

Abstract
Leprosy is a chronic disease characterized by skin and peripheral nerve pathology and immune responses that fail to control Mycobacterium leprae. Toll-interacting protein (TOLLIP) regulates Toll-like receptor (TLR) and interleukin 1 receptor (IL-1R) signaling against mycobacteria. We analyzed messenger RNA (mRNA) expression of candidate immune genes in skin biopsy specimens from 85 individuals with leprosy. TOLLIP mRNA was highly and specifically correlated with IL-1R antagonist (IL-1Ra). In a case-control gene-association study with 477 cases and 1021 controls in Nepal, TOLLIP single-nucleotide polymorphism rs3793964 TT genotype was associated with increased susceptibility to leprosy (recessive, P = 1.4 × 10−3) and with increased skin expression of TOLLIP and IL-1Ra. Stimulation of TOLLIP-deficient monocytes with M. leprae produced significantly less IL-1Ra (P < .001), compared with control. These data suggest that M. leprae upregulates IL-1Ra by a TOLLIP-dependent mechanism. Inhibition of TOLLIP may decrease an individual's susceptibility to leprosy and offer a novel therapeutic target for IL-1–dependent diseases.
Keywords: leprosy, Mycobacterium leprae, IL-1, IL-1Ra, IL-1 receptor, TLR regulation, TOLLIP, Skin immunity, immune evasion, genetics
Leprosy is one of the world's oldest recorded diseases yet remains a public health problem, with >200 000 new cases in 2013 [1, 2]. Development of new therapeutics to shorten the duration of treatment and reduce inflammatory side effects involved in neuropathology could help accomplish these goals and significantly reduce morbidity. The immune pathogenesis of leprosy remains poorly understood, partly owing to experimental challenges [3, 4], including slow mycobacterial growth, challenging bacterial culture protocols, and limitations within animal models [5, 6].
Leprosy is characterized by a range of clinical and immune responses that depend upon host genetics and environmental factors. Histologic features of these lesions are classified into 5 categories: tuberculoid, borderline tuberculoid, borderline, borderline lepromatous, and lepromatous [7]. Classically, tuberculoid and borderline tuberculoid leprosy (hereafter, “tuberculoid leprosy”) skin lesions are immunologically characterized with elevated T-helper type 1 (TH1) memory responses, with well-circumscribed granulomatous inflammation and few bacilli detectable. Borderline lepromatous and lepromatous (hereafter, “lepromatous leprosy”) skin lesions demonstrate TH2 immune responses, with ill-defined granulomas and many bacilli within dermal foamy macrophages.
One of the defining characteristics of M. leprae as an infectious pathogen is its ability to evade host immunity. M. leprae can persist subclinically for decades, evading both innate and antigen-specific immunity. To avoid immune detection, M. leprae inhibits activation and maturation of dendritic cells, induces microRNAs that downregulate host immunity, and encodes cell wall lipids that impair immune responses [8–11]. Cell wall lipids in M. leprae further suppress T-cell differentiation and activation [12]. Additionally, M. leprae acts to inhibit apoptosis in myeloid cells by manipulating multiple critical transcription factors [13]. However, the complete range of factors that permit leprosy to remain quiescent within the skin for years are still under investigation.
Host genetics play a critical role in the pathogenesis of leprosy, with most people demonstrating innate resistance to M. leprae infection [14–17]. Multiple lines of evidence support this hypothesis, including twin [18], linkage [19], and gene association studies [16, 20]. A recent genome-wide association study found several major leprosy susceptibility loci within genes related to induction of inflammasomes and interleukin 1 (IL-1) signaling [21, 22]. Evasion of IL-1 may improve M. leprae survival in macrophages and nerves. IL-1 protects the host during the initial stages of Mycobacterium tuberculosis infection and correlates with pulmonary tuberculosis severity [23]. IL-1 signaling is tightly regulated at multiple levels, including by IL-1 receptor antagonist (IL-1Ra; also abbreviated “IL1RN,” by the Human Genome Organisation), which binds IL-1 receptor (IL-1R) without inducing signaling [24, 25]. Unlike IL-1β, which is induced primarily via inflammasomes, IL-1Ra is induced by varied stimuli, including lipopolysaccharide (LPS), immunoglobulin G complexes, and IL-1 itself [26, 27]. M. leprae may preferentially induce antiinflammatory cytokines [28–30], and a better understanding of how IL-1 alters leprosy pathogenesis may illuminate novel host-directed therapeutic strategies.
Toll-interacting protein (TOLLIP) regulates Toll-like receptor (TLR) and IL-1R signaling in a complex fashion that is only partially understood [31, 32]. TOLLIP regulates an antiinflammatory bias in the cytokine response after TLR2 and TLR4 signaling, characterized by increased interleukin 10 (IL-10) and decreased interleukin 6 expression in peripheral blood monocytes [33]. We discovered that TOLLIP genetic variants are strongly associated with pulmonary and meningeal tuberculosis in an adult Vietnamese population [33]. Although previous data suggest that TOLLIP regulates IL-1R signaling by altering its location within endosomes [34], the specific mechanism by which it mediates risk against mycobacteria is unclear.
In this article, we demonstrate that TOLLIP and IL-1Ra expression are highly correlated in skin leprosy lesions and show that genetic variation in TOLLIP is associated with leprosy susceptibility, TOLLIP messenger RNA (mRNA) expression, and IL-1Ra expression. We also describe that M. leprae induces IL-1Ra via a TOLLIP-dependent mechanism. Together, these data define a novel mechanism for M. leprae immune evasion by selective induction of IL-1Ra, increasing the dose of IL-1 required to induce potent antimycobacterial immunity in the skin.
MATERIALS AND METHODS
Ethics Statement
All human subjects gave their informed consent to participate in the studies. No children were enrolled in this study. Informed consent was obtained orally, as well as via written communication, owing to the high rates of illiteracy in the populations studied. Subjects who were unable to read and write provided a thumbprint as a proof of consent, while those who could read and write provided a signature. The Nepal Health Research Council and the University of Washington institutional review boards approved all informed consent documents and procedures, according to Department of Health and Human Services guidelines.
Study Population
Peripheral blood and skin biopsy specimens were obtained from patients at Anandaban Hospital in Kathmandu, Nepal. The cases comprised individuals of 8 different ethnic and religious groups, including Vaishya, Chhetri, Brahmin, and Sudra. A total of 477 healthy adult controls were compared to 1021 individuals with leprosy in the candidate gene case-control study. Unrelated controls were recruited from the same ethnic population and geographic region of Nepal. Controls were healthy individuals who had never had tuberculosis, had no history of leprosy in the family, and were living in a leprosy-endemic area. Leprosy diagnosis and classification were determined after a comprehensive clinical examination, slit skin smear, and skin biopsy assessment, using the Ridley–Jopling classification system [7]. We also evaluated cutaneous immune responses in 85 prospectively enrolled subjects with leprosy. This cohort included 38 individuals with tuberculoid leprosy, 3 with borderline leprosy, and 44 with lepromatous leprosy. Thirty-six patients had a type 1 immune reaction, and 9, all with lepromatous leprosy, had a type II immune reaction at the time of biopsy. Bacillary index was measured and correlated strongly with clinical polarity of leprosy presentation (Supplementary Table 1). Although BCG vaccination for the participants was not recorded, many individuals in Nepal have received this vaccine. One of the biopsy samples was used for histological diagnosis and immunohistochemical analysis, and the other was used for mRNA isolation. Biopsy specimens were fixed, mounted in paraffin, and stained with Fite stain, hematoxylin, and eosin for light microscopy viewing by experienced leprosy pathologists at the Research and Leprosy Centre, Schieffelin Institute of Health (Karigiri, India).
Genotyping and SNP Selection
DNA from subjects in Nepal was obtained by extraction from whole blood, using Nucleon BACC2 Genomic DNA (Amersham Lifesciences) and Roche High-Pure PCR template extraction kits. We identified haplotype-tagging SNPs from Han Chinese in Beijing populations from the International HapMap Project (available at: http://www.hapmap.org) and the Genome Variation Server. We searched a region on chromosome 11p15.5, 10 kb upstream and downstream of TOLLIP, for tagged SNPs with an R2 linkage disequilibrium of >0.8 and minor allele frequency of >0.05. Genotyping was performed with Sequenom's MassARRAY. Selected SNPs were confirmed using Taqman genotyping technology (Applied Biosystems). The Stata/Intercooled, version 13.0, software program PWLD (StataCorp) was used to calculate R2 linkage disequilibrium between the polymorphisms. SNPs were excluded from further analysis if Hardy–Weinberg equilibrium in control samples had a P value of < .001.
Statistics
For cutaneous expression correlation, the nonparametric Spearman rank correlation test statistic (ρ) was used for a correlation coefficient and was generated using R, version 3.0.1. Hierarchical clustering of the Spearman ρ statistics was performed using the complete-linkage method, which clusters results of individual tests on the basis of the maximum distance between test results. The Mann–Whitney U test was used to make comparisons of the cytokine and mRNA responses between groups of individuals, as small sample sizes prevented the assumption of a normal distribution. It was also used to compare cytokine mRNA levels between genotypes. A P value of <.05 was considered significant in the initial analysis. Thresholds for achieving statistical significance were adjusted using a Bonferroni correction for multiple comparisons. Comparisons between cytokine responses in cell lines were made using 2-sided Student t tests.
Supplemental Methods
For further description of molecular materials and methods, please see the Supplementary Materials and Methods.
RESULTS
Correlation of TOLLIP and IL-1Ra Expression in Leprosy Skin Biopsy Specimens
To examine the TOLLIP-dependent immune response in humans, we examined whether cutaneous mRNA levels of TOLLIP were associated with expression of other immune molecules in 85 Nepalese patients with leprosy in a range of clinical states (38 with tuberculoid leprosy and 47 with borderline or lepromatous leprosy; Supplementary Table 1). These molecules included cytokines, chemokines, and lineage markers of T cells, monocytes, and macrophages that are important for mycobacterial control, as described recently [35]. Using hierarchical clustering, we found that TOLLIP expression most strongly correlated with IL-1RN (IL-1Ra; ρ = 0.934) and IL-18 (ρ = 0.18; Figure 1A–C). TOLLIP expression was not associated with IL1B (Figure 1D) or any other gene transcripts that were present in independent clusters, including (1) FOXP3, NLRC4, CCL18, CD209, CD14, and IL10; (2) IL1B, IL6, CD22, IL23A, IL12B, TNF, CD3D, and CIITA; (3) IFNA1, IFNA8, IFNB1, and CCL17; (4) IL13, IL22, IL29, and IL4; (5) IL17A and IL21; and (6) IL27, IFNG, and IL12A. Because the degree of correlation of TOLLIP with IL-1Ra was much greater than that of TOLLIP with IL18, we declined to further evaluate the role of TOLLIP expression on IL18. We did not observe any statistically significant association between TOLLIP or IL-1Ra mRNA expression levels and leprosy polarity. We performed immunohistochemical analysis to examine IL-1Ra and TOLLIP protein expression in 50 skin biopsy specimens, taken to represent the full range of clinical disease seen. Both TOLLIP and IL-1Ra staining were found predominantly in the cytoplasm of dermal histiocytes. TOLLIP and IL-1Ra protein staining intensity correlated significantly with mRNA (Figure 2A–F, which includes representative samples with high [Figure 2A] and low [Figure 2B] protein staining; P = .0031 and < .0001, by linear regression, for IL-1Ra and TOLLIP correlation, respectively; Figure 2C and 2D). Protein expression of TOLLIP and IL-1Ra were correlated significantly (P = .0446, by linear regression; data not shown). Negative controls demonstrated no background staining (Supplementary Figure 1). Together, these data suggest that TOLLIP and IL-1Ra expression are highly correlated in cutaneous leprosy lesions.
Figure 1.

Hierarchical clustering of Toll-interacting protein (TOLLIP) with cytokines in leprosy skin biopsy specimens. Heat map of 32 immune genes and TOLLIP in skin biopsy specimens from individuals with leprosy. Correlation was performed in 85 skin biopsy specimens from individuals with leprosy encompassing all categories of disease and including those with immune reactions. Statistical analyses were performed using a nonparametric Spearman rank correlation test. A, TOLLIP formed a cluster with interleukin 1 receptor antagonist (IL-1Ra; ρ = 0.93) and interleukin 18 (IL-18; ρ = 0.18). Heat map values extend from red to blue, with the reddest values denoting a ρ of −1, indicating a strong negative correlation between expression levels, and the bluest values representing a ρ of 1, indicating a strong positive correlation between expression values. B, Correlation scatterplot of messenger RNA (mRNA) expression of TOLLIP and IL-1Ra. C, Correlation scatterplot of mRNA expression of TOLLIP and IL-18, which was not as strong as that for IL-1Ra. D, Correlation scatterplot of TOLLIP with IL-1β, which was not clustered with TOLLIP. mRNA expression is shown in log10 scale after normalization with GAPDH. This figure is available in black and white in print and in color online.
Figure 2.

Toll-interacting protein (TOLLIP) and interleukin 1 receptor antagonist (IL-1Ra) immunohistochemical findings. Skin biopsy specimens were obtained from lesions in individuals with leprosy and stained for TOLLIP and IL-1Ra. A and B, A skin biopsy specimen from a representative individual with 3+ TOLLIP and 3+ IL-1Ra staining (A) and 1+ TOLLIP and 1+ IL-1Ra staining (B). C, Plot of the IL-1Ra messenger RNA level, normalized to GAPDH, compared to the IL-1Ra immunohistochemical grade staining score (1+–3+). IL-Ra protein expression and mRNA expression were correlated by linear regression. *P = .0031. D, The TOLLIP mRNA level, normalized to GAPDH, was compared to the TOLLIP immunohistochemical grade staining score (1+–3+). TOLLIP protein expression and mRNA expression were correlated by linear regression. **P < .0001.
TOLLIP Variant rs3793964 Is Associated With Leprosy Susceptibility
To assess the role of TOLLIP genetic variation in cutaneous immune responses and susceptibility to leprosy, we performed a candidate gene case-control study. We examined whether 6 haplotype-tagging TOLLIP SNPs were associated with susceptibility to leprosy in 1021 leprosy cases and 477 healthy controls (Figure 3A and 3B, Supplementary Table 2, and Table 1). Three polymorphisms were associated with susceptibility to leprosy (rs5743942, rs3793964, and rs3829223; P = .02, .000022, and .005, respectively, by the allelic trend test). These SNPs had low levels of linkage disequilibrium between each other (R2, 0.02–0.11). The association of rs3793964 with leprosy remained significant after a conservative Bonferroni correction (adjusted P = .000132). SNP rs3793964 and leprosy susceptibility best fit a recessive model, with genotype TT associated with an increased risk of leprosy, compared with genotypes CC/TC (odds ratio [OR], 1.76; 95% confidence interval [CI], 1.23–2.55; P = .0014; Table 1). The association of SNP of rs3793964 remained statistically significant when corrected for age, sex, and ethnicity (OR, 1.40; 95% CI, 1.18–1.66) by logistic regression. Using publicly available data from a leprosy genome-wide association study, we attempted to validate our genetic findings [22]. We found that TOLLIP SNP rs3793964 was associated with an increased risk for developing leprosy in a concordant fashion with our discovery cohort (allelic OR, 1.186; 95% CI, 1.006–1.397; P = .0389, by the allelic trend test; data available at the National Center for Biotechnology Information Database of Genotypes and Phenotypes [accession number pha002872]). As secondary analyses, we found that TOLLIP variants were not associated with leprosy polarity (tuberculoid leprosy vs borderline or lepromatous leprosy), type 1 immune reaction (reversal reaction), or type 2 immune reaction (erythema nodosum leprosum; Supplementary Tables 4–6). Together, these data suggest that the TOLLIP SNP rs3793964 is associated with susceptibility to leprosy in Nepal and China.
Figure 3.

Toll-interacting protein (TOLLIP) variants, linkage disequilibrium, and association with skin messenger RNA (mRNA) expression. Haplotype-tagging TOLLIP single-nucleotide polymorphisms (SNPs) studied were selected from the Chinese Han (CHB) population in HapMap. A, TOLLIP gene chromosomal location and SNP map. B, Linkage disequilibrium of selected haplotype-tagging SNPs in the study population. Shaded boxes describe minor allele frequency of SNP and open boxes show R2 linkage disequilibrium values. C, Log10 interleukin 1 receptor antagonist (IL-1Ra) skin mRNA expression, stratified by TOLLIP SNP rs3793964 genotype. D, Log10 TOLLIP skin mRNA expression, stratified by TOLLIP SNP rs3793964 genotype. Statistical significance was determined via a generalized linear model. Lines represent median values of each sample. *P = .018 and **P = .022.
Table 1.
Association of Toll-Interacting Protein (TOLLIP) Polymorphisms With Leprosy Susceptibility
| SNP (Allelea), Group | AA (Frequency) |
Aa (Frequency) |
aa (Frequency) |
Trend P |
Dom P | OR (95% CI) | Rec P |
OR (95% CI) | HWE P |
|---|---|---|---|---|---|---|---|---|---|
| rs5743867 (C/T) | |||||||||
| Leprosy cases | 649 (0.72) | 234 (0.26) | 21 (0.02) | .123 | .12 | 1.24 (0.94–1.62) | .89 | 1.16 (0.51–2.91) | .566 |
| Controls | 412 (0.76) | 120 (0.22) | 10 (0.02) | … | … | ||||
| rs5743890 (A/G) | |||||||||
| Leprosy cases | 812 (0.92) | 70 (0.08) | 1 (0.001) | .800 | .914 | 1.04 (0.66–1.64) | … | .398 | |
| Controls | 490 (0.93) | 39 (0.07) | 1 (0.002) | … | … | ||||
| rs5743899 (A/G) | |||||||||
| Leprosy cases | 493 (0.56) | 331 (0.38) | 53 (0.06) | .936 | .99 | 0.99 (0.78–1.26) | .90 | 1.07 (0.64–1.82) | .608 |
| Controls | 303 (0.57) | 197 (0.37) | 28 (0.05) | … | … | ||||
| rs5743942 (C/T) | |||||||||
| Leprosy cases | 373 (0.44) | 361 (0.42) | 118 (0.14) | .0186 | .01 | 0.72 (0.57–0.92) | .32 | 0.85 (0.61–1.19) | .962 |
| Controls | 198 (0.37) | 249 (0.47) | 82 (0.16) | … | … | ||||
| rs3793964 (C/T) | |||||||||
| Leprosy cases | 331 (0.38) | 381 (0.44) | 152 (0.18) | .000022 | .0002 | 1.55 (1.22–1.98) | .0014 | 1.76 (1.23–2.55) | .198 |
| Controls | 249 (0.47) | 219 (0.42) | 57 (0.11) | … | … | ||||
| rs3829223 (C/T) | |||||||||
| Leprosy cases | 299 (0.34) | 417 (0.48) | 160 (0.18) | .00549 | .034 | 0.76 (0.58–0.98) | .017 | 0.71 (0.53–0.95) | .361 |
| Controls | 147 (0.27) | 262 (0.49) | 122 (0.23) | … | … |
Abbreviations: CI, confidence interval; Dom, dominant genetic model; HWE, Hardy–Weinberg equilibrium; OR, odds ratio; Rec, recessive genetic model; SNP, single-nucleotide polymorphism; Trend, allelic trend test.
a The first allele is the major allele (A), and the second is the minor allele (a).
TOLLIP SNP rs3793964 Is Associated With TOLLIP and IL-1Ra Cutaneous mRNA Expression
To examine the pattern of inheritance of the genetic association between TOLLIP SNP rs3793964 and leprosy, we stratified our skin biopsy samples by genotype and compared TOLLIP and IL-1Ra mRNA expression, using a genotypic model. The presence of the T allele of rs3793964 was significantly associated with increased TOLLIP mRNA and IL-1Ra mRNA (P = .018 and P = .022, respectively, by a generalized linear model). Furthermore, the data were most consistent with a recessive model; the genotype TT of rs3793964 was associated with higher levels of skin TOLLIP mRNA than genotypes CC/CT (median number of copies, 28 249 for TT vs 5714 for CC/CT; P = .011, by the Mann–Whitney test; Figure 3C). Similarly, the TOLLIP rs3793964 TT genotype was associated with higher IL-1Ra mRNA levels than the CC/TC genotype in a recessive model (median number of copies, 141 579 for TT vs 50 118 for CC/CT; P = .060, by the Mann–Whitney test; Figure 3D). These data suggest that genotype TT of rs3793964 is associated with increased TOLLIP and IL-1Ra mRNA in skin tissue.
M. leprae Selectively Induces IL-1Ra via a TOLLIP-Dependent Mechanism
We next evaluated potential mechanisms by which TOLLIP regulates IL-1Ra in monocytes. We stimulated human peripheral blood monocytes and THP-1 cells with 20 µg/mL whole irradiated M. leprae (iMLep) and found that IL-1Ra was strongly induced (Figure 4A and Supplementary Figure 2). Interestingly, this induction was partially selective, as iMLep did not induce several other proinflammatory cytokines, including IL-1β, IL-6, and TNF (Figure 4A). By contrast, stimulation with 10 ng/mL LPS was less selective and induced a different pattern of cytokines, with increased IL-6 and TNF levels but a decreased IL-1Ra level, compared with iMLep (Figure 4A). IL-1β was only weakly induced by either stimulus. We found similar patterns of cytokine secretion in 20 µg iMLep-stimulated THP-1 cells (Supplementary Figure 2). This observation is consistent with data from prior studies [28].
Figure 4.

Toll-interacting protein (TOLLIP) regulation of interleukin 1 receptor antagonist (IL-1Ra) expression after Mycobacterium leprae monocyte stimulation. Cytokine responses in peripheral blood monocytes from a healthy volunteer after stimulation with lipopolysaccharide (LPS) and whole irradiated M. leprae (iMLep). A, Interleukin 1 receptor antagonist (IL-1Ra), IL-1β, interleukin 6 (IL-6), tumor necrosis factor (TNF), and interleukin 8 (IL-8) cytokine production after 20 µg/mL iMLep and 10 ng/mL LPS stimulation of 2 × 105 human peripheral blood mononuclear cells from a healthy donor. Data are representative of 2 experiments. B–D, 105 TOLLIP-deficient (TOLLIP-KO) or empty vector (EV) THP-1 monocytes were stimulated with medium, 20 µg/mL iMLep, or 250 ng/mL PAM2 CysKKKK (TLR2/6 ligand) for 24 hours, followed by measurement of IL-1Ra (B), TNF (C), and IL-8 (D) production. Error bars show standard errors of the mean for 3 technical replicates from this experiment, and these data are representative of 2 independent experiments. *P < .05 and **P < .001, by the 2-sided Student t test.
To examine TOLLIP's role in IL-1Ra induction, we created a TOLLIP-deficient THP-1 cell line (TOLLIP-KO), using CRISPR/Cas9 gene editing. We confirmed knockout by sequencing and Western blot analyses (Supplementary Figure 3; sequencing data not shown). After stimulation with iMLep, TOLLIP-KO monocytes produced less IL-1Ra as compared to control cells transfected with an empty vector (P = .0024; Figure 4B). TOLLIP-KO monocytes stimulated with iMLep had no difference in TNF levels, compared with control cells (Figure 4C). Consistent with TOLLIP's effect as a TLR2 regulator, stimulation of TOLLIP-KO monocytes with PAM2CysKKKK (250 ng/mL; TLR2/6) led to significantly increased TNF and IL-8 levels (P = .043 and <.0001, respectively; Figure 4C and 4D).
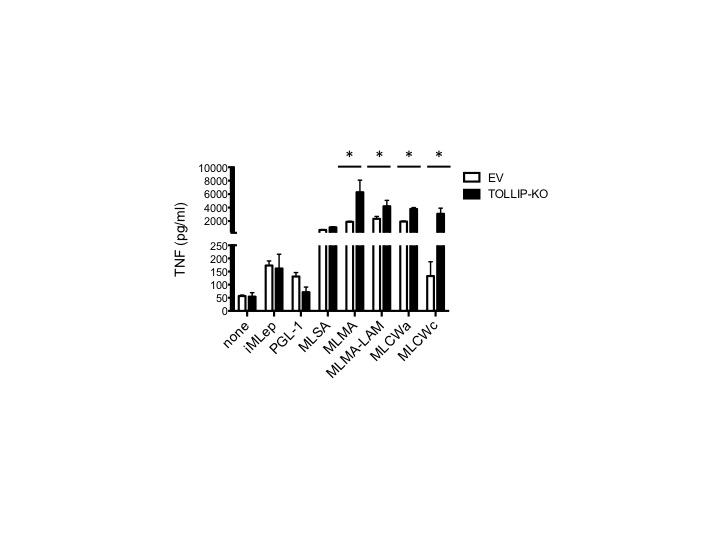
To determine whether the effect of TOLLIP on IL-1Ra expression was due to TLR or IL-1R activation by M. leprae, we stimulated EV and TOLLIP-KO THP-1 monocytes with several pure TLR agonists (PAM2, PAM3, LPS, and FliC) and IL-1β and measured cytokine responses after 24 hours of stimulation. Stimulation of TOLLIP-KO cells with PAM2, PAM3, and LPS led to similar secretion of IL-1Ra and significantly increased TNF in TOLLIP-KO, compared with findings for EV cells (P < .05; Figure 5A and 5B). Next, we determined whether specific fractions of M. leprae stimulated TOLLIP-dependent induction of IL-1Ra. We compared iMLep, M. leprae cytosol (MLSA), M. leprae cell membrane (MLMA), M. leprae cell membrane with lipoarabinomannan removed (MLMA – LAM), PGL-1 conjugated to human serum albumin, M. leprae cell wall (MLCWa), and M. leprae cell wall core (MLCWc). In contrast to iMLep, different fractions of M. leprae induced higher IL-1Ra secretion in TOLLIP-KO cells, compared with EV (P < .0001; Figure 5C). Further, TOLLIP-KO and EV cells stimulated with iMLep induced similar amounts of TNF, but multiple fractional preparations of M. leprae–stimulated TOLLIP cells induced increased TNF levels, compared with control (iMlep, P = .98; MLMA, MLMA – LAM, MLCWa, and MLCWc, P < .05; Supplementary Figure 4). Last, we infected our EV and TOLLIP-KO cells with live M. leprae and incubated them overnight at 33°C. TOLLIP deficiency was significantly associated with decreased IL-1Ra levels (P = .04; Figure 5D). Key findings were replicated using a second primer sequence to knock out TOLLIP gene function (data not shown). Together, these data suggest that whole M. leprae but not other TLR ligands or fractions of M. leprae induces IL-1Ra secretion in monocytes by a TOLLIP-dependent mechanism.
Figure 5.

Toll-interacting protein (TOLLIP)–dependent interleukin 1 receptor antagonist (IL-1Ra) secretion in monocytes requires whole Mycobacterium leprae. TOLLIP and empty vector (EV) control THP-1 cells were stimulated for 24 hours, followed by collection of supernatants and measurement of cytokine concentrations by enzyme-linked immunosorbent assay. A and B, Secreted IL-1Ra (A) and tumor necrosis factor (TNF; B) concentrations after stimulating 105 TOLLIP or EV monocytes with 80 µg/mL whole irradiated M. leprae (iMLep), 250 ng/mL PAM2, 250 ng/mL PAM3, 10 ng/mL LPS, 50 ng/mL Salmonella typhimurium flagellin FliC, or 100 ng/mL recombinant IL-1β for 24 hours. C, Secreted IL-1Ra concentrations after stimulating 105 TOLLIP or EV THP-1 monocytes with 80 µg/mL of the following M. leprae cellular fractions: iMLep, phenolic glycolipid 1 conjugated to human serum albumin (PGL1), M. leprae cytosolic fractions (MLSA), M. leprae cell membrane (MLMA), M. leprae cell membrane with lipoarabinomannan removed (MLSA – LAM), M. leprae cell wall (MLCWa), or M. leprae cell wall core (MLCWc). D, Secreted IL-1Ra concentrations after stimulating 105 TOLLIP-deficient THP-1 cell line (TOLLIP-KO) or EV THP-1 monocytes with iMLep or live M. leprae (multiplicity of infection, 10) derived from footpads of nude mice and incubated overnight at 33°C. Error bars show standard errors of the mean of 3 technical replicates from this experiment, which is representative of 2 independent experiments. *P < .05 and **P < .001, by the 2-sided Student t test.
DISCUSSION
In this study, we demonstrated that TOLLIP and IL-1Ra expression were highly correlated in leprosy skin biopsy specimens. TOLLIP polymorphisms were associated with increased TOLLIP and IL-1Ra skin expression and increased susceptibility to leprosy. Finally, M. leprae induced IL-1Ra in monocytes by a TOLLIP-dependent mechanism. Together, these observations suggest a strategy by which M. leprae evades host immunity in tissues through TOLLIP-dependent upregulation of IL-1Ra.
TOLLIP may influence one or more of several mechanisms that M. leprae uses to evade host antimicrobial responses [36]. M. leprae is a poor inducer of several classic proinflammatory cytokines, including IL-6, TNF, and IL-1β [29, 37]. By contrast, M. leprae is a strong inducer of antiinflammatory cytokines including IL-1Ra [28, 38, 39]. We previously found that TOLLIP negatively regulates proinflammatory IL-6 and TNF while positively regulating antiinflammatory IL-10 after TLR2 stimulation in peripheral blood monocytes [33]. Our current study links TOLLIP to M. leprae's ability to induce IL-1Ra, another potent antiinflammatory cytokine, in cutaneous tissue. These data are consistent with a model in which M. leprae induces IL-1Ra and thereby inhibits activation of IL-1R signaling in myeloid cells. Individuals with genetically regulated increased TOLLIP expression induce more cutaneous IL-1Ra, which is associated with increased susceptibility to leprosy (Supplementary Figure 4). Interestingly, there was no association of TOLLIP with disease polarity, despite the fact that the lepromatous and tuberculoid poles have different cytokine profiles. Our findings suggest that TOLLIP may regulate early steps in leprosy pathogenesis, whereas other pathways regulate polarization.
Although whole M. leprae induces TOLLIP-dependent IL-1Ra in monocytes, fractional preparations of M. leprae do not. One or more potential mechanisms may explain our observation. M. leprae may secrete a soluble factor like Mycobacterium ulcerans mycolactone, which inhibits production of cytokine mRNA in macrophages by preventing protein translocation into the endoplasmic reticulum [40, 41]. TOLLIP promotes trafficking of proteins through the endoplasmic reticulum and alters cytokine production via a posttranslational mechanism, suggesting that a similar factor may play a role in altering immune responses to M. leprae, absent in our fractions [42]. Alternately, whole M. leprae may activate PI3 kinase signaling via the phagolysosome and induce IL-1Ra secretion. Previous studies indicate that PI3K pathway activation may mediate IL-1Ra upregulation after M. leprae infection of monocytes [28]. Furthermore, recent work suggests that IRAK-1 activates and TOLLIP inhibits PI3K pathways after LPS stimulation, depending upon the dose administered [43]. Finally, TOLLIP regulates autophagy through its LC3-binding and CUE ubiquitin-binding domains [44]. Taken together, these observations suggest possible mechanisms by which TOLLIP selectively influences IL-1Ra activity in the skin.
Our study has several limitations. First, we examined leprosy skin biopsy specimens that were in a chronic disease state not necessarily reflective of the early postinfection period. Therefore, our findings may be biased toward measurement of chronic inflammatory markers, rather than toward the factors that may be most critical immediately after infection. However, we demonstrated a critical early finding in vitro: TOLLIP activates IL-1Ra preferentially after M. leprae infects monocytes. Our data also demonstrate that M. leprae detectably influences host immune responses during the chronic stages of infection and in vitro, even if M. leprae is dead. Residual M. leprae can remain as persistent antigen in host tissues for years after effective multidrug therapy, placing skin and nerves at increased risk for development of leprosy reactions, neuropathy, and disability. Second, our genetic association data may have biases due to ethnic heterogeneity within the case-control cohorts. Candidate gene association studies are subject to confounding due to population substructure [45]. We adjusted our analysis for ethnicity and sex by logistic regression and found no significant effects on our findings. We validated the association between rs3793964T allele and leprosy in an ethnically distinct cohort, using publicly available data. As a consequence of the haplotype-tagging SNP method to assess for genetic associations in TOLLIP, our findings may be due to genes adjacent to TOLLIP. One such gene, MUC5B, has been associated with idiopathic pulmonary fibrosis but does not have any known function in skin and is not expressed in dermal, adipose, or lymph tissue [46]. We hypothesize that the SNPs associated with outcomes in our study are in linkage disequilibrium with a functionally active causal variant in the TOLLIP gene that alters its tissue mRNA expression by altering promoter, transcription factor, or microRNA binding.
The ability of TOLLIP to induce IL-1Ra and block IL-1 signaling is a potential therapeutic target in infectious and inflammatory disease. Recent data suggest that IL-1 and inflammasome-related genes may be major susceptibility genes for leprosy [21, 22, 16]. Further studies in M. tuberculosis models suggest that IL-1 has an important role in the initial control of disease but that excessive levels can lead to increased susceptibility and tuberculosis severity [23, 47]. An inhibitor of TOLLIP may reduce IL-1Ra levels and increase killing of M. leprae during the initial stages of infection. This host-directed therapeutic medication could be used to prevent leprosy transmission, treat early disease, or be used in conjunction with current therapies to reduce leprosy treatment time. Alternately, activation of TOLLIP, administration of recombinant IL-1Ra, or blockade of genes downstream of TOLLIP may reduce harmful immune reactions and leprosy neuropathology [48, 49]. This work may be generalizable to modulating immunity in other infections, autoimmune diseases, or cancers in which IL-1 or control of IL-1 is critical for optimal immune responses.
Supplementary Data
Supplementary materials are available at http://jid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. We thank Kelly Hudkins and Charles Alpers for their work at the University of Washington Immunohistochemistry Core Facility; Dan Stetson, for his invaluable technical help with CRISPR/Cas9 gene editing; the patients and staff at Anandaban Hospital, for the clinical work associated with this study; Mr Kapil Neupane, Bishwa Sapkota, Chaman Ranjit, and Pratibha Thapa, for technical assistance; Dr Lakshmi Rajan and Dr Joyce Ponnaiya of the Research and Leprosy Centre, Schieffelin Institute of Health (Karigiri, India), for their expert histopathological services; and Ramanuj Lahiri at the National Hanson's Disease Program, for his generous preparation of live M. leprae.
Disclaimer. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (grant K08 AI102971 [to J. A. S.], K08 AI080952 [to W. R. B.], and K24 AI089794 [to T. R. H.]), the Heiser Program for Research in Tuberculosis and Leprosy (to W. R. B.), the Burroughs Wellcome Fund (to T. R. H.), and The Leprosy Mission International (to D. A. H.).
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Institute for Health Metrics and Evaluation. Global burden of leprosy. http://www.healthdata.org/search-gbd-data?s=leprosy Accessed 15 May 2015.
- 2.World Health Organization. Global leprosy update, 2013; reducing disease burden. Wkly Epidemiol Rec 2014; 89:389–400. [PubMed] [Google Scholar]
- 3.Geluk A, Bobosha K, van der Ploeg-van Schip JJ et al. . New biomarkers with relevance to leprosy diagnosis applicable in areas hyperendemic for leprosy. J Immunol 2012; 188:4782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duthie MS, Ireton GC, Kanaujia GV et al. . Selection of antigens and development of prototype tests for point-of-care leprosy diagnosis. Clin Vaccine Immunol 2008; 15:1590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lahiri R, Randhawa B, Franken KL et al. . Development of a mouse food pad model for detection of sub clinical leprosy. Lepr Rev 2011; 82:432–44. [PubMed] [Google Scholar]
- 6.Pena M, Geluk A, Van Der Ploeg-Van Schip JJ, Franken KL, Sharma R, Truman R. Cytokine responses to Mycobacterium leprae unique proteins differentiate between Mycobacterium leprae infected and naive armadillos. Lepr Rev 2011; 82:422–31. [PubMed] [Google Scholar]
- 7.Ridley DS, Jopling WH. Classification of leprosy according to immunity. A five-group system. Int J Lepr Other Mycobact Dis 1966; 34:255–73. [PubMed] [Google Scholar]
- 8.Cole ST, Eiglmeier K, Parkhill J et al. . Massive gene decay in the leprosy bacillus. Nature 2001; 409:1007–11. [DOI] [PubMed] [Google Scholar]
- 9.Murray RA, Siddiqui MR, Mendillo M, Krahenbuhl J, Kaplan G. Mycobacterium leprae inhibits dendritic cell activation and maturation. J Immunol 2007; 178:338–44. [DOI] [PubMed] [Google Scholar]
- 10.Liu PT, Wheelwright M, Teles R et al. . MicroRNA-21 targets the vitamin D-dependent antimicrobial pathway in leprosy. Nat Med 2012; 18:267–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabouret G, Astarie-Dequeker C, Demangel C et al. . Mycobacterium leprae phenolglycolipid-1 expressed by engineered M. bovis BCG modulates early interaction with human phagocytes. PLoS Pathog 2010; 6:e1001159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moura AC, Mariano M. Lipids from Mycobacterium leprae cell wall suppress T-cell activation in vivo and in vitro. Immunology 1997; 92:429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasan Z, Ashraf M, Tayyebi A, Hussain R. M. leprae inhibits apoptosis in THP-1 cells by downregulation of Bad and Bak and upregulation of Mcl-1 gene expression. BMC Microbiol 2006; 6:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alter A, Grant A, Abel L, Alcais A, Schurr E. Leprosy as a genetic disease. Mamm Genome 2011; 22:19–31. [DOI] [PubMed] [Google Scholar]
- 15.Gupte MD, Vallishayee RS, Anantharaman DS et al. . Comparative leprosy vaccine trial in south India. Indian J Lepr 1998; 70:369–88. [PubMed] [Google Scholar]
- 16.Misch EA, Berrington WR, Vary JC, Hawn TR. Leprosy and the human genome. Microbiol Mol Biol Rev 2010; 74:589–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Britton WJ, Lockwood DN. Leprosy. Lancet 2004; 363:1209–19. [DOI] [PubMed] [Google Scholar]
- 18.Chackravartti MR, Vogel F. A twin study on leprosy. Stuttgart, Germany: Georg Thieme Publishers, 1973. [Google Scholar]
- 19.Mira MT, Alcais A, Nguyen VT et al. . Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 2004; 427:636–40. [DOI] [PubMed] [Google Scholar]
- 20.Miller EN, Jamieson SE, Joberty C et al. . Genome-wide scans for leprosy and tuberculosis susceptibility genes in Brazilians. Genes Immun 2004; 5:63–7. [DOI] [PubMed] [Google Scholar]
- 21.Liu H, Irwanto A, Fu X et al. . Discovery of six new susceptibility loci and analysis of pleiotropic effects in leprosy. Nat Genet 2015; 47:267–71. [DOI] [PubMed] [Google Scholar]
- 22.Zhang FR, Huang W, Chen SM et al. . Genomewide association study of leprosy. N Engl J Med 2009; 361:2609–18. [DOI] [PubMed] [Google Scholar]
- 23.Mayer-Barber KD, Andrade BB, Oland SD et al. . Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014; 511:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arend WP, Malyak M, Guthridge CJ, Gabay C. Interleukin-1 receptor antagonist: role in biology. Annu Rev Immunol 1998; 16:27–55. [DOI] [PubMed] [Google Scholar]
- 25.Granowitz EV, Clark BD, Mancilla J, Dinarello CA. Interleukin-1 receptor antagonist competitively inhibits the binding of interleukin-1 to the type II interleukin-1 receptor. J Biol Chem 1991; 266:14147–50. [PubMed] [Google Scholar]
- 26.Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 2008; 223:20–38. [DOI] [PubMed] [Google Scholar]
- 27.Poutsiaka DD, Clark BD, Vannier E, Dinarello CA. Production of interleukin-1 receptor antagonist and interleukin-1 beta by peripheral blood mononuclear cells is differentially regulated. Blood 1991; 78:1275–81. [PubMed] [Google Scholar]
- 28.Sinsimer D, Fallows D, Peixoto B, Krahenbuhl J, Kaplan G, Manca C. Mycobacterium leprae actively modulates the cytokine response in naive human monocytes. Infect Immun 2010; 78:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki K, Fukutomi Y, Matsuoka M et al. . Differential production of interleukin 1 (IL-1), IL-6, tumor necrosis factor, and IL-1 receptor antagonist by human monocytes stimulated with Mycobacterium leprae and M. bovis BCG. Int J Lepr Other Mycobact Dis 1993; 61:609–18. [PubMed] [Google Scholar]
- 30.Yamamura M, Uyemura K, Deans RJ et al. . Defining protective responses to pathogens: cytokine profiles in leprosy lesions. Science 1991; 254:277–9. [DOI] [PubMed] [Google Scholar]
- 31.Burns K, Clatworthy J, Martin L et al. . Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat Cell Biol 2000; 2:346–51. [DOI] [PubMed] [Google Scholar]
- 32.Zhang G. Negative regulation of Toll-like receptor-mediated signaling by TOLLIP. J Biol Chem 2002; 277:7059–65. [DOI] [PubMed] [Google Scholar]
- 33.Shah JA, Vary JC, Chau TT et al. . Human TOLLIP regulates TLR2 and TLR4 signaling and its polymorphisms are associated with susceptibility to tuberculosis. J Immunol 2012; 189:1737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brissoni B, Agostini L, Kropf M et al. . Intracellular trafficking of interieukin-1 receptor I requires Tollip. Curr Biol 2006; 16:2265–70. [DOI] [PubMed] [Google Scholar]
- 35.Berrington WR, Kunwar CB, Neupane K et al. . Differential dermal expression of CCL17 and CCL18 in tuberculoid and lepromatous leprosy. PLoS Negl Trop Dis 2014; 8:e3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scollard DM, Adams LB, Gillis TP, Krahenbuhl JL, Truman RW, Williams DL. The continuing challenges of leprosy. Clin Microbiol Rev 2006; 19:338–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hashimoto K, Maeda Y, Kimura H et al. . Mycobacterium leprae infection in monocyte-derived dendritic cells and its influence on antigen-presenting function. Infect Immun 2002; 70:5167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Costa RD, Mendonca VA, Lyon S et al. . Evaluation of the expression of interleukin 1 beta (IL-1beta) and interleukin 1 receptor antagonist (IL-1Ra) in leprosy patients. Rev Soc Bras Med Trop 2008; 41(suppl 2):99–103. [DOI] [PubMed] [Google Scholar]
- 39.Fafutis-Morris M, Guillen-Vargas CM, Navarro-Fierro S, Morales-Ortiz R, Armendariz-Borunda J. Serum IL-1ra is elevated in lepromatous leprosy patients. Int J Lepr Other Mycobact Dis 1999; 67:287–91. [PubMed] [Google Scholar]
- 40.Simmonds RE, Lali FV, Smallie T, Small PL, Foxwell BM. Mycolactone inhibits monocyte cytokine production by a posttranscriptional mechanism. J Immunol 2009; 182:2194–202. [DOI] [PubMed] [Google Scholar]
- 41.Hall BS, Hill K, McKenna M et al. . The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog 2014; 10:e1004061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Didierlaurent A, Brissoni B, Velin D et al. . Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Mol Cell Biol 2006; 26:735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng H, Maitra U, Morris M, Li L. Molecular mechanism responsible for the priming of macrophage activation. J Biol Chem 2013; 288:3897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu K, Psakhye I, Jentsch S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014; 158:549–63. [DOI] [PubMed] [Google Scholar]
- 45.Cordell HJ, Clayton DG. Genetic association studies. Lancet 2005; 366:1121–31. [DOI] [PubMed] [Google Scholar]
- 46.Noth I, Zhang Y, Ma SF et al. . Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013; 1:309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang G, Zhou B, Li S et al. . Allele-specific induction of IL-1beta expression by C/EBPbeta and PU.1 contributes to increased tuberculosis susceptibility. PLoS Pathog 2014; 10:e1004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cavalli G, Dinarello CA. Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatology (Oxford) 2015; 54:2134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arend WP. The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev 2002; 13:323–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}