

Abstract
Background
The failing human heart is characterized by metabolic abnormalities, but these defects remains incompletely understood. In animal models of HF there is a switch from a predominance of fatty acid utilization to the more oxygen-sparing carbohydrate metabolism. Recent studies have reported decreases in myocardial lipid content, but inclusion of diabetics and nondiabetics obscures the distinction of adapations to metabolic derangements from adaptations to heart failure per se.
Methods and Results
We performed both unbiased and targeted myocardial lipid surveys using liquid chromatography-mass spectroscopy in non-diabetic, lean, predominantly non-ischemic advanced HF patients at the time of heart transplantation or LVAD implantation. We identified significantly decreased concentrations of the majority of myocardial lipid intermediates, including long-chain acylcarnitines, the primary subset of energetic lipid substrate for mitochondrial fatty acid oxidation. We report for the first time significantly reduced levels of intermediate and anaplerotic acyl-CoA species incorporated into Krebs cycle, while the myocardial concentration of acetyl-CoA was significantly increased in end-stage heart failure. In contrast, we observed an increased abundance of ketogenic β-hydroxybutyryl CoA, in association with increased myocardial utilization of β-hydroxybutyrate. We observed a significant increase in the expression of the gene encoding succinyl-CoA: 3oxoacid-CoA transferase (SCOT), the rate limiting enzyme for myocardial oxidation of βOHB and acetoacetate.
Conclusions
These findings indicate increased ketone utilization in the severely failing human heart independent of diabetes, support the role of ketone bodies as an alternative fuel and myocardial ketone oxidation as a key metabolic adaptation in the failing human heart.
Keywords: Ketone Metabolism, metabolism, lipid metabolites, heart failure, cardiomyopathy
Introduction
Under normal conditions, the turnover of ATP in the human heart is ~6Kg or the equivalent of 12 times its own weight, in order to perform the daily work of circulating seven tons of blood. Heart failure (HF) is a progressive condition where the heart cannot meet this physiologic demand. Despite advances in the treatment of this syndrome, our understanding of the energy metabolic mechanisms limiting cardiac pump function remains incomplete.
Although there is consensus that HF is characterized by metabolic abnormalities, the nature of these defects remains controversial. Under normal circumstances, fatty acids (FAs) are the predominant energetic substrate for the heart, with β-oxidation providing 50–70% of myocardial ATP need.1 After transport into cardiomyocytes, FAs are imported into mitochondria, via acylcarnitine intermediates, converted to acyl-Coenzyme A (CoA) thioesters, then oxidized via β-oxidation or re-esterified into triglycerides (TG) and stored.1 The state of metabolism in the failing human heart has been more difficult to discern. In animal models of HF there is a quantitative switch from a predominance of fatty acid utilization to the more oxygen-sparing carbohydrate metabolism.2 Consistent with this altered substrate utilization, Chokshi et al. reported decreases in myocardial triglyceride and overall fatty acid content but did not report specifically on changes in myocardial acylcarnitine content.2 In complementary studies, Gupte et al. reported substantially decreased medium- and shortchain acylcarnitines, including C2 acylcarnitine, a surrogate for acetyl-CoA, but did not report differences in the medium and longchain acylcarnitine species, especially those that are derived from FAs that are predominant in the human diet (palmitate and oleic acid).3
In spite of these observations, an integrated signature of metabolism in the non-diabetic failing human heart has not been defined. A particularly important factor confounding elucidation of the metabolic signature of human HF per se is the inclusion of metabolic comorbidities (diabetes, obesity, insulin resistance) that may affect myocardial metabolism and substrate utilization independent of HF. Accordingly, we performed both unbiased and targeted myocardial lipid survey using liquid chromatography-mass spectroscopy in non-diabetic, advanced HF patients at the time of orthotopic heart transplantation or LVAD implantation. In these non-diabetic patients, we identified significantly decreased concentrations of the majority of myocardial lipids, including long-chain acylcarnitines, the primary subset of energetic lipid substrate for mitochondrial fatty acid oxidation, in advanced HF. We also report for the first time significantly reduced levels of long-chain acyl-CoA species incorporated into Krebs cycle. In contrast, we observed an increased abundance of ketogenic β-hydroxybutyryl CoA, in association with a decrease in myocardial β-hydroxybutyrate and increased circulating ketones suggesting increased use of ketones as fuel in the severely failing human myocardium from non-diabetics. The novel observation of metabolite and gene expression signatures consistent with increased ketone oxidation in the chronically failing heart combined with alterations in the abundance of acetyl-CoA (increased) and succinyl-CoA (decreased) supports a hypothesis that use of ketone bodies as an alternate fuel could influence carbon flux contributing to the energy metabolic derangements in HF.
Materials and Methods
Human Heart Procurement
Whole human hearts were procured from two separate patient groups; non-failing brain dead organ donors with no history of HF (NF), and end stage HF transplant patients’ hearts (F) that were obtained at the time of orthotopic heart transplantation. All hearts received in-situ cold cardioplegia and were placed on wet ice in 4 °C Krebs-Henseleit Buffer. Transmural left ventricular samples, excluding epicardial fat, were snap frozen in liquid nitrogen and stored at −80 °C. All study procedures were approved by the University of Pennsylvania Hospital Institutional Review Board, and prospective informed consent for research use of heart tissue was obtained from all transplant recipients and next-of-kin in the case of organ donors
Patient Cohort
The cases include n=15 patients with chronic dilated non-ischemic cardiomyopathy. NF controls (n=20) were selected to match on age, no history of diabetes mellitus (DM) and non-obese status by body mass index criteria (BMI less than 30). A spectrum of donor heart non-failing phenotypes was available and included 8 subjects with significantly increased left ventricular mass indicative of left ventricular hypertrophy (measured ex-vivo at the time of tissue procurement and indexed to body surface area).4
The cohort consisted of patients with chronic, severe AHA/ACC Stage D HF. Transmural samples of the left ventricular myocardium were obtained from the apex at the time of LVAD implantation (n=7) or the free wall at the time of heart transplantation without LVAD (n=8). Exclusion criteria for the failing cohort included diabetes, obesity (BMI more than 30), myocarditis (viral, autoimmune), infiltrative or hypertrophic cardiomyopathy, hepatitis B, C, or HIV, or severe renal disease. Non-failing control samples were derived from hearts of brain dead organ donors with no history of heart failure, no history of diabetes mellitus, and no history of obesity (total N=20). Subjects in the NF cohort with an LVEF<45% were excluded. The initial untargeted lipidomics for discovery were performed with myocardial samples derived from 7 end-stage failing patients and 10 non-failing donors. The subsequent validation with targeted and untargeted lipidomics were performed on 8 additional patients with the same inclusion and exclusion criteria and 10 additional NF donor subjects.
Lipodomic Profiling
Methods are described in detail in the Supplement. Briefly, approximately 50 mg (+/− 0.1 mg) heart was extracted by a modified Folch liquid-liquid extraction and analyzed with liquid chromatography-nano electrospray high resolution mass spectrometry (LC-nanoHRMS) with minor modifications from previous methods 5, 6 using [2H213C2]-Cer-(d18:1/C18:0) (Avanti Polar Lipids, Alablaster, AL) and [13C4]-palmitoylcarnitine (Sigma) as internal standards. Acyl-CoA species were extracted and analyzed by stable isotope dilution-LC-tandem mass spectrometry (SID-LC-MS/MS) as previously described7, 8 using internal standards generated from [13C315N1]-pantothenate (Isosciences, King of Prussia, PA) by pan6 deficient yeast culture for long chain species9 or in Hepa1c1c7 cells for short chain species.10
RNA Isolation and cDNA Amplification
RNA was isolated from human left ventricle samples by using the Qiagen miRNeasy Mini kit (Qiagen Inc., Germantown, MD) with on-column DNAse treatment. RNA quantity was measured by Broad Range RNA assay for the Qubit (Invitrogen). Two μg of RNA was reverse transcribed using the ABI® High Capacity cDNA Reverse Transcription Kit. The cDNA was measured using the Single Stranded DNA kit for the Qubit then diluted to 10ng/μl. Four μl of the diluted cDNA from each patient was added to a mix of TaqMan Universal Master Mix along with appropriate TaqMan assay. Probes and primers used are listed in (Supplemental Table 1). All samples and reagents were mixed and dispensed using the Biomek 4000 (Beckman Coulter).
Quantitative Real Time PCR
Real-time analysis was carried out on a 7900HT Real-time PCR System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s recommended protocol. Raw cycle threshold (Ct) values were calculated using SDS 2.4 and RQ manager 1.2 software (Applied Biosystems) applying automatic baselines and threshold. The resulting value is the ΔCt. The difference in ΔCt (ΔΔCt) was then calculated by subtracting the average ΔCt of the endogenous control RPL5 from the ΔCt of target genes.
Ketone Body Assay
Commercially available colorimetric assay (Cayman Chemicals, item #700190, Ann Arbor, MI) were utilized according to the manufactures directions to determine total blood and myocardial β-hydroxybutyrate (β-HB). Systemic venous blood and myocardium were collected from n=24 patients (n=12 NF donors n=12 DCM). The assay was performed in triplicate, and compared to the supplied standard. The 96-well assays contained either blood or myocardium and 50 μl of sample was added per well to 50 μl of developer solution and incubated at 25 °C in the dark. Absorbance was read at 450 nm. A protein bicinchoninic acid assay (BCA) was performed to determine protein-corrected ketone body concentrations in the myocardium whereas the serum samples were normalized by volume.
Non-Esterified Fatty Acid Assay
A commercially available colorimetric assay (Wako Diagnostics HR Series NEFA-HR, product numbers: 999–34691, 991–34891, 993–35191) was employed. In brief Serum from the previous 24 patients was utilized to characterize their Non-Esterified Fatty Acid (NEFA) content. The samples were normalized by volume and absorbance was read at 550 nm to determine NEFA in serum samples.
Statistical Analysis
A univariate statistical approach with no correction for multiple comparisons was chosen for discovery metabolomics. As Franceschi et al. have demonstrated, multiple comparisons corrections in metabolomics do not prevent false positives completely, and induce a large penalty in false negatives.11 Since the purpose of the discovery experiment was to enable further validation experiments (using the gold standard stable isotope dilution LC-MS), we considered the risk of type I errors which would be then corrected by more analytically appropriate experiments to be low. Type II errors however would preclude the application of any validation experiments, thus resulting in an erroneous finding which would not be corrected by our experimental approach.12 Nevertheless, to test the robustness of our results, we applied a false-discovery rate adjustment in the lipidomic analysis of the validation cohort and report these results in the Supplemental Material. Further, multivariate statistical methods would be unsuited for our task due to limited sample sizes imposed by the nature of the tissue, and the difficulty of analytical quality control in untargeted experiments versus the stringent quality control afforded by stable isotope internal standards. Analysts were blinded to heart classification during extraction, then unblinded for data analysis using SIEVE (Thermo Scientific), Metaboanalyst 2.0,13 and GraphPad v5. To identify differential abundance in myocardial metabolite and gene expression levels, a p-value of less than 0.05 was considered significant, as derived from Welch’s unpaired t-test or the non-parametric Mann-Whitney test.
Results
Global Lipidomics Reveals a Reduced Abundance of Lipids in DCM
Our discovery cohort consisted of 7 non-diabetic end-stage failing versus 10 non-diabetic, non-failing donor hearts. Based on results from the discovery cohort, a validation set of heart tissue was analyzed by targeted and untargeted stable isotope dilution (SID) lipidomic analysis by nanoUPLC-HRMS in positive ion mode with 8 failing hearts and 9 non-failing hearts from non-obese, non-diabetic subjects (Supplemental Table 2). The baseline characteristics including myocardial structure and function of the combined discovery and validation cohorts, stratified by non-diabetic failing and non-failing status, are summarized in Table 1.
Table 1.
Subject characteristics. The Failing (CHF) group includes myocardial procurement at time of heart transplant (N=8) and at time of LVAD implantation (N=7). BMI, body mass index; NF, non-failing, DCM, dilated cardiomyopathy, ICM, ischemic cardiomyopathy; LVEF, left ventricular ejection fraction; BSA, body surface area.
| Average | Non-Failing (n=20) | Failing(CHF, n=15) | LVAD n=7 (included in Failing) | P value |
|---|---|---|---|---|
| Age | 54.1+/−11.92 | 50.47+/−11.76 | 50 +/−10.6 | 0.58 |
| Sex | 12 male 8 female | 13 Male 2 Female | 6 Male 1 Female | n/a |
| BMI (kg/m2) | 25.87+/−3.75 | 25.65+/−5.30 | 29.78+/− 6.52 | 0.149 |
| Etiology | 20 NF | 13 DCM 2 ICM | 5 DCM 2 ICM | n/a |
| LVEF (%) | 59.03+/−16.44 | 17.5 +/−9.9 | 21.67+/− 12.58 | *<0.005 |
| Heart Weight Index (g/m2) | 201.66+/−17.93 | 267.21+/−49.12 | n/a | *<0.005 |
| BSA (m2) | 1.89+/−0.21 | 1.98+/−0.21 | 2.07 +/− 0.23 | 0.143 |
Heatmap projection of the total 4,186 features detected by LC-HRMS lipid species indicated an overall decrease in fatty acid species in the failing hearts. To interrogate the differences between failing and non-failing hearts, differential features were sorted by an unpaired t-test with Welch’s correction followed by manual inspection to examine peak shape and correct automated peak integration. The remaining 63 differentially abundant features were plotted again on a heatmap, demonstrating a striking depletion of lipid species abundance in the non-diabetic, failing hearts (Figure 1).
Figure 1.
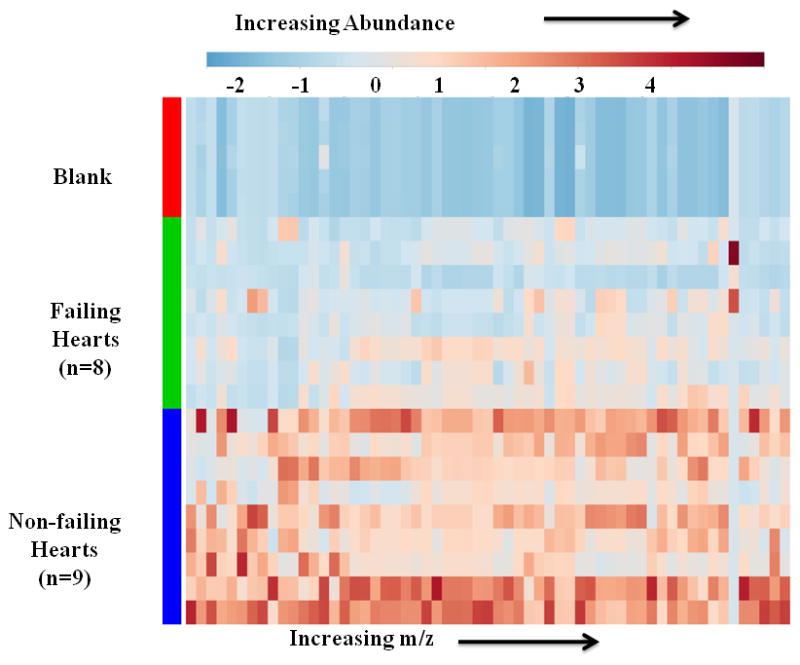
LC-HRMS lipidomic analysis reveals a depletion of lipid features from failing human hearts. Heatmap plot of 63 differentially abundant LC-HRMS lipid features from failing and non-failing hearts. Differential features were sorted by p-value < 0.01 using Welch’s unpaired t-test and manual curation for chromatographic peak shape and integration. Features that were differentially abundant between failing and non-failing hearts were mostly depleted in the failing hearts.
Based on the findings of differentially abundant fatty acid intermediates or derivatives in the discovery cohort, analysis of the acylcarnitine profile from the human heart tissues was undertaken by SID-LC-HRMS using [13C4]-palmitoylcarnitine as a stable isotope internal standard to adjust for extraction and analysis. Assignment of chromatographic peaks to acylcarnitine species was done by an accurate mass of ≤3 ppm, retention time consistent with an acylcarnitine in the chromatographic method (< 10 min), and MS/MS fragmentation pattern (Supplemental Figure 1). The identity of the peak corresponding to palmitoylcarnitine was also confirmed by co-elution with the [13C4]-palmitoylcarnitine internal standard. SID-LC-HRMS analysis revealed a greater than 10-fold reduction in palmitoylcarntine in failing hearts (mean non-failing vs. failing, 2.617 vs. 0.2076, p = 0.0025) (Figure 2A, Supplemental Figure 2). Quantitation of other acylcarnitines by LC-HRMS revealed a depletion of similar magnitude in failing hearts of acylcarnitines from hexanoylcarnitine (C6) to docosohexaenoylcarnitine (C22:6) (p < 0.0001) (Figure 2B).
Figure 2.


SID-LC-HRMS analysis indicates a reduction in acylcarnitine species in failing versus non-failing hearts. Significant reductions, by Mann-Whitney non-parametric testing, were seen in (A) palmitoylcarnitine, (B) the detected acylcarnitine profile for non-failing (hatched) versus failing (solid black) heart tissue.
Quantitation of Acyl-CoA Species and KREBS cycle Intermediates Reflects Metabolic Dysregulation in DCM
Quantification of acyl-CoAs by SID-LC-MS/MS in the myocardium from non-diabetic HF patients, as compared to non-diabetic non-failing subjects, revealed a significant decrease in succinyl-CoA (avg 10.5 vs. 17.7 pmol/mg, p = 0.023), propionyl-CoA (avg. 0.9 versus 1.8 pmol/mg, p=0.02), and an increase in β-hydroxybutyryl (β-HB)-CoA (avg. 0.57 versus 0.29 pmol/mg, p =0.015) (Table 2). This decrease in succinyl-CoA was observed while an increase in acetyl-CoA was found in end-stage failing myocardium (avg 15.0 vs. 7.7 pmol/mg, p=0.028). The ratio of myocardial succinyl-CoA to acetyl-CoA, a potential marker of KREBS cycle, is significantly decreased in end-stage HF (0.84 versus 1.93, p= 0.003) (Figure 3A), and the absolute level of βHB-CoA, a ketogenic substrate derived when fatty acid oxidation rates exceed the tricarboxylic acid cycle, was 2-fold higher in the failing group (Figure 3B). We also performed absolute myocardial quantitation of succinate, derived from succinyl-CoA and fumarate, derived from the oxidation of succinate—metabolites incorporated in the second phase of the Krebs cycle. A non-significant trend for decreased succinate (97.4 vs 55.6 ng/mg, p=0.105) and a significant decreased level of fumarate (17.4 vs 26.4 ng/mg, p=0.01) was identified in end-stage failing myocardium, as compared to non-failing (Supplemental Figure 3). Medium to long chain acyl-CoAs showed no or very weak differences between failing and non-failing subjects, except dodecanoyl-CoA (lauroyl-CoA) which was depleted in the end-stage failing heart (Table 2).
Table 2.
Total short chain and medium chain acyl-CoA profile of non-failing versus failing hearts quantified by SID-LC-MS/MS.
| Non-failing | Failing | |||
|---|---|---|---|---|
| Acyl-CoA | pmol/mg | SEM | pmol/mg | SEM |
| CoASH | 0.32 | 0.03 | 0.19 | 0.04 |
| Acetyl | 7.74 | 1.45 | 14.98 | 5.53 |
| Propionoyl | 1.76 | 0.20 | 0.89 | 0.18 |
| Succinyl | 17.74 | 2.35 | 10.54 | 2.22 |
| Butanoyl | 3.04 | 0.92 | 2.28 | 0.32 |
| Malonyl | 1.83 | 0.19 | 1.95 | 0.20 |
| 3-Hydroxybutanoyl | 0.29 | 0.05 | 0.57 | 0.13 |
| Hexanoyl | 0.16 | 0.04 | 0.16 | 0.04 |
| Octanoyl | 0.11 | 0.03 | 0.09 | 0.02 |
| Decanoyl | 0.13 | 0.02 | 0.09 | 0.02 |
| Dodecanoyl | 0.12 | 0.03 | 0.06 | 0.01 |
Figure 3.


Acyl-CoA quantitation by SID-LC-MS/MS analysis demonstrates metabolic alterations. (A) The ratio of succinyl-CoA/acetyl-CoA is significantly decreased in failing myocardium (B) A significant change in failing hearts of short chain acyl-CoA species, including the ketogenic 3-hydroxybutanoyl-CoA (β-HB), is observed as compared to non-failing. A Mann-Whitney non-parametric test performed for each of these comparisons.
Decreased Myocardial Lipids is Not Due to Reduced Peripheral Substrate Availability in HF
The myocardial utilization of substrate is correlated with peripheral availability, especially in the case of lipids and ketone bodies. If the identification of a significant decrease in energetic myocardial lipids were reflective of the peripheral milieu, a decreased concentration of plasma free fatty acids would be present in the patients with end-stage HF. However, we have identified a significant increase in the plasma concentration of non-esterified free fatty acids (NEFAs) in non-diabetic patients with advanced HF at the time of cardiac transplantation as compared to the non-failing subjects (Figure 4: mean 1.03 mM versus 0.56 mM, p=0.016 by unpaired t-test with Welch’s correction).
Figure 4.

Non-Esterified Fatty Acids (NEFAs) in non-diabetic patients. NEFA in non-failing and non-diabetic DCM patients demonstrate significantly increased circulating free fatty acids in the advanced heart failure cohort (Mann Whitney non-parametric test).
Downregulation of Lipid Storage and Carnitine Transport Genes in Advanced HF
The selected panel for quantification with rtPCR included genes implicated in a broad range of lipid metabolism--transport, storage, and energetics. Supplemental Table 3 is the list of all genes assayed, fold change expressed as the myocardial expression in failing relative to non-failing. All genes assayed were relatively abundantly expressed in myocardium in comparison to the housekeeping gene RPL5. Differential expression was plotted against RPL5 and significance was determined utilizing Welch’s corrected unpaired t-test and False Discovery Rate (FDR) less than p = 0.05 (Figure 5). Among the genes significantly decreased in the DCM myocardium were the high affinity carnitine transporter, SLC22A5 (p = 0.003), perilipin 2 (p < 0.0001), PLA2G2A (p=0.0003) and the phospholipid transport protein PTLP (p = 0.001). In comparison to non-failing, we observed no significant differences in the myocardial expression of key regulators of fatty acid β-oxidation, carnitine palmitoyltransferases (CPT1a, CPT1b, CPT2) or PPARα the peroxisome proliferator-activated receptor alpha (Supplemental Table 3).
Figure 5.

Myocardial Gene Expression. Adjusted for the housekeeping gene RPL5, myocardial gene expression is reported as fold change in 21 failing samples relative to transcript abundance in 21 non-failing samples. The transcripts with differential gene expression by parametric (t-test with Welch’s correction) testing include Phospholipase A2 (PLA2G2A), Perilipin 2 (PLIN2), Phospholipid Transfer Protein (PLTP), peroxisome proliferator-activated receptor γ coactivator (PGC-1αFold Change 1.49, p=0.0008, Solute Carrier Family 22 (organic cation /carnitine transporter) member 5 (SLC22A5).
Evidence for Utilization of Ketone Bodies as an Alternative Fuel in Advanced HF
We measured paired serum and myocardial β-hydroxybutyrate in subjects with DCM and non-failing donor subjects. There was an increased concentration of serum ketone bodies present in the DCM group (mean 145μM in DCM vs. 19 μM in the NF group, p=0.02), but myocardial ketone bodies were decreased in the DCM patients (mean 110 μM in DCM vs. 179 μM in NF, p=0.005, Figure 6). The ratio of serum to myocardial ketone bodies, an index of the utilization of β-hydroxybutyrate in the heart, is significantly increased in patients with end-stage HF (Ratio in Failing=1.48 vs. 0.22 in NF, p=0.019).
Figure 6.

Paired serum and myocardial measurements of β-hydroxybutyrate. There is a marked increase in systemic blood in DCM patients whereas there is a marked decrease of β-hydroxybutyrate within the failing myocardium from the paired data, implicating a process of increased myocardial ketone utilization. The p-values reflect a Mann-Whitney non-parametric test.
To determine if the gene regulatory program driving the utilization of ketone bodies is activated in HF, we examined the myocardial expression of genes implicated in processing of exogenous ketone bodies to acetyl-CoA (Figure 7). We identified increased expression of 3-D-hydroxybutyrate dehydrogenases (BDH1 and BHD2), enzymes involved in the conversion of ketone bodies back into acetyl-CoA for combustion and energy production via the Krebs cycle. We also identified a significant increase in the levels of mRNA encoding the rate-limiting enzyme of ketone body metabolism, 3-oxoacid-CoA-transferase (OXCT1). Conversely, hydroxymethylglutaryl-CoA synthase (HMGCS), the rate-limiting enzyme for primarily hepatic production of ketones from acetyl-CoA (ketogenesis) is significantly downregulated in the myocardium of DCM.
Figure 7.


Increased expression of the genes implicated in ketone oxidation was identified, including β-hydroxybutyrate dehydrogenase Type 1 and 2 (BDH1 and BDH2, p=0.01 for both) and 3-oxoacid CoA transferase 1(OXCT1) also known as succinyl-CoA:3-oxoacid CoA Transferase (SCOT, p=0.0006) in advanced heart failure (N=21 samples), relative to non-failing (N=21 samples). Note that the enzyme involved in ketogenesis, 3-hydroxy-3 methylglutaryl-CoA synthase (HMGCS2) is decreased in the myocardium of end-stage failing patients (p=0.006). The illustration in the top panel demonstrates the proposed link between the decreased pool of succinyl-CoA which is necessary for Krebs (TCA) cycling and the presence of ketone oxidation as the rate limiting enzyme OXCT1 (also known as SCOT) requires succinyl-CoA as a CoA donor for acetoacetate to yield acetoacetyl-CoA. Increased myocardial ketone oxidation could also explain the increased pool of Acetyl-CoA which was identified in end-stage failing myocardium, especially with the disruption in the Krebs cycle.
Expression of SCOT (OXCT1) is Correlated with Myocardial Succinyl-CoA in DCM
Recognizing that OXCT1 (also known as SCOT) is essential for conversion of acetoacetyl-CoA into two acetyl-CoA molecules that can enter the tricarboxylic acid cycle for oxidation and ATP production14, we examined the relationship between SCOT and myocardial succinyl-CoA. This analysis identified a very significant positive correlation only in the DCM group (Figure 8). There was no correlation observed in the non-failing non-diabetic subjects.
Figure 8.
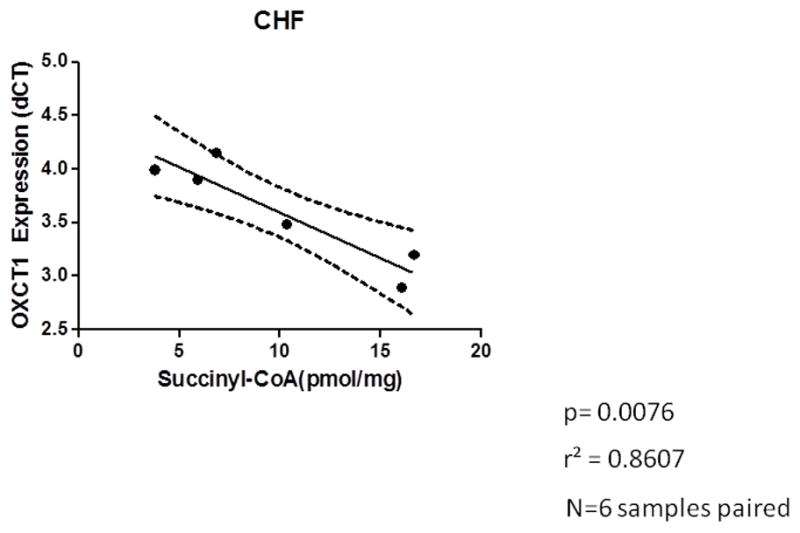
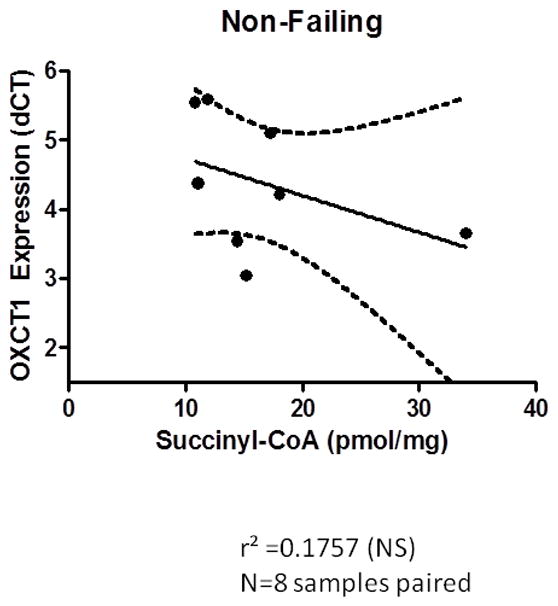
a A positive correlation of increased OCXT1 gene expression when succinyl-CoA is abundant. Expression is reciprocal as it is reported as delta ct. p=0.0076 r2 =0.8607 n=6. A Pearson correlation was calculated and the p-value reported is calculated from an F test with the null hypothesis that the overall slope is zero. b No correlation between the expression of SCOT and the concentration of succinyl-CoA is identified in the non-failing myocardium.
Myocardial Lipid Signatures of Pathological Hypertrophy vs. Failure
In a subset of 8 non-failing (LVEF>55%) subjects with myocardial hypertrophy by indexed heart weight to body surface criteria, we investigated whether the changes in lipid species and expression of ketolytic oxidation genes that were observed in end-stage failing myocardium would also be present in the hearts with left ventricular hypertrophy (LVH). In this comparison, we observed no lipid species concordance between the LVH and DCM groups. The only significant difference in the abundance of myocardial lipid species--as compared to the non-hypertrophied group--was identified in the end-stage failing group of patients. No difference in any of the medium and long chain acylcarnitines was present between the hypertrophied and non-hypertrophied non-failing hearts. In fact, our principal component analysis (PCA) of the three groups—failing, LVH, and non-failing—is depictive of the separation in myocardial lipid abundance that is driven by a marked decrease in the end-stage failing group only (see Heatmap and PCA plot in Supplemental Figure 4 and 5, respectively). An unsupervised cluster analysis (Supplemental Figure 6) of all the probed lipid species (4,186) correctly separated the clinical phenotypes into two groups: end-stage failing patients versus non-failing hearts from non-diabetic donors with and without left ventricular hypertrophy. Subject 1305 who was not insulin requiring but did have a history of using oral glycemic agents, was the only subject who was not classified correctly. Similar to what we observed with the abundance of lipid species, the expression of myocardial genes encoding the enzymes for ketone oxidation were not significantly different in LVH (Supplemental Figure 7) with the differentially increased expression only observed in end-stage failing myocardium.
Discussion
In this case control study of non-diabetic advanced heart failure, we report a significantly decreased abundance in end-stage failing myocardium of medium to long-chain acylcarnitines, the primary subset of energetic lipid substrate for mitochondrial fatty acid oxidation. Despite an increased level of acetyl-CoA in the myocardium, we observed significantly reduced levels of second span Krebs cycle intermediates including succinyl-CoA, fumarate, and the anaplerotic propionyl-CoA. In contrast, we identified signals suggesting increased use of ketones as fuel in non-diabetic failing myocardium: an increased abundance of ketogenic β-hydroxybutyryl CoA and a decreased level of myocardial β-hydroxybutyrate despite increased circulating levels of ketones. Finally, it appears that these metabolic characteristics emerge in the severely failing hearts, but not in non-failing donor hearts with significant pathological left ventricular hypertrophy. Thus, these data provide a summary of the metabolic signature or transition that is characteristic of a more advanced state of chronic human HF independent of diabetes. Previous studies identifying excess lipotoxic species in human myocardium suggested a link—which had already been established in animal models of lipotoxicity15--between excess myocardial lipids and myocardial insulin resistance. Even recently, these studies included both diabetic and non-diabetic patients and targeted a restricted set of lipid species: diacylglycerols and ceramides.2 Because diabetic patients are known to have distinct metabolic adaptations to support myocardial energetic demands irrespective of their HF status, the contributions of heart failure vs. diabetes to toxic lipid species are difficult to define in these prior studies. By including an unbiased lipidomic survey of end-stage failing myocardium and non-failing hearts in non-diabetic subjects, the present study addresses deficiencies of prior studies.
Decreased Myocardial Acylcarnitines in Advanced Human HF
Despite increased levels of circulating non-esterified free fatty acids (NEFAs) in serum and a significant increase in the gene expression of the main transporter of albumin-bound NEFAs—the cardiomyocyte specific fatty acid transport protein (SLC27A6, Supplemental Figure 8)—the myocardial concentration of acylcarnitines is significantly decreased in the end-stage failing heart. Unlike the recent report by Lai, et al. in a mouse model of hypertrophy and early HF16 which identified increased levels of many acylcarnitine species as the distinguishing metabolite signature of the failing group, our data in non-diabetic advanced human HF are consistent with significantly reduced abundance of myocardial acylcarnitines, especially the ones which are esterified from the major free fatty acids in the human diet—palmitic and oleic acid (palmitoylcarnitine and palmitoleoylcarnitine). Beyond species differences, a possible explanation of the discrepancy between our data and the metabolite signature by Lai et al., is the stage and severity of HF that has been implicated as an important factor in determining lipid substrate utilization.17, 18
The decrease in myocardial acylcarnitines cannot be explained by changes in the outer or inner mitochondrial membrane carnitine palmitoyltransferases, CPT-1 or CPT-2, for several reasons. First expression of CPT-1a, CPT-1b, or CPT-2 were not decreased in end-stage failing myocardium, nor was the expression of the regulator PPARα. In fact, expression of the peroxisome proliferator-activated receptor γ coactivator (PGC-1α orchestrating mitochondrial biogenesis and fatty acid oxidation, was found to be increased by 1.5 fold in end-stage failing myocardium (Figure 5) as previously reported.19, 20 Moreover, the highly regulated metabolite malonyl-CoA which inhibits CPT-1 was not increased in the myocardium of non-diabetic advanced HF patients (Table 2). Finally, the abundance of long-chain acyl-CoA species were not increased in the failing heart, which would be expected with myocardial suppression of the carnitine palmitoyltransferase system.
Our data do support another potential mechanism to explain decreased myocardial acylcarnitines in advanced HF: the decreased abundance of the plasmalemmal carrier OCTN2 (SLC22A5: fold change 0.54, p=0.00046, see figure 5) responsible for cellular carnitine uptake. OCTN2 is regulated by PPARα There is strong evidence from murine PPARα agonist and PPARα KO models that OCTN2 expression contributes to tissue, including myocardial, carnitine levels.21 This mechanism deserves further investigation, given the known association of primary carnitine deficiency due to mutations in the OCTN2 gene with cardiomyopathic failure22 and the reduced expression of OCTN2 in endomyocardial biopsy specimens of patients with chronic DCM.23 Our results were unequivocal in identifying marked and consistent reductions in myocardial acylcarnitines in advanced HF across a broad spectrum of carbon chain lengths (Figure 2B, Supplemental Figure 2). Transgenic models with impaired FAO such as the LCAD deficient and PPARα knock out mice have each demonstrated increased accumulation of long-chain acylcarnitines in the myocardium. 24, 25 Together, these results raise the question about whether in the more advanced stages of HF—where increased myocardial insulin resistance has been observed2— there is increased reliance on lipid and ketone body substrates to generate acetyl-CoA.17
Evidence for Disruption in the Krebs Cycle in Advanced HF
Our study identifies an impairment in the Krebs cycle linking substrate oxidation to oxidative phosphorylation, providing initial evidence for a mechanism of altered mitochondrial metabolism in human HF already reported in animal models of cardiac injury and failure.26, 27 Specifically, the increased myocardial acetyl-CoA coupled with decreased levels of metabolites incorporated in the second span of the Krebs cycle (succinyl-CoA, succinate and fumarate) suggest the end-stage failing human myocardium has deficiencies in the Krebs cycle. This also includes a depleted pool of intermediate metabolites which feed the Krebs cycle through anaplerosis (propionyl-CoA), supporting recent observations that a disruption in Krebs cycle activity is an important signature of progression to HF.3, 26 Although our data is supportive of a possible “bottleneck” at the level of acetyl-CoA which disrupts the flux of substrate oxidation, we do not have evidence to determine if the increased acetyl-CoA is derived from glycolytic flux or increased oxidative flux of FFAs or ketones.
The ratio of [succinyl-CoA] to [acetyl-CoA], which is significantly decreased in the end-stage failing myocardium, may also depict an imbalance of the intracellular levels of important regulatory intermediates which regulate mitochondrial and other cellular metabolism through succinylation and acetylation.28–30 The importance of maintaining adequate levels of succinyl CoA that are commensurate with cellular energetic demands is noteworthy since succinylation in many key mitochondrial pathways (including FA oxidation, branched-chain amino acid catabolism and the Krebs cycle) is primarily a non-enzymatic process which could be dependent on compartmentalized concentrations of succinyl-CoA.29 In a similar manner, increasing the intracellular pools of acetyl-CoA could increase protein acetylation, which is also known to have broad regulatory effects on cellular metabolism.
In contrast to the decrease in succinyl CoA, absolute levels of β-hydroxybutyryl-CoA (βHB-CoA), a ketogenic substrate derived when fatty acid oxidation rates exceed the tricarboxylic acid cycle, were higher in the failing group (Figure 3B).31, 32 FA oxidation to βHB-CoA may constitute a compensatory metabolic alteration that takes place in the setting of energetic failure by maintaining ATP production through the FADH2-dependent complex II respiration.32 Alternatively, the increased oxidation of ketones could also explain, when FAO is substrate limited, the increase in myocardial β-Hydroxybutyryl-CoA (also known as 3-hydroxy-butanoyl-CoA—see Figure 7), which we identified in the end-stage failing group. Furthermore, βHB-CoA formed by increased oxidation of fatty acids or ketones is metabolized to form acetoacetyl-CoA, which undergoes thiolase-mediated breakdown to acetyl-CoA. In this manner, the acetyl-CoA pool and cellular bioenergetics are maintained in the setting of advanced HF, where myocardial insulin resistance could inhibit the flux of glucose-derived acetyl-CoA.2
Evidence for Increased Myocardial Utilization of Ketones in HF
Our data suggest that the severely failing myocardium is utilizing the ketone body β-hydroxybutyrate (βOHB). In line with previous studies, our study found increased concentration of βOHB in the serum of advanced HF patients with no history of diabetes mellitus.33 Despite this increase in peripheral βOHB, we identified a decrease in myocardial βOHB in end-stage HF. This finding, along with a significant increase in the gene expression of BDH1, BDH2, and the rate limiting enzyme succinyl-CoA: 3oxoacid-CoA transferase (SCOT, encoded by nuclear OXCT1) involved in the myocardial oxidation of βOHB, supports a hypothesis of increased utilization of this ketone body in the myocardium of end-stage HF. In turn, utilization of βOHB might therefore explain the increased acetyl-CoA levels from increased myocardial oxidation of ketone bodies derived from a peripheral ketotic milieu in advanced HF.
Chronic HF is characterized by the upregulation of neurohormonal factors, such as catecholamines and natriuretic peptides, which are known to activate adipocyte lipolysis34, 35 and release non-esterified free fatty acids (NEFAs). Our data demonstrated a two-fold increase in the serum concentration of free fatty acids in non-diabetic HF patients at the time of cardiac transplantation (Figure 4) as well as a significant increase in the serum concentration of β-hydroxybutyrate (Figure 6). We postulate that the increased circulating levels of NEFAs drive hepatic ketogenesis to synthesize ketones and, in the setting of an energetically compromised state, a transition occurs in the failing myocardium which allows the utilization of ketones to be oxidized as an important substrate, resulting in the decreased myocardial levels of the metabolized ketone β-hydroxybutyrate despite relatively high circulating ketone levels in end-stage HF.
Decreased myocardial succinyl-CoA levels may also be explained by the increased oxidation of ketones—driven in part by the increased abundance and possibly increased activation of SCOT which utilizes succinyl-CoA as a CoA donor in this rate limiting step of ketolysis. The high degree of correlation between the expression of SCOT and the concentration of succinyl-CoA found only in the myocardium of end-stage failing patients (Figure 8) is highly suggestive that the myocardial levels of succinyl-CoA are associated with the abundance of SCOT. Prior studies in animals using the isolated working perfused heart model have demonstrated that the utilization of ketones in the form of acetoacetate is associated with a decreased pool of free or unesterified coenzyme A (CoASH) with the sequestration of free CoA as acetoacetyl-CoA and acetyl-CoA and result in a depletion of succinyl-CoA both by inhibition of 2-oxoglutarate dehydrogenase with insufficient CoASH or the increased activation of acetoacetate by the Succinyl CoA:3-oxoacid-CoA transferase (SCOT, see figure 7).36, 37 Our data suggests the end-stage failing heart is recapitulating this metabolic signature of utilizing ketones at the expense of a decreased myocardial concentration of unesterified CoA. The importance of ketone oxidation as a metabolic adaptation in the heart was recently demonstrated in a cardiomyocyte-specific knock out model of SCOT, where mitochondrial abnormalities and myocardial dysfunction were induced after increased load with transverse aortic constriction in the SCOT-knockout mice.38 Further studies will be needed to define whether oxidation of ketones in advanced HF is energetically adaptive and up to what point that is the case.
Limitations
Our data provide a set of metabolic “snapshots” of highly relevant lipid pathways but do not yield any definitive measurements to derive the flux of carbohydrate, lipid, or ketone metabolism in the myocardium of end-stage HF. Nevertheless, the snapshot of a decreased myocardial concentration of β-hydroxybutyrate in the setting of an increased serum concentration, coupled with the increased gene expression of all the ketolytic oxidizing enzymes BDH1, BDH2 and OXCT1 (SCOT) suggest increased myocardial substrate utilization of ketones in advanced human HF. Under non-failing conditions, myocardial expression of SCOT and ketone oxidation are actually suppressed when the heart is exposed to a peripheral ketotic milieu.39 This distinction of the metabolic adaptation to a ketogenic environment in the non-failing heart supports a hypothesis of metabolic reprogramming which allows for increased myocardial ketone utilization in advanced HF. Future studies are necessary to confirm through in vivo flux analyses if the failing human heart is actively and preferentially oxidizing ketones in its energy deprived state.
Although we confirmed in a second cohort the depletion of lipid substrates characterizing the failing myocardium in the advanced stage of HF, we have not performed paired analyses in subjects who have demonstrated some degree of recovery of their myocardial function with therapies such as LVAD to observe if these disruptions in lipid metabolism are reversible. Furthermore, we have no data in patients with a less advanced form of this syndrome –such as ACC/AHA Stage B or C patients.
Although we had only two female subjects in the failing group of patients, we observed the same pattern of lipid abundance, significant decrease in myocardial acylcarnitines, when the male subjects were analyzed separately. Future studies should determine if there is a gender interaction which may differentiate the metabolic adaptation between male and female patients with advanced HF.
Conclusion
We have identified an abnormal lipid signature in the myocardium of non-diabetic, end-stage HF patients that is marked by a severe deficit in a broad range of myocardial acylcarnitines, an increase in acetyl-CoA, and a decrease in Krebs cycle intermediates including succinyl-CoA. Our metabolite and gene expression data support increased myocardial utilization of βOHB in end-stage HF and a potential link between increased ketone utilization and the abundance and function of regulatory intermediates such as acetyl-CoA and succinyl-CoA. Whether the disruptions in FA and ketone metabolism highlighted here are adaptive or maladaptive remains to be determined, but these findings nevertheless support the need to consider species-dependent, etiology-dependent and severity-dependent distinctions as we elucidate the metabolic characteristics of the failing human heart and devise therapeutic strategies for improving the regulation of myocardial energetics.
Supplementary Material
Clinical Perspectives.
Human HF is a progressive syndrome which remains associated with significant mortality. Our understanding of disease progression within the hemodynamic and neurohormonal paradigms have been elucidated to a great extent, allowing therapeutic advances that have improved symptoms and survival. However, the metabolic adaptations of the failing human heart remain elusive, precluding the development of therapeutic strategies which can enhance the efficiency and energetics of failing myocardium. Furthermore, our understanding of the transition from hypertrophy to failure is also rudimentary in humans, including the metabolic changes which take place. In this report, we describe the characteristic lipid signature of end-stage failing myocardium in advanced heart failure, marked by: 1). A significant decrease in long –chain acylcarnitines; 2) A decrease in the myocardial metabolite pool of downstream TCA cycle intermediates including succinyl-CoA, despite increased myocardial Acetyl-CoA; 3)Increased myocardial utilization of β-hydroxybutyrate and upregulation of genes implicated in myocardial ketone oxidation including the succinyl-CoA transferase (SCOT). Based on these novel observations, our enhanced understanding of the metabolic signature in non-diabetic heart failure may allow translation into therapeutic strategies which could decelerate disease progression or even reverse the cardiomyopathic burden in our patients with chronic heart failure.
Acknowledgments
Funding Sources: This research was supported by the U.S. National Institutes of Health grants R01 HL089847 and R01 HL105993 to K.B. M. J. E. Rame is supported by the American Heart Association Scientist Development Grant (0730375N). Support for this study also came from a contribution by Janet H. Winston and Fran Levy.
Footnotes
Disclosures: None.
References
- 1.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 2.Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, Milting H, Chung CS, Jorde U, Naka Y, Mancini DM, Goldberg IJ, Schulze PC. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation. 2012;125:2844–2853. doi: 10.1161/CIRCULATIONAHA.111.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupte AA, Hamilton DJ, Cordero-Reyes AM, Youker KA, Yin Z, Estep JD, Stevens RD, Wenner B, Ilkayeva O, Loebe M, Peterson LE, Lyon CJ, Wong ST, Newgard CB, Torre-Amione G, Taegtmeyer H, Hsueh WA. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ Cardiovasc Genet. 2014;7:266–276. doi: 10.1161/CIRCGENETICS.113.000404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brumback LC, Kronmal R, Heckbert SR, Ni H, Hundley WG, Lima JA, Bluemke DA. Body size adjustments for left ventricular mass by cardiovascular magnetic resonance and their impact on left ventricular hypertrophy classification. Int J Cardiovasc Imaging. 2010;26:459–468. doi: 10.1007/s10554-010-9584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bird SS, Marur VR, Sniatynski MJ, Greenberg HK, Kristal BS. Serum lipidomics profiling using lc-ms and high-energy collisional dissociation fragmentation: Focus on triglyceride detection and characterization. Anal Chem. 2011;83:6648–6657. doi: 10.1021/ac201195d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snyder NW, Khezam M, Mesaros CA, Worth A, Blair IA. Untargeted metabolomics from biological sources using ultraperformance liquid chromatography-high resolution mass spectrometry (uplc-hrms) J Vis Exp. 2013;75:50433. doi: 10.3791/50433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basu SS, Mesaros C, Gelhaus SL, Blair IA. Stable isotope labeling by essential nutrients in cell culture for preparation of labeled coenzyme a and its thioesters. Anal Chem. 2011;83:1363–1369. doi: 10.1021/ac1027353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snyder NW, Basu SS, Zhou Z, Worth AJ, Blair IA. Stable isotope dilution liquid chromatography/mass spectrometry analysis of cellular and tissue medium-and long-chain acyl-coenzyme a thioesters. Rapid Commun Mass Spectrom. 2014;28:1840–1848. doi: 10.1002/rcm.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snyder NW, Tombline G, Worth AJ, Parry RC, Silvers JA, Gillespie KP, Basu SS, Millen J, Goldfarb DS, Blair IA. Production of stable isotope-labeled acyl-coenzyme a thioesters by yeast stable isotope labeling by essential nutrients in cell culture. Anal Biochem. 2015;474:59–65. doi: 10.1016/j.ab.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basu SS, Blair IA. Silec: A protocol for generating and using isotopically labeled coenzyme a mass spectrometry standards. Nat Protoc. 2012;7:1–11. doi: 10.1038/nprot.2011.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franceschi P, Masuero D, Vrhovsek U, Mattivi F, Wehrens R. A benchmark spike-in data set for biomarker identification in metabolomics. J Chemometrics. 2012;26:16–24. [Google Scholar]
- 12.Saccenti E, Hoefsloot HC, Smilde AK, Westerhuis JA, Hendriks MM. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics. 2014;10:361–374. [Google Scholar]
- 13.Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS. Metaboanalyst 2.0-a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012;40:W127–W133. doi: 10.1093/nar/gks374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: On the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids. 2004;70:243–251. doi: 10.1016/j.plefa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 16.Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: A multisystems approach. Circ Heart Fail. 2014;7:1022–1031. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obrzut S, Tiongson J, Jamshidi N, Phan HM, Hoh C, Birgersdotter-Green U. Assessment of metabolic phenotypes in patients with non-ischemic dilated cardiomyopathy undergoing cardiac resynchronization therapy. J Cardiovasc Transl Res. 2010;3:643–651. doi: 10.1007/s12265-010-9223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuunanen H, Engblom E, Naum A, Scheinin M, Nagren K, Airaksinen J, Nuutila P, Iozzo P, Ukkonen H, Knuuti J. Decreased myocardial free fatty acid uptake in patients with idiopathic dilated cardiomyopathy: Evidence of relationship with insulin resistance and left ventricular dysfunction. J Card Fail. 2006;12:644–652. doi: 10.1016/j.cardfail.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Ahuja P, Wanagat J, Wang Z, Wang Y, Liem DA, Ping P, Antoshechkin IA, Margulies KB, Maclellan WR. Divergent mitochondrial biogenesis responses in human cardiomyopathy. Circulation. 2013;127:1957–1967. doi: 10.1161/CIRCULATIONAHA.112.001219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. Pgc-1alpha and erralpha target gene downregulation is a signature of the failing human heart. J Mol Cell Card. 2009;46:201–212. doi: 10.1016/j.yjmcc.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Vlies N, Ferdinandusse S, Turkenburg M, Wanders RJ, Vaz FM. Ppar alpha-activation results in enhanced carnitine biosynthesis and octn2-mediated hepatic carnitine accumulation. Biochim Biophys Acta. 2007;1767:1134–1142. doi: 10.1016/j.bbabio.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Ye J, Ganapathy V, Longo N. Mutations in the organic cation/carnitine transporter octn2 in primary carnitine deficiency. Proc Natl Acad Sci U S A. 1999;96:2356–2360. doi: 10.1073/pnas.96.5.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grube M, Ameling S, Noutsias M, Kock K, Triebel I, Bonitz K, Meissner K, Jedlitschky G, Herda LR, Reinthaler M, Rohde M, Hoffmann W, Kuhl U, Schultheiss HP, Volker U, Felix SB, Klingel K, Kandolf R, Kroemer HK. Selective regulation of cardiac organic cation transporter novel type 2 (octn2) in dilated cardiomyopathy. Am J Pathol. 2011;178:2547–2559. doi: 10.1016/j.ajpath.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makowski L, Noland RC, Koves TR, Xing W, Ilkayeva OR, Muehlbauer MJ, Stevens RD, Muoio DM. Metabolic profiling of pparalpha−/− mice reveals defects in carnitine and amino acid homeostasis that are partially reversed by oral carnitine supplementation. FASEB J. 2009;23:586–604. doi: 10.1096/fj.08-119420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Vlies N, Tian L, Overmars H, Bootsma AH, Kulik W, Wanders RJ, Wood PA, Vaz FM. Characterization of carnitine and fatty acid metabolism in the long-chain acyl-coa dehydrogenase-deficient mouse. Biochem J. 2005;387:185–193. doi: 10.1042/BJ20041489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dodd MS, Atherton HJ, Carr CA, Stuckey DJ, West JA, Griffin JL, Radda GK, Clarke K, Heather LC, Tyler DJ. Impaired in vivo mitochondrial krebs cycle activity after myocardial infarction assessed using hyperpolarized magnetic resonance spectroscopy. Circ Cardiovasc Imaging. 2014;7:895–904. doi: 10.1161/CIRCIMAGING.114.001857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schroeder MA, Lau AZ, Chen AP, Gu Y, Nagendran J, Barry J, Hu X, Dyck JR, Tyler DJ, Clarke K, Connelly KA, Wright GA, Cunningham CH. Hyperpolarized (13)c magnetic resonance reveals early- and late-onset changes to in vivo pyruvate metabolism in the failing heart. Eur J Heart Fail. 2013;15:130–140. doi: 10.1093/eurjhf/hfs192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM, Jacobson MP, Verdin E. Sirt3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl coa synthase 2 and regulates ketone body production. Cell Metab. 2010;12:654–661. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B, Uppala R, Fitch M, Riiff T, Zhu L, Zhou J, Mulhern D, Stevens RD, Ilkayeva OR, Newgard CB, Jacobson MP, Hellerstein M, Goetzman ES, Gibson BW, Verdin E. Sirt5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013;18:920–933. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, Gibson BW. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of sirt3 in metabolic pathways. Proc Natl Acad Sci U S A. 2013;110:6601–6606. doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basu SS, Blair IA. Rotenone-mediated changes in intracellular coenzyme a thioester levels: Implications for mitochondrial dysfunction. Chem Res Toxicol. 2011;24:1630–1632. doi: 10.1021/tx200366j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Worth AJ, Basu SS, Snyder NW, Mesaros C, Blair IA. Inhibition of neuronal cell mitochondrial complex i with rotenone increases lipid β-oxidation supporting acetyl-coenzyme a levels. J Biol Chem. 2014 doi: 10.1074/jbc.M114.591354. jbc. M114. 591354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lommi J, Kupari M, Koskinen P, Naveri H, Leinonen H, Pulkki K, Harkonen M. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol. 1996;28:665–672. doi: 10.1016/0735-1097(96)00214-8. [DOI] [PubMed] [Google Scholar]
- 34.Lafontan M, Berlan M, Stich V, Crampes F, Riviere D, De Glisezinski I, Sengenes C, Galitzky J. Recent data on the regulation of lipolysis by catecholamines and natriuretic peptides. Ann Endocrinol (Paris) 2002;63:86–90. [PubMed] [Google Scholar]
- 35.Lafontan M, Moro C, Berlan M, Crampes F, Sengenes C, Galitzky J. Control of lipolysis by natriuretic peptides and cyclic gmp. Trends Endocrinol Metab. 2008;19:130–137. doi: 10.1016/j.tem.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Russell RR, 3rd, Taegtmeyer H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J Clin Invest. 1991;87:384–390. doi: 10.1172/JCI115008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Russell RR, 3rd, Taegtmeyer H. Coenzyme a sequestration in rat hearts oxidizing ketone bodies. J Clin Invest. 1992;89:968–973. doi: 10.1172/JCI115679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schugar RC, Moll AR, Andre d’Avignon D, Weinheimer CJ, Kovacs A, Crawford PA. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol Metab. 2014;3:754–769. doi: 10.1016/j.molmet.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wentz AE, d’Avignon DA, Weber ML, Cotter DG, Doherty JM, Kerns R, Nagarajan R, Reddy N, Sambandam N, Crawford PA. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J Biol Chem. 2010;285:24447–24456. doi: 10.1074/jbc.M110.100651. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.