

Abstract
Aims/hypothesis
Obesity and consequent insulin resistance are known risk factors for type 2 diabetes. A compensatory increase in beta cell function and mass in response to insulin resistance permits maintenance of normal glucose homeostasis, whereas failure to do so results in beta cell failure and type 2 diabetes. Recent evidence suggests that the circadian system is essential for proper metabolic control and regulation of beta cell function. We set out to address the hypothesis that the beta cell circadian clock is essential for the appropriate functional and morphological beta cell response to insulin resistance.
Methods
We employed conditional deletion of the Bmal1 (also known as Arntl) gene (encoding a key circadian clock transcription factor) in beta cells using the tamoxifen-inducible CreERT recombination system. Upon adulthood, Bmal1 deletion in beta cells was achieved and mice were exposed to either chow or high fat diet (HFD). Changes in diurnal glycaemia, glucose tolerance and insulin secretion were longitudinally monitored in vivo and islet morphology and turnover assessed by immunofluorescence. Isolated islet experiments in vitro were performed to delineate changes in beta cell function and transcriptional regulation of cell proliferation.
Results
Adult Bmal1 deletion in beta cells resulted in failed metabolic adaptation to HFD characterised by fasting and diurnal hyperglycaemia, glucose intolerance and loss of glucose-stimulated insulin secretion. Importantly, HFD-induced beta cell expansion was absent following beta cell Bmal1 deletion indicating impaired beta cell proliferative and regenerative potential, which was confirmed by assessment of transcriptional profiles in isolated islets.
Conclusion/interpretation
Results of the study suggest that the beta cell circadian clock is a novel regulator of compensatory beta cell expansion and function in response to increased insulin demand associated with diet-induced obesity.
Keywords: Beta cell, Beta cell expansion, Beta cell failure, BMAL1, Circadian clock, Clock genes, High fat diet
Introduction
Insulin resistance, typically attributed to obesity, is a well-established risk factor for type 2 diabetes [1]. However, most obese insulin resistant individuals are able to compensate for increased insulin demand [2] due to an adaptive increase in insulin secretion [3–5] and beta cell mass [6, 7]. In contrast, individuals that go on to develop type 2 diabetes display impaired adaptation characterised by beta cell dysfunction and failed beta cell expansion [6, 8]. These observations raise the question, what molecular mechanisms underlie failed metabolic and beta cell adaptation to insulin resistance in type 2 diabetes?
Studies show that circadian clocks play an increasingly appreciated role in metabolic control, regulation of cell proliferation and control of beta cell function [9–11]. The mammalian circadian clock mechanism is a highly conserved network of several ‘clock’ genes and proteins that coordinate rhythmic processes (~24 h) at behavioural, physiological and cellular levels [12]. In short, the positive limb of this circuit includes genes Clock and Bmal1 (also known as Arntl), which encode basic helix loop helix period (PER)-aryl hydrocarbon receptor nuclear translocator (ARNT)-single-minded proteins that form activator complexes and initiate transcription by binding to promoter regions of target genes [13]. Per1/2/3 and Cry1/2 genes comprise the negative limb of the clock gene feedback loop where PER and cryptochrome proteins form heterodimers and inhibit transcriptional activation by circadian locomotor output cycles kaput (CLOCK)–brain and muscle ARNT-like 1 (BMAL1), allowing a new circadian cycle to repeat [14]. Given that functional CLOCK–BMAL1 activator complexes are essential for circadian function and transcriptional control of clock-controlled genes, genetic disruption of Bmal1 compromises cellular clock function [15], metabolic control and beta cell function [16].
Circadian disruption is becoming increasingly prevalent in today’s society. Thus, growing attention has been placed on understanding the role of circadian clocks in pathogenesis of type 2 diabetes [17]. Indeed, conditions associated with circadian rhythm disruption increase the risk for type 2 diabetes in humans, mediated partly through deleterious effects on the beta cell [18–24]. Accordingly, beta cell-specific clock gene deletion compromises insulin secretion and promotes development of diabetes [22, 23]. the role of circadian clocks in beta cell expansion and metabolic adaptation to insulin resistance remains unexplored. Therefore, in the current study, we employed conditional deletion of Bmal1 gene in beta cells by using tamoxifen-inducible CreERT mediated recombination system to test the hypothesis that an intact beta cell circadian clock is essential for successful metabolic and beta cell adaptation to diet-induced obesity.
Methods
Animals
Mice homozygous for the floxed Bmal1 gene (B6.129S4 [Cg]-Arntltm1Weit/J, The Jackson Laboratory, Bar Harbor, ME, USA) [25] were crossed with mice with a tamoxifen-inducible Cre-mediated recombination system driven by the rat Insulin2 promoter (STOCK Tg [Ins2-cre/ ERT] 1Dam/J, The Jackson Laboratory) [26]. Resulting offspring were genotyped to identify mice that are homozygous for the deletion of Bmal1 gene in beta cells (β-Bmal1−/−), heterozygous (β-Bmal1+/−) or controls (β-Bmal1+/+). In total, 93 mice (45 males and 48 females) were used in this study including 31 controls (β-Bmal1+/+), 20 heterozygous (β-Bmal1+/−), 24 homozygous knockouts (β-Bmal1−/−) and 18 male C57BL6 mice (for circadian gene array analyses in islets). Mice were housed at University of California, Los Angeles (UCLA) and Mayo Clinic, Rochester animal facilities under standard 12 h light/12 h dark (LD) cycle. Lights on is denoted as Zeitgeber time (ZT) 0 and lights off as ZT 12. All procedures were approved by UCLA and Mayo Clinic Institutional Animal Care and Use Committee. Animals were assigned to experiments randomly based on genotype and sex, and experimenters were not blind to group assignment and outcome assessment. No data were excluded from the report.
Study design
Refer to Electronic Supplementary Material (ESM) Fig. 1. Mice were fed chow diet (24.7% energy from protein, 62.1% energy from carbohydrate, 13.2% energy from fat; 5053; LabDiet, St Louis, MO, USA) throughout or switched to a high fat diet (HFD) (20% energy from protein, 20% energy from carbohydrate, 60% energy from fat; D12492; Research Diets, New Brunswick, NJ, USA) at 2 months of age. For conditional deletion of Bmal1, mice received three i.p. injections each containing 4 mg tamoxifen (Sigma-Aldrich, St Louis, MO, USA) on alternate days at 2 months.
Behavioural monitoring of circadian activity
Mice were housed individually in cages outfitted with an optical beam sensor system to monitor circadian behavioural rhythms in activity (Respironics, Murrysville, PA, USA) and monitored for 10 days in LD followed by 10 days in constant darkness (DD).
Circadian expression of clock gene mRNA in pancreatic islets
For assessment of circadian mRNA expression in islets, β-Bmal1 mice aged 3.5 months were euthanised at ZT 0, 8 and 16, and C57BL6 mice aged 2 months were euthanised at ZT 0, 4, 8, 12, 16 and 20 to isolate islets using standard collagenase method. Total RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and subjected to whole genome array analysis (Arraystar, Rockville, MD, USA).
Metabolic in vivo studies
Circadian metabolic profiles, glucose tolerance and insulin tolerance were assessed as described in detail in ESM Methods.
Immunofluorescence and immunohistochemistry
Mice were euthanised and pancreas immediately harvested and fixed in 4% paraformaldehyde. Immunohistochemical analysis was used for quantification of beta and alpha cell mass whereas immunofluorescent analysis was used for determination of beta cell replication, apoptosis, and beta cell and hypothalamic expression of BMAL1 (see ESM Methods).
Measurements of beta cell function and proliferative potential in isolated islets
Mice aged 3.5 months were euthanised and islets isolated using standard collagenase method. Subsequently, in vitro studies to assess beta cell function and proliferative potential were conducted as described in detail in ESM Methods.
Analytical methods
Blood glucose was measured using FreeStyle Lite Blood Glucose Monitoring system (Abbott Laboratories, Abbott Park, IL, USA). Plasma insulin was measured using ELISA (Alpco Diagnostics, Salem, NH, USA).
Statistical analysis and calculations
Activity recordings were analysed using ClockLab software (Actimetrics, Wilmette, IL, USA). Circadian gene expression data was analysed by Short time-series expression miner (STEM) as described [27]. Statistical analysis was performed using ANOVA with post hoc tests wherever appropriate (GraphPad Prism v.6.0, San Diego, CA, USA). Data are presented as means ± SEM and assumed statistically significant at p<0.05.
Results
All β-Bmal1 genotypes displayed comparable circadian activity under standard LD and DD, suggesting absence of potential effects of Bmal1 deletion on the suprachiasmatic nucleus clock (Fig. 1a–f). Specifically, no significant changes in circadian period, power of rhythm or average daily locomotor activity were observed under ‘free-run’ DD conditions among the three genotypes (p>0.05, Fig 1e). Bmal1 mRNA assessed in whole islet lysates, was substantially suppressed in ß -Bmal1−/− (~80%, p<0.001) and in heterozygous ß -Bmal1+/− (~50%, p<0.01) mice compared with control β-Bmal1+/+ animals (Fig. 1j). Immunofluorescence confirmed the absence of BMAL1 protein in β-Bmal1−/− mice islets; however, BMAL1 was detectable in islets from heterozygous β-Bmal1+/− mice (Fig. 1k). In addition, BMAL1 protein was expressed in alpha cells as well as in the hypothalamus of β-Bmal1−/− mice, consistent with the beta cell-specific nature of Bmal1 deletion (ESM Figs 2, 3).
Fig. 1.

Behavioural circadian rhythms are not altered in β-Bmal1−/− mice. Behavioural activity of representative (a) β-Bmal1+/+, (b) β-Bmal1+/− and (c) β-Bmal1−/− mice monitored for 10 days in standard LD cycle followed by 10 days in DD. Shaded areas represent periods of dark. Mean circadian activity (expressed as counts/30 min) across the 24 h circadian day in (d) β-Bmal1+/+, (e) β-Bmal1+/− and (f) β-Bmal1−/− mice. Each bar represents mean ± SEM (n=3 per group, per time point). Shaded areas represent periods of dark. (g) Circadian free running period (in DD), (h) strength of circadian rhythm denoted by fast Fourier transform (FFT) values, and (i) average daily activity of β-Bmal1 mice. Bars represent mean ± SEM (n=3 per group). (j) Normalised Bmal1 mRNA expression from whole islet lysates across the 24 h circadian day averaged for each of the three genotypes (n=3) to reflect mean overall levels of Bmal1 mRNA. Values are relative fold change with β-Bmal1+/+ at ZT 0 set as 1. Statistical significance is denoted by **p<0.01 and ***p<0.001 (k) Examples of islets stained by immunofluorescence for insulin (green) and BMAL1 (red), and imaged at ×63 magnification in mice euthanised at ZT 4 (light cycle) and 16 (dark cycle) in the respective genotypes. (g–j) β-Bmal1+/+, white bars; β-Bmal1+/−, grey bars; β-Bmal1−/−, black bars
Body weight was not significantly different among three genotypes prior to or after tamoxifen administration and on either chow or HFD (ESM Fig. 4). Furthermore, 8 weeks of HFD resulted in induction of insulin resistance, the extent of which was comparable among all three β-Bmal1 genotypes (ESM Fig. 5). Fasting glucose was comparable among the three genotypes prior to tamoxifen administration and remained unchanged for 8 weeks following tamoxifen administration in mice fed chow (p>0.05 vs all groups, Fig. 2a). In contrast, HFD led to a progressive induction of fasting hyperglycaemia in β-Bmal1−/− mice (p<0.05 vs baseline and vs all other genotypes at 4–8 weeks post tamoxifen, Fig. 2b). Similarly, non-fasted diurnally sampled glucose concentrations in β-Bmal1−/− (HFD) mice were also elevated compared with all groups (p<0.05 vs all groups, Fig. 2d). Importantly, while there was no significant relationship between HFD-induced body weight gain and fasting glucose in β-Bmal1+/+ and β-Bmal1+/− mice, increase in body weight predicted the subsequent rise in fasting glycaemia in β-Bmal1−/− mice (r=0.8, p<0.001), suggestive of impaired metabolic adaptation to HFD (ESM Fig. 6).
Fig. 2.
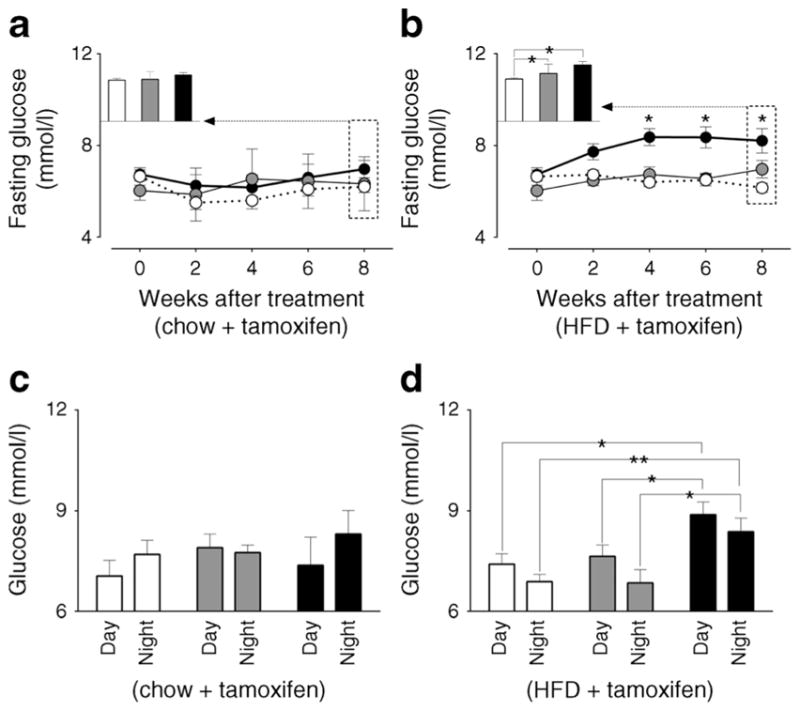
Effects of adult beta cell-specific Bmal1 deletion on fasting and diurnal glycaemia in vivo following exposure to either chow or HFD. Graphs representing longitudinal changes in fasting glucose in different β-Bmal1 mice at baseline (time-0), and 2, 4, 6 and 8 weeks after tamox-ifen in mice fed (a) chow or (b) HFD. (a, b) Each data point represents mean ± SEM (n=5–7 per group per time point). Statistical significance is denoted by *p<0.05 compared with own baseline. Inserts represent mean fasting glucose in respective genotypes at week 8. Bar graphs representing diurnal plasma glucose under ad libitum fed conditions following 8-week exposure to (c) chow or (d) HFD. (c, d) Graphs represent mean ± SEM (n=5–7 per group). Statistical significance is denoted by *p<0.05 and **p<0.01. (a–d) β-Bmal1+/+, white circles/bars; β-Bmal1+/−, grey circles/bars; β-Bmal1−/−, black circles/bars
Next we examined longitudinal changes in glucose tolerance and in vivo glucose-stimulated insulin secretion (GSIS) at baseline and after 2, 4, 6, and 8 weeks of tamoxifen in mice exposed to either chow or HFD (Figs 3, 4). Loss of beta cell Bmal1 led to glucose intolerance associated with diminished GSIS even under regular chow (p<0.05 vs β-Bmal1+/+ from 2–8 weeks post tamoxifen, Figs 3a,b, 4a,b). Under HFD, β-Bmal1−/− mice exhibited further deterioration in glucose tolerance and profound loss of GSIS (Figs 3c,d, 4c,d). Importantly, exposure to HFD resulted in development of glucose intolerance (p<0.05 vs β-Bmal1+/+ at 6–8 weeks post tamoxifen, Figs 3c,d, 4c,d) and diminished GSIS in heterozygous β-Bmal1+/− mice suggesting that haploinsufficiency of Bmal1 compromised metabolic function and islet adaptation in context of HFD-induced increase in insulin demand. Notably, induction of glucose intolerance in β-Bmal1+/− mice was only present after tamoxifen administration thus confirming specificity of tamoxifen-inducible Bmal1 deletion (ESM Fig. 7).
Fig. 3.

Effects of adult beta cell-specific Bmal1 deletion on glucose tolerance in vivo following exposure to either chow or HFD. Graphs depicting sampled blood glucose and corresponding glucose AUC during 90 min i.p. glucose tolerance tests conducted at baseline and 2, 4, 6 and 8 weeks after tamoxifen in β-Bmal1+/+ (white circles/bars), β-Bmal1+/− (grey circles/bars) and β-Bmal1−/− (black circles/bars) mice exposed to 8 weeks of (a, b) chow or (c, d) HFD. (a–d) Data expressed as mean ± SEM (n=5–7 per group). Statistical significance is denoted by *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. Baseline data are common for chow and HFD groups
Fig. 4.

Effects of adult beta cell-specific Bmal1 deletion on GSIS in vivo following exposure to either chow or HFD. Graphs depicting sampled plasma insulin and corresponding insulin response expressed as AUC insulin/AUC glucose during the first 30 min of 90 min i.p. glucose tolerance tests conducted at baseline and 2, 4, 6 and 8 weeks after tamoxifen in β-Bmal1+/+ (white circles/bars), β-Bmal1+/− (grey circles/bars) and β-Bmal1−/− (black circles/bars) mice exposed to 8 weeks of (a,b) chow or (c,d) HFD. (a–d) Data expressed as mean ± SEM (n=5–7 per group). Statistical significance is denoted by *p<0.05 and **p<0.01. Baseline data is common for chow and HFD groups
We next assessed the role of beta cell Bmal1 in HFD-induced beta cell expansion, proliferation and survival (Figs 5, 6). Importantly, alpha cell mass was comparable across the three genotypes either on chow or HFD (Fig. 5, 6b). In contrast, HFD provoked ~50% increase in beta cell mass in control β-Bmal1+/+ mice (p<0.01, chow vs HFD, Figs 5, 6a). This increase was associated with augmented islet size (p<0.05, chow vs HFD, Figs 5, 6c), islet density (p<0.01, chow vs HFD, Figs 5, 6d) and approximately threefold increase in the frequency of beta cell proliferation (p<0.05, chow vs HFD, Figs 5, 6e), with no change in pancreatic weight (ESM Fig. 8). In contrast, both β-Bmal1+/− and β-Bmal1−/− mice failed to adaptively increase beta cell mass, islet size, islet density and beta cell proliferation in response to HFD (p>0.05, chow vs HFD, Figs 5, 6). Moreover, while there was no significant increase (p=0.27) in beta cell apopto-sis in control β-Bmal1+/+ mice on HFD, the frequency of beta cell apoptosis increased several fold in β-Bmal1−/− mice upon exposure to HFD (p<0.05, chow vs HFD; Fig. 6f, ESM Fig. 9).
Fig. 5.

Effects of adult beta cell-specific Bmal1 deletion on islet morphology following exposure to either chow or HFD. (a, b) Representative examples of scanned whole pancreas sections (magnification ×5) stained for insulin by immunohistochemistry in brown and by immunofluorescence in green from β-Bmal1+/+, β-Bmal1+/− and β-Bmal1−/− mice exposed to 8 weeks of chow or HFD (scale bars, 1,000 μm). (c, d) Representative examples of islets stained by immunofluorescence for insulin (Ins, green), glucagon (Glu, red) and nuclei (DAPI, blue) (c), and insulin (green), replication marker Ki67 (red) and nuclei (DAPI, blue) (d) imaged at ×20 magnification from β-Bmal1+/+, β-Bmal1+/− and β-Bmal1−/− mice exposed to 8 weeks of chow or HFD (scale bars, 50 μm)
Fig. 6.

Effects of adult beta cell-specific Bmal1 deletion on beta cell mass, proliferation and apoptosis in vivo following exposure to either chow or HFD. Bar graphs depicting (a) beta cell mass, (b) alpha cell mass, (c) average islet size, (d) islet density, (e) frequency of beta cell replication and (f) apoptosis in β-Bmal1+/+, β-Bmal1+/− and β-Bmal1−/−mice exposed to 8 weeks of either chow (white bars) or HFD (black bars). (a–f) Graphs represent mean ± SEM (n=5–7 per group). Statistical significance is denoted by *p<0.05, and **p<0.01
In the next series of studies, we set out to confirm that impairments in beta cell function and beta cell turnover observed in vivo in β-Bmal1−/− mice are attributed to Bmal1 deletion rather than a secondary effect due to changes in metabolic milieu associated with diabetic phenotype (Figs 7, 8). Consistent with in vivo observations, loss of Bmal1 resulted in a significant decline of GSIS (p<0.05 vs β-Bmal1+/+, Fig. 7) shown in isolated islets by islet perifusion. Notably, this decline in GSIS was attributed in part to loss of pulsatile pattern of insulin release as well as elevated basal insulin secretion (Fig. 7).
Fig. 7.

Effects of adult beta cell-specific Bmal1 deletion on GSIS in vitro in isolated islets. (a) Representative pulsatile and (b) mean ± SEM (n=5 per group) insulin secretion profiles sampled at 4 mmol/l (0–40 min) and 16 mmol/l glucose (40–80 min) in islets from β-Bmal1+/+ (thin line, white circles) and β-Bmal1−/− (thick line, black circles) mice. (c) Corresponding mean ± SEM (n=5 per group) rates of insulin secretion calculated as AUC from insulin concentrations obtained during islet perifusion in islets from β-Bmal1+/+ and β-Bmal1−/− mice. White bars, 4 mmol/l glucose; black bars, 16 mmol/l glucose. Statistical significance is denoted by *p<0.05
Fig. 8.

Adult beta cell-specific Bmal1 deletion impairs proliferative potential of islets in vitro and wild-type mouse islets show circadian oscillations of genes involved in cell proliferation in vivo (a). Scatter plot showing log10 normalised expression of 84 cell cycle genes in Exendin-4-treated islets from β-Bmal1−/− mice compared with β-Bmal1+/+. The central line indicates unchanged gene expression. Dotted boundary lines represent fold regulation cut-off set at twofold. Red circles, upregulated genes; green circles, downregulated genes; black circles, unchanged genes. Heat map showing graphical representation of fold regulation expression data of selected 31 genes in islets from β-Bmal1+/+ (n=3) and β-Bmal1−/− (n=3) mice treated with Exendin-4. Bar graph under heat map depicts fold change with error (95% CI) of the 31 genes sequentially shown in the heat map. Statistical significance is denoted by *p<0.05 and †p<0.09. (b) Graph showing normalised expression of 23 cell cycle genes that show significant circadian oscillations (p=3.4×10−5) in islets from C57BL6 mice (n=3–4 per time point) isolated at 4 h intervals around the 24 h LD cycle. Shaded area represents ‘lights off’ portion of the day. Heat map depicts log2 normalised expression data of the 23 rhythmic cell proliferation genes. (a, b) Genes marked with blue circles are significantly altered in β-Bmal1−/− islets in vitro as well as show significant rhythmic oscillations in wild-type islets in vivo. Max., maximum; Min., minimum
To provide evidence for the direct involvement of Bmal1 in transcriptional regulation of cell proliferation in beta cells, we treated isolated islets from β-Bmal1+/+ and β-Bmal1−/− mice with a known inducer of beta cell proliferation/cell cycle progression (glucagon-like peptide-1 pathway agonist, Exendin-4) and subsequently assessed transcriptional profiles of cell cycle activators/inhibitors by targeted PCR array (Fig. 8a). In accordance with failed beta cell expansion in vivo, islets isolated from β-Bmal1−/− mice displayed impaired transcriptional control of cell cycle regulation in vitro highlighted by diminished expression of notable cell cycle activators (e.g. Bcl2, Brca1, Ccnb2, Cdk6, Cdc20), as well as increased expression of notable cell cycle inhibitors (e.g. Wee1, Cdkn2b, Rbl2, Hus1, Gadd45a). Finally, we conducted genome-wide transcriptional analysis of islets isolated from wild-type C57BL6 mice at 4 h intervals throughout the 24 h LD cycle (Fig. 8b), designed to detect presence of circadian oscillations in key cell cycle related transcripts in vivo. We subsequently analysed circadian gene expression data by STEM [27] and demonstrated significant circadian rhythms (p=3.4×10−5, Fig. 8b) in a cluster of genes regulating cell cycle progression, notably enriched with transcripts shown to be significantly affected by Bmal1 deletion in beta cells (Fig. 8b).
Discussion
In this study, we sought to gain insights into molecular mechanisms underlying compensatory vs maladaptive beta cell response to diet-induced obesity. Consistent with previous reports, exposure to HFD in mice with an intact circadian clock led to compensatory expansion of beta cell mass due to increased beta cell proliferation, islet size and islet density, resulting in preservation of normal beta cell function and glucose homeostasis. In contrast, beta cell-specific adult deletion of Bmal1 resulted in maladaptive beta cell response to HFD, characterised by failed beta cell expansion due to diminished cell proliferation, increased apoptosis and loss of GSIS. Our work demonstrates that beta cell Bmal1 is an important regulator of beta cell compensatory growth, survival and function in response to increased insulin demand associated with diet-induced obesity.
Circadian disruption, achieved either by exposure to aberrant LD cycles or deletions of key circadian clock components, has been shown to impart deleterious consequences upon glucose homeostasis and beta cell function [22–24, 28]. Our current results are consistent with previous work and extend these observations by demonstrating that adult beta cell-specific deletion of Bmal1 leads to loss of GSIS (both in vivo and in vitro) and impaired beta cell functional and morphological adaptation to HFD. The use of adult Bmal1 deletion in our study is important given the role of circadian clocks in embryonic development and maturation [10].
Recent studies have shed light on potential mechanisms of impaired GSIS in circadian clock-mutant mice [22, 23]. These include (1) impairments in insulin vesicle trafficking and exocytosis; (2) diminished mitochondrial function and ATP production; and (3) increased susceptibility to oxidative stress due to dysregulation of BMAL1-mediated expression of Nrf2 (also known as Nfe2l2), (encoding a key oxidative stress response transcription factor) [22, 23]. Given that beta cell exposure to increased metabolic demand is associated with production of reactive oxygen species and propensity for oxidative stress [29], an impaired ability to respond to oxidative stress in beta cell Bmal1 null mice likely contributed to overt beta cell failure observed in our study. Interestingly in our study under HFD conditions, even haploinsufficiency of Bmal1 manifested in glucose intolerance and beta cell dysfunction further underscoring the important role of Bmal1 in beta cell adaptation to metabolic stress.
A key observation in our study is that loss of beta cell Bmal1 results in failed expansion of beta cell mass in response to diet-induced obesity in mice. Studies in rodents and humans show that obesity is associated with compensatory increase in beta cell mass, the extent of which positively correlates with the degree of insulin resistance [6, 7, 30, 31]. Importantly, failure of beta cell mass expansion in the context of obesity is associated with loss of glycaemic control and progression to type 2 diabetes [6, 30]. What mechanisms underlie successful vs failed beta cell expansion in response to insulin resistance? This question remains a topic of debate partly because mechanisms driving beta cell expansion appear to differ in humans and rodents [32]. Studies in human pancreas report that beta cell expansion during obesity/insulin resistance does not coincide with increased frequency of beta cell proliferation [7, 31]. These data suggest that obesity-driven beta cell formation in humans relies on enhanced beta cell neogenesis and/ or transdifferentiation [31]. However, lack of pancreas access in humans and inability to conduct longitudinal studies hinders the ability to precisely delineate mechanisms of beta cell expansion in humans. Future studies will be needed to address whether beta cell Bmal1 expression is important for modulating beta cell expansion in humans.
The circadian clock has long been appreciated as an important regulator of cell cycle control, cellular response to DNA damage, as well as cell fate differentiation and stem cell function [10, 33]. Our studies are consistent with these observations and demonstrate, for the first time, that Bmal1-deficient beta cells display diminished proliferative potential. Indeed, key circadian clock components (CLOCK–BMAL1) appear to exert direct control over cell division through transcriptional control (‘gating’) of cell cycle checkpoint proteins [34, 35]. In our study, we observed that loss of beta cell Bmal1 resulted in attenuated expression of cell cycle activators (e.g. Bcl2, Brca1, Ccnb2, Cdk6, Cdc-20) and a parallel increased expression of cell cycle inhibitors (e.g. Wee1, Cdkn2b, Rbl2, Hus1, Gadd45a), a number of which have been shown to harbour circadian E-box promoter sequences [35–38]. Consistent with these observations, we reported significant circadian rhythms in islet mRNA expression of genes regulating cell cycle progression, notably enriched with transcripts modulated by Bmal1 deletion in beta cells.
Another intriguing observation in our study is that Bmal1-deficient beta cells demonstrate increased rate of beta cell apoptosis and DNA damage detected by increased TUNEL immunoreactivity in response to HFD. This observation coincided with altered expression of transcripts involved in regulation of cell cycle arrest and apoptosis in response to DNA damage (e.g. Hus1, Brca1, Gadd45a) in β-Bmal1−/− mice [39]. The circadian clock has long been known to regulate cellular response to DNA damage through modulation of cellular DNA repair, cell cycle checkpoint progression, as well as control of cell cycle arrest and apoptosis [40]. Thus, it is plausible to hypothesise that loss of Bmal1 in beta cells may compromise the ability of beta cells to respond to oxidative stress-induced DNA damage leading to increased propensity for beta cell apoptosis.
It is important to acknowledge some limitations of our study. First, although common beta cell deleter mouse lines (e.g. Ins2-Cre and Ins2-CreERT) provide important tools for exploration of beta cell-specific gene functions, these mouse lines have been shown to express islet-specific human growth hormone (hGH), which impacts regulation of beta cell function and glycaemic control [41]. We confirmed the presence of hGH expression in islets of Ins2-CreERT mice (data not shown) that could account for some observed differences in progression of glucose intolerance and hyperglycaemia in response to HFD compared with previously published data [42]. Second, to isolate effects of Bmal1 deletion on cell cycle gene expression (independent of changes in metabolic milieu in vivo), we opted to perform in vitro experiments in isolated islets acutely treated with a known activator of beta cell proliferation. Although our in vitro results are consistent with in vivo observations of diminished beta cell proliferation and increased beta cell apoptosis, it will be important in future to assess changes in beta cell transcriptional profiles in vivo in lean and obese β-Bmal1−/− mice.
In conclusion, epidemiological studies report clear associations between circadian disruption and increased susceptibility to obesity, the metabolic syndrome and type 2 diabetes [18, 19, 43, 44]. Our results suggest that the beta cell circadian clock (via Bmal1) is an important transcriptional regulator of the compensatory beta cell expansion, survival and function in response to increased insulin demand associated with diet-induced obesity. Further work is warranted to understand the role of circadian clocks as modulators of human beta cell function, expansion and survival in obesity and type 2 diabetes.
Supplementary Material
Acknowledgments
The authors thank P.C. Butler and C.S. Colwell (University of California Los Angeles, Los Angeles, CA, USA) for helpful discussions and insightful comments. The authors are also grateful to members of the Scarisbrick lab: M. Radulovic and L. Kleppe (Mayo Clinic, Rochester, MN, USA) for help with frozen sectioning of mouse brain. Some of the data were included in an oral presentation abstract at the 75th ADA Scientific Sessions, 5–9 June 2015, Boston, MA, USA.
Funding We acknowledge funding support from the National Institutes of Health (DK098468 to AVM), and the Center for Regenerative Medicine (Mayo Clinic, Rochester, MN, USA).
Abbreviations
- ARNT
Aryl hydrocarbon receptor nuclear translocator
- BMAL1
Brain and muscle ARNT-like 1
- CLOCK
Circadian locomotor output cycles kaput
- DD
Constant darkness
- GSIS
Glucose-stimulated insulin secretion
- HFD
High fat diet
- hGH
Human growth hormone
- LD
Light (12 h)/dark (12 h)
- PER
Period
- STEM
Short time-series expression miner
- UCLA
University of California Los Angeles
- ZT
Zeitgeber time
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00125-015-3859-2) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Duality of interest The authors declare that they have no duality of interest associated with this manuscript.
Contribution statement KR contributed to the conception and design, acquisition of data, analysis and interpretation of data, drafting and critically revising the manuscript. TWH contributed to the conception and design, acquisition of data, and drafting the manuscript. AVM contributed to the conception and design, analysis and interpretation of data, drafting and critically revising the manuscript. AVM is the guarantor of this work, had full access to all data in the study, and takes responsibility for the integrity and accuracy of data analysis. All authors approved the final version.
References
- 1.Yki-Jarvinen H. Role of insulin resistance in the pathogenesis of NIDDM. Diabetologia. 1995;38:1378–1388. doi: 10.1007/BF00400597. [DOI] [PubMed] [Google Scholar]
- 2.Polonsky KS, Given BD, Van Cauter E. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest. 1988;81:442–448. doi: 10.1172/JCI113339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polonsky KS, Given BD, Hirsch L, et al. Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest. 1988;81:435–441. doi: 10.1172/JCI113338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 5.Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest. 1981;68:1456–1467. doi: 10.1172/JCI110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 7.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. beta-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36:111–117. doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kahn SE. Clinical review 135: the importance of beta-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. 2001;86:4047–4058. doi: 10.1210/jcem.86.9.7713. [DOI] [PubMed] [Google Scholar]
- 9.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown SA. Circadian clock-mediated control of stem cell division and differentiation: beyond night and day. Development. 2014;141:3105–3111. doi: 10.1242/dev.104851. [DOI] [PubMed] [Google Scholar]
- 11.Rakshit K, Thomas AP, Matveyenko AV. Does disruption of circadian rhythms contribute to beta-cell failure in type 2 diabetes? Curr Diab Rep. 2014;14:474. doi: 10.1007/s11892-014-0474-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–775. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gekakis N, Staknis D, Nguyen HB, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 14.Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 15.Bunger MK, Wilsbacher LD, Moran SM, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rudic RD, McNamara P, Curtis AM, et al. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2004;2:e377. doi: 10.1371/journal.pbio.0020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silva CM, Sato S, Margolis RN. No time to lose: workshop on circadian rhythms and metabolic disease. Genes Dev. 24:1456–1464. doi: 10.1101/gad.1948310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin YC, Hsiao TJ, Chen PC. Persistent rotating shift-work exposure accelerates development of metabolic syndrome among middle-aged female employees: a five-year follow-up. Chronobiol Int. 2009;26:740–755. doi: 10.1080/07420520902929029. [DOI] [PubMed] [Google Scholar]
- 19.Pan A, Schernhammer ES, Sun Q, Hu FB. Rotating night shift work and risk of type 2 diabetes: two prospective cohort studies in women. PLoS Med. 2011;8:e1001141. doi: 10.1371/journal.pmed.1001141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suwazono Y, Dochi M, Oishi M, Tanaka K, Kobayashi E, Sakata K. Shiftwork and impaired glucose metabolism: a 14-year cohort study on 7104 male workers. Chronobiol Int. 2009;26:926–941. doi: 10.1080/07420520903044422. [DOI] [PubMed] [Google Scholar]
- 21.Buxton OM, Cain SW, O'Connor SP, et al. Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci Transl Med. 2012;4:129ra143. doi: 10.1126/scitranslmed.3003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee J, Moulik M, Fang Z, et al. Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced beta-cell failure in mice. Mol Cell Biol. 2013;33:2327–2338. doi: 10.1128/MCB.01421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marcheva B, Ramsey KM, Buhr ED, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian J, Block GD, Colwell CS, Matveyenko AV. Consequences of exposure to light at night on the pancreatic islet circadian clock and function in rats. Diabetes. 2013;62:3469–3478. doi: 10.2337/db12-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Storch KF, Paz C, Signorovitch J, et al. Intrinsic circadian clock of the mammalian retina: importance for retinal processing of visual information. Cell. 2007;130:730–741. doi: 10.1016/j.cell.2007.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 27.Ernst J, Bar-Joseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinforma. 2006;7:191. doi: 10.1186/1471-2105-7-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gale JE, Cox HI, Qian J, Block GD, Colwell CS, Matveyenko AV. Disruption of circadian rhythms accelerates development of diabetes through pancreatic beta-cell loss and dysfunction. J Biol Rhythm. 2011;26:423–433. doi: 10.1177/0748730411416341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev. 2007;29:351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matveyenko AV, Gurlo T, Daval M, Butler AE, Butler PC. Successful versus failed adaptation to high-fat diet-induced insulin resistance: the role of IAPP-induced beta-cell endoplasmic reticulum stress. Diabetes. 2009;58:906–916. doi: 10.2337/db08-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mezza T, Muscogiuri G, Sorice GP, et al. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes. 2014;63:994–1007. doi: 10.2337/db13-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meier JJ, Bhushan A, Butler PC. The potential for stem cell therapy in diabetes. Pediatr Res. 2006;59:65R–73R. doi: 10.1203/01.pdr.0000206857.38581.49. [DOI] [PubMed] [Google Scholar]
- 33.Masri S, Cervantes M, Sassone-Corsi P. The circadian clock and cell cycle: interconnected biological circuits. Curr Opin Cell Biol. 2013;25:730–734. doi: 10.1016/j.ceb.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grechez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J Biol Chem. 2008;283:4535–4542. doi: 10.1074/jbc.M705576200. [DOI] [PubMed] [Google Scholar]
- 35.Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003;302:255–259. doi: 10.1126/science.1086271. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y, Xu J, Borowicz S, Collins C, Huo D, Olopade OI. c-Myc activates BRCA1 gene expression through distal promoter elements in breast cancer cells. BMC Cancer. 2011;11:246. doi: 10.1186/1471-2407-11-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunt T, Sassone-Corsi P. Riding tandem: circadian clocks and the cell cycle. Cell. 2007;129:461–464. doi: 10.1016/j.cell.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 38.Mateyak MK, Obaya AJ, Sedivy JM. c-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol Cell Biol. 1999;19:4672–4683. doi: 10.1128/mcb.19.7.4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 40.Sancar A, Lindsey-Boltz LA, Kang TH, Reardon JT, Lee JH, Ozturk N. Circadian clock control of the cellular response to DNA damage. FEBS Lett. 2010;584:2618–2625. doi: 10.1016/j.febslet.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brouwers B, de Faudeur G, Osipovich AB, et al. Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab. 2014;20:979–990. doi: 10.1016/j.cmet.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montgomery MK, Hallahan NL, Brown SH, et al. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia. 2013;56:1129–1139. doi: 10.1007/s00125-013-2846-8. [DOI] [PubMed] [Google Scholar]
- 43.Kroenke CH, Spiegelman D, Manson J, Schernhammer ES, Colditz GA, Kawachi I. Work characteristics and incidence of type 2 diabetes in women. Am J Epidemiol. 2007;165:175–183. doi: 10.1093/aje/kwj355. [DOI] [PubMed] [Google Scholar]
- 44.Mikuni E, Ohoshi T, Hayashi K, Miyamura K. Glucose intolerance in an employed population. Tohoku J Exp Med. 1983;141(Suppl):S251–S256. doi: 10.1620/tjem.141.suppl_251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.