

Abstract
GPCR are ubiquitous in mammalian cells and present intricate mechanisms for cellular signaling and communication. Mechanistically, GPCR signaling was identified to occur vectorially through heterotrimeric G proteins that are negatively regulated by GRK and arrestin effectors. Emerging evidence highlights additional roles for GRK and Arrestin partners, and establishes the existence of interconnected feedback pathways that collectively define GPCR signaling. GPCR influence cellular dynamics and can mediate pathologic development, such as cancer and cardiovascular remolding. Hence, a better understanding of their overall signal regulation is of great translational interest and research continues to exploit the pharmacologic potential for modulating their activity.
1. Introduction
Cellular differentiation is a hallmark of multicellular organisms that is essential for specialization at the cell, tissue, and organ level. Specialization promotes organismal development and survival by permitting cells to differentiate into cellular lineages that possess unique properties, such as sensation, secretion, absorption, or conductivity [1]. Multicellular organisms coordinate these activities to sustain homeostasis. Cell surface receptors and signaling molecules represent a pivotal mechanism for overcoming this challenge by facilitating cellular communication [2, 3]. The Seven Transmembrane Span Receptors (7TMR) / G Protein-Coupled Receptor (GPCR) family represents the largest superfamily of cell surface receptors, with over 800 distinct receptors encoded within the human genome [4]. This review highlights the biologic role and regulation of 7TMR/GPCR and their interacting proteins, while reviewing their structure, classification, and mechanism of action.
2. 7TMR/GPCR Signaling Overview
2.1 Diversity
7TMR/GPCR are activated by diverse extracellular signals, such as proteins, peptides, lipids, small molecules, ions, and photons. These receptors transduce extracellular cues into intracellular signals to facilitate cell responses. 7TMR/GPCR and their diversity underpin their ability to regulate highly varied physiological functions responsible for sensation, adaptation, and survival [4–7]. In the context of sensation, ocular processing is initiated by four visual 7TMR/GPCR, gustation uses several sweet, bitter and umami (glutamate) GPCR; and olfactory sensation is initiated by several hundred 7TMR/GPCR [7–10]. In addition, over 150 distinct GPCR and their cognate ligands regulate critical biophysical processes, such as endocrine physiology, immunomodulation, and neurotransmission/neuromodulation. Despite extensive research over the past 3 decades, over 100 “orphan” receptors remain that have either no known endogenous ligand or unclear physiological functions [11].
2.2 Structure and classification
The 7TMR/GPCR family name reflects its member’s structure and function. This receptor family is structurally defined by a seven alpha-helix transmembrane span topology [4, 7, 12]. The amino terminus and three transmembrane connecting loops are positioned extracellularly, while the carboxy terminus and three additional transmembrane connecting loops are located intracellularly. In addition to structural homology, this receptor family largely shares a common mechanism of action: GPCR activation initiates signal transduction across the cell membrane, which activates an intracellular heterotrimeric GTP-binding proteins (G proteins) [5].
The 7TMR structural superfamily is divided into three canonical receptor families and other non-canonical families [13]. These families are defined by conserved sequence elements located primarily in the receptor transmembrane region. Overall, these classifications are subdivided into canonical (family A-C) and non-canonical groupings. Unlike family A-C receptors, non-canonical 7TMR appear to primarily utilize mechanisms other than G proteins for signal propagation. However, numerous recent reports argue that specific non-canonical 7TMR can indeed activate G proteins and be regulated like GPCRs, in addition to their non-canonical activities [14]. Family A represents the largest canonical subfamily and is defined by homology with the visual receptor rhodopsin and the β2-adrenergic receptor (β2AR). Family B is defined by sequence homology to secretin and glucagon receptors. Expression of family B receptors is localized largely to gastrointestinal and neuronal tissue. The smallest canonical 7TMR subfamily, family C receptors are neuromodulatory metabotropic glutamate and GABAB receptors. This family is expressed in both the peripheral and central nervous systems and is responsible for processing diverse signals, such as pain, anxiety, and memory formation. Non-canonical receptors include those of the Frizzled family that recognize Wnt ligands, or the Hedgehog co-receptor Smoothened. These receptors exert their effect through G protein independent mechanisms [13].
2.3 G protein signaling mechanism
G protein activation is dependent primarily upon GPCR stimulation. GPCRs remain mostly inactive on the cell surface until bound by a cognate activating agonist and initiate G protein activation. A GPCR’s corresponding G protein generally remains inactive until GPCR activation. In their inactive conformation, G proteins exist in a heterotrimeric state: the constituent α-subunit, bound to GDP, is tightly associated with the βγ-subunit dimer. GPCRs act as guanine nucleotide exchange factors (GEF) for G proteins when agonist-activated [15–17]. Consequently, GPCR activation stimulates the exchange of GDP for GTP on the α-subunit. GDP/GTP exchange on the α-subunit stimulates the dissociation of activated α•GTP from the βγ-subunits. Activated α•GTP then binds to downstream signaling effectors, such as adenylyl cyclase or phospholipase C, to alter their activity to increase levels of intracellular second messengers, such as cyclic AMP (cAMP) or inositol tris-phosphate (IP3) [17, 18]. In addition to α•GTP, unbound βγ-subunits bind to and activate their own effectors, including G protein-regulated Inward Rectifying K (GIRK) channels and protein kinases, such as GRK2 and PAK [19–24].
For deactivation, the α-subunit encodes an intrinsic GTPase that hydrolyzes GTP to GDP to inactivate its ability to propagate the signal cascade [15, 16]. Additional GTPase-activating proteins accelerate this deactivation, such as regulator of G protein signaling (RGS) family accessory proteins [25, 26]. When GTP bound to an α-subunit is hydrolyzed to GDP, the α•GDP dissociates from its effectors and re-associates with βγ-subunits, thereby reassembling an inactive G protein heterotrimer. The dissociation of the α-subunit and βγ-subunit from their effectors effectively ends the propagation of the signal cascade by deactivation of the effector [15–17].
G protein mediated GPCR signaling is a highly amplified system. Upon activation, GPCR’s guanine nucleotides exchange activity last long enough to induce activation of numerous individual G proteins. Activated G proteins provide further signal amplification owing to α-subunit’s ability to produce many second messenger molecules (e.g., cAMP, IP3) during the time it remains bound to an effector enzyme. Consequently, small amounts of a hormone released from a distant part of a multicellular organism can profoundly influence the function of a distant target cell.
2.4 G protein classification
G proteins are classified based upon their α subunit structure and function, and fall into four subclasses. Each G protein subclass has distinct downstream effectors and produces unique signaling effects on the cell. First, the stimulatory G proteins (Gs) stimulate adenylyl cyclase to convert ATP into cyclic AMP (cAMP). Elevated cAMP results in the activation of cAMP-regulated proteins, such as, specific kinases (protein kinase A, PKA), cyclic nucleotide-gated channels, and the Epac GEF for Rap small GTP-binding proteins [5, 15, 16, 27]. Second, the inhibitory heterotrimeric Gi proteins mechanistically oppose Gs activity by inhibitiong adenylyl cyclase, which reduces intracellular cAMP levels [15, 16, 28]. Furthermore, the βγ-subunits of Gi proteins can exert cellular effects by regulating GIRK channels and other effectors. Transducins (Gt) represent specific Gi family members and are stimulated by light-activated rhodopsin to regulate retinal cGMP phosphodiesterase [29]. Similarly, gustducin (Ggust) is a Gi family member stimulated by tastant receptors in taste buds. Third, Gq proteins activate phospholipase C-β enzymes upon stimulation by cognate GPCRs. Phospholipase C-β hydrolyzes plasma membrane phosphatidyl inositol lipids, releasing IP3 and diacyl glycerol that act to increase intracellular calcium and protein kinase C activity [28]. Fourth, the G12 proteins activate GEFs for the RhoA small GTP-binding protein, activating it to regulate the cytoskeleton. [28, 30, 31].
2.5 GPCR functional classification
In addition to classifications based upon sequence similarity (section 2.2), GPCRs can be categorized by their cognate G protein and the downstream pathways they regulate [32]. Interestingly, while some GPCRs appear to exclusively couple with one class of G protein (e.g., “Gs-coupled receptor” or “Gq-coupled receptor”), other GPCR can activate G proteins from two or more classes . Not all G proteins within a class are equivalent: receptors can have a preference for specific subtypes of G proteins within a class. In particular, evidence using mice deficient in individual Gi family members has made it clear that a receptor may couple to only one member of the Gi subfamily even when other members are available. This signaling diversity is further increased by the fact that there are five β-subunits and twelve γ-subunits that can theoretically assemble (with the α-subunits) several hundred combinations, and both receptors and βγ-subunit-activated effectors can prefer specific α+βγ combinations. Despite this complexity, the general principles of G protein mediated signaling hold true [28, 33].
GPCRs can also be categorized according to their ligands [32, 34]. Receptors that bind the same ligand are generally also similar based upon other classification systems. For instance, receptors that bind the same ligand are usually very highly related and form distinct subgroups by homology clustering. For example, receptors for glycoprotein hormones (e.g., LH, FSH) are structurally similar to each other, as are receptors for the biogenic amines (e.g., serotonin, dopamine, adrenaline). While many unique ligand – receptor pairs exist, some endogenous ligands can bind with high affinity to multiple receptor subtypes (e.g., 12 GPCRs activated by serotonin, 5 by acetylcholine). Overall, this complexity of GPCRs and ligands increases the specificity of desired target organ effects.
3. 7TMR/GPCR Dynamics
As noted, receptors are generally inactive in the absence of their activating agonist. Once a specific 7TMR/GPCR is stimulated, it activates downstream intracellular signaling pathways in response to agonist binding. However, this model vastly oversimplifies the dynamics of 7TMR/GPCR mediated signaling. This section seeks to define the mechanisms that regulate the signaling process.
3.1 Receptor desensitization
7TMR/GPCR-regulated systems exhibit graded responses upon activation. The amount of ligand signal defines the downstream signal; more agonist promotes more response. Because GPCR systems are highly amplified a small amount of ligand occupying a fraction of receptors, may induce a maximal response. However, each receptor system displays a memory of prior activation that influences its ability to be stimulated in the future. The fraction of receptors that must be activated to achieve a full effect varies based on how active the system has been recently. Cells with receptors that have been active recently respond by becoming less sensitive to further stimulation, while cells that have not been stimulated for some time become more sensitive to future stimulation. This phenomenon is termed desensitization [22, 35–37]. Desensitization serves to prevent overstimulation of target cells in the face of high agonist concentrations, while the reverse process of resensitization allows cells to still recognize and respond to low amounts of agonist [38–40]. Overall, these interlinked processes allow a cell to maintain sensitivity to a ligand in conditions of either high and low signaling tone.
The ability to alter GPCR sensitivity is critical for cellular responses such as chemotaxis, wherein a cell responds to a gradient of chemoattractant agonist. Cells must interpret differences in agonist concentration that vary by a few percent difference from one side of the cell to other to effectively migrate towards the source of a ligand, such as a site of infection. Receptor desensitization mechanisms allow cells to decipher the cues of a shallow ligand gradient through differential signaling sensitivity to define the ‘front’ versus the ‘rear’ of a migrating cell [41]. Immune cells utilize such a mechanism in order to migrate towards injured tissues following gradients of tissue-released GPCR ligand chemokines [42].
Overstimulated receptors require higher levels of agonist to produce equal downstream effects. This is clinically exemplified in heart failure, wherein poor contractility of the heart drives catecholamine release to act on cardiac adrenergic receptors to increase contractility. As heart receptors adapt to the high levels of stimulation, this positive feedback overdrive eventually becomes ineffective and actually counterproductive, as it promotes further pathological remodeling of the heart [28, 43, 44].
3.2 Receptor sequestration
Receptor levels on the cell surface are dynamic and change in response to agonist stimulation. Receptors are removed from the cell surface upon activation, and return to the cell surface after agonist levels subside. Short-term endocytotic removal of receptors from the cell surface is termed sequestration [45]. Different receptor types utilize distinct pathways for removal from the cell surface, such as clathrin-coated pits or caveolae-mediated endocytosis. Once internalized, receptors traffic in intracellular vesicles through a series of intracellular vesicular compartments that eventually lead to release of the receptor back onto the cell surface in an activatable state [46, 47].
3.3 Receptor downregulation
Long-term over-activation of a GPCR can lead to a substantial reduction in cell surface receptor levels. This is termed down-regulation and can result in substantial reductions in receptors present at the cell surface. Mechanistically, down-regulation is initially comparable to receptor sequestration. However, unlike sequestration, down-regulation targets internalized receptors for lysosomal degradation. Thus, recovery of normal receptor level at the cell surface requires new receptor synthesis [48, 49].
3.4 Constitutive activity
Receptors possess varying degrees of agonist-independent activity, termed constitutive activity, for activating downstream signaling pathways [50]. Constitutive activity levels are receptor specific and depend on receptor expression level. Receptors with low constitutive activity exhibit the highest degree of ligand-stimulated activity, whereas agonist activation of receptors with high constitutive activity may only promote a two-fold increase in signal upon agonist stimulation, due to their high basal level of activity. Constitutive activity is hypothesized to occur as a consequence of the molecular dynamics of the receptor protein: receptors constantly transition among multiple conformations, and a subset of these conformations are capable of inducing G protein activation, thus producing constitutive activity. A receptor with high constitutive activity thus has more low-energy states that favor G protein activation than a receptor with low constitutive activity. [51–54].
3.5 Agonist and antagonist dynamics
GPCR conformational states determine their ability to activate coupled G proteins. Agonists stabilize active GPCR conformations to induce signaling, while antagonists stabilize inactive conformational states to prevent signaling. Agonists are classified based upon the degree to which they activate GPCRs. A full agonist promotes maximal GPCR activation (maximal as compared to the native ligand), while a partial agonist activates GPCR activity to a lesser degree even at the highest concentrations of that ligand. In contrast, an antagonist inhibits activation by either occupying the binding site for the natural ligand (orthosteric site) or another site (allosteric site) that precludes subsequent agonist mediated GPCR activation [55, 56]. Furthermore, agonists can alter receptor constitutive activity. Inverse or negative agonists silence the activity of constitutively active receptors by suppressing their basal constitutive activity, through preventing spontaneous transitions to active states. In contrast, neutral antagonists bind a receptor without altering the ability to transition among inactive and active states, thus permitting basal activity to continue [57, 58]. Overall, molecules that bind to a receptor can affect GPCR function along a continuum, from full activation through complete inactivation.
4. Signal Termination: GRK, Arrestin, and G protein switching
Termination of GPCR signaling is mediated by multiple mechanisms. Signaling at the receptor can be terminated by degrading the ligand, as is the case for most peptide agonists, or by removing the ligand from the vicinity of the receptor, as through reuptake of the biogenic amine dopamine from synapses into presynaptic neurons for re-storage in vesicles for later release. The intrinsic GTPase activity of G proteins and its acceleration by GTPase-activating proteins of the RGS protein family (section 2.3) limits the lifetime of downstream signaling once an agonist is no longer present at sufficient concentration at the receptor to drive new G protein activation. But in addition to these general modes of regulation, GPCRs themselves are subject to direct regulation by a two part mechanism consisting of G protein-coupled receptor kinases (GRKs) and arrestins. These two classes of cellular regulatory proteins cooperatively function to terminate GPCR signaling, as well as by mechanisms that alter receptor coupling to G proteins.
4.1 Regulated GPCR-G protein coupling and G protein switching
GPCR affinity for specific G protein subtypes is influenced by several factors, including receptor structure and posttranslational modification. Individual GPCRs may dynamically couple amongst several G protein subtypes. Indeed, receptors may experience a phenomena termed “G protein switching”, wherein a GPCR’s affinity for a particular G protein subtype alters in response to cellular dynamics, such as receptor posttranslational modifications. The β2AR demonstrates G protein switching, as it dynamically couples with both Gs and Gi proteins [33, 59]. β2AR G protein switching occurs as a negative feedback regulatory mechanism mediated by PKA phosphorylation of the receptor. Upon initial stimulation, β2AR activates the Gs protein to increase adenylate cyclase signaling and PKA activity to propagate the cellular response. In addition to eliciting second messenger responses, PKA phosphorylates site-specific residues on β2AR [60]. PKA phosphorylation of β2AR alters the receptor coupling from Gs to Gi, and activated Gi subsequently deactivates Gs protein and results in a decrease in cAMP production and PKA activity [33, 61]. Effectively, PKA phosphorylation of β2AR and G protein switching mediates a negative feedback loop with respect to the initial Gs signal activation.
4.2 GRK signal termination
GPCR kinases (GRKs) are protein serine/threonine kinases of the protein kinase A/G/C (AGC) subfamily. This kinase family initiates GPCR signal termination by phosphorylating ligand-bound GPCR. These kinases recognize the active conformation of GPCR and phosphorylate serine and threonine residues within the carboxy terminal tail and third intracellular loop of the receptor. GPCR phosphorylation itself generally does not decouple the activated GPCR from its cognate G protein. Instead, this phosphorylation triggers the second step of signal termination, by marking activated receptors for binding of arrestin proteins to the phospho-receptor [36, 62–64].
The GRK family consists of seven members that are categorized into three subfamilies. GRK1 subfamily consists of GRK1 (rhodopsin kinase) and GRK7 (cone kinase); the GRK2 subfamily is composed of GRK2 and GRK3; and the GRK4 subfamily is formed by GRK4, GRK5 and GRK6 [45, 63]. The GRK1 kinases are predominately localized to retinal tissue and influences visual acquisition by regulating visual GPCR. The GRK2 subfamily, in contrast, is ubiquitously expressed and possess a unique βγ-subunit binding domain. The βγ-subunit binding domain of this subfamily permits translocation of these GRKs to the inner leaflet of the plasma membrane upon interaction with the βγ-subunit released by a receptor-activated G protein. In the GRK4 subfamily, GRK5 and GRK6 are expressed in many tissues, while GRK4 expression is primarily localized to the testes. Within the GRK family, GRKs 2, 3, 5, and 6 are the most widely expressed isoforms that are responsible for the bulk of receptor phosphorylation throughout the body.
A large mismatch exists between the number of GPCRs and the number of GRKs expressed within an organism [36, 62–64]. Inherit within this mismatch, GRKs are relatively promiscuous in recognizing receptor. While there appear to be some GRK preferences among receptor types, specificity is largely driven by the context of GPCR activation and by relative GRK concentrations in a cell of interest, rather than by recognition of any consensus phosphorylation site [65]. GRKs exhibit some preference for phosphorylation sites on receptors, but many studies have shown that GRKs will find other sites to phosphorylate on a receptor if their preferred sites are unavailable, such as after receptor mutagenesis.
4.3 Arrestin signal termination
Arrestins constitute a scaffold protein family that initiate the second part of GPCR signal termination [38, 45]. Arrestins recognize active GPCR and their binding affinity is augmented by GRK mediated phosphorylation. Arrestins have varying affinities for different phosphorylation patterns. Highly phosphorylated serine/threonine clusters induce stronger arrestin-GPCR binding. The arrestin family consists of four proteins, arrestin1-4. Arrestin1 and arrestin4 are unique for their involvement in visual processing. These proteins are expressed in the rods and cones of retinal tissue, wherein they bind to phospho-rhodopsin and phospho-iodopsin to regulate GPCR mediated visual perception [63, 66, 67]. Unlike the visual arrestins, Arrestin2 (aka βarrestin1) and arrestin3 (aka βarrestin2) are expressed in most tissue types [63].
Arrestins promote GPCR signal termination by two distinct, but complementary mechanisms: receptor desensitization and receptor sequestration. These scaffold proteins mediate receptor desensitization by directly inhibiting G protein - GPCR coupling [45]. Mechanistically, arrestin-bound GPCRs are sterically blocked from interacting with their cognate G protein [38]. This occurs almost immediately after receptor activation, since G proteins, GRKs, and arrestins compete for binding to the receptor in its active state. Thus the relative abundance of these three classes of proteins in a cell determine much of the kinetics of receptor signaling. Though GRK-mediated receptor phosphorylation may partially accomplish G protein decoupling, the binding of arrestins to a GRK-phosphorylated GPCR significantly augments this signal blockage mechanism [35, 38, 46]. Efficient arrestin binding results in homologous desensitization, or the turning off of those specific receptors that have been recently stimulated.
Arrestin-mediated receptor trafficking from the cell surface, or sequestration, occurs minutes to hours after agonist stimulation. Arrestins act as adaptors to associate bound receptors with cellular endocytic machinery proteins to mediate GPCR internalization into vesicles as specific cargo. Arrestin binding to phosphorylated GPCRs promotes arrestin interaction of adaptin and clathrin, two proteins responsible for mediating receptor internalization [68, 69]. GPCR internalization sequesters the activated receptors from further access to agonists outside the cell and effectively reduces the number of receptors present on the cell surface for ligand stimulation.
After mediating GPCR internalization, arrestins may either dissociate or remain bound to a receptor as it begins to transit through intracellular vesicular compartments, depending on arrestin affinity for the phospho-receptor [70]. Early dissociation of arrestin from a weak-binding GPCR keeps the receptor on a quick itinerary leading to receptor recycling, wherein the endocytosed GPCR quickly returns to the cell surface to engage in further ligand binding. Failure of the arrestin-GPCR complex to dissociate early marks that receptor for further processing through intracellular compartments that result in either receptor degradation or delayed receptor recycling [22, 49]. Among other factors, the conformational state of the receptor and the pattern of GRK-mediated phosphorylation collectively influence arrestin-GPCR binding dynamics.
GRK/arrestin signal regulation is stoichiometric, with one arrestin molecule able to bind to and regulate only one GPCR. Low receptor ligand occupancy can induce full G protein signaling for an extended period with low desensitization, since few receptors can activate many G proteins through amplification, but also leads to only a few receptors at a time being removed from a functional state on the cell surface. The endocrine system demonstrates this phenomenon, where agonists released into the systemic circulation are highly diluted within the vasculature that carries them to their target tissues where they can have sustained effects. In contrast, high receptor ligand occupancy will induce an intense burst of G protein activity, followed by profound GRK/arrestin mediated desensitization. This type of regulation is most evident at neuronal synapses, where a neurotransmitter agonist is released in large quantities into synapses, and signaling occurs in bursts rather than constantly.
4.4 Arrestin GPCR feedback regulation
Arrestins additionally regulate GPCR activity through non-adapter based feedback regulatory mechanisms. The β2AR system is regulated by arrestin through feedback regulation. β2AR activation yields an increase in PKA activity secondary to cAMP production. Phosphodiesterase (PDE) opposes this activity by degrading cAMP. Upon β2AR activation, arrestin scaffolds with PDE4D and promotes its translocation to the plasma membrane [71]. PDE4D translocation and activation negatively regulates the β2AR cascade by degrading the substrate necessary for PKA activation. Interestingly, observations demonstrate that these events additionally inhibit PKA mediated phosphorylation of active β2AR [71]. Hindrance of PKA-mediated β2AR phosphorylation inhibits G protein switching [33, 61]. Arrestins additionally partake in Gq-coupled M1 muscarinic receptor feedback regulation [72]. DAG and IP3 are formed upon Gq stimulation. Diacylglycerol kinases (DGKs) terminate Gq signaling by phosphorylating the second messenger DAG. Similar to PDE4D interactions in the β2AR-Gs system, arrestins scaffold with and activate DGKs to degrade DAG. Effectively, arrestin promotes Gq negative feedback regulation by activation removal of the second messenger DAG.
5. GRK and Arrestin-Mediated Signaling
Arrestins and GRKs also function in GPCR signal termination-independent processes. They serve as molecular scaffolds that can alter the activity of partners including protein kinases, transcription factors, and other cellular signal modulators. GRKs can regulate the activity of many cytoplasmic and nuclear non-GPCR substrates through both scaffold- and kinase activity-dependent functions.
5.1 Non-canonical GRK activity
Accumulating evidence highlights a non-canonical GRK function in regulating cellular processes independent of GPCR signal termination. Non-canonical GRK activity extends to regulating non-GPCR substrates, cytoskeletal dynamics, protein catalytic activity and other diverse functions. Non-GPCR GRK substrates are documented in several cellular compartments, including the plasma membrane, cytoplasm, and nucleus. This kinase family regulates non-GPCR substrates in both kinase-dependent and -independent fashion [36, 62, 63].
GRK2 and GRK3 have two distinct receptor-independent functions in regulating G proteins. As noted previously, GRK2 family members bind to G protein βγ-subunits released by receptor-activated G proteins [36, 63]. Since βγ-subunits are plasma membrane anchored, this recruits GRK2 to the plasma membrane in the vicinity of activated receptors. Although βγ-subunit binding does not directly alter GRK2 kinase catalytic activity, GRK2 can in fact be considered a βγ-subunit effector because this efficient localization greatly enhances the effectiveness of the kinase. In addition, GRK2 competes for G protein βγ-subunits with other βγ-stimulated effectors, and this domain has been used widely as a βγ-subunit scavenger to assess Gα versus Gβγ functions [20, 73]. Disease states that increase or decrease GRK2 levels thus alter Gβγ signaling tone. The second G protein function is that GRK2 and GRK3 also bind to activated Gq α-subunits through their RGS-like domain. Again, activated Gq does not appear to alter GRK2 function, but GRK2 does compete with PLC-β and other effectors for activated Gq subunits.
GRK-mediated phosphorylation regulates non-GPCR receptor activation. For instance, the platelet-derived growth factor receptor β (PDGFRβ) is a single-transmembrane span tyrosine kinase receptor responsible for mediating cellular proliferation and survival signals upon ligand stimulation. GRK2 negatively regulates PDGFRβ kinase activity by phosphorylating specific serine residues, thus decreasing PDGFRβ-mediated phosphorylation of its substrates [74, 75]. Similarly GRK5/6 positively regulate the tyrosine kinase receptor, insulin-like growth factor-1 receptor (IGF-1R). Indeed, IGF1-mediated ERK and AKT activation is diminished in cells when either GRK5/6 expression is decreased [76].
GRKs regulate cytoskeletal dynamics through GPCR-dependent and - independent mechanisms. The ezrin–radixin–moesin (ERM) protein family constitutes three GRK-regulated proteins that mediate cytoskeletal rearrangement [77, 78]. ERM proteins localize to the cytoplasm and promote cytoskeletal rearrangement by facilitating crosstalk between plasma membrane-bound proteins and F-actin [79]. GRK-mediated phosphorylation on specific ERM residues conformationally activates ERM to yield F-actin polymerization. GRK mediated ERM activation promotes cytoskeleton remodeling and cell adhesions [62]. Interestingly, GRK2 mediates muscarinic M1 receptor membrane ruffling through ezrin phosphorylation [62, 78]. Furthermore, GRK5 mediated moesin phosphorylation promotes cellular invasion and migration [77]. Additionally, GRKs influence microtubule (MT) processing, which functions in cytoskeletal polarization and migration [80]. MT dynamics are regulated by post-translational modifications, such as acetylation and phosphorylation [81]. Indeed, GRKs phosphorylate tublin, the basic monomer of MT [82]. GRKs phosphorylate and activate histone deacetylase 6 (HDAC6) to deacetylate α-tubulin, an MT subunit. GRK mediated HDAC6 deacetylation promotes MT polymerization [80].
This kinase family additionally regulates nuclear proteins involved in diverse functions, such as gene transcription and epigenetics. Exeplar transcription factor NF-κB undergoes GRK5-mediated regulation. IκB family members negatively regulate NF-κB by binding and sequestering NF-κB in the cytoplasm. IκB kinase (IKK) phosphorylation of IκB family members promotes NF-κB dissociation and subsequent IκB family member degradation. GRK5 additionally mediates IκB family member degradation and NF-κB activation by phosphorylating this protein family upon TNFα stimulation [83]. Interestingly, additional reports argue that GRK5 can oppose NF-κB activity through mediating IκBα nuclear accumulation [84]. Furthermore, GRK5 augments nuclear factor of activated T cells (NFAT) transcriptional activity in an apparent kinase independent manner, potentially by some molecular scaffold activity [85].
5.2 Arrestin-mediated signaling
Arrestin-mediated GPCR signal termination constitutes their defining function. Available evidence, however, highlights that arrestins additionally mediate signal transduction, cellular organization, cytoskeletal dynamics, and other cellular processes through molecular scaffold activity [45]. Molecular scaffolds regulate the subcellular spatiotemporal distribution of proteins, promote complex formation by bridging interacting components, and reduce off target effects by increasing reaction fidelity. Proteomic screens identified novel arrestin interactors in diverse pathways, such as signal transduction, motor processing, metabolomics, and cytoskeletal arrangement [22]. This scaffold ability increases mechanistic impact of arrestins on cell physiology in both GPCR-dependent and -independent states [22, 38].
Arrestins extend the GPCR signaling pathway beyond G proteins by scaffolding with kinases. GPCR/arrestin-mediated scaffolding occurs secondary to GPCR agonist activation [86]. For instance, arrestin-bound GPCR promotes tyrosine kinase Src recruitment . Arrestin-bound GPCR enhances the catalytic activity of Src to further diversify cellular effects, including promoting further GPCR signaling. Src-bound arrestin potentiates GPCR signaling by targeting GRKs for degradation, thus inhibiting the initial step of GPCR signal termination [70]. Furthermore, the Src-arrestin complex extends GPCR signaling pathways by promoting G protein-independent signaling. This complex trans-activates other pathways, such as the pro-mitogenic EGF receptor that can activate ERK to initiate pro-mitogenic transcriptional activity [23, 87].
Arrestins exert a contextual influence on kinases. Their scaffolding ability can either directly or indirectly alter kinase activity. Src-independent arrestin ERK activation demonstrates direct arrestin mediated regulation. Arrestin2 and 3 each complex with ERK independent of Src [22]. However, Arrestin2 and 3 maintain opposing functions. Arrestin2 inhibits ERK activation, while arrestin3 augments the kinase activity [22]. In contrast, arrestin indirectly regulates the transcription factor NF-κB by directly altering the stability of a regulatory scaffold protein. Arrestin indirectly promotes NF-κB inhibition by stabilizing IκBα. Arrestin-IκBα scaffolding prevents IKK mediated phosphorylation and subsequent NF-κB release. Consequently, arrestin indirectly regulates NF-κB by stabilizing the negative regulator protein responsible for its cytoplasmic localization [88].
Scaffold proteins regulate the spatiotemporal distribution of cellular components ERK activation post GPCR activation supports this conclusion. GPCR stimulation promotes ERK activation via G protein mediated mechanisms. Activation by this pathway results in nuclear translocation of ERK and subsequent activation of downstream transcription factors. Arrestin mediates an alternative pathway: ERK can form a complex with GPCR-bound arrestin via a G protein-independent mechanism. ERK recruitment to the plasma membrane results in its presence in clathrin-coated pits together with GPCR and arrestin. Vesicle-found ERK is sequestered in the cytoplasm where it activates a separate set of effectors distinct from the subset of G proteinactivated ERK substrates [22, 89, 90].
5.3 GPCR biased signaling
GPCR ligand activation induces receptor conformational changes, which stimulate downstream effector molecules. The G protein and arrestin signal pathways present two exclusive effector molecules influenced by GPCR structural dynamics. Active GPCR conformations are dynamic and exist in several structural forms, with partially non-redundant functions. Indeed, certain GPCR conformations specifically promote either a G protein or arrestin signaling pathway [91]. Ligand-receptor dynamics reflect this conformational complexity. Balanced ligands permit all potential active GPCR conformations, while biased ligands only permit GPCR to assume a subset of potential active structures [34, 92, 93]. In contrast to balanced ligands, biased ligands induce conformations that permit the specific activation of either G protein or arrestin based signaling [34, 92, 93]. For example, carvedilol, an FDA approved beta-blocker, demonstrates biased activity in the cardiovascular system. This therapeutic induces GPCR conformations that selectively activate arrestin pathway, without stimulating G protein activity [94].
Figure 1. Classical 7TMR/GPCR signaling.
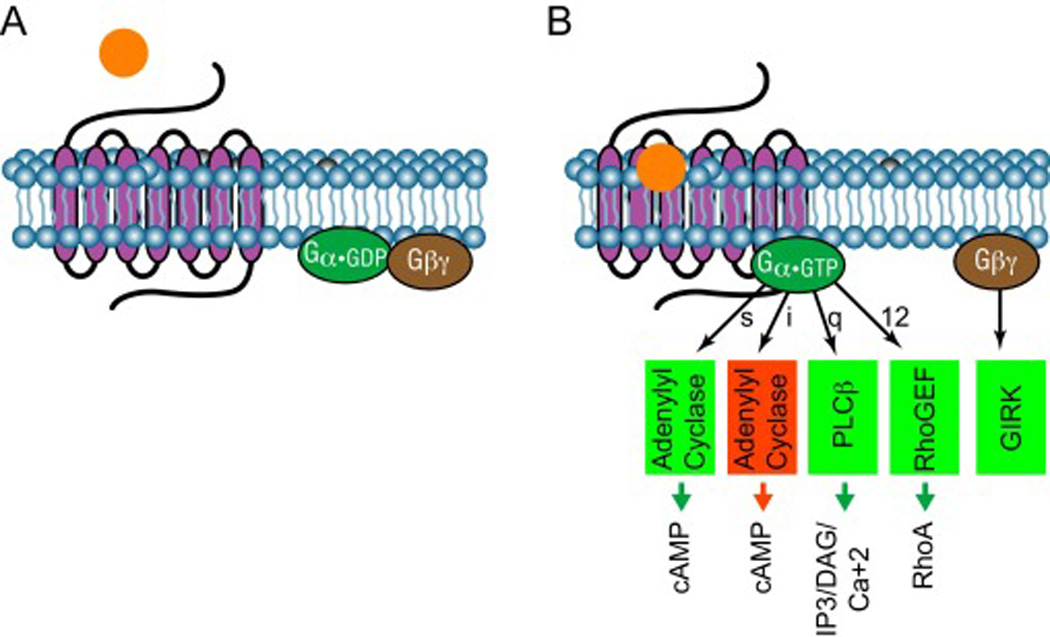
A) Inactive G proteins exist in a heterotrimeric state: the constituent α-subunit, bound to GDP, is associated with a βγ-subunit dimer. B) GPCR activation promotes GDP/GTP exchange on the α-subunit, which stimulates the dissociation of activated α•GTP from the βγ-subunits. Gsα•GTP and Gqα••GTP respectively activate adenylyl cyclase and phospholipase C, which increase intracellular second messengers cyclic AMP (cAMP) and inositol tris-phosphate (IP3). Giα•GTP inhibits adenylyl cyclase thereby decreasing cAMP accumulation.
Figure 2. 7TMR/GPCR signal regulation.
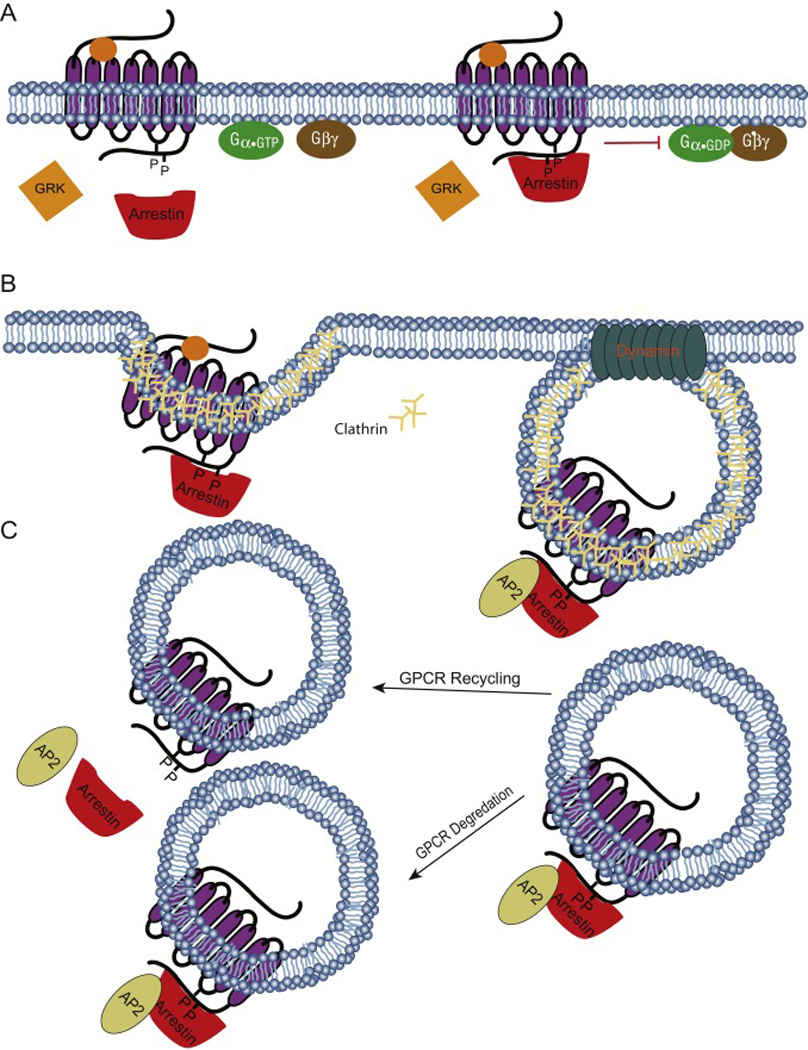
A) Receptor desensitization. GRKs phosphorylate the C-terminal of activated GPCRs. Arrestin binds to the phosphorylated receptor that is then sterically blocked from interacting with cognate G protein. B) Receptor sequestration. Arrestin serve as an adaptor and recruit endocytic machinery proteins to mediate GPCR internalization into vesicles. GPCR internalization sequesters the activated receptors from further stimulation. C) Arrestin may dissociate or remain bound to the receptor in vesicle. Early dissociation of arrestin promotes receptor recycling. Failure of the arrestin-GPCR complex to dissociate marks that receptor for further processing that results in either receptor degradation or delayed receptor recycling.
Figure 3. GPCR feedback regulation.
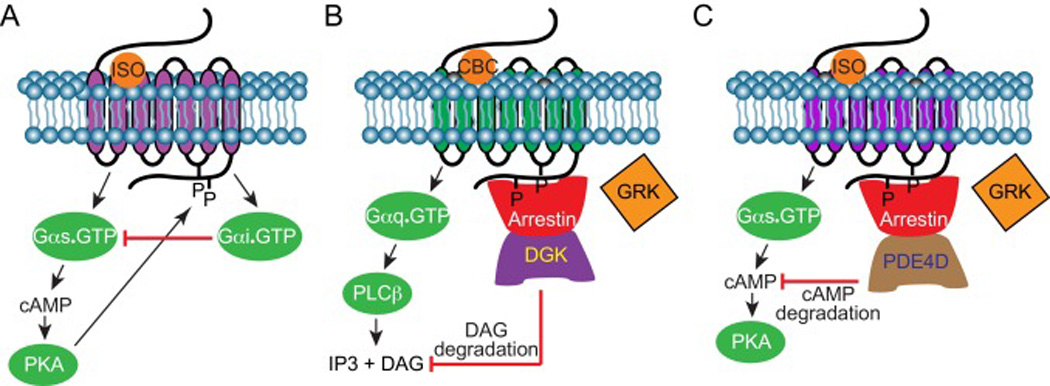
A) G protein switching. Upon initial stimulation, β2AR activates the Gs protein to increase adenylate cyclase signaling and PKA activity to propagate the cellular response. PKA initiates G protein switching by phosphorylating site-specific residues on β2AR. PKA phosphorylation of β2AR alters the receptor coupling from Gs to Gi. Activated Gi yields a decrease in cAMP production and PKA activity. B) Arrestins partake in Gq-coupled M1 muscarinic receptor feedback regulation. Arrestin scaffolds with diacylglycerol kinases (DGKs) and recruits them to the plasma membrane. These events activate DGKs to degrade DAG, promoting arrestin-mediated Gq negative feedback regulation. C) Upon β2AR activation, arrestin scaffolds with PDE4D and promotes its translocation to the plasma membrane. PDE4D translocation and activation negatively regulates the β2AR-cAMP cascade by degrading the substrate necessary for PKA activation
Figure 4. Arrestin mediated signaling.
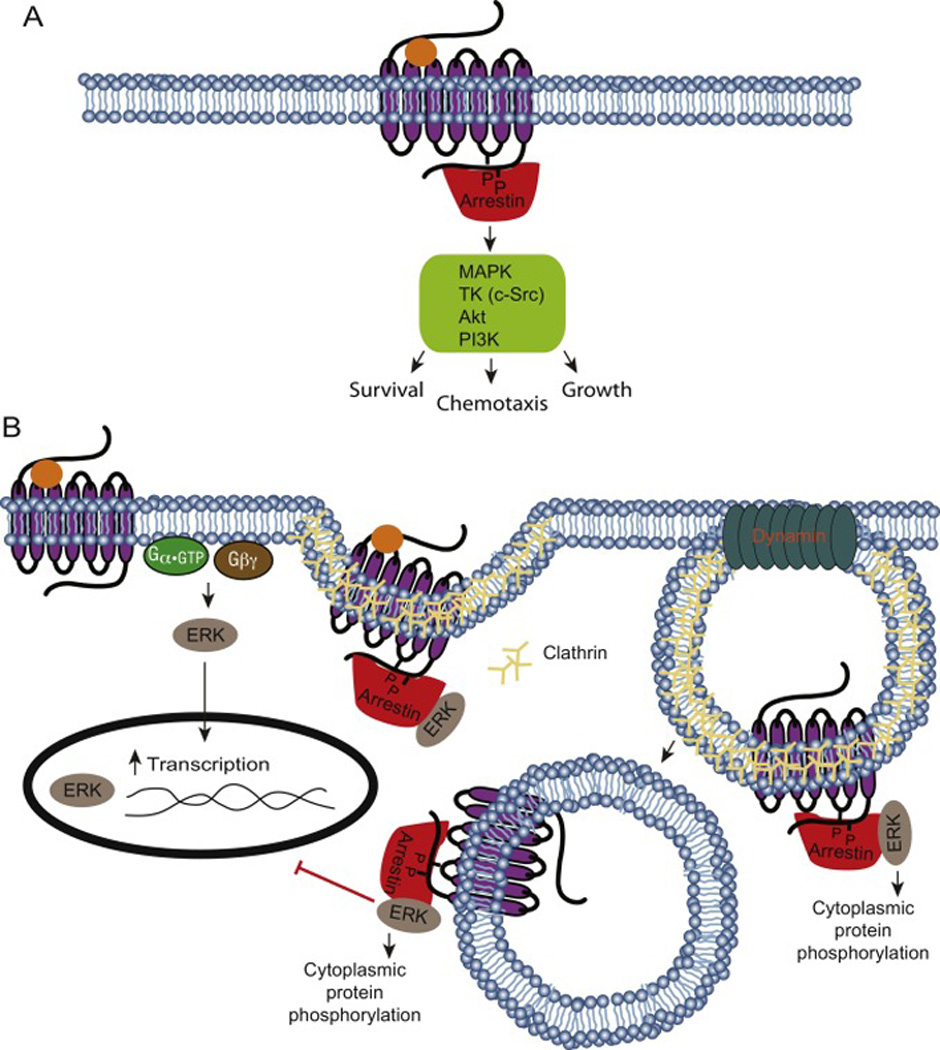
A) Arrestins extend the GPCR signaling pathway beyond G proteins by scaffolding with kinases. GPCR ligand activation induces receptor conformational changes that can result in arrestin signaling activation. Arrestin mediated signaling is independent of G protein coupling and can regulate diverse cellular processes. B) Arrestins regulate protein spatiotemporal distribution. GPCR stimulation promotes ERK activation via G protein mediated mechanisms. This pathway promotes nuclear translocation of ERK and subsequent activation of downstream transcription factors. Arrestin mediates an alternative pathway wherein ERK complexes with GPCR-bound arrestin. Vesicle-bound ERK is sequestered in the cytoplasm and activates a separate set of effectors.
Perspective.
GPCR-mediated signal transduction presents an intricate mechanism for controlling cellular activities and facilitating communication. Diverse processes ranging from cardiovascular remodeling to endocrine signaling utilize GPCR activity for eliciting cellular responses. Pharmacotherapeutics target GPCR pathways for clinical interventions due to their applicability in controlling clinically-relevant pathways. Indeed, over 50% of pharmacologic interventions target GPCR. However, GPCR’s intricate and diversified mechanisms of activity contribute towards unwanted side effects. Consequently, fundamental advances in GPCR biology directly influence novel pharmacologic development void of deleterious effects. Indeed, gained knowledge in GPCR structure and signal transduction augments the opportunity for smart drug development seeking to produce receptor signal-specific therapies without off-target effects. Identifying GPCR conformation states that selectively activate either arrestin or G protein signaling networks presents an additional control to prevent drug deleterious side effects. While GPCR present a heavily targeted family, less than 10% of GPCR currently are targeted for interventions. This offers the opportunity to identify and target new GPCRs. Overall, the advancement of GPCR research presents profound clinical applications inherit within their influence of cellular dynamics. Recognizing GPCR regulatory feed-back loops expands opportunities to harness specific signals for therapeutic interventions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Works Cited
- 1.Owens DM, Lumpkin EA. Diversification and specialization of touch receptors in skin. Cold Spring Harb Perspect Med. 2014;4(6) doi: 10.1101/cshperspect.a013656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawson T. Specificity in signal transduction: From phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116(2):191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi T, et al. BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proc Natl Acad Sci USA. 2005;102(50):18023–18027. doi: 10.1073/pnas.0503617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3(9):639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 6.Liu Q, et al. Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell. 2009;139(7):1353–1365. doi: 10.1016/j.cell.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palczewski K, Orban T. From atomic structures to neuronal functions of G protein coupled receptors. Annu Rev Neurosci. 2013;36:139–164. doi: 10.1146/annurev-neuro-062012-170313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo DG, Xue T, Yau KW. How vision begins: An odyssey. Proc Natl Acad Sci USA. 2008;105(29):9855–9862. doi: 10.1073/pnas.0708405105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaudhari N, Roper SD. The cell biology of taste. J Cell Biol. 2010;190(3):285–296. doi: 10.1083/jcb.201003144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spehr M, Munger SD. Olfactory receptors: G protein-coupled receptors and beyond. J Neurochem. 2009;109(6):1570–1583. doi: 10.1111/j.1471-4159.2009.06085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Civelli O, et al. G protein-coupled receptor deorphanizations. Annu Rev Pharmacol Toxicol. 2013;53:127–146. doi: 10.1146/annurev-pharmtox-010611-134548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobilka BK. G protein-coupled receptor structure and activation. Biochim Biophys Acta-Biomemb. 2007;1768(4):794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fredriksson R, et al. The G protein-coupled receptors in the human genome form five main families: Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63(6):1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 14.Tu XL, et al. Noncanonical Wnt signaling through G protein-linked PKC delta activation promotes bone formation. Dev Cell. 2007;12(1):113–127. doi: 10.1016/j.devcel.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freissmuth M, Casey PJ, Gilman AG. G proteins control diverse pathways of transmembrane signaling. FASEB J. 1989;3(10):2125–2131. [PubMed] [Google Scholar]
- 16.Fields TA, Casey PJ. Signalling functions and biochemical properties of pertussis toxin-resistant G proteins. Biochem J. 1997;321:561–571. doi: 10.1042/bj3210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oldham WM, Hamm HE. Heterotrimeric G protein activation by G protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9(1):60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 18.Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296(5573):1636–1639. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- 19.Huang CL, et al. Evidence that direct binding of Gβγ to the Girk1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron. 1995;15(5):1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- 20.Daaka Y, et al. Receptor and Gβγ isoform-specific interactions with G proteincoupled receptor kinases. Proc Natl Acad Sci USA. 1997;94(6):2180–2185. doi: 10.1073/pnas.94.6.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menard RE, Mattingly RR. Gβγ subunits stimulate p21-activated kinase 1 (PAK1) through activation of PI3-kinase and Akt but act independently of Rac1/Cdc42. FEBS Lett. 2004;556(1–3):187–192. doi: 10.1016/s0014-5793(03)01406-6. [DOI] [PubMed] [Google Scholar]
- 22.DeWire SM, et al. β-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 23.Daaka Y. G proteins in cancer: The prostate cancer paradigm. Sci STKE. 2004;2004(216):re2. doi: 10.1126/stke.2162004re2. [DOI] [PubMed] [Google Scholar]
- 24.Dorsam RT, Gutkind JS. G protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7(2):79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- 25.Dohlman HG, Thorner J. RGS proteins and signaling by heterotrimeric G proteins. J Biol Chem. 1997;272(7):3871–3874. doi: 10.1074/jbc.272.7.3871. [DOI] [PubMed] [Google Scholar]
- 26.Berman DM, Gilman AG. Mammalian RGS proteins: Barbarians at the gate. J Biol Chem. 1998;273(3):1269–1272. doi: 10.1074/jbc.273.3.1269. [DOI] [PubMed] [Google Scholar]
- 27.Zwartkruis FJT, Bos JL. Ras and Rap1: Two highly related small GTPases with distinct function. Exp Cell Res. 1999;253(1):157–165. doi: 10.1006/excr.1999.4695. [DOI] [PubMed] [Google Scholar]
- 28.Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85(4):1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 29.Arshavsky VY, Lamb TD, Pugh EN. G proteins and phototransduction. Annu Rev Physiol. 2002;64:153–187. doi: 10.1146/annurev.physiol.64.082701.102229. [DOI] [PubMed] [Google Scholar]
- 30.Siehler S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br J Pharmacol. 2009;158(1):41–49. doi: 10.1111/j.1476-5381.2009.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vazquez-Prado J, et al. Chimeric Gαi2/Gα13 proteins reveal the structural requirements for the binding and activation of the RGS-like (RGL)-containing Rho guanine nucleotide exchange factors (GEFs) by Gα13. J Biol Chem. 2004;279(52):54283–54290. doi: 10.1074/jbc.M410594200. [DOI] [PubMed] [Google Scholar]
- 32.Qian B, et al. Depicting a protein's two faces: GPCR classification by phylogenetic tree-based HMMs. FEBS Lett. 2003;554(1–2):95–99. doi: 10.1016/s0014-5793(03)01112-8. [DOI] [PubMed] [Google Scholar]
- 33.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390(6655):88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 34.Kenakin T. The measurement of receptor signaling bias. Method Mol Biol. 2015;1335:163–176. doi: 10.1007/978-1-4939-2914-6_11. [DOI] [PubMed] [Google Scholar]
- 35.Hausdorff WP, Caron MG, Lefkowitz RJ. Turning off the signal: Desensitization of β-adrenergic-receptor function. FASEB J. 1990;4(11):2881–2889. [PubMed] [Google Scholar]
- 36.Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annu Rev Biochem. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- 37.Pao CS, Benovic JL. Phosphorylation-independent desensitization of G protein-coupled receptors? Sci STKE. 2002;2002(153):pe42. doi: 10.1126/stke.2002.153.pe42. [DOI] [PubMed] [Google Scholar]
- 38.Gainetdinov RR, et al. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- 39.Perry SJ, Lefkowitz RJ. Arresting developments in heptahelical receptor signaling and regulation. Trend Cell Biol. 2002;12(3):130–138. doi: 10.1016/s0962-8924(01)02239-5. [DOI] [PubMed] [Google Scholar]
- 40.Ferguson SSG, et al. Molecular mechanisms of G protein-coupled receptor desensitization and resensitization. Life Sci. 1998;62(17–18):1561–1565. doi: 10.1016/s0024-3205(98)00107-6. [DOI] [PubMed] [Google Scholar]
- 41.Xu JS, et al. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 2003;114(2):201–214. doi: 10.1016/s0092-8674(03)00555-5. [DOI] [PubMed] [Google Scholar]
- 42.Liu Z, et al. TLR4 signaling augments monocyte chemotaxis by regulating G protein-coupled receptor kinase 2 translocation. J Immunol. 2013;191(5):2847–2847. doi: 10.4049/jimmunol.1300790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rockman HA, Koch WJ, Lefkowitz RJ. Seven transmembrane-spanning receptors and heart function. Nature. 2002;415(6868):206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 44.Salazar NC, Chen J, Rockman HA. Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim Biophys Acta-Biomemb. 2007;1768(4):1006–1018. doi: 10.1016/j.bbamem.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore CAC, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 46.Drake MT, Shenoy SK, Lefkowitz RJ. Trafficking of G protein-coupled receptors. Circ Res. 2006;99(6):570–582. doi: 10.1161/01.RES.0000242563.47507.ce. [DOI] [PubMed] [Google Scholar]
- 47.Krueger KM, et al. The role of sequestration in G protein-coupled receptor resensitization: Regulation of β2-adrenergic receptor dephosphorylation by vesicular acidification. J Biol Chem. 1997;272(1):5–8. doi: 10.1074/jbc.272.1.5. [DOI] [PubMed] [Google Scholar]
- 48.Tsao P, Cao T, von Zastrow M. Role of endocytosis in mediating downregulation of G protein-coupled receptors. Trend Pharmacol Sci. 2001;22(2):91–96. doi: 10.1016/s0165-6147(00)01620-5. [DOI] [PubMed] [Google Scholar]
- 49.Leftowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308(5721):512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 50.Allen LF, et al. G protein-coupled receptor genes as protooncogenes: Constitutively activating mutation of the α1B-adrenergic receptor enhances mitogenesis and tumorigenicity. Proc Natl Acad Sci USA. 1991;88(24):11354–11358. doi: 10.1073/pnas.88.24.11354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yasuda N, et al. Agonist-independent constitutive activity of angiotensin II receptor promotes cardiac remodeling in mice. Hypertension. 2012;59(5):E51–E51. doi: 10.1161/HYPERTENSIONAHA.111.175208. [DOI] [PubMed] [Google Scholar]
- 52.Milligan G. Constitutive activity and inverse agonists of G protein-coupled receptors: A current perspective. Mol Pharmacol. 2003;64(6):1271–1276. doi: 10.1124/mol.64.6.1271. [DOI] [PubMed] [Google Scholar]
- 53.Kjelsberg MA, et al. Constitutive activation of the α1b-adrenergic receptor by all amino-acid substitutions at a single site: Evidence for a region which constrains receptor activation. J Biol Chem. 1992;267(3):1430–1433. [PubMed] [Google Scholar]
- 54.Decaillot FM, et al. Opioid receptor random mutagenesis reveals a mechanism for G protein-coupled receptor activation. Nat Struct Biol. 2003;10(8):629–636. doi: 10.1038/nsb950. [DOI] [PubMed] [Google Scholar]
- 55.Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat Rev Drug Disc. 2013;12(8):630–644. doi: 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- 56.Seifert R, et al. Functional differences between full and partial agonists: Evidence for ligand-specific receptor conformations. J Pharmacol Exp Therap. 2001;297(3):1218–1226. [PubMed] [Google Scholar]
- 57.Zhu J, et al. Inverse agonism and neutral antagonism at a constitutively active α1a adrenoceptor. Br J Pharmacol. 2000;131(3):546–552. doi: 10.1038/sj.bjp.0703584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shryock JC, Ozeck MJ, Belardinelli L. Inverse agonists and neutral antagonists of recombinant human A1 adenosine receptors stably expressed in Chinese hamster ovary cells. Mol Pharmacol. 1998;53(5):886–893. [PubMed] [Google Scholar]
- 59.Zamah AM, et al. Protein kinase A-mediated phosphorylation of the β2-adrenergic receptor regulates its coupling to Gs and Gi: Demonstration in a reconstituted system. J Biol Chem. 2002;277(34):31249–31256. doi: 10.1074/jbc.M202753200. [DOI] [PubMed] [Google Scholar]
- 60.Okamoto T, et al. Identification of a Gs activator region of the β2-adrenergic receptor that is autoregulated via protein kinase A-dependent phosphorylation. Cell. 1991;67(4):723–730. doi: 10.1016/0092-8674(91)90067-9. [DOI] [PubMed] [Google Scholar]
- 61.Baillie GS, et al. β-arrestin mediated PDE4 cAMP phosphodiesterase recruitment regulates β-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci USA. 2003;100(3):940–945. doi: 10.1073/pnas.262787199. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Gurevich EV, et al. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol Therap. 2012;133(1):40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- 64.Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- 65.Nobles KN, et al. Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal. 2011;4(185):ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murakami A, et al. X-arrestin: A new retinal arrestin mapping to the x-chromosome. FEBS Lett. 1993;334(2):203–209. doi: 10.1016/0014-5793(93)81712-9. [DOI] [PubMed] [Google Scholar]
- 67.Shinohara T, et al. Primary and secondary structure of bovine retinal S-antigen (48-Kda Protein) Proc Natl Acad Sci USA. 1987;84(20):6975–6979. doi: 10.1073/pnas.84.20.6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laporte SA, et al. The β2-adrenergic receptor/ β-arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96(7):3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goodman OB, Jr, et al. β-arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature. 1996;383(6599):447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 70.Luttrell LM, Lefkowitz RJ. The role of β-arrestins in the termination and transduction of G protein-coupled receptor signals. J Cell Sci. 2002;115(3):455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 71.Perry SJ, et al. Targeting of cyclic AMP degradation to β2-adrenergic receptors by β-arrestins. Science. 2002;298(5594):834–836. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 72.Nelson CD, et al. Targeting of diacylglycerol degradation to M1 muscarinic receptors by β-arrestins. Science. 2007;315(5812):663–666. doi: 10.1126/science.1134562. [DOI] [PubMed] [Google Scholar]
- 73.Ford CE, et al. Molecular basis for interactions of G protein βγ subunits with effectors. Science. 1998;280(5367):1271–1274. doi: 10.1126/science.280.5367.1271. [DOI] [PubMed] [Google Scholar]
- 74.Wu JH, et al. The platelet-derived growth factor receptor-beta phosphorylates and activates G protein-coupled receptor kinase 2. J Bio Chem. 2005;280(35):31027–31035. doi: 10.1074/jbc.M501473200. [DOI] [PubMed] [Google Scholar]
- 75.Hildreth KL, et al. Phosphorylation of the platelet-derived growth factor receptor-beta by G protein-coupled receptor kinase 2 reduces receptor signaling and interaction with the Na+/H+ exchanger regulatory factor. J Bio Chem. 2004;279(40):41775–41782. doi: 10.1074/jbc.M403274200. [DOI] [PubMed] [Google Scholar]
- 76.Zheng HY, et al. Selective recruitment of G protein-coupled receptor kinases (GRKs) controls signaling of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci USA. 2012;109(18):7055–7060. doi: 10.1073/pnas.1118359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chakraborty PK, et al. G protein-coupled receptor kinase GRK5 phosphorylates moesin and regulates metastasis in prostate cancer. Cancer Res. 2014;74(13):3489–3500. doi: 10.1158/0008-5472.CAN-13-2708. [DOI] [PubMed] [Google Scholar]
- 78.Cant SH, Pitcher JA. G protein-coupled receptor kinase 2-mediated phosphorylation of ezrin is required for G protein-coupled receptor-dependent reorganization of the actin cytoskeleton. Mol Biol Cell. 2005;16(7):3088–3099. doi: 10.1091/mbc.E04-10-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neisch AL, Fehon RG. Ezrin, radixin and moesin: Key regulators of membrane-cortex interactions and signaling. Curr Opin Cell Biol. 2011;23(4):377–382. doi: 10.1016/j.ceb.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lafarga V, et al. A novel GRK2/HDAC6 interaction modulates cell spreading and motility. EMBO J. 2012;31(4):856–869. doi: 10.1038/emboj.2011.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Etienne-Manneville S. Microtubules in cell migration. Annu Rev Cell Dev Biol. 2013;29:471–499. doi: 10.1146/annurev-cellbio-101011-155711. [DOI] [PubMed] [Google Scholar]
- 82.Pitcher JA, et al. The G protein-coupled receptor kinase 2 is a microtubule-associated protein kinase that phosphorylates tubulin. J Biol Chem. 1998;273(20):12316–12324. doi: 10.1074/jbc.273.20.12316. [DOI] [PubMed] [Google Scholar]
- 83.Islam KN, et al. Regulation of nuclear factor κB (NF-κB) in the nucleus of cardiomyocytes by G protein-coupled receptor kinase 5 (GRK5) J Biol Chem. 2013;288(50):35683–35689. doi: 10.1074/jbc.M113.529347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sorriento D, et al. The G protein-coupled receptor kinase 5 inhibits NF-κB transcriptional activity by inducing nuclear accumulation of IκBα. Proc Natl Acad Sci USA. 2008;105(46):17818–17823. doi: 10.1073/pnas.0804446105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hullmann JE, et al. GRK5-mediated exacerbation of pathological cardiac hypertrophy involves facilitation of nuclear NFAT activity. Circ Res. 2014;115(12) doi: 10.1161/CIRCRESAHA.116.304475. p. 976-U128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tohgo A, et al. β-arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277(11):9429–9436. doi: 10.1074/jbc.M106457200. [DOI] [PubMed] [Google Scholar]
- 87.Luttrell LM, et al. β-arrestin-dependent formation of β2-adrenergic receptor Src protein kinase complexes. Science. 1999;283(5402):655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 88.Witherow DS, et al. β-arrestin inhibits NF-κB activity by means of its interaction with the NF-κB inhibitor IκBα. Proc Natl Acad Sci USA. 2004;101(23):8603–8607. doi: 10.1073/pnas.0402851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tohgo A, et al. The stability of the G protein-coupled receptor-β-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278(8):6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 90.Luttrell LM, et al. Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc Natl Acad Sci USA. 2001;98(5):2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reiter E, et al. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.DeWire SM, Violin JD. Biased ligands for better cardiovascular drugs dissecting G protein-coupled receptor pharmacology. Circ Res. 2011;109(2):205–216. doi: 10.1161/CIRCRESAHA.110.231308. [DOI] [PubMed] [Google Scholar]
- 93.Violin JD, et al. Biased ligands at G protein-coupled receptors: Promise and progress. Trend Pharmacol Sci. 2014;35(9):489–489. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 94.Wisler JW, et al. A unique mechanism of β-blocker action: Carvedilol stimulates β-arrestin signaling. Proc Natl Acad Sci USA. 2007;104(42):16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]