_16306 | FOXM-5190a_16326 | FOXM_5190a-SIX1 | FOXM_5190a-SIX8 | FOXM_5190a-SIX9 | FOXM5190a-SIX13 |
|---|---|---|---|---|---|---|---|---|---|---|
| Protein length (aa) | 199 | 264 | 91 | 144 | 131 | 111 | 279 | 141 | 122 | 292 |
| Mature protein length (aa) | 186 | 247 | 72 | 128 | 112 | 95 | 263 | 126 | 105 | 276 |
| Cysteine count in mature protein | 7 | 8 | 8 | 6 | 10 | 9 | 8 | 2 | 6 | 6 |
| % Cysteine (of aa length) | 3.5 | 3 | 8.8 | 4.2 | 8.9 | 8.1 | 2.8 | 1.4 | 5.7 | 2.1 |
| Predicted to be secreted by SignalP | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| WoLFPsort predicted location | extr | extr | extr | extr | extr | extr | extr | extr | extr | extr, mito |
| Phobius predicted signal peptide | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| TMHMM predicted transmembrane count | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pfam domains | - | - | - | LysM | - | - | - | - | - | - |
| Number of aligned RNAseq reads overlapping gene model from combined 2d in planta libraries | 24 | 1039 | 89 | 378 | 10 | 369 | 2546 | 828 | 246 | 440 |
| Average depth of RNAseq coverage per base of gene | 4 | 123 | 27 | 88 | 3 | 76 | 354 | 141 | 33 | 61 |
| Gene length (bp) | 655 | 844 | 324 | 432 | 458 | 488 | 720 | 587 | 728 | 720 |
| Proportion of gene model supported by RNAseq | 0.96 | 1 | 1 | 1 | 0.69 | 1 | 0.99 | 1 | 1 | 1 |
| Scaffold | 122 | 322 | 364 | 461 | 485 | 531 | 3157, 696, 1167 | 420 | 270 | 306, 3812 |
| Scaffold length (bp) | 28,293 | 2356 | 1489 | 971 | 914 | 815 | 219, 647, 416 | 1129 | 4106 | 3,471, 204 |
| Predicted scaffold type | Dispensable | Dispensable | Dispensable | Dispensable | Dispensable | Dispensable | Dispensable | Dispensable | Dispensable | Dispensable |
| Orthology | Has orthologs in other ff.spp. | Has orthologs in other ff.spp. | Single ortholog unique to Fom5190a | Has orthologs in other ff.spp. | Has orthologs in other ff.spp. | Has orthologs in other ff.spp. | Has orthologs in other ff.spp. | Has orthologs in other ff.spp. | Has orthologs in other ff.spp. | Has orthologs in other ff.spp. |
Further manual inspection of the annotation and level of RNA-seq expression of the 19 SSPs encoded on Fom-5190a dispensable scaffolds identified a subset of 10 genes that were prioritised for further initial investigation as effector candidates (which included the SIX gene homologs) (Table 5). Protein orthology analysis supported only one of these proteins (FOXM_5190a_16257) as lineage-specific to Fom-5190a with the others sharing best BLASTP matches of 42–99 % similarity to other F. oxysporum or F. fujikuroi proteins (Additional file 22). Of the SIX homologs, FOXM_5190a_SIX1 was most similar to that identified in Fop-37622 (87 %) and the FOXM-5190a_SIX8 best BLASTP match was FOC-38_SIX8, differing by only one amino acid (99.3 % identity) suggesting that these genes, in particular SIX8, may have a common origin. Fom-5190a SIX9 and SIX13 homologs shared less conservation to legume pathogenic ff. spp., with best matches to Fol (42 %) and F. oxysporum f. sp. melonis (74 %) respectively. Two other effector candidates FOXM_5190a_16301 and FOXM_5190a_16306, which contain no characterised domains, were also most similar to proteins from legume-infecting ff. spp., Fop-37622 (95 %) or Foc-38-1 (99 %) respectively. While these ten effector candidates constitute a shortlist for further investigation, the overall Fom-5190a pathogenicity gene set may be much larger with 183 of the 580 predicted SSPs detected as expressed in planta at 2 dpi.
Because our analysis highlighted the potential importance of SIX gene homologs during Fom-5190a infection we searched for these proteins in the other F. oxysporum ff. spp. and sp. (summarised in Table 6). We identified several homologs of Fol SIX genes in all the legume-infecting ff. spp. The Foc-38-1 assembly contained homologs of SIX5, 8, 11, 13 and 14, whilst Fop-37622 contained SIX1, 9, 13 and 14 (Table 6). All of the SIX homologs were encoded on scaffolds predicted to be dispensable except Foc-38-1_SIX8 which is encoded on a scaffold with similarity to Fol core chromosome 5. There are many SIX8 genes in Fol but none occur on core chromosome 5 (Table 6) [18]. While it is possible the Foc-38-1-SIX8 -encoding genomic region was mis-assembled, self-alignment of Illumina generated genomic read data supports the current assembly. There is the potential for transposon-mediated translocation of genes from dispensable regions into the core genome to have occurred. Thus it may be that the location of this gene has been shuffled in Foc-38-1, facilitated by the adjacent mimps and other TEs (Foc-SIX8 is located on the end of Scaffold 138 next to a ‘RESTLESS’-like transposase).
Table 6.
SIX gene presence on chromosomes/scaffolds in published Fusarium speciesa
| F. oxysporum f. sp. lycopersici | F. oxysprum f. sp. medicaginis Fom-5190a | F. oxysporum f. sp. ciceris 38-1 | F. oxysporum f. sp. pisi 37622b | F. oxysporum f. sp. brassica Fo5176 | F. oxysporum f. sp. cubense II5 | F. oxysporum f. sp. melonis 26406_1 | F. solani (N. haematococca) | F. virguliforme Mont-1 | F. graminearum 1-4 | F. verticillioides | F. fujikuroi IMI 58289 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| six1 | 14 (2nd half dup. on chr 14) | split over 696 and 1167 (partial dup. 241) | - | 1.281, 1.315 | contig00620 | 1.92, 1.101 | 1.500 | - | - | - | - | - |
| six2 | 14 | - | - | - | - | 1.95 | - | - | - | - | 5 | 06 |
| six3 | 14 | - | - | - | - | - | - | - | - | - | - | - |
| six4 | - | - | - | - | contig01435 | 1.102 | - | - | - | - | - | - |
| six4b | _ | - | - | - | contig04359 | _ | - | - | - | - | - | - |
| six5 | 14 (partial match Chr 5) | - | 1215 | partial match 1.4 | - | - | 1.4 | - | - | - | - | - |
| six6 | 14 | - | - | - | - | 1.102 | 1.226 | - | - | - | - | - |
| six7 | 14 | - | - | - | - | - | - | - | - | - | - | - |
| six8 | 2, 3 (3 copies), 6 (2 copies), 7, 14 (2 copies) | 420 | 0138 | - | contig03501 | 1.127 | - | - | - | - | - | - |
| six9 | 14 | 270 | - | 1.43 | contig02779 | 1.86, 1.70 | - | - | - | - | - | - |
| six10 | 14 | - | - | - | - | - | - | - | - | - | - | - |
| six11 | 14 | - | 1126 | - | - | - | 1.532 | - | - | - | - | - |
| six12 | 14 | - | - | - | - | - | - | - | - | - | - | - |
| six13 | 6 (2 copies) | 306 (split), 3812, 306 | 1143 | 1.90 | - | 1.216 | 1.222 | - | - | - | - | - |
| six14 | 14 (1st half dup.) | - | 0905 | 1.191 | - | - | - | - | - | - | 2 | - |
aID of scaffold or contig showing TBLASTN matches with an Expect (E) value score below 10−5, dup duplicate. b Fop-37622 contained annotated homologs of SIX1 (FOVG_19815, FOVG_19730 and SIX9 (FOVG_17008) and matches by TBLASTN to unannotated versions of SIX13 and SIX14 which were manually annotated for this study (Additional file 23) and SIX5 where the coding sequence was interrupted by stop codons
Interestingly, only SIX13 was found in all the legume infecting ff. spp. and this SIX gene is the only one in Fol (race 2) not found on CDC 14, but instead on CDC 6 [18]. As orthologs of this protein were also detected in ff. spp. infecting melons and banana it appears unlikely to have a role in legume host-specificity but may play a role in pathogenicity. We also observed SIX gene homologs in two other Fusarium species, F. verticilloides (SIX2 and SIX14) and F. fujikuroi (SIX2) [85] (Table 6). The presence of SIX genes outside the species F. oxysporum has previously been observed in F. verticilloides (SIX2) [9] and F. foetens (SIX1) [62]. SIX1, SIX8, SIX9 and SIX13 homologs were identified in at least 5 out of the 7 F. oxysporum species analysed in Table 6, suggesting these proteins may play conserved roles in pathogenicity but not host-specificity, unless small amino acid changes govern their interaction with host proteins. Top BLASTP matches for SIX protein homologs in Foc-38-1 and Fop-37622 show that SIX8 is highly conserved between Fom-5190a and Foc-38-1, SIX13 between all three legume-infecting ff. spp. and SIX14 between pea- and chickpea-infecting isolates (Additional files 22 and 23). Phylogenetic relationships between the SIX genes encoded by Fom-5190a that were also present in other ff. spp. (Additional file 24) suggests that the relationship between proteins encoded on predicted dispensable scaffolds differs from that of the conserved core proteins (Additional file 10). This is not unexpected if dispensable genomic regions are indeed readily exchanged amongst different isolates [9], whilst core regions remain relatively stable or if gene content of dispensable regions is undergoing more frequent mutation and rearrangement facilitated by repetitive elements. This finding is supported by a recent study [80] that showed incongruent phylogenies between SIX genes (1 and 6) and the house-keeping gene EF-1α and additionally presented evidence of potential vertical transmission of SIX6 between related isolates. It is most likely that the SIX genes have a common ancestry either laterally or vertically and we can speculate that minor sequence differences contributing to their alternate phylogeny may be the result of host-driven selection.
A recent study examining the landscape of the Fol pathogenicity chromosomes identified small clusters of SIX genes which were associated with a class of DNA transposons known as MITEs (Miniature Inverted-repeat Transposable Elements) [18]. These MITEs, include an upstream (within 1500 bp) incomplete fragment of the Impala transposon sequence (miniature Impala or ‘mimp’) [18] and often an additional downstream miniature Fot5 transposon (mFot5) [18]. Their presence in gene-flanking sequences has also been used as a criterion to support the prediction of effector genes in ff. spp. infecting tomato and melons [10, 18]. We therefore searched for the presence of these TEs or their inverted repeats around the predicted legume-infecting ff. spp. SIX genes and effector candidates. The SIX13 homolog residing on Fom-5190a Scaffold 306 is flanked by a partial Impala 430 bp upstream. In the Foc-38-1 assembly several SIX gene homologs had matches to mimps upstream (SIX5, SIX8 and SIX14), and SIX13 was flanked by a downstream complete Fot5 transposase which may facilitate movement of this gene. In the case of Foc-38-1_SIX13 and the Fom-5190a SIX genes where mimps were not identified upstream, the SIX gene homologs resided on short-length scaffolds with little surrounding sequence assembled, inhibiting the search for upstream or flanking intergenic sequences. In Fop-37622 upstream mimps were only identified close to SIX1 (FOVG_19815), SIX9 and SIX13 although the region upstream of Fop-37622 SIX14 had undefined sequence hampering identification of a possible mimp.
qRT-PCR validation of Fom-5190a effector candidates during host infection
To validate our RNAseq data and determine expression of the ten shortlisted effector candidates over a longer period of infection, we examined via qRT-PCR their expression in vitro and over a 1–7 day in planta time-course in susceptible M. truncatula plants (Fig. 2). By 10 dpi, most infected plants had visible wilting symptoms and the majority of infected plants were killed by 21 dpi (Additional file 25a). Increasing fungal biomass over the course of infection was indicated by an increase in the amount of fungal ITS relative to plant ITS detected via qRT-PCR as the infection progressed (Additional file 25b).
Fig. 2.
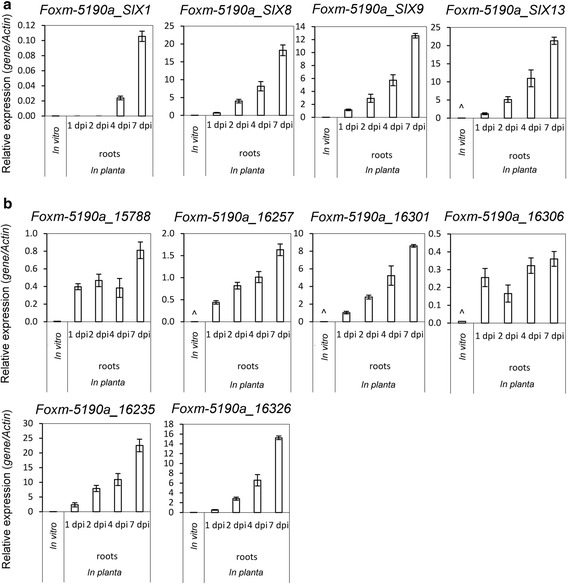
Expression of candidate pathogenicity genes in vitro versus in planta. a Expression of SIX genes and b candidate pathogenicity genes as determined by qRT-PCR in in vitro samples and M. truncatula DZA315 root samples harvested at 1, 2, 4 and 7 days post inoculation (dpi) with Fom-5190a. In vitro samples are averages ± SE of 3 biological replicates. In planta samples are averages ± SE of 4 biological replicates each consisting of pools of 10 seedlings. Gene expression levels are relative to the fungal actin gene (FOXM-5190a_13365). Note: ^ No detectable expression in 1 or 2 out of the 3 in vitro replicates
Many fungal effector proteins are only expressed in planta and cannot be detected in vitro, or if so, at very low levels [57, 86] (reviewed in [87]). All of the Fom-5190a SIX gene homologs were expressed in planta and showed lower or no expression in vitro, exhibiting a pattern of increased expression over the course of the infection (1–7 days, Fig. 2a), peaking at 7 dpi with fold-inductions over in vitro ranging from 1050 to over 60,000. The other six predicted effector genes prioritised for initial follow up studies in Fom-5190a shared this expression pattern, albeit to various levels of induction (Fig. 2b). After FOXM-5190a-SIX13, FOXM-5190a-16235 and FOXM-5190a-16326 showed the largest fold-inductions in expression in planta versus in vitro, and both of their encoded proteins exhibited similar levels of identity across several F. oxysporum ff. spp. (Additional file 22). The Fom_5190a lineage specific gene FOXM_5190a_16257, showed strong expression in planta increasing over the course of infection but no or very little expression in vitro. This protein had an upstream mimp and no similarity to any proteins in the non-redundant database at NCBI (threshold e ≤ 1×10−5) making it a strong candidate for a host specific effector. However a Hidden Markov Model analysis identified a 38 aa region within the 91 aa protein sharing 58 % identity to a region from a hypothetical Colletotrichum orbiculare MAFF 240422 protein (Cob_00676) and other pathogenic fungi from the Ascomycota such as Pseudocercospora fijiensis, Claviceps purpurea and Sphaerulina musiva. An iterative search [88] of these sequences for distinct regions of similarity identified a motif resembling a zinc finger domain, suggesting FOXM-5190a_16257 may target host DNA sequences. The hypothetical FOXM_5190a_16306 protein which shares 99 % amino acid identity to FOC38_16051 had 27-fold up-regulated expression by 1 dpi, compared to in vitro, and showed a slight further increase in expression over the sampled time-course. Another candidate, FOXM_5190a_16301, whose product shares 95 % aa identity with a Fop_37622 protein (FOVG_19456) encodes a LysM domain and has some similarity to Ecp7 - a small, cysteine-rich, secreted effector protein identified in Cladosporium fulvum (syn. Passalora fulva) of unknown function [89] - which has homologs in several fungal species. This protein is an ortholog of a recently identified F. oxysporum f. sp. melonis candidate effector (FOM_19260), which has an upstream mimp and shares a promoter with another candidate effector [10]. However in Fom-5190 the lack of upstream assembled sequence meant we were unable to identify a mimp or potentially co-regulated gene. These genes and the others identified from the RNA expression analysis have been prioritised for further investigation of their roles in pathogenicity and host specificity.
Conclusions
The addition of the genomic sequences of the legume-infecting F. oxysporum ff. spp. presented here adds to the accumulated bioinformatics resources for Fusarium oxysporum formae speciales and helps provide a powerful knowledge-base for predicting lineage-specific genes involved in host-specific pathogenicity. Analysis of pathogenicity-related CDC gene content conserved amongst the legume-infecting Fusarium oxysporum ff. spp. identified several Fom-5190a scaffolds enriched in genes with known or potential roles in pathogenicity, in particular carbohydrate active enzymes, cytochrome p450s, MFS and ABC transporters, fungal transcription factors as well as newly predicted effectors. While the source of these conserved gene sequences is yet to be elucidated, it is evident that parts of the potential dispensable chromosomes greater than just the repetitive regions are shuffled around within the genome of each f. sp., with those changes at least in Fom-5190a possibly associated with active transposable elements. As transposons can be a major source of genetic recombination in an asexual species, this may have contributed to the increased assembly size of Foc-38-1 and potentially the evolution of a large number of races in this f. sp.. Interestingly, the predicted dispensable scaffolds in Foc-38-1 and Fom-5190a shared more identity with Fop-37622 than with each other, and little with another legume-infecting Fusarium species (F. solani). They also shared to a similar degree, CDC content with ff. spp. that are pathogenic on non-legume plant species including Arabidopsis and melon, but not with Fol.
Combining this observation with the differing presence of SIX gene homologs across these ff. spp., points towards different sources for their origins of pathogenicity and suggests that pathogenicity on legumes is a complex phenotype. It is apparent that legume-pathogenicity is not simply governed by a small set of conserved genes retained from an ancestral species that are specific to legume-infecting isolates. It is possible, though unlikely based on our analyses, that the respective legume host-specific pathogenicity genes of the three legume-infecting ff. spp. may have the same origin but have since diverged significantly. Previous studies have shown that there can be multiple origins for pathogenicity on a given host within a F. oxysporum f. sp. and individual isolates phenotypically classified as a particular f. sp. can be more closely related to isolates belonging to a different f. sp. [90, 91]. We speculate that the origin of legume host-specific pathogenicity is not likely to have arisen from recent horizontal transfer events, as this would have resulted in greater sequence similarities than we observed. Whether the pathogenicity components were transferred as a whole chromosome from other F. oxysporum ff. spp. and subsequently reshuffled, mutated or partially lost remains to be elucidated, but will perhaps be revealed with the sequencing of additional legume-infecting ff. spp. and races.
For the model legume-infecting f. sp. Fom-5190a, we shortlisted a set of candidate effectors four of which showed greatest similarity to proteins from another legume pathogenic ff. spp., suggestive of a conserved role in legume pathogenicity. Initial verification via expression analysis of these candidates supports this approach and lays the framework that will facilitate functional characterisation of these candidates in subsequent studies, for the ultimate application of this knowledge towards the development of Fusarium wilt resistance in economically important legume crops.
Methods
Isolate sources
F. oxysporum f. sp. medicaginis (Weimer) W.C. Snyder & H.N. Hansen, (Fom-5190a, BRIP 5190a/IMI 172838, collection number 19911) was isolated from wilting leaves in a commercial field of Medicago sativa by John. A. Irwin in Boonah (QLD, Australia) in 1973 and is not known to infect other legume species. F. oxysporum Schlecht.: Fr. f. sp. ciceris (Padwick) Matuo and K. Sato (Foc-38-1), represents the most virulent race of this forma specialis (race 1) and was isolated from Cicer arietinum (chickpea) in Patancheru (Hyderabad, India). F. oxysporum f. sp. pisi (Fop-37622) was obtained from J.M. Kraft (USDA-ARS, Prosser, Washington, USA) via Hans VanEtten. It was determined by Dr. Kraft to be race 5.
Fusarium growth conditions and DNA extraction
Foc-38-1 and Fom-5190a DNA extraction was performed using a cetyltrimethylammonium bromide (CTAB) based method as per Gao et al. [92]. Foc-38-1 was grown in potato dextrose broth in 250 ml flasks and incubated in a rotary shaker at 120 rpm at 25 °C. Fom-5190a was grown in a petri dish containing one-half-strength potato dextrose broth for 7 days at 22 °C. Mycelia were harvested by filtering through Miracloth, and washed repeatedly with sterile distilled water to remove excess of salts adhering to it. One gram of mycelium was crushed in liquid nitrogen prior to DNA extraction.
Genome assembly
Fom-5190a and Foc-38-1 draft genome assemblies were assembled from paired-end and mate-paired Illumina 100 bp reads (Additional file 3). For Fom-5190a and Foc-38-1 assemblies, paired-end Illumina reads were trimmed of contaminating adapter sequences using Cutadapt 1.1 [93]. Reads less than 25 bp in length, after trimming, were discarded. Overlapping reads were merged using Flash 1.2.2 [94]. Fom-5190a 454 reads were trimmed/filtered using Mothur [95] to assess quality, remove homopolymers and convert raw SFF data to fasta and qual formats. Custom perl scripts were used to recognise titanium linkers and split sff reads into paired fastq format. For each isolate an initial assembly was created using Soapdenovo v2.04 [96] utilising merged and paired-end reads at the optimised kmer length of 27 for Fom-5190a and 19 for Foc-38-1. The resultant assemblies (derived from paired-end data only) were further scaffolded with both paired-end and mate-paired libraries, progressing from shortest to largest insert size using 5 successive rounds of SSPACE 2.0 [97] per mate-pair library. For Fom-5190a, 454 reads were incorporated following the 3 kb mate-pairs. Between iterations of SSPACE, scaffold gaps were filled by performing 5 rounds of GAPCLOSER (soapdenovo/1.05-gc1.12) [96]. Scaffolds obtained after all scaffolding and gap-closing was completed were filtered to remove those less than 200 bp for Fom-5190a and the assembly sequences were re-ordered by decreasing length. For SSPACE scaffolding of Foc-38-1 a minimum scaffold size of 1000 bp was used to eliminate potential assembly errors due to ‘shadow-library’ contamination (unfiltered paired-end fragments) in the Illumina mate-paired library. The completeness of the Fom-5190a and Foc-38-1 genome assemblies and their representation of their respective gene contents was assessed with CEGMA v 2.4 [46]. The whole-genome sequence of Fop-37622 was assembled using ALLPATHS-LG (version R37753) run with default parameters (kmer size of 96) [98]. Mitochondrial sequences were removed by searching against an NCBI mitochondrial database. Ab initio gene models were created combining predictions from GeneMarkES [99], GeneId [100], Augustus [101], GlimmerHMM [102] and SNAP [103], in conjunction with strand-specific PASA alignment [104] and GeneWise features from BLAST [105] against the UniRef90 database [106]. The gene models were further updated with RNAseq datasets. The resulting annotation was filtered to remove spurious genes that overlap with transposons [107].
Sequence conservation analysis
Chromosome sequences of F. oxysporum f. sp. lycopersici (Fol) [GenBank: CM000589-603] [9] and F. solani (syn. Nectria haematococca) [11] (constructed from [GenBank: GG698896-GG699104], ordered and joined by 100 bp of “N” bases), which have been previously characterised into core and accessory chromosomes, were masked for de novo-predicted repetitive DNA sequences using RepeatMasker v4.0.5 [108]. Repeat-masked Fol and F. solani chromosome sequences were then compared to the genome assembly sequences of other published/publicly available Fusarium spp. via MUMmer v3.1 (PROmer --mum, delta-filter) [55]. The percentage of the un-masked length of each chromosome that was covered by one or more PROmer matches was compared to high-quality reference sequences in which CDCs have been well defined - Fol and F. solani - via BEDTools CoverageBed [109] and visualised using Circos v0.67-1 [110] (Fig. 1 and Additional file 9).
Prediction of non-core scaffolds
Scaffolds representing Fom-5190a, Fop-37622 and Foc-38-1 dispensable chromosomes were predicted based on MUMmer v3.0 (promer --mum, delta-filter -g) [55] alignments to repeat-masked sequences of Fol and F. solani of which genome assemblies for both species contain full length chromosome sequences that have been previously characterized as core and accessory chromosomes [9]. Foc-38-1, Fop-37622 and Fom-5190a scaffolds with > = 30 % of their length covered by unique promer matches to core chromosomes of Fol and F. solani (i.e. excluding Fol CDCs 3, 6, 14 and 15 [9] or F. solani CDCs 14, 15 and 17 [11]) were assigned as core scaffolds and all others were considered lineage specific and thus potentially part of a CDC.
Annotation of genome features
Protein-coding gene regions of Fom-5190a and Foc-38-1 were initially predicted de novo via GeneMark-ES v 20120203 [99] using a minimum contig length of 200 bp. Protein sequences from publicly available Fusarium spp. genome projects, PHI-base [111, 112] and secreted-in-xylem (SIX) protein sequences obtained from GenBank were used to refine the GeneMark-ES predicted annotations via EVidenceModeller v 2012-06-25 [113]. Regions of both assemblies homologous to SIX proteins by TBLASTN (e-value threshold 1e-5) [105] were manually annotated based on homology and RNAseq data if not previously predicted.
In order to ensure that genes that played potential roles in pathogenicity and host specificity were correctly annotated following automated gene annotation, the assemblies were examined for matches to genes known to be involved in pathogenicity in other fungi [18, 84, 111, 112, 114]. Genes of interest that had not been annotated correctly were manually annotated based on homology and the RNA-seq data.
Repetitive DNA regions were predicted within genome assemblies of Fom-5190a, Foc-38-1 and other publicly available Fusarium spp. (Additional file 8) by both de novo prediction and comparison with the RepBase database of known fungal repeat sequences [115]. Repeat families were predicted de novo using RepeatScout v1.0.5 [116] (default parameters), the outputs of which were clustered for redundant or fragmented repeat families via Cap3 (-h 70 -z 1 -p 70) [117]. The non-redundant de novo repeat families were mapped to genome assemblies via RepeatMasker v4.0.5 [108] (crossmatch, -no_is -s) to determine the novel repetitive DNA content of each genome. To estimate the relative proportions of known transposon classes and sub-classes, each Fusarium genome sequence was also searched via RepeatMasker v4.0.5 (parameters: -no_is -qq) for matches to RepBase v20140131 [115].
The genome assemblies of Fom-5190a, Foc-38-1 and other publicly-available Fusarium spp. were also searched for non-coding RNA (ncRNA) using the cmsearch program from infernal 1.1rc4 (search mode) [118] using the Rfam 11.0 database [119–121]. Additionally, ribosomal RNA (rRNA) regions were predicted using RNAmmer 1.2 [122] and transfer RNA (tRNA) genes were predicted using tRNAscan v 1.3 [123].
Annotation of protein functions
Within the protein translations of gene annotations of Fom-5190a, Fop-37622 and Foc-38-1, conserved amino-acid domains were identified using HMMER v 3.0 [124], against the PFAM-A database (v 27.0) (gathering cut-offs) and InterProScan v4.8 [125, 126]. Carbohydrate-active enzyme (CAZyme) (www.cazy.org) [67, 68] annotations were assigned to protein sequences via dbCAN [127] and HMMER v3.0 [124] with default settings. BLAST (version 2.2.26) [105] searches were performed at a significance score threshold of 1e−5 unless otherwise specified.
Orthologs of genes known to be involved in pathogenicity in other species (PHIbase) or F. oxysporum f. sp. (SIX genes) were identified via reciprocal BLAST analysis of both the predicted proteins and the scaffolds (1e−5) Predicted Fom-5190a and Foc-38-1 proteins and genomic scaffolds were also compared to 2309 protein sequences from the Pathogen Host Interaction database (PHIbase- version 3.5) [111, 112] which have been experimentally tested for roles in pathogenicity. Matches were considered only for reciprocal BLAST matches below an expectation value of 1×10−5. Genes potentially involved in the synthesis of secondary metabolites were identified using SMURF [128]. The potential localisation of predicted proteins was analysed via WoLFP SORT [129] and Phobius [130].
The proteins of the five reference Fusarium genomes (Fo5176, Fol, F. gaminearum, F. solani, F. verticilloides) and those of Fom-5190a, Foc-38-1 and Fop-37622 were classified for the purpose of this study as small secreted proteins (SSPs), based on criteria previously used by Ohm and colleagues [131]. These criteria include prediction of secretion by SignalP v.4.1b [132], with one or less N terminal transmembrane domains as predicted by TMHMM v. 2.0c [133] although the length cut-off was increased from 200 to 300 aa to include known effector proteins identified in Fusarium sp. such as the SIX proteins.
Statistical examination for over- or under-representation of protein functional attributes
The number of genes with specific functional attributes (Pfam domains [65]) was compared between predicted core and dispensable scaffolds and compared using Fisher’s exact test. Those that were increased on dispensable scaffolds with a significance threshold of p ≤ 0.05 are listed in Additional files 15, 16 and 17.
Orthology
Proteinortho v4.26 [134] was used to detect orthologs of Fom-5190a, Fop-37622 and Foc-38-1 compared with 41 isolates of the following fungal species: Alternaria brassicicola, Ashbya gossypii, Blumeria gramminis f. hordei, Botrytis cinerea, Cladosporium fulvum (syn. Passalora fulva), Coccidiodes immitis, Fusarium acuminatum, Fusarium culmorum, Fusarium graminearum, Fusarium fujikuroi, Fusarium incarnartum-Fusarium equiseti species complex, Fusarium oxysporum, Fusarium oxysporum f. sp. conglutinans, Fusarium oxysporum f. sp. cubense, Fusarium oxysporum f. sp. lycopersici, Fusarium oxysporum f. sp. melonis, Fusarium oxysporum f. sp. pisi, Fusarium oxysporum f. sp. radicis-lycopersici, Fusarium oxysporum f. sp. raphani, Fusarium oxysporum f. sp. vasinfectum, Fusarium pseudograminearum, Fusarium solani (syn. Nectria haematococca), Fusarium verticilliodes (syn. Gibberella fujikuroi), Grosmania clavigera, Leptosphaeria maculans, Magnaporthe oryzae (syn. grisea), Mycosphaerella graminicola (syn. Zymoseptoria tritici), Neurospora crassa, Parastagonospora nodorum, Podospora anserina, Saccharomyces cerevisiae, Sordaria macrospora, Trichoderma reesei, Tuber melanosporum, Uncinocarpus reesii, Verticillium dahliae. Details of the specific isolates and data sets used are provided in Additional file 5. Orthologs were determined via reciprocal BLASTP using parameters: −e = 1e−5, alg.conn. = 0.1, coverage = 0.5, percent_identity = 25, adaptive_similarity = 0.75, retaining both pairs and singletons (Additional file 6). These isolates were selected based on their close relation to the genus Fusarium or the fact that they either shared a similar host range or infection mode or had a very diverse one. F. virguliforme and F. circinatum were used for comparison at the whole genome sequence level only.
Phylogeny
From the Proteinortho predictions, 100 proteins were randomly chosen that had predicted one-to-one orthologies across all Fusarium sp. genome assemblies and protein sequences were concatenated, all of the orthologs in f. sp. medicaginis were encoded on predicted core scaffolds. Multiple sequence alignments were calculated using Clustal Omega version 1.2.1 [135] and phylogenetic trees constructed using RAxML version 8.1.20 [136]. Workflows were automated using the ete build function of the ETE toolkit [137] and trees were drawn using ete view with branch support values shown.
Sample preparation for RNA-seq and qRT-PCR
For F. oxysporum inoculations of M. truncatula the isolate Fom-5190a was maintained on sterile filter paper and grown on one-half-strength potato dextrose agar. For spore production, 3 agar plugs were removed to inoculate flasks containing 100 mL of one-half-strength potato dextrose broth and grown for 3 days at 28 °C/100 rpm. The inoculum was drained through Miracloth (Calbiochem, San Diego), centrifuged to pellet spores, and resuspended in sterile distilled water before quantification with a haemocytometer. The spore concentration was adjusted to 1 × 106 spores mL−1 in sterile distilled water and used for plant inoculations. The M. truncatula accession DZA315.15 susceptible to Fom-5190a (J. Lichtenzveig unpublished, this work) was germinated on damp filter paper, transplanted into 30 mm Jiffy-7 peat pellets and grown under a short-day light regime (8-h light/16-h dark) in a growth room set at 22 °C. After 2 weeks, roots protruding from the peat pellets were removed. Peat pellets were then inoculated by placing them in a petri dish of spore suspension for 5 min, followed by a further 1 mL of spore suspension added to the base of the hypocotyl. Inoculated pellets were transferred to growth trays lined with a plastic sheet and a thin layer of damp vermiculite, covered with a clear plastic dome to maintain humidity, and incubated under a long-daylight regime (16-h light/8-h dark) growth room set at 28 °C.
RNA isolation
For qRT-PCR and RNA-seq experiments on Fom-5190a infected M. truncatula accession DZA315.15, root tissue was collected from 10 plants per replicate at 1, 2, 4 and 7 days post inoculation and pooled for RNA extraction. For Fom-5190a in vitro samples, mycelia was grown in a petri dish containing one-half-strength potato dextrose broth for 7 days at 22 °C and mycelia harvested by filtering through Miracloth. Three or four separate biological replicates were taken for all experiments, then frozen in liquid nitrogen, and stored at −80 °C. RNA extraction was performed using a Trizol extraction followed by DNase treatment using TURBO DNase (Ambion). RNA samples were cleaned via RNeasy mini spin columns (Qiagen).
qRT-PCR
Following RNA isolation and DNase treatment, complementary DNA synthesis was performed using SuperscriptIII reverse transcriptase (Invitrogen) with oligo (dT) (Invitrogen) and RNasin (Promega) with 1 μg of input RNA. qRT-PCR was performed using SsoFast EvaGreen Supermix (Bio-Rad) on a CFX384 (Bio-Rad) system. Thermoycling and melt-curve conditions were as described by Oñate-Sánchez et al. [138]. Absolute gene expression levels relative to F. oxysporum actin were used for each complementary DNA sample using the equation: relative ratio gene of interest/Actin = (Egene-Ct gene)/(EActin-Ct Actin) where Ct is the cycle threshold value. Medicago root samples were verified for even abundance of plant input material using the M. truncatula B-tubulin reference gene [139] which was found to be within ± 1 Ct across all samples. Primer sequences are listed in Additional file 26.
RNA-seq library construction, Illumina sequencing and read-mapping
RNA integrity was confirmed using the Agilent 2100 Bioanalyser Plant Nano system (Agilent Biotechnologies). Stranded Illumina TruSeq libraries were generated from 1 μg of total RNA and sequenced (100 bp paired end reads) on an Illumina HiSeq platform by the Australian Genome Research Facility (AGRF). 51–60 million reads were generated for each sample. RNA-seq paired-end reads were trimmed for low-quality base-calls and Illumina adapter sequences via Cutadapt (v1.1, parameters: −quality-cutoff 30 --overlap 10 --times 3 –minimum-length 25) [93]. Reads trimmed to less than 25 bp were discarded and remaining reads sorted into pairs and singleton reads. RNA-seq reads were mapped to the Fom-5190a genome assembly via Tophat2 (v2.0.9, parameters: −b2-very-sensitive -r 80 --mate-std-dev 40 -i 20 -I 4000 -g 20 --report-secondary-alignments --report-discordant-pair-alignments -m 0 --min-coverage-intron 20 --microexon-search --library-type fr-firststrand) [140].
Data sources and acknowledgements
This research was undertaken with the assistance of resources from the Australian Genome Research Facility (AGRF) and the National Computational Infrastructure Specialised Facility for Bioinformatics (NCI-SF Bioinformatics), which are both supported by the Australian Government. The work was supported by iVEC through the use of advanced computing resources located at the Pawsey Supercomputing Centre. Data used for comparative analysis was obtained from NCBI, unpublished Fusarium data sets obtained from “Fusarium Comparative Sequencing Project, Broad Institute of Harvard and MIT (http://www.broadinstitute.org/)”, Broad Institute Genomics Platform, Feb 2014. F. solani and F. fujikuoroi data was obtained from the JGI Genome portal (http://genome.jgi.doe.gov/), all data sources are outlined in Additional file 4. We also thank Elaine Smith for excellent technical assistance and Dr Donald Gardiner for critical reading of the manuscript and useful suggestions.
Availability of supporting data
Trimmed sequencing data for Fom-5190a was deposited into the NCBI/GenBank database under BioProject number PRJNA294248 (http://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA294248). This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession LSNI00000000. The version described in this paper is version LSNI01000000. Raw sequence data and the assembly for Foc-38-1 were deposited into the NCBI/GenBank database under BioProject numbers PRJNA282695 and PRJNA188291 (http://www.ncbi.nlm.nih.gov/bioproject/?term=282695, http://www.ncbi.nlm.nih.gov/bioproject/PRJNA188291). The genome and annotation of the Fop-37622 were deposited at DDBJ/EMBL/GenBank under the accession number AGBI00000000.1 (http://www.ncbi.nlm.nih.gov/nuccore/AGBI00000000).
Additional files
Summary of conditionally-dispensable chromosomes in Ascomycete plant pathogens. (DOCX 36 kb)
F. oxysporum f. sp. medicaginis ( Fom- 5190a) and F. oxysporum f. sp. ciceris ( Foc -38-1) disease symptoms. (a) Fom-5190a disease symptoms on Medicago truncatula. The left image shows the Fusarium wilt susceptible M. truncatula accession DZA315 at 10 days post inoculation (dpi) displaying wilting leaf symptoms. The centre image shows an uprooted plant from the same experiment at 18 dpi showing stunted, necrotic roots compared to a mock inoculated seedling (right image) at the same time-point. (b) F. oxysporum f. sp. ciceris (Foc-38-1) disease symptoms on Cicer arietinum. The left and centre images show the Fusarium wilt susceptible C. arietinum accession JG 62 at 9 and 18 days post inoculation respectively, with wilting leaf symptoms prominent. A mock/control inoculated seedling (right image) is shown as a reference. (TIF 8644 kb)
Sequencing data used for genome assemblies. (DOCX 13 kb)
Protein/gene set comparisons across Fusarium sp. (DOCX 11 kb)
Summary of genome and protein sequence resources used for analysis. (XLSX 20 kb)
Orthology analysis. (XLSX 23360 kb)
G:C content analysis by scaffold/chromosome for legume-infecting ff. spp., and Fol and F. solani. (XLSX 338 kb)
Repeat analysis. (XLSX 20 kb)
F. solani chromosomes highlighting features of CDCs in comparison to core chromosomes. Outer ring-F. solani chromosomes CDCs highlighted in red. Inner rings: (a) gene density in 100 kb windows, (b) repeat density in 100 kb windows, (c) GC content in 50 k bp windows range 45-55 %, (d) Region of F. solani chromosomes overlapped by Fom-5190a sequences, (e) Foc-38-1, (f) F. oxysporum f. sp. pisi-37622 HDV247, (g) F. oxysporum f. sp. brassica Fo5176, (h) F. oxysporum f. sp. melonis, (i) F. oxysporum f. sp. lycopersici, (j) F. fujikuori, (k) F. verticilliodes, (l) F. virguliforme, and (m) F. graminearum. (TIFF 1823 kb)
Fusarium sequences conserved with core and dispensable chromosomes of Fol and F_solani. (XLSX 40 kb)
Phylogenetic tree illustrating the relationship between the Fusarium sp. isolates used in this study Branch support values are shown in red. This tree illustrates that the legume-infecting isolates do not appear to be more closely related to each other than to other ff. spp. (PDF 21 kb)
Ortholog found only in legume-infecting Fusarium sp. (DOCX 11 kb)
Dispensable scaffold predictions in Fom -5190, Foc -38-1 and Fop -37622. (XLS 2255 kb)
Comparison of differences in length between core and lineage specific scaffolds across Fusarium species. (DOCX 12 kb)
Pfam domains more abundant on predicted dispensable scaffolds in Fom -5190a. (DOCX 18 kb)
Pfam domains more abundant on predicted dispensable scaffolds in Foc -38-1. (DOCX 14 kb)
Pfam domains more abundant on predicted dispensable scaffolds in Fop -37622. (DOCX 16 kb)
Proteins with similarities to known fungal pathogenicity genes and their expression at 2 dpi in Fom -5190a. (DOCX 16 kb)
Properties of predicted Fom -5190a genes/proteins. (XLSX 5099 kb)
Properties of predicted Foc -38-1 genes/proteins. (XLSX 6173 kb)
Properties of predicted Fop-37622 genes/proteins. (XLSX 4021 kb)
Best BLASTP matches of Fom -5190a candidate effector proteins and SIX protein orthologs versus NCBI non-redundant protein database (Feb 2015). (DOCX 15 kb)
Best BLASTP matches of Foc 38-1 and Fop -37622 SIX protein orthologs and predicted Fop _SIX13 and [ _SIX14 aa sequences. (DOCX 18 kb)
{kind=link}
Phylogenetic trees illustrating the relationship between SIX proteins detected as encoded in Fusarium oxysporum f. sp. medicaginis and other ff. spp. (a) SIX1, (b) SIX8, (c) SIX9 and (d) SIX13. The relationship between the SIX proteins suggests a greater similarity between those from the legume–infecting ff. spp. than the phylogenetic analysis based on core proteins identified. (PNG 165 kb)
Disease symptoms and relative abundance of Fom -5190a in infected M. truncatula DZA315 root samples. (a) Disease symptoms of DZA315 plants at 14 days post treatment with Fom-5190a or a control (mock) treatment. (b) Relative Fom-5190a fungal abundance was determined by qRT-PCR expression of Fom-5190a_18S relative to M. truncatula_18S expression in M. truncatula DZA315 root samples harvested at 1, 2, 4 and 7 days post inoculation (dpi). Samples are averages ± SE of 4 biological replicates consisting of pools of 10 seedlings. (TIF 2196 kb)
Primer sequences used for qRT-PCR. (DOCX 12 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AW and JH performed NGS QC, assembled and annotated the Fom and Foc genome sequences, AW, JH and LT conducted the comparative analysis. MS and SA contributed to generation of the Foc genome sequence. LT conducted Fom experiments and prepared DNA and RNA for sequencing. MS and RKV initiated and conducted the Foc experiments, and MS and RS prepared DNA for Foc sequencing. LT and SB performed and analysed the qRT-PCR transcript data. AW and GG assembled and analysed the RNA-seq data. HK, TS, SY and LM produced the genome assembly of Fop. JL provided data and with LK sequenced the Fom isolate. JS conducted the protein phylogeny analysis. BK and JA assisted with Fom experiments. RG assisted and SP provided inputs in Foc experiments. AW, JH and LT wrote the manuscript, KS, RV, MS, RS, JL, BK, JA, LK, JS, HK and LM edited the manuscript. KS, HK, LM and RV conceived the study. All authors read and approved the manuscript.
Contributor Information
Angela H. Williams, Email: Angela.Williams2@csiro.au
Mamta Sharma, Email: mamta.sharma@CGIAR.ORG.
Louise F. Thatcher, Email: Louise.Thatcher@csiro.au
Sarwar Azam, Email: shrisarwarazam@gmail.com.
James K. Hane, Email: james.hane@curtin.edu.au
Jana Sperschneider, Email: Jana.Sperschneider@csiro.au.
Brendan N. Kidd, Email: Brendan.Kidd@csiro.au
Jonathan P. Anderson, Email: Jonathan.Anderson@csiro.au
Raju Ghosh, Email: r.ghosh@cgiar.org.
Gagan Garg, Email: Gagan.Garg@csiro.au.
Judith Lichtenzveig, Email: Judith.Lichtenzveig@curtin.edu.au.
H. Corby Kistler, Email: hckist@umn.edu.
Terrance Shea, Email: tshea@broadinstitute.org.
Sarah Young, Email: sarah@broadinstitute.org.
Sally-Anne G. Buck, Email: 21130902@student.uwa.edu.au
Lars G. Kamphuis, Email: Lars.Kamphuis@csiro.au
Rachit Saxena, Email: R.Saxena@cgiar.org.
Suresh Pande, Email: s.pande@cgiar.org.
Li-Jun Ma, Email: lijun@biochem.umass.edu.
Rajeev K. Varshney, Email: R.K.Varshney@CGIAR.ORG
Karam B. Singh, Email: Karam.Singh@csiro.au
References
- 1.Michielse CB, Rep M. Pathogen profile update: Fusarium oxysporum. Mol Plant Pathol. 2009;10(3):311–24. doi: 10.1111/j.1364-3703.2009.00538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fourie G, Steenkamp ET, Ploetz RC, Gordon TR, Viljoen A. Current status of the taxonomic position of Fusarium oxysporum formae specialis cubense within the Fusarium oxysporum complex. Infec Genet Evol. 2011;11(3):533–42. doi: 10.1016/j.meegid.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Dean R, Van Kan JA, Pretorius ZA, Hammond‐Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J. The Top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol. 2012;13(4):414–30. doi: 10.1111/j.1364-3703.2011.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alabouvette C, Olivain C, Migheli Q, Steinberg C. Microbiological control of soil-borne phytopathogenic fungi with special emphasis on wilt-inducing Fusarium oxysporum. New Phytol. 2009;184(3):529–44. doi: 10.1111/j.1469-8137.2009.03014.x. [DOI] [PubMed] [Google Scholar]
- 5.Baayen RP, O’Donnell K, Bonants PJ, Cigelnik E, Kroon LP, Roebroeck EJ, Waalwijk C. Gene Genealogies and AFLP Analyses in the Fusarium oxysporum Complex Identify Monophyletic and Nonmonophyletic Formae Speciales Causing Wilt and Rot Disease. Phytopathology. 2000;90(8):891–900. doi: 10.1094/PHYTO.2000.90.8.891. [DOI] [PubMed] [Google Scholar]
- 6.Leslie JF. Fungal vegetative compatibility. Annu Rev Phytopathol. 1993;31(1):127–50. doi: 10.1146/annurev.py.31.090193.001015. [DOI] [PubMed] [Google Scholar]
- 7.Stergiopoulos I, de Wit PJ. Fungal effector proteins. Annu Rev Phytopathol. 2009;47:233–63. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- 8.Kamoun S. Groovy times: filamentous pathogen effectors revealed. Curr Opin Plant Biol. 2007;10(4):358–65. doi: 10.1016/j.pbi.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 9.Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464(7287):367–73. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt SM, Lukasiewicz J, Farrer R, van Dam P, Bertoldo C, Rep M. Comparative genomics of Fusarium oxysporum f. sp. melonis reveals the secreted protein recognized by the Fom-2 resistance gene in melon. New Phytologist. 2015;209(1):307–18. doi: 10.1111/nph.13584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, Grimwood J, Schmutz J, Taga M, White GJ, Zhou S, et al. The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 2009;5(8):e1000618. doi: 10.1371/journal.pgen.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Covert SF, Enkerli J, Miao VP, VanEtten HD. A gene for maackiain detoxification from a dispensable chromosome of Nectria haematococca. Mol Gen Genet. 1996;251(4):397–406. doi: 10.1007/BF02172367. [DOI] [PubMed] [Google Scholar]
- 13.Miao VP, Covert SF, VanEtten HD. A fungal gene for antibiotic resistance on a dispensable (“B”) chromosome. Science. 1991;254(5039):1773–6. doi: 10.1126/science.1763326. [DOI] [PubMed] [Google Scholar]
- 14.Zolan ME. Chromosome-length polymorphism in fungi. Microbiol Rev. 1995;59(4):686–98. doi: 10.1128/mr.59.4.686-698.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balesdent MH, Fudal I, Ollivier B, Bally P, Grandaubert J, Eber F, Chèvre AM, Leflon M, Rouxel T. The dispensable chromosome of Leptosphaeria maculans shelters an effector gene conferring avirulence towards Brassica rapa. New Phytol. 2013;198(3):887–98. doi: 10.1111/nph.12178. [DOI] [PubMed] [Google Scholar]
- 16.Mehrabi R, Bahkali AH, Abd-Elsalam KA, Moslem M, Ben M’Barek S, Gohari AM, Jashni MK, Stergiopoulos I, Kema GHJ, de Wit PJGM. Horizontal gene and chromosome transfer in plant pathogenic fungi affecting host range. FEMS Microbiol Rev. 2011;35(3):542–54. doi: 10.1111/j.1574-6976.2010.00263.x. [DOI] [PubMed] [Google Scholar]
- 17.Cuomo CA, Güldener U, Xu J-R, Trail F, Turgeon BG, Di Pietro A, Walton JD, Ma L-J, Baker SE, Rep M. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science. 2007;317(5843):1400–2. doi: 10.1126/science.1143708. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt SM, Houterman PM, Schreiver I, Ma L, Amyotte S, Chellappan B, Boeren S, Takken FL, Rep M. MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genomics. 2013;14(1):119. doi: 10.1186/1471-2164-14-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houterman PM, Speijer D, Dekker HL, de Koster CG, Cornelissen BJ, Rep M. The mixed xylem sap proteome of Fusarium oxysporum-infected tomato plants. Mol Plant Pathol. 2007;8(2):215–21. doi: 10.1111/j.1364-3703.2007.00384.x. [DOI] [PubMed] [Google Scholar]
- 20.Houterman PM, Cornelissen BJ, Rep M. Suppression of plant resistance gene-based immunity by a fungal effector. PLoS Pathog. 2008;4(5):e1000061. doi: 10.1371/journal.ppat.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houterman PM, Ma L, Van Ooijen G, De Vroomen MJ, Cornelissen BJ, Takken FL, Rep M. The effector protein Avr2 of the xylem‐colonizing fungus Fusarium oxysporum activates the tomato resistance protein I‐2 intracellularly. Plant J. 2009;58(6):970–8. doi: 10.1111/j.1365-313X.2009.03838.x. [DOI] [PubMed] [Google Scholar]
- 22.Rep M, Van Der Does HC, Meijer M, Van Wijk R, Houterman PM, Dekker HL, De Koster CG, Cornelissen BJ. A small, cysteine‐rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I‐3‐mediated resistance in tomato. Mol Microbiol. 2004;53(5):1373–83. doi: 10.1111/j.1365-2958.2004.04177.x. [DOI] [PubMed] [Google Scholar]
- 23.Gawehns F, Houterman P, Ichou FA, Michielse C, Hijdra M, Cornelissen B, Rep M, Takken F. The Fusarium oxysporum Effector Six6 Contributes to Virulence and Suppresses I-2-Mediated Cell Death. Mol Plant Microbe Interact. 2014;27(4):336–48. doi: 10.1094/MPMI-11-13-0330-R. [DOI] [PubMed] [Google Scholar]
- 24.Ma L, Houterman PM, Gawehns F, Cao L, Sillo F, Richter H, Clavijo-Ortiz MJ, Schmidt SM, Boeren S, Vervoort J, et al. The AVR2–SIX5 gene pair is required to activate I-2-mediated immunity in tomato. New Phytol. 2015;208(2):507–18. doi: 10.1111/nph.13455. [DOI] [PubMed] [Google Scholar]
- 25.Nene Y. Multiple-disease resistance in grain legumes. Annu Rev Phytopathol. 1988;26(1):203–17. doi: 10.1146/annurev.py.26.090188.001223. [DOI] [Google Scholar]
- 26.Rubiales D, Fondevilla S, Chen W, Gentzbittel L, Higgins TJ, Castillejo MA, Singh KB, Rispail N. Achievements and challenges in legume breeding for pest and disease resistance. Crit Rev Plant Sci. 2015;34(1–3):195–236. doi: 10.1080/07352689.2014.898445. [DOI] [Google Scholar]
- 27.Rubiales D, Mikic A. Introduction: Legumes in sustainable agriculture. Crit Rev Plant Sci. 2015;34(1–3):2–3. doi: 10.1080/07352689.2014.897896. [DOI] [Google Scholar]
- 28.Emberger G, Welty R. Evaluation of virulence of Fusarium oxysporum f. sp. medicaginis and Fusarium Wilt resistance in alfalfa. Plant Dis. 1983;67(1):94–8. doi: 10.1094/PD-67-94. [DOI] [Google Scholar]
- 29.Jiménez-Díaz RM, Basallote-Urebra MJ, Rapoport H. Colonization and pathogenesis in chickpeas infected by races of Fusarium oxysporum f. sp. cicero. In: Tjamos EC, Beckman CH, editors. Vascular wilt diseases of plants. Springer Verlag; 1989. p. 113–21.
- 30.Navas-Cortés JA, Hau B, Jiménez-Díaz RM. Yield loss in chickpeas in relation to development of Fusarium wilt epidemics. Phytopathology. 2000;90(11):1269–78. doi: 10.1094/PHYTO.2000.90.11.1269. [DOI] [PubMed] [Google Scholar]
- 31.Landa BB, Navas-Cortés JA, del Mar J-GM, Katan J, Retig B, Jiménez-Díaz RM. Temperature response of chickpea cultivars to races of Fusarium oxysporum f. sp. ciceris, causal agent of Fusarium wilt. Plant Dis. 2006;90(3):365–74. doi: 10.1094/PD-90-0365. [DOI] [PubMed] [Google Scholar]
- 32.Ramírez-Suero M, Khanshour A, Martinez Y, Rickauer M. A study on the susceptibility of the model legume plant Medicago truncatula to the soil-borne pathogen Fusarium oxysporum. Eur J Plant Pathol. 2010;126(4):517–30. doi: 10.1007/s10658-009-9560-x. [DOI] [Google Scholar]
- 33.Abera M, Sakhuja PK, Fininsa C, Ahmed S. Status of chickpea fusarium wilt (Fusarium oxysporum f. sp. ciceris) in northwestern Ethiopia. Archives Phytopathology Plant Protect. 2011;44(13):1261–72. doi: 10.1080/03235408.2010.490410. [DOI] [Google Scholar]
- 34.Trapero-Casas A, Jiménez-Díaz RM. Fungal wilt and root rot diseases of chickpea in southern Spain. Phytopathology. 1985;75(1):146–1151. [Google Scholar]
- 35.Haware M, Nene Y, Rajeshwari R. Eradication of Fusarium oxysporum f. sp. ciceri transmitted in chickpea seed. Phytopathology. 1978;68(9):1364–7. doi: 10.1094/Phyto-68-1364. [DOI] [Google Scholar]
- 36.Haware M, Nene Y. Races of Fusarium oxysporum f. sp. ciceri. Plant Dis. 1982;66(9):809–10. doi: 10.1094/PD-66-809. [DOI] [Google Scholar]
- 37.Jiménez‐Gasco M, Milgroom M, Jiménez‐Díaz R. Gene genealogies support Fusarium oxysporum f. sp. ciceris as a monophyletic group. Plant Pathol. 2002;51(1):72–7. doi: 10.1046/j.0032-0862.2001.00610.x-i1. [DOI] [Google Scholar]
- 38.Chatterjee M, Gupta S, Bhar A, Chakraborti D, Basu D, Das S. Analysis of root proteome unravels differential molecular responses during compatible and incompatible interaction between chickpea (Cicer arietinum L.) and Fusarium oxysporum f. sp. ciceri Race1 (Foc1) BMC Genomics. 2014;15(1):949. doi: 10.1186/1471-2164-15-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiménez-Gasco MM, Milgroom MG, Jiménez-Díaz RM. Stepwise evolution of races in Fusarium oxysporum f. sp. ciceris inferred from fingerprinting with repetitive DNA sequences. Phytopathology. 2004;94(3):228–35. doi: 10.1094/PHYTO.2004.94.3.228. [DOI] [PubMed] [Google Scholar]
- 40.Halila M, Strange R. Identification of the causal agent of wilt of chickpea in Tunisia as Fusarium oxysporum f. sp. ciceri race 0. Phytopathol Mediterr. 1996;67–74.
- 41.Haware M, Jimenez-Diaz R, Amin K, Phillips J, Halila H: Integrated management of wilt and root rots of chickpea. Chickpea in the Nineties: Proceedings of the second international work shop on chickpea improvement, Patancheru, India 1990:129–137.
- 42.Infantino A, Kharrat M, Riccioni L, Coyne C, McPhee K, Grünwald N. Screening techniques and sources of resistance to root diseases in cool season food legumes. Euphytica. 2006;147(1–2):201–21. doi: 10.1007/s10681-006-6963-z. [DOI] [Google Scholar]
- 43.Arcioni S, Damiani F, Pezzotti M, Lupotto E. Legumes and Oilseed Crops I. Berlin Heidelberg: Springer-Verlag; 1990. Alfalfa, lucerne (Medicago spp.) pp. 197–241. [Google Scholar]
- 44.Cook DR. Medicago truncatula—a model in the making!: Commentary. Curr Opin Plant Biol. 1999;2(4):301–4. doi: 10.1016/S1369-5266(99)80053-3. [DOI] [PubMed] [Google Scholar]
- 45.Cash D: Chapter 1. Global Status and Development Trends of Alfalfa. Alfalfa Management Guide for Ningxia United Nations Food and Agriculture Organization 2009:1–2.
- 46.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23(9):1061–7. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 47.Rouxel T, Grandaubert J, Hane JK, Hoede C, van de Wouw AP, Couloux A, Dominguez V, Anthouard V, Bally P, Bourras S, et al. Effector diversification within compartments of the Leptosphaeria maculans genome affected by Repeat-Induced Point mutations. Nat Commun. 2011;2:202. doi: 10.1038/ncomms1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodwin SB, M’Barek SB, Dhillon B, Wittenberg AH, Crane CF, Hane JK, Foster AJ, Van der Lee TA, Grimwood J, Aerts A, et al. Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 2011;7(6):e1002070. doi: 10.1371/journal.pgen.1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oliver R. Genomic tillage and the harvest of fungal phytopathogens. New Phytol. 2012;196(4):1015–23. doi: 10.1111/j.1469-8137.2012.04330.x. [DOI] [PubMed] [Google Scholar]
- 50.Stukenbrock EH. Evolution, selection and isolation: a genomic view of speciation in fungal plant pathogens. New Phytol. 2013;199(4):895–907. doi: 10.1111/nph.12374. [DOI] [PubMed] [Google Scholar]
- 51.Wöstemeyer J, Kreibich A. Repetitive DNA elements in fungi (Mycota): impact on genomic architecture and evolution. Curr Genet. 2002;41(4):189–98. doi: 10.1007/s00294-002-0306-y. [DOI] [PubMed] [Google Scholar]
- 52.Han Y, Liu X, Benny U, Kistler HC, VanEtten HD. Genes determining pathogenicity to pea are clustered on a supernumerary chromosome in the fungal plant pathogen Nectria haematococca. Plant J. 2001;25(3):305–14. doi: 10.1046/j.1365-313x.2001.00969.x. [DOI] [PubMed] [Google Scholar]
- 53.Covey PA, Kuwitzky B, Hanson M, Webb K. Multilocus analysis using putative fungal effectors to describe a population of Fusarium oxysporum from sugar beet. Phytopathology. 2014;104(8):886–96. doi: 10.1094/PHYTO-09-13-0248-R. [DOI] [PubMed] [Google Scholar]
- 54.Langin T, Capy P, Daboussi M-J. The transposable element impala, a fungal member of the Tc1-mariner superfamily. Mol Gen Genet. 1995;246(1):19–28. doi: 10.1007/BF00290129. [DOI] [PubMed] [Google Scholar]
- 55.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Covert SF. Supernumerary chromosomes in filamentous fungi. Curr Genet. 1998;33(5):311–9. doi: 10.1007/s002940050342. [DOI] [PubMed] [Google Scholar]
- 57.Thatcher LF, Gardiner DM, Kazan K, Manners JM. A highly conserved effector in Fusarium oxysporum is required for full virulence on Arabidopsis. Mol Plant Microbe Interact. 2012;25(2):180–90. doi: 10.1094/MPMI-08-11-0212. [DOI] [PubMed] [Google Scholar]
- 58.Chakrabarti A, Rep M, Wang B, Ashton A, Dodds P, Ellis J. Variation in potential effector genes distinguishing Australian and non‐Australian isolates of the cotton wilt pathogen Fusarium oxysporum f. sp. vasinfectum. Plant Pathol. 2011;60(2):232–43. doi: 10.1111/j.1365-3059.2010.02363.x. [DOI] [Google Scholar]
- 59.Fraser‐Smith S, Czislowski E, Meldrum R, Zander M, O’Neill W, Balali G, Aitken E. Sequence variation in the putative effector gene SIX8 facilitates molecular differentiation of Fusarium oxysporum f. sp. cubense. Plant Pathol. 2014;63(5):1044–1052. doi: 10.1111/ppa.12184. [DOI] [Google Scholar]
- 60.Lievens B, Houterman PM, Rep M. Effector gene screening allows unambiguous identification of Fusarium oxysporum f. sp. lycopersici races and discrimination from other formae speciales. FEMS Microbiol Lett. 2009;300(2):201–15. doi: 10.1111/j.1574-6968.2009.01783.x. [DOI] [PubMed] [Google Scholar]
- 61.Meldrum R, Fraser-Smith S, Tran-Nguyen L, Daly A, Aitken E. Presence of putative pathogenicity genes in isolates of Fusarium oxysporum f. sp. cubense from Australia. Australasian Plant Pathol. 2012;41(5):551–7. doi: 10.1007/s13313-012-0122-x. [DOI] [Google Scholar]
- 62.Laurence MH, Summerell BA, Liew ECY. Fusarium oxysporum f. sp. canariensis: evidence for horizontal gene transfer of putative pathogenicity genes. Plant Pathol. 2015;64(5):1068–1075. doi: 10.1111/ppa.12350. [DOI] [Google Scholar]
- 63.Temporini ED, VanEtten HD. An analysis of the phylogenetic distribution of the pea pathogenicity genes of Nectria haematococca MPVI supports the hypothesis of their origin by horizontal transfer and uncovers a potentially new pathogen of garden pea: Neocosmospora boniensis. Curr Genet. 2004;46(1):29–36. doi: 10.1007/s00294-004-0506-8. [DOI] [PubMed] [Google Scholar]
- 64.Coleman JJ, Wasmann CC, Usami T, White GJ, Temporini ED, McCluskey K, VanEtten HD. Characterization of the Gene Encoding Pisatin Demethylase (FoPDA 1) in Fusarium oxysporum. Mol Plant Microbe Interact. 2011;24(12):1482–91. doi: 10.1094/MPMI-05-11-0119. [DOI] [PubMed] [Google Scholar]
- 65.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J. Pfam: the protein families database. Nucleic Acids Res. 2013;42(DI):D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koonin EV, Aravind L. The NACHT family – a new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem Sci. 2000;25(5):223–4. doi: 10.1016/S0968-0004(00)01577-2. [DOI] [PubMed] [Google Scholar]
- 67.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37(suppl 1):D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–5. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gomez-Gomez E, Anaya N, Roncero MI, Hera C. Folyt1, a new member of the hAT family, is active in the genome of the plant pathogen Fusarium oxysporum. Fungal Genet Biol. 1999;27(1):67–76. doi: 10.1006/fgbi.1999.1132. [DOI] [PubMed] [Google Scholar]
- 70.Kessmann H, Edwards R, Geno PW, Dixon RA. Stress responses in alfalfa (Medicago sativa L.) V. Constitutive and elicitor-induced accumulation of isoflavonoid conjugates in cell suspension cultures. Plant Physiol. 1990;94(1):227–32. doi: 10.1104/pp.94.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Delserone LM, McCluskey K, Matthews D, VanEtten H. Pisatin demethylation by fungal pathogens and nonpathogens of pea: association with pisatin tolerance and virulence. Physiol Mol Plant Pathol. 1999;55(6):317–26. doi: 10.1006/pmpp.1999.0237. [DOI] [Google Scholar]
- 72.Higgins VJ. Role of the phytoalexin medicarpin in three leaf spot diseases of alfalfa. Physiol Plant Pathol. 1972;2(3):289–300. doi: 10.1016/0048-4059(72)90012-4. [DOI] [Google Scholar]
- 73.Duczek LJ, Higgins VJ. Effect of treatment with the phytoalexins medicarpin and maackiain on fungal growth in vitro and in vivo. Can J Bot. 1976;54(23):2620–9. doi: 10.1139/b76-282. [DOI] [Google Scholar]
- 74.Denny TP, VanEtten HD. Tolerance by Nectria haematococca MP VI of the chickpea (Cicer arietinum) phytoalexins medicarpin and maackiain. Physiol Plant Pathol. 1981;19(3):419–37. doi: 10.1016/S0048-4059(81)80073-2. [DOI] [Google Scholar]
- 75.Delserone LM, Matthews DE, VanEtten HD. Differential toxicity of enantiomers of maackiain and pisatin to phytopathogenic fungi. Phytochemistry. 1992;31(11):3813–9. doi: 10.1016/S0031-9422(00)97534-4. [DOI] [Google Scholar]
- 76.Wasmann C, VanEtten HD. Transformation-mediated chromosome loss and disruption of a gene for pisatin demethylase decrease the virulence of Nectria haematococca on pea. Mol Plant Microbe Interact. 1996;9(9):793–803. doi: 10.1094/MPMI-9-0793. [DOI] [Google Scholar]
- 77.Reimmann C, VanEtten HD. Cloning and characterization of the PDA6-1 gene encoding a fungal cytochrome P-450 which detoxifies the phytoalexin pisatin from garden pea. Gene. 1994;146(2):221–6. doi: 10.1016/0378-1119(94)90296-8. [DOI] [PubMed] [Google Scholar]
- 78.Weltring KM, Turgeon BG, Yoder OC, VanEtten HD. Isolation of a phytoalexin-detoxification gene from the plant pathogenic fungus Nectria haematococca by detecting its expression in Aspergillus nidulans. Gene. 1988;68(2):335–44. doi: 10.1016/0378-1119(88)90036-4. [DOI] [PubMed] [Google Scholar]
- 79.Liu X, Inlow M, VanEtten HD. Expression profiles of pea pathogenicity (PEP) genes in vivo and in vitro, characterization of the flanking regions of the PEP cluster and evidence that the PEP cluster region resulted from horizontal gene transfer in the fungal pathogen Nectria haematococca. Curr Genet. 2003;44(2):95–103. doi: 10.1007/s00294-003-0428-x. [DOI] [PubMed] [Google Scholar]
- 80.Rocha LO, Laurence MH, Ludowici VA, Puno VI, Lim CC, Tesoriero LA, Summerell BA, Liew ECY: Putative effector genes detected in Fusarium oxysporum from natural ecosystems of Australia. Plant Pathol. 2015:online early view 6 Nov Doi:10.1111/ppa.12472.
- 81.Milani NA, Lawrence DP, Elizabeth Arnold A, VanEtten HD. Origin of pisatin demethylase (PDA) in the genus Fusarium. Fungal Genet Biol. 2012;49(11):933–42. doi: 10.1016/j.fgb.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 82.Enkerli J, Bhatt G, Covert SF. Nht1, a transposable element cloned from a dispensable chromosome in Nectria haematococca. Mol Plant Microbe Interact. 1997;10(6):742–9. doi: 10.1094/MPMI.1997.10.6.742. [DOI] [PubMed] [Google Scholar]
- 83.de Wit PJ, Mehrabi R, van den Burg HA, Stergiopoulos I. Fungal effector proteins: past, present and future. Mol Plant Pathol. 2009;10(6):735–47. doi: 10.1111/j.1364-3703.2009.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rep M. Small proteins of plant‐pathogenic fungi secreted during host colonization. FEMS Microbiol Lett. 2005;253(1):19–27. doi: 10.1016/j.femsle.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 85.Wiemann P, Sieber CMK, von Bargen KW, Studt L, Niehaus E-M, Espino JJ, Huß K, Michielse CB, Albermann S, Wagner D, et al. Deciphering the Cryptic Genome: Genome-wide Analyses of the Rice Pathogen Fusarium fujikuroi Reveal Complex Regulation of Secondary Metabolism and Novel Metabolites. PLoS Pathog. 2013;9(6):e1003475. doi: 10.1371/journal.ppat.1003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Parada R, Sakuno E, Mori N, Oka K, Egusa M, Kodama M, Otani H. Alternaria brassicae produces a host-specific protein toxin from germinating spores on host leaves. Phytopathology. 2008;98(4):458–63. doi: 10.1094/PHYTO-98-4-0458. [DOI] [PubMed] [Google Scholar]
- 87.Catanzariti A-M, Jones DA. Effector proteins of extracellular fungal plant pathogens that trigger host resistance. Funct Plant Biol. 2010;37(10):901–6. doi: 10.1071/FP10077. [DOI] [Google Scholar]
- 88.Johnson LS, Eddy SR, Portugaly E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics. 2010;11(1):431. doi: 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bolton MD, Van Esse HP, Vossen JH, De Jonge R, Stergiopoulos I, Stulemeijer IJ, Van Den Berg G, Borrás‐Hidalgo O, Dekker HL, De Koster CG. The novel Cladosporium fulvum lysin motif effector Ecp6 is a virulence factor with orthologues in other fungal species. Mol Microbiol. 2008;69(1):119–36. doi: 10.1111/j.1365-2958.2008.06270.x. [DOI] [PubMed] [Google Scholar]
- 90.Kistler H, Rep M, Ma L. Structural dynamics of Fusarium genomes. In: Brown DW, Proctor RH, editors. Fusarium: genomics, molecular and cellular biology. Norwich: Horizon Scientific Press; 2013. [Google Scholar]
- 91.Lievens B, Rep M, Thomma BP. Recent developments in the molecular discrimination of formae speciales of Fusarium oxysporum. Pest Manag Sci. 2008;64(8):781–8. doi: 10.1002/ps.1564. [DOI] [PubMed] [Google Scholar]
- 92.Gao L-L, Hane JK, Kamphuis LG, Foley R, Shi B-J, Atkins CA, Singh KB. Development of genomic resources for the narrow-leafed lupin (Lupinus angustifolius): construction of a bacterial artificial chromosome (BAC) library and BAC-end sequencing. BMC Genomics. 2011;12(1):521. doi: 10.1186/1471-2164-12-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–2. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 94.Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–63. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl Environ Microbiol. 2009;75(23):7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1(1):18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27(4):578–9. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 98.Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci. 2011;108(4):1513–8. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18(12):1979–90. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guigó R, Knudsen S, Drake N, Smith T. Prediction of gene structure. J Mol Biol. 1992;226(1):141–157. doi: 10.1016/0022-2836(92)90130-C. [DOI] [PubMed] [Google Scholar]
- 101.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(suppl 2):ii215–ii225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 102.Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20(16):2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- 103.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5(1):1–9. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, Jr, Hannick LI, Maiti R, Ronning CM, Rusch DB, Town CD, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31(19):5654–5666. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 106.Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, Consortium tU UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31(6):926–932. doi: 10.1093/bioinformatics/btu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ma L-J, Shea T, Young S, Zeng Q, Kistler HC. Genome Sequence of Fusarium oxysporum f. sp. melonis Strain NRRL 26406, a Fungus Causing Wilt Disease on Melon. Genome Announc. 2014;2(4):e00730–14. doi: 10.1128/genomeA.00730-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Smit A, Hubley R, Green P. RepeatMasker Open-3.0. URL: http://www.repeatmasker.org, 1996–2010.
- 109.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Winnenburg R, Baldwin TK, Urban M, Rawlings C, Köhler J, Hammond-Kosack KE. PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 2006;34(suppl 1):D459–64. doi: 10.1093/nar/gkj047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Winnenburg R, Urban M, Beacham A, Baldwin TK, Holland S, Lindeberg M, Hansen H, Rawlings C, Hammond-Kosack KE, Köhler J. PHI-base update: additions to the pathogen–host interaction database. Nucleic Acids Res. 2008;36(suppl 1):D572–6. doi: 10.1093/nar/gkm858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9(1):R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pazzagli L, Cappugi G, Manao G, Camici G, Santini A, Scala A. Purification, characterization, and amino acid sequence of cerato-platanin, a new phytotoxic protein from Ceratocystis fimbriata f. sp. platani. J Biol Chem. 1999;274(35):24959–64. doi: 10.1074/jbc.274.35.24959. [DOI] [PubMed] [Google Scholar]
- 115.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1–4):462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 116.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(suppl 1):i351–8. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 117.Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9(9):868–77. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25(10):1335–7. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37(suppl 1):D136–40. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31(1):439–41. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33(suppl 1):D121–4. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–8. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):0955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server Issue):W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33(suppl 2):W116–20. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zdobnov EM, Apweiler R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17(9):847–8. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 127.Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40(W1):W445–51. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND. SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47(9):736–41. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Horton P, Park K-J, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35(suppl 2):W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Käll L, Krogh A, Sonnhammer ELL. A Combined Transmembrane Topology and Signal Peptide Prediction Method. J Mol Biol. 2004;338(5):1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 131.Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC, Dhillon B, Glaser F. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 2012;8(12):e1003037. doi: 10.1371/journal.ppat.1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 133.Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 134.Lechner M, Findeiß S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: Detection of (Co-) orthologs in large-scale analysis. BMC Bioinformatics. 2011;12(1):124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539–9. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Huerta-Cepas J, Dopazo J, Gabaldon T. ETE: a python Environment for Tree Exploration. BMC Bioinformatics. 2010;11(1):24. doi: 10.1186/1471-2105-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Oñate-Sánchez L, Anderson JP, Young J, Singh KB. AtERF14, a member of the ERF family of transcription factors, plays a nonredundant role in plant defense. Plant Physiol. 2007;143(1):400–9. doi: 10.1104/pp.106.086637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gao L-L, Anderson JP, Klingler JP, Nair RM, Edwards OR, Singh KB. Involvement of the octadecanoid pathway in bluegreen aphid resistance in Medicago truncatula. Mol Plant Microbe Interact. 2007;20(1):82–93. doi: 10.1094/MPMI-20-0082. [DOI] [PubMed] [Google Scholar]
- 140.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]