

Conspectus
Our long-time niche in synthetic biological inorganic chemistry has been to design ligands and generate coordination complexes of copper and/or iron ions, those reacting with dioxygen (O2) and/or nitrogen oxides (e.g., nitric oxide (NO(g)) and nitrite (NO2−)). As inspiration for this work, we turn to mitochondrial cytochrome c oxidase which is responsible for dioxygen consumption and is also the predominant target for NO(g) and nitrite within mitochondria. In this Account, we highlight recent advances in studying synthetic heme/Cu complexes in two respects. First, there is the design, synthesis and characterization of new O2-adducts whose further study will add insights into O2-reductive cleavage chemistry. Second, we describe how related heme/Cu constructs reduce nitrite ion to NO(g) or the reverse, oxidize NO(g) to nitrite. The reactions of nitrogen oxides occur as part of CcO’s function, which is intimately tied to cellular O2-balance.
We had first discovered that reduced heme/Cu compounds react with O2 giving μ-oxo heme-FeIII-O-CuII(L) products; their properties are discussed. The O-atom is derived from dioxygen and interrogations of these systems led to the construction and characterization of three distinctive classes of heme-peroxo-complexes, two high-spin and one low-spin species.
Recent investigations include a new approach to the synthesis of low-spin heme-peroxo-Cu complexes, employing a “naked” synthon, where the copper ligand denticity and geometric types can be varied. The result is a collection of such complexes; spectroscopic and structural features (by DFT calculations) are described. Some of these compounds are reactive toward reductants/protons effecting subsequent O-O cleavage. This points to how subtle improvements in ligand environment lead to a desired local structure and resulting optimized reactivity, as known to occur at enzyme active-sites.
The other sector of research is focused on heme/Cu assemblies mediating the redox interplay between nitrite and NO(g). In the nitrite reductase chemistry, the cupric center serves as a Lewis acid while the heme is the redox active center providing the electron. The orientation of nitrite in approaching the ferrous heme center and N-atom binding are important. Also, detailed spectroscopic and kinetic studies of the NO(g) oxidase chemistry, in excellent agreement with theoretical calculations, reveal the intermediates and key mechanistic steps. Thus, we suggest that both chemical and biochemical heme/Cu mediated nitrite reductase and NO(g) oxidase chemistry require N-atom binding to a ferrous heme along with cupric ion O-atom coordination, proceeding via a three-membered O-Fe-N chelate ring transition state. These important mechanistic features of heme/Cu systems interconverting NO(g) and nitrite are discussed for the first time.
1. Introduction
Mitochondrial cytochrome c oxidase (CcO) is the terminal component of the respiratory chain that carries out the enzymatic four-electron reduction of dioxygen (O2) to water (H2O). This enzyme is essential to respiration, providing the energy to produce a proton gradient across the membrane, which supplies the driving force for ATP synthesis. The binding of O2 and its subsequent reduction to water take place at the CcO binuclear active-site consisting of heme-a3 and CuB ligated by three histidine (His) residues with a unique linkage of one His to a nearby tyrosine residue. During catalytic turnover, the FeII/CuI center binds and reduces O2, sequentially forming superoxide (A), (hydro)peroxide and ferryl (PM, after O-O cleavage, thought to be assisted by tyrosine H-atom injection) intermediates (Figure 1), followed by stepwise protonation-reduction giving water and regeneration of the FeII/CuI site.1–3 The O-O bond reductive cleavage event is a crucial but complicated process; it depends on electron-transfer and protonation steps, and its full understanding has implications well beyond CcO.4
Figure 1.

Cytochrome c oxidase reduces dioxygen via the proposed intermediates shown (top). This heme-a3/CuB binuclear active-site also functions in NO2−/NO(g) interconversion in a regulatory role modulating O2-balance (bottom).
This same heme-a3/CuB binuclear center is also a major site of respiratory regulation as it interacts with nitrite (NO2−) and nitric oxide (NO) to modulate cellular O2-balance (Figure 1). In tissues experiencing hypoxia, when the O2 concentration decreases below physiological levels,5 nitrite serves as an acceptor for accumulating electrons at the CcO binuclear active-site and the enzymatic one-electron reduction of nitrite (NO2−) takes place generating nitric oxide. The NO(g) produced by CcO reversibly interacts with the same center, and thus, decrease its affinity to O2-reduction resulting in cellular O2 accumulation. Some of mitochondrially produced NO(g) also participates in the up-regulation of hypoxic nuclear genes (hypoxic signaling).5–6 In turn, at mitochondrial sites recovering from hypoxia, local accumulation of O2 progressively triggers one-electron oxidation of NO(g) back to NO2− ion, the latter held in reserve to be reconverted when needed. This process serves as an adaptive mechanism eliminating NO(g)-mediated respiratory inhibition.7–8
In this Account, we describe our recent findings on both subjects. A great emphasis has been placed on design and generation of relevant O2-adducts, i.e., heme-Fe-(O22−)-Cu(L) complexes, as a segue to future chemistries involving the critical O-O cleavage process. We also highlight our investigations on the interconversion of NO(g) and nitrite mediated by heme/Cu assemblies. Our work is thus aimed at introducing synthetic heme/Cu binuclear systems capable of performing chemical transformations of relevant small molecules, those occurring at the CcO active-site. Our goal is to provide fundamental insights into essential factors contributing to these redox processes.
2. Initial Work on Heme-FeII…CuI(L)/O2 Chemistry; FeIII-Oxo-CuII Complexes
Our initial attempts at modeling the CcO heme-a3/CuB dioxygen chemistry led to discovery of μ-oxo heme-FeIII-O-CuII(L) compounds forming from reaction of O2 with equimolar quantities of the corresponding reduced heme and copper mononuclear complexes; the μ-oxo ligand derives from O2. Interestingly, despite the known substantial thermodynamic stability of μ-oxo iron dimer heme-FeIII-O-FeIII-heme, here such μ-oxo heme-FeIII-O-CuII(L) complexes are the kinetically stable products which can also be generated by acid-base reaction of oxidized metal complexes, i.e., reaction of heme-FeIII-OH and [(L)CuII] plus a base.9–10
We recently added new crystal structures, [(TMPP)FeIII-O-CuII(tmpa)]+ and [(F8)FeIII-O-CuII(MePY2)]+ (Chart 1), to our library of such compounds, which conform to previous findings in that the Fe-O-Cu core in the former compound with its tetradentate chelate for cupric center, TMPA, is near-linear while the latter complex bearing a tridentate copper(II) ligand, MePY2, has a very bent μ-oxo moiety (Chart 1). They also exhibit the very red-shifted Soret bands (λmax = 443 and 438 nm, respectively) characteristic of the μ-oxo compounds relative to that of classical high-spin ferric hemes, probably due to a weaker affinity of Cu(II) for the bridging oxo, thus the greater degree of π charge donation available onto ferric center by the oxo group. Also, [(TMPP)FeIII-O-CuII(tmpa)]+ is the first example of the heme-FeIII-O-CuII(L) class which contains the strongly donating TMPP porphyrinate. Notably, comparison of [(TMPP)FeIII-O-CuII(tmpa)]+ and its closely related analog [(F8)FeIII-O-CuII(tmpa)]+ in which only the porphyrinate identity varies, reveals that the cupric centers adapts very different geometries; for the former the tmpa-Cu N4O-ligation occurs within a nearly perfect trigonal bipyramidal (TBP) geometry (τ = 0.9) while for the latter the copper center adjusts to a distorted square pyramidal (SP) coordination (τ = 0.3, Chart 1). Steric constraints imposed by o-fluorine substituents of the meso-phenyl groups on the attached porphyrin ring in F8 ligand dictate structural properties of the cupric center resulting in deviation from the normal TBP geometry seen for [(tmpa)CuII(X)]n+ (X = H2O, MeCN, Cl−, NO2−) compounds.11
Chart 1.

Molecular Structures of μ-oxo [(porphyrinate)FeIII-O-CuII(L)]+ Complexes. tmpa tris(2-pyridylmethylamine), MePY2 bis(2-pyridyl-ethyl)methylamine F8 tetrakis(2,6-diflurophenyl)-porphyrinate(2-), TMPP tetrakis(4-methoxyphenyl)porphyrinate(2-).
In all μ-oxo compounds, the bridging oxo moiety is very basic and can be reversibly protonated to give the protonated acid-base partner, heme-FeIII-(OH)-CuII(L). For some cases corresponding μ-hydroxo analogues have been characterized and pKa values reported.12–13 In general, protonation of the Fe-O-Cu core results in bending and re-hybridization of the bridging oxygen moiety, accompanied by bond elongation.
3. High-spin Heme-Peroxo-Copper Complexes
In view of our interest in studying the CcO active-site O2-reduction chemistry (Figure 1), Fe/Cu/O2 intermediates preceding the formation of μ-oxo heme-FeIII-O-CuII(L) compounds have been primary targets for study. A first generation example is the metastable heme-peroxo-copper complex [(F8)FeIII-(O22−)-CuII(tmpa)]+ (HS-TMPA, Scheme 1), formed from O2-bubbling through cold solutions of a 1:1 mixture of [(tmpa)CuI(MeCN)]+/(F8)FeII.14 Structural and spectroscopic studies on HS-TMPA provided evidence for a bridging peroxide unit bound in a side-on fashion to the high-spin iron(III) center and end-on to the copper(II) center.15 Key observations were that: (i) 2H-NMR spectroscopy revealed HS-TMPA to possess antiferromagnetically coupled (through the peroxide-bridge) high-spin iron(III) and copper(II) ions, leading to an S = 2 system; (ii) resonance Raman spectroscopic (rRaman) measurements identified O-O, Fe-O and Cu-O vibrations (Scheme 1); (iii) X-ray absorption fine structure (EXAFS) provided Cu-O, Fe-O and Fe…Cu contributions at 1.87, 1.94 and 3.72 Å. These spectroscopic/structural features correspond to data from the only crystallographically characterized heme-peroxo-copper system.16
Scheme 1.

As to the formation of HS-TMPA, stopped-flow UV-vis measurements revealed an iron(III)-superoxo intermediate [(L)(F8)FeIII-(O2. −)] (L = solvent, i.e. EtCN) initially formed. Indeed, addition of 1 equiv [(tmpa)CuI(MeCN)]+ to this leads to HS-TMPA. Scheme 1 depicts alternative synthetic routes giving HS-TMPA. With the expectation that significant (or even small) ligand variations can dramatically alter structure and other properties, we also synthesized heme-(O2)-copper assemblies using tridentate chelates for copper. A well-studied example is [(F8)FeIII-(O22−)-CuII(AN)]+ (HS-AN, AN is 3,3′-iminobis(N,N-dimethylpropylamine, Scheme 1; EXAFS, Fe…Cu = 3.63 Å). Further analyses led to the conclusion that HS-AN and other analogs with tridentate ligands for copper possess a side-on peroxide coordination mode (i.e., μ-η2:η2) to both metal ions (Scheme 1).17–18
4. Low-Spin Heme-Peroxo-Copper Complex LS-AN
A crucial advance for our research came when we reported the generation of the low-spin (LS) [(DCHIm)(F8)FeIII-(O22−)-CuII(AN)]+ (LS-AN),19 likely a close analogue of the putative heme-peroxo-copper species formed in CcO. Addition of an axial base ligand (DCHIm = 1,5-dicyclohexylimidazole) to HS-AN leads to the new species LS-AN, which presented unusually strong low-energy UV-vis features (Scheme 1). A change in the spin-state is associated with an alternation in the bridging mode of the peroxide ligand, forming an end-on FeIII-(μ-η1:η1-O22−)-CuII core. 2H-NMR spectroscopy indicates an overall S = 0 spin-state, derived from antiferromagnetic coupling of a low-spin Fe(III) (S =½) to the S = ½ Cu(II). Two isotope-sensitive features at 796 and 586 cm−1 are assigned to O-O and Fe-O vibrations, respectively, based on rRaman. Note the large change in νO-O in comparing high- and low-spin AN-analogs. EXAFS measurements on LS-AN give Cu-O, Fe-O and Fe…Cu distances of 1.98, 1.81 and 4.01 Å, respectively. Relative to HS-AN, a shortening of the Fe-O bond (1.94 vs 1.81 Å) and elongation of the Fe…Cu distance (3.72 vs 4.01 Å) provide for the new end-on peroxide bridging mode in LS-AN (Scheme 1).
5. “Naked” Fe-Peroxo-Cu Synthon for New Low-Spin Compounds
We recently explored a new synthetic approach,20 following a discovery leading to the high-spin complex [(F8)FeIII-(O22−)-CuII(MeTHF)3]+ (HS-3MeTHF, Scheme 1), a “naked” synthon as a starting point for the generation of an array of heme-peroxo-copper assemblies. Addition of [CuI(CH3CN)4][B(C6F5)4] to a solution of [(MeTHF)(F8)FeIII-(O2. −)] at low temperatures (−90 to −125 ºC) leads to the clean formation of HS-3MeTHF, formulated as possessing a side-on FeIII-(μ-η2:η2-O22−)-CuII core with three solvent molecules surrounding the copper(II) center, due to its spectroscopic similarities with HS-AN, see Scheme 1 for spectroscopic data.
The utility of HS-3MeTHF is illustrated by its use in generation of a new low-spin complex [(DCHIm)(F8)FeIII-(O22−)-CuII(tmpa)]+ (LS-TMPA, Scheme 1). Addition of 1 equiv of the free ligand TMPA to the HS-3MeTHF gives HS-TMPA by replacement of the labile solvent ligands to copper. A further step comes from addition of 1 equiv DCHIm to HS-TMPA, giving the new compound LS-TMPA. Like LS-AN, LS-TMPA also possesses unusual low-energy UV-vis features (Scheme 1). 2H-NMR spectroscopy confirmed the change in spin-state, as the pyrrole signal shifted from 99 ppm for HS-TMPA (S = 2) to 5 ppm for LS-TMPA (S = 0). The change in the spin-state is again linked to alteration in the peroxide coordination mode. Note the data given in Scheme 1 shows that O-O, Fe-O and Cu-O stretching frequencies for LS-TMPA are systematically higher than those for HS-TMPA, especially the νFe-O value. This appears to be the first case in which the effect of changing the spin-state on all relevant vibrations, O-O, Fe-O, and Cu-O, could be evaluated.20
Most interestingly, we found that simple monodentate ligand donors such as imidazoles, native CcO active-site ligands, could be added to the solvato “naked” complex HS-3MeTHF to generate new low-spin heme-(O2)-copper assemblies. Addition of excess of DCHIm to HS-3MeTHF at −125 ºC, led to the formation of a new species with UV-vis features very similar to those found for LS-AN. Titration experiments provided evidence that 4 equiv DCHIm were required for its formation, formulating the new low-spin peroxo as [(DCHIm)(F8)FeIII-(O22−)-CuII(DCHIm)3]+ (LS-3DCHIm) with one imidazole ligand coordinated to the heme and three to the cupric ion. 2H-NMR spectroscopic characterization corroborated the S = 0 complex ground state while rRaman revealed the existence of a particularly high O-O stretching vibration, νO-O = 876 cm−1 (Scheme 1).20
When LS-3DCHIm is warmed to −90 ºC in the presence of excess DCHIm, a new species with shifted UV-vis features forms (Scheme 1). The same species is also obtained when 5 equiv DCHIm are added to HS-3MeTHF generated at −90 ºC. Cooling this new complex with four imidazole donors for the copper ion, [(DCHIm)(F8)FeIII-(O22−)-CuII(DCHIm)4]+ (LS-4DCHIm, Scheme 1) does not lead to the reversible formation of LS-3DCHIm; thus, LS-4DCHIm is the thermodynamic product, formed after the generation of the kinetic product, LS-3DCHIm. 2H-NMR spectroscopy indicates that these two complexes are distinctly different (Scheme 1). However, LS-4DCHIm has distinctive but surprisingly similar vibrational features to those found for LS-3DCHIm, νFe-O = 594 cm−1 and νO-O = 876 cm−1; coordination of an extra imidazole does not induce dramatic changes in the Fe-O and O-O bond strengths. Interestingly, the compounds LS-3DCHIm and LS-4DCHIm can also be used as starting material for the generation of other low-spin compounds. In fact, addition of 1 equiv TMPA to LS-3DCHIm or LS-4DCHIm results in imidazole ligand displacement, giving LS-TMPA.20
6. Structural Variations and Comparison of Heme-Peroxo-Copper Cores
As described above, we have synthesized a series of heme-peroxo-Cu(L) complexes with varying ligands (L) to Cu(II) ion and observed that spectroscopic parameters for L = TMPA vs AN vs (3 or 4) DCHIm exhibit distinctive differences. Thus, there are clear differences in structure and bonding (electronic-structure) amongst the compounds. With Ed Solomon’s group at Stanford University, DFT calculations have been carried out in order to determine finer details as concerns structural and vibrational characteristics of these compounds.20 One focus was to compare and contrast the (HS-AN)/(LS-AN) pair. We see that axial base coordination (DCHIm) promotes isomerization of the side-on FeIII-(μ-η2:η2-O22−)-CuII core in HS-AN, giving an end-on FeIII-(μ-η1:η1-O22−)-CuII structure in LS-AN, responsible for the change in spin-state. A significant reorganization of the peroxo core has occurred, as evidenced by the increased Fe…Cu distance (+0.36 Å) and shortened Fe-O (-0.20 Å), O-O (-0.06 Å), and Cu-O (-0.09 Å) bond lengths (Figure 2). These changes stem from: (i) the transition to an end-on geometry in which the Fe and Cu ions bind only one oxygen atom and (ii) the low-spin nature of heme-iron(III). The O-O distance shortening is small, but can be explained by an increase in O22− (π*) donation into the Fe(dz2) orbital which increases the O-O bond strength. Similarly, DFT calculations on the (HS-TMPA)/(LS-TMPA) pair, leads to similarly observed trends (Figure 2).20 DCHIm coordination as a heme axial base ligand produces rearrangement of the peroxo core with a notorious increase in the Fe-Cu distance (+0.77 Å) but shortened Fe-O (-0.20 Å) and O-O (-0.06 Å) distances (Figure 2, bottom right). However, in this case, only a slight change in the geometry around copper center is observed (Cu-O distance shortened 0.02 Å) which is explained by the retention of the peroxide coordination mode (i.e., end-on to Cu) upon heme-Fe spin change.
Figure 2.

DFT optimized structures for HS-AN, LS-AN, HS-TMPA and LS-TMPA complexes. Adapted with permission from ref. 20 Copyright (2015) American Chemical Society.
We have previously mentioned21 that heme-peroxo-copper reactivity is enhanced in the low-spin-state. Thus, comparison of LS-AN and LS-TMPA is worthy of discussion. They both possess end-on FeIII-(μ-η1:η1-O22−)-CuII low-spin cores, however their structural differences (Figure 2) are prominent. A significantly greater Fe…Cu distance (+0.50 Å) and much larger ∠Cu-O-O (+22º) are found for LS-TMPA. As shown before, these variances are also reflected in the complexes’ distinctive spectroscopic features (UV-vis, rRaman). Ongoing computational studies will include analyses of LS-3DCHIm vs LS-4DCHIm and comparison to the other low-spin complexes. These complexes possess the same νO-O values (876 cm−1) (vide supra), but this frequency is much higher (> 64 cm−1) than what is found for LS-AN or LS-TMPA.20
As seen from the structural and spectroscopic information as displayed in Scheme 1 and Figure 2, significant variations are observed as a function of the nature of the specific ligand to copper ion and the heme spin-state. Peroxo-bridged heme/Cu species are possible CcO reaction intermediates forming between Oxy and PM (Figure 1);19–20,22–23 we regard them as a near ideal starting point for studying O-O reductive cleavage reactivity.24 Further, initial studies reveal that LS-3DCHIm and LS-4DCHIm, along with other compounds (Scheme 1), possess markedly different behavior with respect to O–O cleavage upon protonation and/or reduction. Our long-term goal is to understand these reactivity differences in terms of heme-peroxo-Cu complex structure/bonding variations, and the results will impact on CcO chemistry and/or other processes where O-O reductive cleavage occurs (see Introduction).
7. Initial Work on Nitrite and NO(g) Interconversion
In biological systems, the production of NO(g) as a signaling agent, e.g., in smooth muscle cell relaxation, is critically important. Aside from NO(g) synthesis by cyt. P-450 like NO-synthases which produce NO(g) via L-arginine oxidative chemistry,25 nitrite also serves as a critical NO(g) source (also see below). The cellular balance of NO(g) is very tightly regulated and mechanisms for its removal include NO(g)-reductase (NOR) coupling NO(g) molecules to give N2O, and NO(g)-dioxygenase (NOD) producing nitrate as a benign NO(g) oxidation product.25 NO(g) is also “removed” via oxidation to nitrite, to later be converted back to nitric oxide when needed. The blood copper protein ceruloplasmin oxidizes NO(g) to nitrite,26 as does cytochrome c oxidase.
In fact, our heme/Cu assemblies as heme-a3/CuB CcO active-site models are not only capable of O2-reduction, but they possess redox activity toward nitrogen oxides including nitrite and NO(g) interconversions. To help shed light on the molecular mechanism of this redox interplay between these two molecules and the roles that the metal centers may play, we have employed our binuclear synthetic systems. In our initial report,27 we introduced a synthetic heme/Cu assembly that in its partially reduced form facilitates nitrite reduction to NO(g). Thus, addition of the cupric-nitrite complex [(tmpa)CuII(NO2)]+ to 2 equiv of the ferrous heme (F8)FeII (λmax = 421 nm) resulted in formation of a one-to-one mixture of the ferrous heme-nitrosyl complex (F8)FeII(NO) (λmax = 399 nm) and the μ-oxo compound [(F8)FeIII-O-CuII(tmpa)]+ (λmax = 435 nm, Scheme 2), readily confirmed by UV-vis, EPR and IR (νNO = 1688 cm−1) spectroscopies. Here, the second equivalent of ferrous heme was not involved in the redox chemistry and only trapped the NO(g) produced. Our electrochemical measurements showed that in this heme/Cu assembly, the cuprous complex [(tmpa)CuI(MeCN)]+ (-0.42 V vs Fc+/0) is a chemically stronger reductant compared to the ferrous heme (F8)FeII (-0.20 V vs Fc+/0). However, based on our control experiments, neither of these reduced compounds, nor the combination of the two, is capable of reducing nitrite to NO(g). Thus, our initial study27 highlighted two primary findings: (i) the ferrous heme is the redox active center providing the electron (ii) while the cupric center serves as a Lewis acid facilitating nitrite (N-O) bond cleavage. This overall chemical transformation can also be viewed as one-electron reduction of nitrite (formally NO+), which accompanies a transfer of an oxygen atom derived from the nitrite, finally forming an oxo-bridge FeIII-O-CuII product.
Scheme 2.

Heme/Cu Assembly-Mediated Interconversion of Nitrite and Nitric Oxide.
Interestingly, in turn, we showed that the fully oxidized form of the same assembly as a μ-oxo compound [(F8)FeIII-O-CuII(tmpa)]+ (λmax = 435 nm) could oxidize NO(g) back to nitrite (Scheme 2).27 Addition of NO(g) to the μ-oxo complex [(F8)FeIII-O-CuII(tmpa)]+ resulted in reformation of the cupric-nitrite complex [(tmpa)CuII(NO2)]+; the reduced heme (F8)FeII formed from this NO(g) oxidase chemistry is trapped by a second equiv of NO(g) to give (F8)FeII(NO) (λmax = 399 nm). The results of product analysis using UV-vis, EPR and IR spectroscopies and capillary electrophoresis (nitrite analysis, 95% yield) confirmed the nature of the products and proposed stoichiometric reaction. For comparison, the μ-hydroxo complex [(F8)FeIII-(OH)-CuII(tmpa)]2+ was also tested for “NO(g) oxidase” chemistry, but addition of NO(g) resulted in no nitrite production; the μ-oxo complex [(F8)FeIII-O-CuII(tmpa)]+ is unique in its ability to effect this redox and oxo transfer reaction.
8. Investigation on Nitrite Reductase Chemistry Mediated by Heme/Cu Assemblies
To further investigate the key factors involved in the nitrite reductase chemistry the nature of the heme and/or the copper ligand were synthetically modified (Scheme 3).28
Scheme 3.

Nitrite Reductase Chemistry Mediated by Heme/Cu Assemblies. Top: General reaction equation. Bottom: structures of copper and heme complexes used in our study.
8.1. Pophyrinate Variation
Toward a better understanding of the role of the reducing ability of the ferrous heme center, a different tetraarylporphyrinate bearing strong electron-donating peripheral groups, TMPP, was employed and its reactivity was compared with the FeII/CuII-nitrite “parent” reaction, the combination of [(tmpa)CuII(NO2)]+ and (F8)FeII, where the heme possesses electron-withdrawing peripheral substituents.28 Product analysis confirmed the same overall redox reaction, converting nitrite to NO(g), thus generating a one-to-one mixture of the μ-oxo compound [(TMPP)FeIII-O-CuII(tmpa)]+ (λmax = 443 nm) and ferrous heme nitrosyl complex (TMPP)FeII(NO) (λmax = 410 nm). Notably, on the basis of UV-vis monitoring the disappearance of the Soret band for the ferrous heme, the reaction proceeds at about the same rate as for the FeII/CuII-nitrite “parent” reaction, pointing to the fact that even with the greater electron-donating ability of the ferrous heme (TMPP)FeII, the nitrite reduction reaction is not accelerated.
8.2. Influence of Copper Coordination Environment on Nitrite Reduction
In order to examine the effect of differing modes of coordination of nitrite to the cupric center, we employed [(AN)CuII(NO2)](CF3SO3), having a tridentate alkylamine chelate rather than a tetradentate pyridyl-alkylamine ligand, as in [(tmpa)CuII(NO2)][B(C6F5)4] (Scheme 3).28 As revealed by X-ray crystallography, these two cupric-nitrite complexes exhibit different binding modes of nitrite to the copper center, O,O′-bidentate binding in the former and O-unidentate coordination in the latter (Note: the outer O-atom is very accessible). Reactions of both cupric-nitrite complexes with either ferrous-heme leads to the same overall redox reaction reducing nitrite to NO(g). Interestingly, we observed that when [(AN)CuII(NO2)]+ was reacted with either (F8)FeII or (TMPP)FeII, the rate of ferrous heme disappearance was roughly twice as fast as the reaction of the same heme with the [(tmpa)CuII(NO2)]+. Thus, faster reduction of nitrite to NO(g) occurs when the nitrito ligand is bidentate vs unidentate coordinated to the cupric center. We concluded that this difference in nitrite coordination mode leads to differing approaches of the cupric-nitrite compound to the reduced heme (i.e. the reduction center) to transiently form a bridged ferrous heme-(nitrite)-cupric complex. For [(AN)CuII(NO2)]+, having O,O′-bidentate nitrite ligation, one could anticipate a facile approach of a nitrite nitrogen atom to the ferrous heme, leading to a nitro (N-ligated) mode of coordination (Figure 3), perhaps in the rate-determining step. This can be followed by an intramolecular electron-transfer from the ferrous heme to the bridging nitrite generating a bridged hydronitrite radical (NO22−) assembly which rapidly converts to NO(g) and a kinetically stable μ-oxo (O2−) complex [(porphyrinate)FeIII-O-CuII(AN)]+ (F8 or TMPP porphyrinate).28
Figure 3.
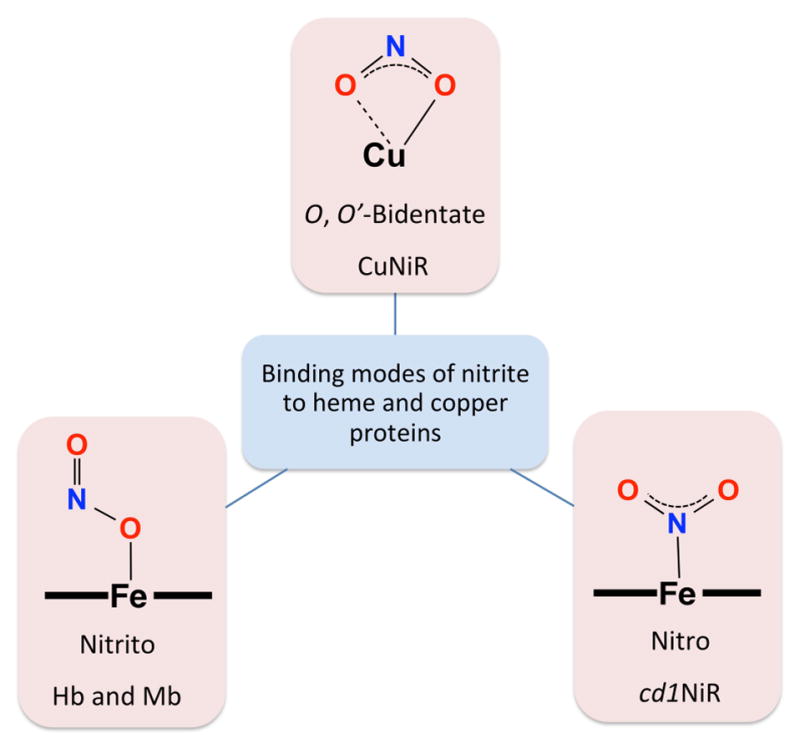
Binding modes of nitrite to heme and copper proteins.
Another possibility is that in both cases, using [(tmpa)CuII(NO2)]+ or [(AN)CuII(NO2)]+, NO2− binds to the ferrous heme must occur through the nitrogen atom in order to lead to a productive reaction, i.e., resulting in nitrite (N-O) bond cleavage and NO(g) formation. This would be consistent with the expectation that soft Fe(II) favors the softer N-atom coordination versus that hard O-atom of nitrite based on “hard-soft” acid-base principles. So, even for [(tmpa)CuII(NO2)]+, an effective reaction may require a relatively slow nitrite group rearrangement/reorientation in order that the nitrogen atom can bind the ferrous heme. Such a rearrangement is not required for the nitrite bound in [(AN)CuII(NO2)]+, thus a faster reaction occurs, all consistent with the observations.
In general, enzymatic nitrite reductase reactivity (i.e. one-electron reduction of nitrite to NO(g)) has been ascribed to two classes of proteins, containing either copper (CuNiR) or heme (e.g., cd1NiR, hemoglobin (Hb) and myoglobin (Mb)) as cofactor.25,29–30 As Figure 3 illustrates, based on available crystal structure data, nitrite binds to copper-containing NiRs in the O,O′-bidentate nitrito mode while it coordinates to cd1NiR via the N-unidentate nitro fashion. It has also been shown that nitrite binding to Hb or Mb occurs through an O-unidentate coordination (nitrito mode).31–32 Interestingly, the interactions of nitrite with ba3-CcO, with both heme and copper active-site ions, have been just recently studied by rRaman,32 revealing the existence of a ferrous heme-nitro species, not previously observed. This finding, thus, supports our original hypothesis/thought28 that iron-nitrogen bond formation, as assisted by cupric center orienting/directing of the nitrite ion, is a crucial step in our heme/Cu assembly nitrite-reductase activity. Further support for this supposition is also provided through our detailed mechanistic study of the reverse reaction i.e., oxidation of NO(g) to nitrite discussed below.
9. Mechanistic Study of Nitric Oxide Oxidase Chemistry Mediated by Heme/Cu Assemblies
As indicated in Scheme 3, we initially27 described that in the FeIII/CuII-NO(g) “parent” reaction a fully oxidized heme/Cu assembly, in the form of the μ-oxo complex [(F8)FeIII-O-CuII(tmpa)]+ which mediates the one-electron oxidation of NO(g) to nitrite. To expand on the initial study, we very recently reported a detailed mechanistic study11 using different μ-oxo [(porphyrinate)FeIII-O-CuII(L)]+ complexes in which the heme or copper chelating ligand is modified (Scheme 4); ligand (L) for the cupric core was either a tripodal tetradentate pyridyl-alkylamine chelate or a related tridentate alkylamine ligand, and where two different tetraarylporphyrinates were employed for the heme core, one with electron-withdrawing peripheral substituents versus one with strong electron-donating groups.
Scheme 4.

Variations of Heme/Cu Assemblies in NO(g) Oxidase Chemistry.
9.1. Copper Coordination Environment Variation
The effects of alterations in copper-ligand denticity and electronics were investigated by comparing the tetradentate chelate TMPA used in the FeIII/CuII-NO(g) oxidase “parent” reaction with new tridentate ligands, L = AN and MePY2 (Scheme 4).11 When employing the tridentate chelates, under the same experimental conditions, oxidation of NO(g) by μ-oxo [(F8)FeIII-O-CuII(L)]+ ensued considerably faster than for the parent analog, but still yielding the Cu(II)-nitrite plus ferrous heme-nitrosyl products.
These observations can be justified based on two factors: (i) previous studies reveal that for μ-oxo heme-FeIII-O-CuII(L) complexes where L is the tridentate ligand AN or MePY2,13 the bridging oxo ion is more basic than for tetradentate ligand, i.e., L = TMPA,12 and (ii) for tridentate ligands, there is a lack of steric hindrance, primarily because the FeIII-O-CuII moiety is bent; further, the coordination number for the copper ion is only four, relative to pentacoordination in the TMPA analog, making the bridging oxo ion more accessible for addition of NO(g) (Chart 1 and Scheme 4).11
9.2. Impact of Peripheral Substituents of the Porphyrinate; Dynamics and Intermediates Involved in NO(g) oxidase by Heme/Cu Assemblies
Modification of the nature of the porphyrinate employed in this chemistry has turned out to provide us with many new mechanistic insights.11 A new μ-oxo complex, [(TMPP)FeIII-O-CuII(tmpa)]+ was synthesized and characterized and in its reaction with NO(g) at RT, there was an instantaneous change (as followed by UV-vis monitoring), giving a mixture of species, which eventually all converted to the expected final products, [(tmpa)CuII(NO2)]+ and (TMPP)FeII(NO).
When this reaction was repeated at −20 °C, a new intermediate (λmax = 433 nm) was detected following addition of NO(g) to the solution of μ-oxo compound.11 Later, this isosbestically converted to the final products in a first order process (Scheme 5, kdissoc. = 6.7 ×10−3 s−1 at −20 °C, ΔH‡ = 41.1 ± 0.1 kJ mol−1 and ΔS‡ = −123 ± 2 J mol−1 K−1). In cryo-spray ionization mass spectrometry (CSI-MS) measurements33 at −60 °C, we could detect this new species which presented itself as a manifold of intense peaks with maximum at m/z = 1217.2847, this corresponding to a bis-NO adduct of the initial μ-oxo compound. With this observation, along with the further studies carried out (vide infra), we could conclude that sequential addition of two NO(g) molecules to the starting [(TMPP)FeIII-O-CuII(tmpa)]+ complex led to the bis-NO adduct, formulated as [(NO)(TMPP)FeII-NO2-CuII(tmpa)]+ complex.11
Scheme 5.

Proposed Reaction Steps for NO(g) Oxidase Chemistry Mediated by μ-oxo Heme-FeIII-O-CuII(L) Complexes Generating Nitrite. Adapted with permission from ref. 11 Copyright (2015) American Chemical Society.
Low-temperature stopped-flow kinetic-spectroscopic measurements33 provided (full) spectral changes for the entire course of the reaction. From the analyses carried out, the spectrum of the mono-NO adduct, which forms prior to formation of the bis-NO intermediate, could be extracted (Scheme 5). We also determined corresponding observed rate constants for the addition of first (k1(obs)) and the second (k2(obs)) NO(g) molecules for a range of low temperatures down to −83 °C and different NO(g) concentrations (ranging between 0.28 to 2.3 mM).11 At all temperatures, linear dependencies (k(obs) = k(on) [NO] + k(off)) of the observed rate constants (k1(obs) and k2(obs)) on NO(g) concentration were observed. An overview of the first and second NO(g) binding kinetics at different temperatures, and thermodynamic parameters, are given in Table 1. Notably, the binding of the second NO(g) is irreversible; addition of the first NO(g) to [(TMPP)FeIII-O-CuII(tmpa)]+ occurs about one order of magnitude faster than binding of the second NO(g) (Scheme 5, Table 1).11,34
Table 1.
Binding Kinetics and Thermodynamics of First and Second NO(g) to [(TMPP)FeIII-O-CuII(tmpa)]+.
| First NO(g) binding | Second NO(g) binding | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| T (°C) | k1(on) (M−1 s−1) | k1(off) (s−1) | k2(on) (M−1 s−1) | |||
| −60 | 6,053 ± 186 | 2.61 ± 0.24 | 706 ± 20 | |||
| −67 | 5,061 ± 244 | 1.47 ± 0.32 | 637 ± 40 | |||
| −74 | 2,356 ± 69 | 1.07 ± 0.09 | 504 ± 40 | |||
| −83 | 1,184 ± 12 | 0.50 ± 0.01 | could not be determined11 | |||
|
| ||||||
| ΔH‡(on) (kJ mol−1) | ΔS‡(on) (J mol−1 K−1) | ΔH‡(off) (kJ mol−1) | ΔS‡(off) (J mol−1 K−1) | ΔH‡(on) (kJ mol−1) | ΔS‡(on) (J mol−1 K−1) | |
|
| ||||||
| 24 ± 3 | −64 ± 10 | 22 ± 2 | −131 ± 9 | 7 ± 2 | −155 ± 8 | |
|
| ||||||
| ΔH° (kJ mol−1) | ΔS° (J mol−1 K−1) | |||||
|
| ||||||
| 2.0 ± 0.4 | 67 ± 15 | |||||
|
| ||||||
The small values of ΔH‡(on) and negative activation entropies (ΔS‡(on)) observed are consistent with the associative nature of the NO(g) binding steps. The negligible activation enthalpy (7 ± 2 kJ mol−1) for the binding of the second NO(g) point to an almost activation-less process. Together with the negative ΔS‡(on) value, one can conclude that the rate-determining step does not involve bond breaking; instead, bond making predominates. These data and conclusions are in excellent agreement with DFT studies33 (Scheme 6) which also describe the sequential addition of NO(g) to the μ-oxo complex.11
Scheme 6.

Schematic Calculated Energy Profile for [(TMPP)FeIII-O-CuII(tmpa)]+ Reaction with NO(g). Adapted with permission from ref. 11 Copyright (2015) American Chemical Society. {ΔE + ZPE in kcal mol−1 including solvent correction at both the BP86/6-31G(d) level and OLYP/6-311+G(d,p) level (given in brackets)}.
The first NO(g) attacks the μ-oxo atom, leading to the mono-NO adduct (Scheme 6). According to DFT calculations, electron-transfer occurs here, formally from NO(g) to the heme, via transient formation of a “triangular” structure. Both the original μ-oxo atom and the NO(g)-derived N-atom bind to the iron center (Scheme 6, TS1). The experimentally observed low activation enthalpy (24 ± 3 kJ/mol) and negative activation entropy derived from the kinetic studies are consistent with this sequence of events.11
As relevant to the addition of the second NO(g) molecule, the DFT results reveal that the SOMO of the bis-NO adduct, “intermediate” is predominantly localized on the Fe-porphyrin moiety, pointing to its ferrous character. Experimental evidence confirming the formulation and properties of the “intermediate” were obtained from: (i) the fact that cooling the products, consisting of a 1:1 mixture of (TMPP)FeII(NO) plus [(tmpa)CuII(NO2)]+, leads to association and reformation of the key 433 nm UV-vis feature of the “intermediate”, where the nitrite bound to copper ion coordinates and thus forms a bridge to the ferrous heme-nitrosyl complex; (ii) analyses of EPR spectra of the “intermediate” indicate the presence of a six-coordinated ferrous-heme nitrosyl bearing a trans N-based ligand (a2,Nitrite = 7.74 G), all linked to an EPR active cupric species in a TBP coordination environment.11
In the last step the “intermediate”, undergoes dissociation in a first-order process to release the finally observed mononuclear adducts, the cupric-nitrite and ferrous heme nitrosyl complexes.
10. Broader Perspectives on Nitrite and NO(g) Interconversion
From our broad investigations on nitrite/NO(g) redox interconversions mediated by heme/Cu centers, we conclude that both (bio)chemical transformations necessitate specific nitrite coordination, N-atom to ferrous-heme and O-atom to cupric ion. In fact, we believe that both chemical transformations proceed via the same intermediates.
Our proposed mechanism for nitrite reduction is represented by the forward (left-to-right) reaction in Scheme 7. For aerobic organisms, O2 is the ultimate electron-acceptor for cellular respiration. Under conditions of stress, and when the O2 concentration drops below a certain level, nitrite starts to compete with the residual O2 for reduction by the CcO binuclear active-site, eventually generating NO(g). The NO(g) thus produced reversibly inhibits the oxygen reduction at the same center and allows for O2 accumulation. Then, in this recovery from hypoxia, the reverse reaction consists of the same steps traversed backward; NO(g) is removed and converted back to nitrite for future use. Now, in normoxia, in the absence of NO(g), the CcO recovers its traditional function, i.e., catalyzing the four-electron reduction of O2 to water.
Scheme 7.

We note that the μ-oxo heme-FeIII-O-CuII(L) complexes studied described here may not exactly represent CcO turnover intermediates or even resting-state structures. However, these μ-oxo compounds should be thought of as representatives of actual enzyme active-site heme/Cu entities, those involved in NO(g) oxidation chemistry, possibly a heme-FeIII-OH…CuII or even a high-valent Compound II type species, heme-FeIV=O…Cu.
11. Summary and Outlook
In this Account, we have described our recent studies using synthetic heme/Cu assemblies, investigating their reactivity with dioxygen and nitrogen oxides. The work involves rational design and implementation of constructs mimicking basic aspects of CcO active-site, which allow for redox transformations of these small molecules (i.e., O2, NO(g), and NO2−).
Our long time goals have been to study metal-dioxygen chemistry at heme/Cu centers, as the details of O2-ligation, resulting coordination geometry and electronic-structure require full elucidation. As clean four-electron four-proton O2-reduction will depend on heme and Cu-ligation, factors such as the exact juxtaposition of these two metal ions, redox potentials (FeIII/II, FeIV/III, and CuII/I), relative basicity of O2-derived fragments, and availability and pKa of proton sources, study of these factors are critical in order to fully understand CcO fuel-cell chemistry, i.e., the O-O cleavage process, that pervading all of (bio)chemistry. Here, we briefly summarized past and very recent findings, which have led to the novel chemistry presented, a new powerful synthetic approach to generate many kinds of (new) heme/Cu constructs, which will allow for the future approaches and studies mentioned here. We have succeeded in the synthesis of heme-O2-Cu complexes possessing distinctively varying properties of ligation, structure, spectroscopic features and bonding. We are poised to examine the intimate details of stoichiometric reductive O-O cleavage reactions as a function of those heme-peroxo-Cu complex properties, versus reducing agent power and proton source pKa.
As we have also shown here, the same or similar heme/Cu constructs are able to readily effect one-electron nitrite reduction or NO(g) oxidation, occurring through O-atom transfer redox reactions. Molecular level structural and mechanistic details of these biologically relevant processes have been determined. The chemistry involved in these two processes is closely related; detailed kinetic-thermodynamic and spectroscopic studies reveal that they both proceed by essentially the same (but in opposite direction) pathways and require the involvement of common intermediates (Scheme 7). Of course, in CcO, these reactions as well involve protons; in our synthetic models, these are not needed as the metal ions instead capture O-atoms or ions. Never-the-less, the studies here for the first time point to how these nitrogen oxide reactions can or do occur at heme/Cu centers, and their relationship to dioxygen chemistry. These provide a basis for understanding the self-regulatory role of CcO in effecting cellular O2-balance, and its correlation to NO(g) and nitrite redox interplay with respect to hypoxia. Aside from further inquiries into heme/Cu/NOx chemical systems, near-future goals include the interrogation of heme/Cu constructs for NO(g) reductive coupling, relevant to bacterial denitrification and NO(g) homeostasis.
Acknowledgments
The authors acknowledge financial support from the U.S.A. National Institutes of Health.
Biographies
Shabnam Hematian, a native of Tehran, Iran received her B.S. degree in chemistry from National University of Iran and M.S. degree in inorganic chemistry from Sharif University of Technology. In 2010, she joined the research group of Kenneth D. Karlin at Johns Hopkins University to work on heme/Cu/nitrogen oxides chemistry. She is currently a postdoctoral fellow at California Institute of Technology with Jonas C. Peters. Her research interests include metal-mediated small molecule activation.
Isaac Garcia-Bosch received his Ph.D. in Chemistry from the University of Girona in 2011 under the mentorship of Miquel Costas and Xavi Ribas. After his postdoctoral studies with Kenneth D. Karlin (Johns Hopkins University) as supported by a Marie Curie IOF Fellowship, he was appointed as Assistant Professor in the Department of Chemistry at Southern Methodist University in 2015. His research is focused on the development of first row metal complexes for O2-reduction, H2O-oxidation and bioinspired synthetic catalysis.
Kenneth D. Karlin is Ira Remsen Professor of Chemistry at John Hopkins University. His copper-dioxygen chemistry interests and research program were developed while at the State University of New York at Albany (1977–1989). He received his Ph.D. degree with Stephen J. Lippard from Columbia University (1975), and he was a NATO postdoctoral Fellow at the University of Cambridge (1975–1977). His research spans biologically inspired coordination chemistry of copper and heme/Cu systems with dioxygen and nitrogen oxides.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Jei-Fei M, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Redox-Coupled Crystal Structure Changes in Bovine Heart Cytochrome c Oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 2.Kim E, Chufán EE, Kamaraj K, Karlin KD. Synthetic Models for Heme-Copper Oxidases. Chem Rev. 2004;104:1077–1133. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 3.Yoshikawa S, Shimada A. Reaction Mechanism of Cytochrome c Oxidase. Chem Rev. 2015;115:1936–1989. doi: 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- 4.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Copper Active Sites in Biology. Chem Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castello PR, David PS, McClure T, Crook Z, Poyton RO. Mitochondrial Cytochrome Oxidase Produces Nitric Oxide under Hypoxic Conditions: Implications for Oxygen Sensing and Hypoxic Signaling in Eukaryotes. Cell Metab. 2006;3:277–287. doi: 10.1016/j.cmet.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Poyton RO, Castello PR, Ball KA, Woo DK, Pan N. Mitochondria and Hypoxic Signaling. A New View. Ann NY Acad Sci. 2009;1177:48–56. doi: 10.1111/j.1749-6632.2009.05046.x. [DOI] [PubMed] [Google Scholar]
- 7.Nicholls P, Sharpe M, Torres J, Wilson MT, Cooper CE. Nitric Oxide as an Effector and a Substrate for Cytochrome c Oxidase. Biochem Soc Trans. 1998;26:S323. doi: 10.1042/bst026s323. [DOI] [PubMed] [Google Scholar]
- 8.Torres J, Sharpe MA, Rosquist A, Cooper CE, Wilson MT. Cytochrome c Oxidase Rapidly Metabolises Nitric Oxide to Nitrite. FEBS Lett. 2000;475:263–266. doi: 10.1016/s0014-5793(00)01682-3. [DOI] [PubMed] [Google Scholar]
- 9.Nanthakumar A, Fox S, Murthy NN, Karlin KD, Ravi N, Huynh BH, Orosz RD, Day EP, Hagen KS, Blackburn NJ. Oxo- and Hydroxo-Bridged (Porphyrin)iron(III)-Copper(II) Species as Cytochrome c Oxidase Models: Acid-Base Interconversions and X-ray Structure of the Fe(III)-(O2−)-Cu(II) Complex. J Am Chem Soc. 1993;115:8513–8514. [Google Scholar]
- 10.Karlin KD, Nanthakumar A, Fox S, Murthy NN, Ravi N, Huynh BH, Orosz RD, Day EP. X-ray Structure and Physical Properties of the Oxo-Bridged Complex [(F8-TPP)Fe-O-Cu(TMPA)]+, F8-TPP = Tetrakis(2,6-difluorophenyl)porphyrinate(2-), TMPA = Tris(2-pyridylmethyl)amine: Modeling the Cyhtochrome c Oxidase Fe-Cu Heterodinuclear Active Site. J Am Chem Soc. 1994;116:4753–4763. [Google Scholar]
- 11.Hematian S, Kenkel I, Shubina TE, Dürr M, Liu JJ, Siegler MA, Ivanovic-Burmazovic I, Karlin KD. Nitrogen Oxide Atom-Transfer Redox Chemistry; Mechanism of NO(g) to Nitrite Conversion Utilizing μ-oxo Heme-FeIII–O–CuII(L) Constructs. J Am Chem Soc. 2015;137:6602–6615. doi: 10.1021/jacs.5b02174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fox S, Nanthakumar A, Wikström M, Karlin KD, Blackburn NJ. XAS Structural Comparisons and Reversibly Interconvertible Oxo- and Hydroxo-Bridged Heme-Copper Oxidase Model Compounds. J Am Chem Soc. 1996;118:24–34. [Google Scholar]
- 13.Kopf MA, Neuhold YM, Zuberbühler AD, Karlin KD. Oxo- and Hydroxo-Bridged Heme-Copper Assemblies Formed from Acid-Base or Metal-Dioxygen Chemistry. Inorg Chem. 1999;38:3093–3102. [Google Scholar]
- 14.Ghiladi RA, Hatwell KR, Karlin KD, Huang H-w, Moënne-Loccoz P, Krebs C, Huynh BH, Marzilli LA, Cotter RJ, Kaderli S, Zuberbühler AD. Dioxygen Reactivity of Mononuclear Heme and Copper Components Yielding A High-Spin Heme-Peroxo-Cu Complex. J Am Chem Soc. 2001;123:6183–6184. doi: 10.1021/ja010602y. [DOI] [PubMed] [Google Scholar]
- 15.Chufán EE, Puiu SC, Karlin KD. Heme-Copper/Dioxygen Adduct Formation, Properties, and Reactivity. Acc Chem Res. 2007;40:563–572. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
- 16.Chishiro T, Shimazaki Y, Tani F, Tachi Y, Naruta Y, Karasawa S, Hayami S, Maeda Y. Isolation and Crystal Structure of a Peroxo-Bridged Heme-Copper Complex. Angew Chem Int Ed. 2003;42:2788–2791. doi: 10.1002/anie.200351415. [DOI] [PubMed] [Google Scholar]
- 17.Kim E, Shearer J, Lu S, Moënne-Loccoz P, Helton ME, Kaderli S, Zuberbühler AD, Karlin KD. Heme/Cu/O2 Reactivity: Change in FeIII-(O22−)-CuII Unit Peroxo Binding Geometry Effected by Tridentate Copper Chelation. J Am Chem Soc. 2004;126:12716–12717. doi: 10.1021/ja045941g. [DOI] [PubMed] [Google Scholar]
- 18.Chufan EE, Mondal B, Gandhi T, Kim E, Rubie ND, Moenne-Loccoz P, Karlin KD. Reactivity Studies on FeIII-(O22−)-CuII Compounds: Influence of the Ligand Architecture and Copper Ligand Denticity. Inorg Chem. 2007;46:6382–6394. doi: 10.1021/ic700363k. [DOI] [PubMed] [Google Scholar]
- 19.Kieber-Emmons MT, Qayyum MF, Li Y, Halime Z, Hodgson KO, Hedman B, Karlin KD, Solomon EI. Spectroscopic Elucidation of a New Heme/Copper Dioxygen Structure Type: Implications for O...O Bond Rupture in Cytochrome c Oxidase. Angew Chem Intl Ed. 2012;51:168–172. doi: 10.1002/anie.201104080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Bosch I, Adam SM, Schaefer AW, Sharma SK, Peterson RL, Solomon EI, Karlin KD. A “Naked” FeIII-(O22−)-CuII Species Allows for Structural and Spectroscopic Tuning of Low-Spin Heme-Peroxo-Cu Complexes. J Am Chem Soc. 2015;137:1032–1035. doi: 10.1021/ja5115198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halime Z, Kieber-Emmons MT, Qayyum MF, Mondal B, Gandhi T, Puiu SC, Chufán EE, Sarjeant AAN, Hodgson KO, Hedman B, Solomon EI, Karlin KD. Heme-Copper-Dioxygen Complexes: Toward Understanding Ligand-Environmental Effects on the Coordination Geometry, Electronic Structure, and Reactivity. Inorg Chem. 2010;49:3629–3645. doi: 10.1021/ic9020993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blomberg MRA, Siegbahn PEM, Wikström M. A metal-bridging mechanism for O-O bond cleavage in cytochrome c oxidase. Inorg, Chem. 2003;42:5231–5243. doi: 10.1021/ic034060s. [DOI] [PubMed] [Google Scholar]
- 23.Fee JA, Case DA, Noodleman L. Toward a Chemical Mechanism of Proton Pumping by the B-Type Cytochrome c Oxidases: Application of Density Functional Theory to Cytochrome ba3 of Thermus thermophilus. J Am Chem Soc. 2008;130:15002–15021. doi: 10.1021/ja803112w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.If one starts with a heme-FeIII-superoxide Cu(I) species (a model for compound A, Figure 1), at least two electrons are still required (e.g., from Cu(I), the active site Tyr or iron) to cleave the O-O bond. Our peroxo complexes are at the stage of having one more electron (from Cu(I)); we are thus poised to study the O-O cleavage step. The ultimate goal is to determine what factors pertaining to (heme)Fe-O2-Cu ligation and structure/bonding, and electron reducing ability (E°) and/or proton acidity (pKa) allow for (or not) O-O reductive cleavage without leakage of H2O2 or hydroxyl radical.
- 25.Lehnert N, Berto TC, Galinato MGI, Goodrich LE. The Role of Heme-Nitrosyls in the Biosynthesis, Transport, Sensing, and Detoxification of Nitric Oxide (NO) in Biological Systems: Enzymes and Model Complexes. In: Kadish KM, Smith K, Guilard R, editors. The Handbook of Porphyrin Science. Vol. 14. World Scientific; Singapore: 2011. pp. 1–247. [Google Scholar]
- 26.Shiva S, Wang X, Ringwood LA, Xu X, Yuditskaya S, Annavajjhala V, Miyajima H, Hogg N, Harris ZL, Gladwin MT. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat Chem Biol. 2006;2:486–493. doi: 10.1038/nchembio813. [DOI] [PubMed] [Google Scholar]
- 27.Hematian S, Siegler MA, Karlin KD. Heme/Copper Assembly Mediated Nitrite and Nitric Oxide Interconversion. J Am Chem Soc. 2012;134:18912–18915. doi: 10.1021/ja3083818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hematian S, Siegler MA, Karlin KD. Nitric Oxide Generation from Heme/Copper Assembly Mediated Nitrite Reductase Activity. J Biol Inorg Chem. 2014;19:515–528. doi: 10.1007/s00775-013-1081-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merkle AC, Lehnert N. Binding and activation of nitrite and nitric oxide by copper nitrite reductase and corresponding model complexes. Dalton Trans. 2012;41:3355–3368. doi: 10.1039/c1dt11049g. [DOI] [PubMed] [Google Scholar]
- 30.Maia LB, Moura JJG. How Biology Handles Nitrite. Chem Rev. 2014;114:5273–5357. doi: 10.1021/cr400518y. [DOI] [PubMed] [Google Scholar]
- 31.Yi J, Heinecke J, Tan H, Ford PC, Richter-Addo GB. The Distal Pocket Histidine Residue in Horse Heart Myoglobin Directs the O-Binding Mode of Nitrite to the Heme Iron. J Am Chem Soc. 2009;131:18119–18128. doi: 10.1021/ja904726q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loullis A, Noor MR, Soulimane T, Pinakoulaki E. The Structure of a Ferrous Heme-Nitro Species in the Binuclear Heme a3/CuB Center of ba3-Cytochrome c Oxidase as Determined by Resonance Raman Spectroscopy. Chem Comm. 2015;51:286–289. doi: 10.1039/c4cc08019j. [DOI] [PubMed] [Google Scholar]
- 33.Collaboration with the University of Erlangen-Nuremberg group. See reference 11.
- 34.In the CcO active-site, the heme possesses an axial proximal histidine ligand, which can be considered to be taking the place of our second NO(g) molecule in our synthetic system.