

Abstract
It is well-established that the administration of streptozotocin accelerates diabetic liver injury as well as type-I diabetes, however the underlying mechanisms are poorly understood. Here we investigated the molecular mechanisms of diabetic liver injury in a model of streptozotocin (STZ)-induced type-I diabetes. STZ administration induced type-1 diabetes and chronic liver injury was associated with increased STAT1, which is implicated in diabetic liver injury by virtue of its ability to promote hepatocyte apoptosis, in the liver and pancreas, which were all strongly inhibited in STAT1−/− mice. Similarly, STZ-induced ATF3, a stress-inducible gene, was completely abolished in the liver of IFN-γ−/− mice, but not in STAT1−/− mice. Inhibition of STAT1 by siRNA or dominant-negative DNA did not affect ATF3 protein expression but blocked IFN-γ-induced ATF3 translocation from the cytosol into the nucleus. In contrast, inhibition of ATF3 by using siRNA diminished STAT1 protein expression and IFN-γ/STZ-induced hepatocyte apoptosis. Furthermore, GST pulldown and co-IP assay showed that STAT1 bound to C-terminal domain of ATF3. Such direct interaction increased the stability of STAT1 by inhibiting its ubiquitination as well as proteasome activity. Our results suggest that STAT1 is a common signaling pathway contributing to STZ-induced diabetes and diabetic liver injury. ATF3 functions as a potent regulator of STAT1 stability, accelerating STZ-induced diabetes and diabetic liver injury.
Keywords: Diabetic liver injury, ATF3, STAT1, Streptozotocin (STZ), Apoptosis
1. Introduction
Epidemiological studies show that diabetic patients are at higher risk of chronic liver disease and hepatocellular carcinoma [1–4]. Among them, non-alcoholic fatty liver disease (NAFLD) is probably the most prevalent liver disorder in type-II diabetes that is usually accompanied by central obesity, insulin resistance and other metabolic abnormalities although whether or how NAFLD and insulin resistance are temporally and mechanistically related is controversial [5–7]. Diabetes and insulin resistance were also identified as important factors associated with an increased risk of advanced liver fibrosis in patients with normal ALT [3,8]. The insulin resistance-associated inflammation, lipogenesis, and fibrogenesis are important mechanisms contributing to the pathogenesis of chronic liver disease in type-II diabetes [6,9,10]. In addition, type-I diabetes is also associated with increased risk of chronic liver injury [11,12], however the underlying mechanisms remain largely unknown.
Administration with streptozotocin (STZ) is a well-established experimental tool for the induction of autoimmune type-I diabetes, which is mediated by hyperglycemia and hypoinsulinemia due to its selective cytotoxicity towards insulin-secreting pancreatic β-cells [13,14]. Interestingly, STZ administration also causes diabetic organ complications in various tissues including the liver, kidney, heart, and brain as well as the pancreas [15–17]. In STZ-administrated mice or rat, cytokines secreted through T-cell activation and hyperglycemia as independent risk factors directly cause liver damage, lending to diabetic liver injury. Although the molecular mechanisms underlying STZ-mediated pancreatic β-cell death have been extensively investigated, how STZ induces diabetic liver injury is still not fully understood. Increasing evidence suggests that activation of IFN-γ/STAT1 plays an essential role in STZ-mediated pancreatic β-cell apoptosis and diabetes because STAT1−/− mice are resistant to multiple low-dose STZ-induced diabetes and β-cell apoptosis [18,19]. Recently, others and we have demonstrated that STAT1 activation by IFN-γ also plays an important role in liver injury and hepatocyte apoptosis in a variety of liver injury models induced by LPS/D-GalN and concanavalin A [20–23]. Thus, we hypothesized that injection of STZ can activate IFN-γ/STAT1 signaling pathway, which subsequently not only induces pancreatic β-cell apoptosis but also induces hepatocyte apoptosis, resulting in type-I diabetes and diabetic liver injury, respectively.
ATF3, a member of the ATF/cAMP-responsive element binding protein subfamily, is a stress-inducible transcriptional repressor as well as a basic region-leucine zipper transcription factor [24]. However, the findings about the role of ATF3 in gene regulation and apoptosis have been controversial [25,26]. Activated ATF3 can homodimerize and repress transcription of various promoters with ATF sites or heterodimerize with bZip proteins, c-jun, Jun B, ATF2, or Gadd153/CHOP10 (C/EBP homologous protein) and activate transcription of target genes [27,28]. Recently, we have reported that ATF3 negatively regulates adiponectin gene expression in obesity and type-II diabetes and that lipotoxicity-increased ATF3 was involved in ROS production and loss of mitochondria membrane potential, resulting in pancreatic β-cell apoptosis [29,30].
In this paper, we demonstrated that during STZ-mediated diabetic liver injury, elevated ATF3 interacted directly with STAT1 and enhanced the stability of the latter, and therefore it may serve as a liver injury-inducing factor via stimulation of the apoptotic functions of STAT1.
2. Materials and methods
2.1. Mice
Eight-week-old male STAT1−/− on 129S6/SvEv background and 129S6/SvEv wild-type mice were originally purchased from Taconic (Germantown, NY). BALB/c-background IFN-γ−/− and BALB/c wild-type mice were purchased from Jackson Laboratory (Bar Harbor, ME). For STZ injection, the mice were injected intraperitoneally for 5 consecutive days with STZ (80 mg/kg; sigma) freshly dissolved in 50 mM sodium citrate buffer (pH 4.5) and sacrificed 14 days post the final STZ injection. Animals were considered as diabetics when blood glucose levels exceeded 250 mg/dl, usually within 7 days from the final injection. Furthermore, anti-IFN-γ (2 μg/200 μl) was i.v. injected into mice. All animal experiments were conducted in accordance with guidelines from the Korean National Institutes of Health Animal Facility.
2.2. Plasmids
Human wild-type ATF3 and mutated ATF3 (ΔC, 1-100) with a C-terminal deletion, cDNA expression vectors were a generous gift from Dr. T Hai (Ohio State University). Human ATF3 and ATF3(ΔC) cDNA were amplified separately by PCR and cloned into pEGFP-C2 and pGEX-4T-1 vectors (Clontech, Mountain View, CA). The pcDNA3-Flag-STAT1 or -DN-STAT1 constructs were generously provided by Dr. B. Gao (NIH/NIAAA). From pcDNA3-Flag-STAT1, GFP-STAT1 was constructed by subcloning into the BamHI/XbaI restriction sites of pEGFP-C1.
2.3. Isolation of primary mouse hepatocytes and islet cells
Mice weighing 20–25 g were anesthetized with pentobarbital sodium (30 mg/kg, IP), and the isolation of primary hepatocytes by using liver perfusion method was followed as described previously [22]. Next, islet cells were isolated from overnight-fasted C57BL/6 mice by the collagenase digestion technique as described previously [39].
2.4. RT-PCR analysis
RT-PCR analysis and the sequences of the primers used for murine IFN-γ and TNF-α are as follows: IFN-γ, (F) 5′-AACGCTACACACTGCATC-3′ and (R) 5′-AGCTCATTGAATGCTTGG-3′; TNF-α, (F) 5′-GGCAGGTC-TACTTTGGAGTCA TTGC-3′ and (R) 5′-ACATTCGAGCCAGTGAATTCGG-3′.
2.5. Immunoassaying
Immunohistochemistry and immunocytochemistry analysis were followed as described previously [39].
2.6. GST pull-down assay
Five hundred microgram of lysates was incubated with 3.0 μg of GST or GST-ATF3 proteins coupled to glutathione Sepharose beads in 300 μL of lysis buffer overnight at 4 °C with continuous rocking as described previously [22].
2.7. Proteasome assays
Cells were transfected with Neo-vector, ATF3, or ATF3(ΔC) cDNA. For 20S proteasome inhibition assays, 5 mM Suc-LLVY-AMC substrates and inhibitor, epoxomicin, which is a rapid, potent and irreversible inhibitor of 20S proteasome chymotrypsin-like activity, in DMSO were added to assay solutions at a final DMSO concentration of 1%. The following assay buffer was used: 20 mM Tris–HCl, pH 8.0, 0.5 mM EDTA (plus 0.035% SDS for Suc-LLVY-AMC assays). 20S proteasome was added to the assay buffer containing substrates, inhibitors, and test sample at a final volume of 100 mL at room temperature (25 °C) in a Dynex (Chantilly, CA) Microfluor II 96-well plate and the fluorescence emission immediately was measured at 460 nm (lex, 360 nm) by using a Cytofluor (Perspective Biosystems, Framingham, MA) fluorescence plate reader for 50 min [31].
2.8. In vitro and in vivo ubiquitination assays
In vitro ubiquitination assays were performed as described previously [27] with some modifications. Recombinant purified GST-STAT1 was preincubated with 250 ng of recombinant full-length or the deleted ATF3 protein at 37 °C for 1 h in a 30 μL reaction buffer 1 containing 50 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 5 mM DTT, 4 mM ATP, 100 μM E1, 10 μM E2, 5 μg His-ubiquitin (E1,E2, and His-ubiquitin, Boston Biochem) and His-ubiquitinated proteins were isolated by incubating at 4 °C for 1 h with 20 μL of Ni-nitrilotriacetate agarose (Qiagen) in a final volume of 200 μL in reaction buffer 2 containing 50 mM sodium phosphate, pH 7.9, 300 mM NaCl, 0.05% Tween 20, and 10 mM imidazole. After low-speed centrifugation (735 ×g), washed Ni-agarose beads containing His-ubiquitinated proteins were eluted and then subjected to Western blotting for ubiquitinated STAT1 using anti-polyubiquitin antibody. In vivo ubiquitination assay was also performed as described previously [26]. Cells were co-transfected with the constant amount of HA-STAT1 (0.5 μg) and His-ubiquitin (0.5 μg), together with Flag-ATF3 or ATF3 (ΔC) (0.5 μg). Forty-eight hours after transfection, cells were treated with 10 μM MG-132 for 6h before being harvested. His-ubiquitin-containing protein complexes were pulled down with Ni-agarose beads, and subsequently resolved by 10% SDS-PAGE, followed by immunoblotting with anti-ubiquitin, anti-HA or anti-Flag antibodies.
2.9. Statistical analysis
For comparing values obtained in three or more groups, one-factor analysis of variance was used, followed by Tukey's post hoc test, and P<0.05 was taken to imply statistical significance.
For additional and detailed materials and methods, see Supplementary documents.
3. Results
3.1. STZ induces IFN-γ and STAT1 in diabetic liver injury
To investigate the exact molecular mechanisms involved in liver injury observed in immune-mediated murine diabetes induced by STZ administration, we have firstly examined the development of diabetes and diabetic liver injury by measuring whole blood glucose levels and pancreas or liver pathology. STZ administration elevated the plasma glucose levels to 434±30 mg/dl after 14 days (Fig. 1A), which was associated with increased insulitis (Fig. 1B, left) and β-cell apoptosis (not shown) and decreased body weight (not shown), ATP production (Fig. 1B, right) and islet cell mass (not shown) compared to control mice. Consistent with hyperglycemia, injection with STZ also elevated serum levels of ALT and AST (Fig. 1C, upper) and massive necrosis and inflammation in the liver, but reduced Ki-67+-positive cells, a marker widely used for cell proliferation (lower). Similarly, primary hepatocytes isolated from STZ-treated mice showed more apoptosis than those isolated from control mice (S1A), correlated with the induction of caspase-3 and PARP cleavage and increased cytochrome c release from mitochondria to cytosol (S1B). Concomitantly, IFN-γ-responsive STAT1 protein, which plays an essential role in apoptosis and abnormality of hepatocytes and pancreatic β-cells (not shown) [20–23], was significantly increased in the liver and pancreas of STZ-injected mice (Fig. 1D, upper), confirmed by immunohistochemistry (lower). Phosphorylated STAT1 was slightly elevated in STZ-treated mice liver compared to that of control mice (Fig. 1D). Serum levels of IFN-γ (Fig. 1E) and hepatic expression of IFN-γ mRNA and proteins (Fig. 1F) were significantly elevated after STZ injection, correlated with the elevation of TNF-α expression. Interestingly, STZ-increased IFN-γ or TNF-α expression may be due to STAT1-dependent T-cell activation because STZ administration significantly increased CD4+CD69+ T cells in hepatic (Fig. 1G) or spleen (not shown) lymphocytes isolated from wild-type mice, but not in STAT1−/− mice. These results suggest that IFN-γ/STAT1 pathway may be involved in STZ-induced diabetic liver injury.
Fig. 1.
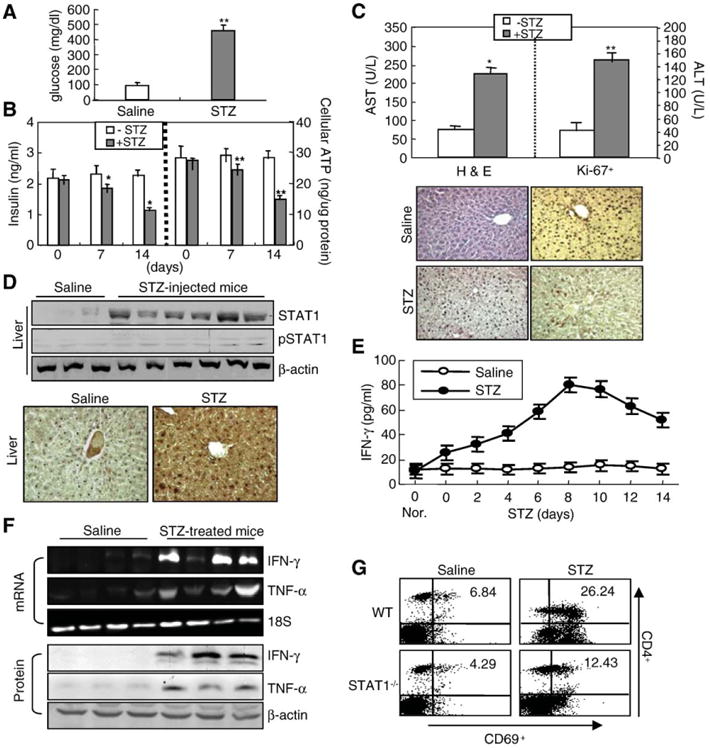
STZ administration induces diabetic liver injury and IFN-γ/STAT1 expression. Control and STZ-treated mice were described in “Materials and methods”. (A) Plasma glucose levels. (B) Insulin and ATP levels were measured. *P<0.01, **P<0.05 in comparison with corresponding STZ-non-treated control groups (0 day). (C) Upper panel: Serum AST and ALT levels. Lower panel: Representative H&E staining and Ki-67 immunostaining of livers from control and STZ-treated mice (×100). (D) Expression of STAT1 and pSTAT1 in the liver (upper, multiple samples in each group). Immunohistochemistry analyses (lower). Enhanced diffuse STAT1 immunostaining (dark brown) was detected (×100). (E) IFN-γ levels were measured by ELISA. After injection of STZ for 5 days, mice were sacrificed at the indicated time points. Values are shown as means±S.E.M. from four mice. (F) Expression of IFN-γ and TNF-α mRNA and protein in the liver analyzed by RT-PCR (upper) and Western blot analyses (lower), respectively. (G) After wild-type and STAT1−/− mice were injected with STZ, spleen lymphocytes were isolated. The surface markers CD4+CD69+ were analyzed by flow cytometry. Representative data are shown. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. STAT1 is essential for STZ-induced diabetic liver injury and diabetes
To determine the role of STAT1 on these events, STAT1−/− mice were used. STAT1−/− mice not expressing STAT1 protein in liver (Fig. 2A) strongly diminished STZ-mediated elevation of serum glucose levels in wild-type mice (Fig. 2B). Furthermore, accelerated AST activity and liver inflammatory responses were attenuated strongly by STAT1 depletion (Fig. 2C). As well, STZ-mediated induction of hepatic necrosis and apoptosis was also diminished in STAT1−/− mice (Fig. 2C, D). Similarly, we have also examined the direct roles of STAT1 depletion on pancreatic β-cell dysfunction. As expected, in wild-type mice, the levels of insulin content and ATP production were decreased significantly by STZ administration, but not in STAT1−/− mice (Fig. 2E). Reduction of pancreatic β-cell mass and increase of TUNEL-positive islet cells observed in STZ-injected wild-type mice were attenuated in STAT1−/− mice (Fig. 2F). Consistent with previous reports [23], IFN-γ induction of apoptosis was also diminished in STAT1−/− hepatocytes. Treatment of IFN-γ or STZ-induced slightly primary hepatocyte apoptosis, while cotreatment with IFN-γ/STZ synergistically induced apoptosis (S2A). Such synergistic induction of hepatocyte apoptosis was diminished in STAT1−/− hepatocytes (S2B) and STAT1 depletion by siRNA (S2C). Although not shown here, STZ induction of apoptosis of islet cells and MIN6N8, pancreatic β-cells, was also strongly attenuated in STAT1−/− mice and STAT1 siRNA-transfected cells.
Fig. 2.
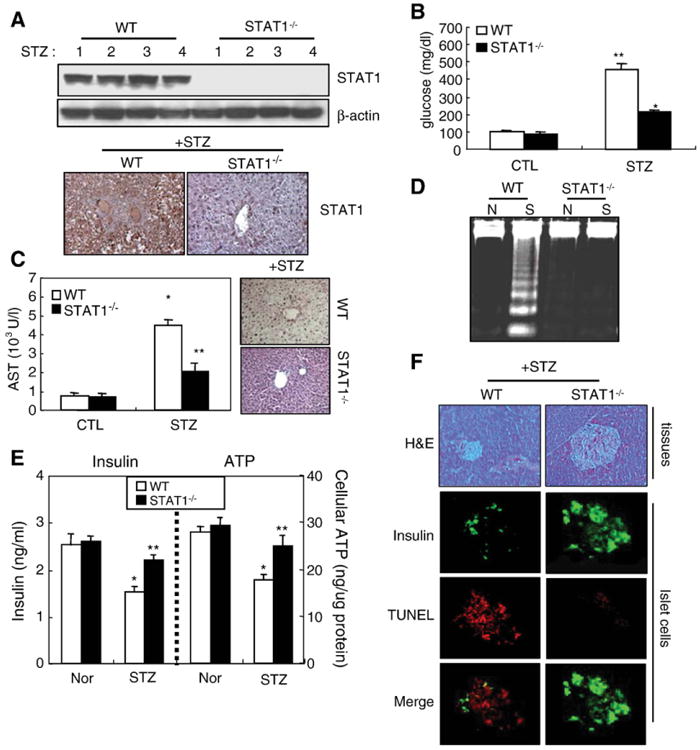
STAT1 is essential for STZ-induced liver injury and diabetes. STZ was administrated into the STAT1+/+ or STAT1−/− mice. (A) STAT1 protein expression (upper: Western blot, lower: IHC for STAT1). (B) Glucose levels. (C) Serum AST levels. *P<0.05, **P<0.01 in comparison with wild-type mice. (D) DNA fragmentation. (E) Insulin content and ATP production in isolated islet cells. *P<0.01, **P<0.05 in comparison with corresponding wild-type control groups. Values in panels are shown as means±S.E.M. from four mice. (F) In pancreas tissues of STAT1+/+ or STAT1−/− mice, photomicrographs of mouse pancreas with H&E staining are shown (×100, left). Immunostaining for insulin (green, positive islet cells) and TUNEL assay were analyzed. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. ATF3 expression and apoptosis induced by STZ and/or IFN-γ are abolished in IFN-γ−/− mice and anti-IFN-γ in vivo injected mice
Our above results suggest that STAT1 may be considered as a common signal pathway contributing to STZ-induced diabetes and diabetic liver injury. Therefore, we first examined the expression of STAT1 downstream genes such as IRF-1 and ATF3 [31]. Fig. 3A showed that expression of hepatic IRF-1 and ATF3 proteins was enhanced in STZ-injected mice compared to control mice. Furthermore, to examine the effects of IFN-γ/STAT1 on the expression of ATF3 and IRF-1 proteins in vitro cell systems, SK-Hep1 cells and primary hepatocytes were treated with IFN-γ, which may play as a stimulus for STAT1 accumulation in STZ-induced liver injury in vivo. IFN-γ induced STAT1 activation 30 min after treatment and increased STAT1, IRF-1, and ATF3 proteins with peak effects occurring at 24–120 h post treatment (Fig. 3B). We have also obtained the same results in STZ-treated pancreas tissues and isolated islet cells or MIN6N8 insulinoma cells (not shown). Next, we examined whether STZ affected IFN-γ induction of STAT1 and ATF3 in primary hepatocytes (Fig. 3C). STZ treatment neither induced STAT1 phosphorylation nor affected IFN-γ activation (IRF-1) of STAT1 phosphorylation at 30 min, whereas IFN-γ alone strongly increased STAT1 phosphorylation. After 24 h, most of STAT1 phosphorylation induced by IFN-γ was completely decreased to the basal levels, but STAT1, IRF-1, and ATF3 proteins were strongly increased. Interestingly, although STZ alone did not induce STAT1 phosphorylation, STZ alone increased STAT1 protein, which was enhanced by cotreatment with IFN-γ. Consistent with the increase of STAT1, ATF3 protein was also increased in STZ alone-treated cells, which was strongly increased by IFN-γ/STZ. Furthermore, IFN-γ induced IRF-1 protein in hepatocytes, while STZ had no effect on IRF-1 expression. In addition, STZ/IFN-γ did not further enhance IRF-1 expression (Fig. 3C), indicating that STZ did not directly regulate STAT1 activation in vitro, it is likely that IFN-γ produced in STZ-treated in vivo mice (Fig. 1) may be directly involved in STAT1 activation and STAT1-mediated liver injury. Therefore, to confirm the role of IFN-γ on STAT1-mediated STZ induction of diabetic liver injury, we used IFN-γ−/− mice and anti-IFN-γ-injected mice. Fig. 3D shows that expression of ATF3, STAT1, and IRF-1 proteins increased significantly in STZ-treated wild-type mice was completely abolished in IFN-γ−/− mice, confirmed by immunohistochemistry, suggesting that ATF3 like STAT1 and IRF-1 may be a downstream gene of IFN-γ signaling. Increased hepatic ATF3 and STAT1 show a strong positive correlation with increase of hepatocyte cell death, as evidenced by increased serum AST levels, which was markedly attenuated in IFN-γ−/− mice (Fig. 3E). In addition, IFN-γ treatment for 72 h strongly induced wild-type hepatocyte apoptosis, but was significantly diminished in IFN-γ−/− mice (Fig. 3F). Furthermore, in saline-injected mice, ATF3, STAT1, and IRF-1 increased strongly by STZ treatment were almost completely abolished by anti-IFN-γ-injected mice (Fig. 3G), suggesting that STZ-induced diabetic liver injury might be mediated by IFN-γ produced in vivo, thereby upregulate STAT1 and ATF3, results in liver injury.
Fig. 3.
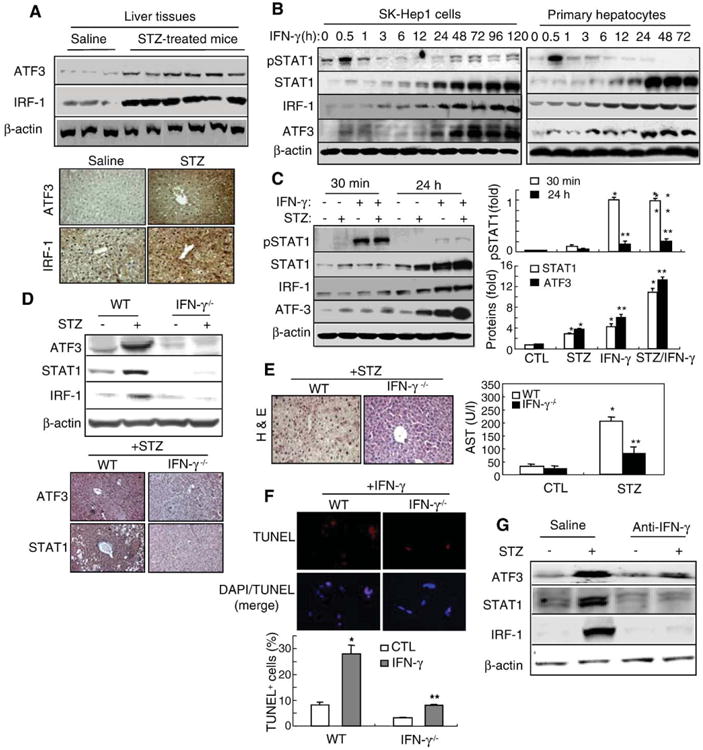
ATF3 expression induced by STZ or IFN-γ was abolished in IFN-γ−/− mice. (A) ATF3 and IRF-1 protein expression are enhanced in STZ-injected mice. Western blot analysis (upper) and immunostaining (×100) (lower). (B) SK-Hep1 and primary hepatocytes were treated with IFN-γ for the indicated time points and then subject to immunoblotting. (C) Primary hepatocytes were treated with IFN-γ and/or STZ for 30 min or 24 h and then subjected to Western blot analysis (left). Relative expression of pSTAT1, STAT1, and ATF3 was quantified (right). Values represent means±S.E.M. from 3 independent experiments. *P<0.05, **P<0.01 in comparison with corresponding control groups. (D) ATF3 and IRF-1 protein expression in STZ-injected wild-type and IFN-γ−/− mice. Western blot analysis (upper) and immunostaining (×100) (lower). (E) H&E staining (left) and AST levels (right) in STZ-treated wild-type and IFN-γ−/− mice. *P<0.01, **P<0.05 in comparison with corresponding non-treated control groups. (F) Isolated primary hepatocytes were treated with IFN-γ and then DAPI staining and TUNEL analyses were performed (upper). TUNEL+-cells were quantified (lower). Values represent means±S.E.M. from 3 independent experiments. *P<0.01, **P<0.05 in comparison with corresponding non-treated control groups. (G) Mice were injected with STZ (60 mg/kg, i.p.) and anti-IFN-γ Ab (2 μg, i.v.). Western blot analysis was performed.
3.4. STAT1 is required for ATF3 nuclear translocation, but not for ATF3 protein expression in hepatocytes
Previous reports showed that IFN-γ induction of ATF3 in monocytes and macrophages is STAT1-dependent [32], we wondered whether STAT1 is also essential for IFN-γ induction of ATF3 in hepatocytes. Interestingly, STZ-mediated induction of hepatic ATF3 protein was not reduced in STAT1−/− mice while induction of IRF-1 was diminished in STAT1−/− mice (Fig. 4A). Blocking of STAT1 activation (pSTAT1) by transfection of dominant-negative (STAT1F) neither affected the expression of basal levels of ATF3 nor affected IFN-γ induction of ATF3, while blocking of pSTAT1 with transfection of STAT1F inhibited IRF-1 expression (Fig. 4B). Interestingly, IFN-γ-induced ATF3 was also colocalized with STAT1 in the nucleus of GFP-STAT1-transfected cells, but not in GFP-STAT1F-transfected cells (Fig. 4C). Furthermore, IFN-γ or STZ/IFN-γ treatment increased ATF3 in both wild-type and STAT1−/− primary hepatocytes (Fig. 4D). However, in STZ/IFN-γ-treated wild-type primary hepatocytes, most of ATF3 was colocalized in the nuclei of TUNEL-positive cells (S2B), but ATF3 increased in STZ/IFN-γ-treated STAT1−/− primary hepatocytes was highly expressed around the nucleus, not in the nucleus (Fig. 4D, upper), confirmed by ATF3 localization analysis (Fig. 4D, lower), suggesting that STAT1 activation may be required for ATF3 nuclear translocation. Moreover, inhibition of STAT1 by STAT1 siRNA neither affected the expression of basal levels of ATF3 nor affected IFN-γ induction of ATF3 (Fig. 4E), correlated with the reduction of IFN-γ-induced ATF3 nuclear translocation (Fig. 4F).
Fig. 4.
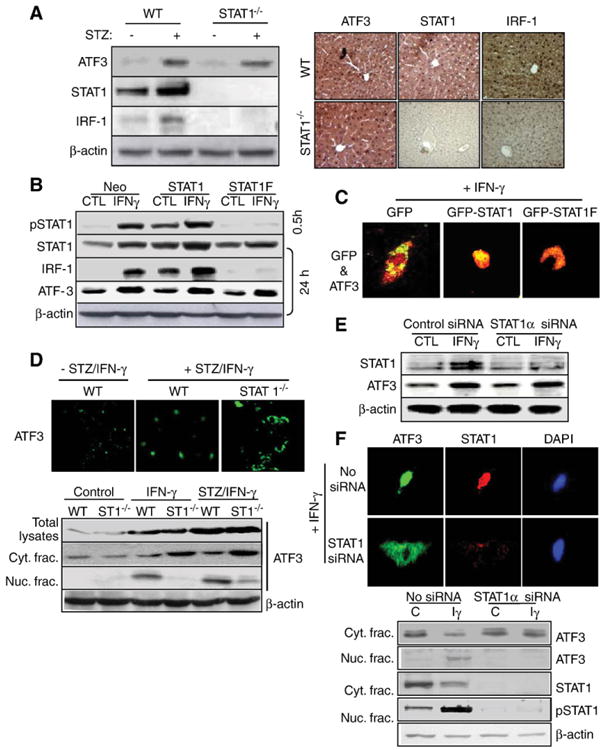
STAT1 is required for ATF3 translocation but not for ATF3 protein expression in hepatocytes. (A) ATF3 and IRF-1 expression in the liver of STAT1−/− mice (upper) and immunohistochemistry analyses for ATF3, STAT1, and IRF-1 (lower). (B) SK-Hep1 cells were transfected with neo control vector, STAT1 expression vector, and dominant-negative STAT1F vector for 48 h, followed by treatment with IFN-γ for 30 min or 24 h. Western blot analyses were performed. (C) After transfection with GFP-empty, GFP-STAT1, and GFP-STAT1F vector, cells were treated with IFN-γ. Immunocytochemistry analysis for ATF3 was performed. Fluorescent microscopic images were taken for GFP (for STAT1) and ATF3 (red), and the final merged images were shown. (D) Primary hepatocytes isolated from wild-type and STAT1−/− hepatocytes were treated with STZ/IFN-γ and then performed immunocytochemistry for ATF3 (green). Fluorescent microscopic images were taken for ATF3 (upper). The isolated cytosolic or nucleic fractions are subjected into the Western blotting (lower). (E) Primary hepatocytes were transfected with STAT1 siRNA and then treated with IFN-γ. Western blot analyses of STAT1 and ATF3. (F) After transfection with STAT1 siRNA, and followed by immunocytochemistry analyses for ATF3 and STAT1 (upper) and Western blot analyses of ATF3 and STAT1 protein expression in nuclear and cytosolic fraction. Iγ indicate IFN-γ. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5. ATF3 accelerates STZ/IFN-γ-mediated hepatocyte apoptosis and isrequired for STAT1 induction
To determine the possibility whether ATF3 can reversibly affect STAT1 expression or its activation, cells were transfected with constructed ATF3 or ATF3(ΔC), a deletion in the C-terminal (101–181) region necessary for interaction with other proteins [24]. ATF3 increased by transfection of GFP-ATF3 enhanced the basal levels and IFN-γ induction of pSTAT1 and STAT1 protein, but not in GFP-ATF3(ΔC)-transfected cells (Fig. 5A). Generally, nuclear localization of ATF3 is likely a prerequisite for its activity [27,28]. Concomitantly, in GFP-ATF3-transfected cells, most of STAT1 activated by IFN-γ was colocalized with GFP-ATF3 in the nucleus, but not in GFP-ATF3(ΔC)-transfected cells (Fig. 5B), supported by experiments using isolated cytosolic and nuclear fractions (Fig. 5C). Furthermore, GFP-ATF3 overexpression also strongly increased IFN-γ induction of apoptosis and apoptosis-related proteins suchascaspase-3 and PARP cleavage, and expression of Bax and p21, but not in GFP-ATF3(ΔC)-transfected cells (Fig. 5D,E). Our results suggest that ATF3, especially the C-terminal region, may play an important role in the functionality of STAT1 such as induction of hepatocyte apoptosis and may function as an upstream regulator of STAT1 induced by IFN-γ. As well, transfection of ATF3 siRNA blocked STZ/IFN-γ-induction of apoptosis in hepatocytes (Figs. 5F, S2C), correlated with reductionof the basal levels and IFN-γ or STZ/IFN-γ-induction of IRF-1 and STAT1 protein and STAT1 activation (Fig. 5G), confirmed by immunocyto-chemistry analysis showing that expression and nuclear translocation of ATF3 and STAT1 by STZ/IFN-γ were significantly decreased in ATF3 siRNA-transfected cells (S3A), correlated with the attenuation of apoptosis-related proteins induced by STZ/IFN-γ (S3B). Furthermore, in cells transfected with flag-tagged ATF3 or ATF3(ΔC) in the presence or absence of GFP-STAT1 or GFP-STATF (S3C), ATF3 or IFN-γ induction of apoptosis was potentiated by cotransfection of GFP-STAT1 (lane 4), but completely abrogated in GFP-STAT1F-transfected cells (lane 6). As well, inhibition of apoptosis in ATF3(ΔC)-transfected cells (lane 3) was restored in part, but not completely, by overexpression of GFP-STAT1 alone (lane 8) compared to GFP-STAT1-transfected cells (lane 5). These results demonstrated that although ATF3 plays as an upstream regulator for STZ/IFN-γ-induction of apoptosis and STAT1 protein expression, eventually activated STAT1 is required and plays as a downstream executive effector for ATF3-mediated apoptosis.
Fig. 5.

C-terminal domain of ATF3 is essential on STAT1 expression and apoptosis induced by IFN-γ. (A) SK-Hep1 cells were transfected with the expression vector encoding for GFP-ATF3 and C-terminal deleted ATF3, GFP-ATF3(ΔC) and analyzed by Western blot. (B) After transfection with GFP-ATF3 or GFP-ATF3(ΔC) and then treated with IFN-γ, immunocytochemistry analysis for ATF3 and STAT1 was performed. Fluorescent microscopic images were taken for GFP (for ATF3) and STAT1 (red), and the final merged images were shown. (C) After transfection, isolated nuclei or cytosolic fraction were subjected to Western blotting. (D) TUNEL assay in ATF3 or ATF3(ΔC)-transfected cells. *P<0.01, **P<0.05 in comparison with corresponding non-treated control groups. (E) Effects of ATF3 on the expression of apoptosis-related proteins. (F) SK-Hep1 cells were transfected with ATF3 siRNA and then treated with IFN-γ/STZ. DNA fragmentation analysis was performed. (G) After transfection with ATF3 siRNA, cells were treated with IFN-γ and STZ/IFN-γ for 30-min (for pSTAT1) or 24 h (for STAT1, IRF-1, and ATF3), followed by Western blot analyses. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.6. ATF3 increased STAT1 stability by inhibiting Ub/proteasome-dependent STAT1 degradation in hepatocytes
To understand the mechanisms underlying ATF3-mediated upregulation of STAT1, we also considered the possibility that ATF3 enhanced STAT1 stability through direct interaction with STAT1. As shown in Fig. 6A, in vivo STZ-injected mice, interaction of immunoprecipitated ATF3 with STAT1 was increased compared to normal mice. Similarly, in vitro binding assay, Flag-ATF3 was also detectable in immuno-complexes of STAT1 captured by anti-GFP antibody (Fig. 6B). In addition, both endogenous STAT1 and ATF3 were immunoprecipitated together in IFN-γ-treated cells but weakly in control cells (Fig. 6C), correlated with the proportional increase of ATF3 and STAT1 by IFN-γ. Concomitantly, GST pull-down assay showed that STAT1 is associated with GST-ATF3 in IFN-γ-treated cells, but not in control cells and GST-ATF3(ΔC) (Fig. 6D,E). However, our data show that the association of ATF3 with STAT1 may not due to the increase of STAT1 protein induced endogenously by IFN-γ or STZ because GST (Fig. 6D, lanes 3–4) or GST-ATF3(ΔC) (Fig. 6E, lanes 6–7) did not associate with STAT1, despite STAT1 was highly induced by IFN-γ. Furthermore, increase of endogenous STAT1 captured by immunoprecipitated ATF3 was also completely abrogated by ATF3-siRNA transfection, which was correlated with decreased levels of STAT1 (Fig. 6F, left). As well, Flag-ATF3, but not Flag-ATF3(ΔC), strongly interacts with STAT1 induced by IFN-γ (Fig. 6F, right), suggesting that ATF3, especially C-terminal region of ATF3, directly binds to STAT1 and subsequently may stabilize the STAT1 protein.
Fig. 6.
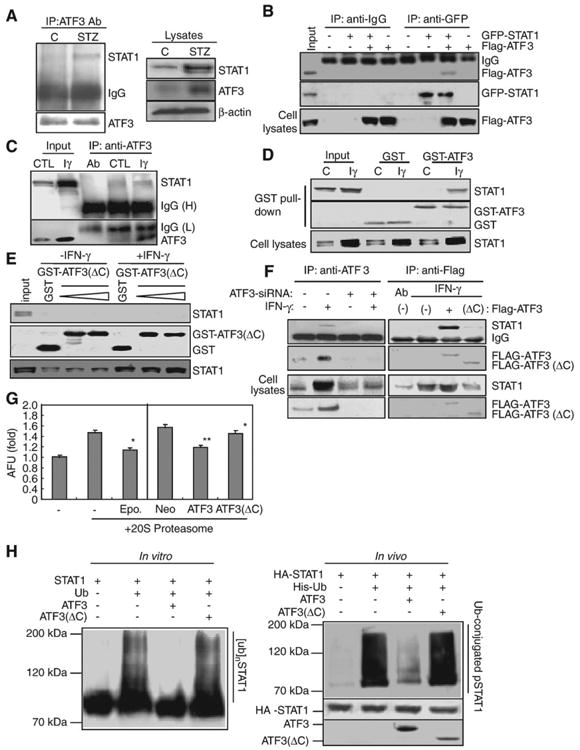
ATF3 increased STAT1 stability by inhibiting Ub/proteasome-dependent STAT1 degradation in hepatocytes. (A) Immunoprecipitation for ATF3 in STZ-injected mice. (B) Cells were transfected with GFP-STAT1 and Flag-ATF3, then immunoprecipitated with anti-GFP. (C) In vivo binding assay. After treatment of IFN-γ, immunocaptured ATF3 was directly interacted with STAT1. (D) GST pull-down assay by using purified GST or GST-ATF3. (E) GST pull-down assay by using purified GST or GST-ATF3(ΔC). (F) Immunocaptured Flag-ATF3, but not ATF3 siRNA (left) or Flag-ATF3(ΔC), directly interacts with STAT1 induced by IFN-γ. (G) 20S proteasome assay. *P<0.01, **P<0.05 in comparison with corresponding control group. (H) In vitro (left) and in vivo (right) ubiquitination assay. GST-STAT1 protein was preincubated with 250 ng of the full-length or the deleted mutant ATF3 protein and then subjected to Western blotting for ubiquitinated STAT1 using anti-polyubiquitin antibody (FK-1) (left). Ubiquitinated products were recovered on nickel-agarose beads and separated by SDS-PAGE, followed by immunoblotting with anti-HA antibody (right).
As the Ub-proteasome system is a one pathway by which STAT1 protein is degraded [33], we wondered whether ATF3 stabilized hepatic STAT1 via affecting STAT1 ubiquitination and the proteolytic activity of 20S proteasome, which was required in degradation of ubiquitinated proteins. Consistent with the inhibition effects of epoxomicin (500 μM), a potent and irreversible inhibitor [32], on the 20S proteasome chymotrypsin-like activity, the proteolytic activities were strongly decreased in ATF3-transfected cells, but not in ATF3(ΔC)-transfected cells (Fig. 6G). Fig 6H, left is an in vitro-reconstituted ubiquitination reaction system performed by utilizing with E1, E2 enzymes and GST-ATF3 or GST-ATF3(ΔC) recombinant proteins to test the effects of ATF3 on STAT1 ubiquitination. Interestingly, in the presence of all the required ubiquitination reaction components, addition of recombinant ATF3 protein, but not ATF3(ΔC) protein, in the ubiquitination reaction completely reduced STAT1 ubiquitination. As well, in vivo ubiquitination assay performed after cotransfection of HA-STAT1 and His-ubiquitin with ATF3 or ATF3 (ΔC) (Fig. 6H, right) shows that the amounts of the ubiquitinated STAT1 were decreased in the presence of ATF3, whereas ATF3(ΔC) inhibited the ubiquitination of STAT1 to a lesser degree. These results strongly suggest that binding of STAT1 to the C-terminal region of ATF3 inhibits STAT1 ubiquitination, thereby involves diabetic liver injury development by increasing the stability of STAT1.
4. Discussion
In this study, we demonstrated 1) that STAT1 is a common signal pathway contributing to STZ-induced diabetes and liver injury. 2) A stress-inducible transcription factor ATF3 interacts directly with STAT1 and enhances the stability of the latter, thereby acting as a critical mediator to stimulate liver injury and apoptosis mediated by STAT1 in immune-mediated diabetes induced by STZ administration. Although in STZ-administrated mice or rat, hyperglycemia as an independent risk factor directly causes liver damage, lending to diabetic liver injury, but exact molecular mechanisms involved in STZ-induced diabetic liver injury were still not clear.
4.1. STAT1, a common signal pathway contributing to STZ-induced diabetes and liver injury
IFN-γ activation of STAT1 has been shown to play an important role in STZ-mediated diabetes [18,19]. Here we provided several evidences suggesting a critical role of STAT1 in STZ-induced diabetic liver injury. First, STZ-induced diabetic liver injury was completely attenuated in STAT1−/− mice. Second, STZ/IFN-γ induction of hepatocyte apoptosis was diminished in STAT1−/− hepatocytes. Third, inhibition of STAT1 protein and activation by siRNA or STAT1F abolished STZ/IFN-γ-mediated hepatocyte apoptosis. Elevation of serum IFN-γ is likely responsible for the induction of STAT1 protein in the liver after STZ administration since IFN-γ has been shown to play an important role in STAT1 protein induction in hepatocytes [20–22]. However, the molecular mechanisms underlying elevation of IFN-γ in STZ-treated mice are not clear although production of cytokines such as IFN-γ, TNF-α, and IL-1 via activation of CD8+ (the initial effector) and CD4+ (the final effector) T-cells in autoimmune diabetes was implicated in β-cell destruction [34–37].
In this paper, we have demonstrated that IFN-γ induced hepatocyte apoptosis via an STAT1-dependent manner (Figs. 2 and 4, and S2), which is consistent with our previous reports [20–23]. Interestingly, treatment with STZ alone slightly induced hepatocyte apoptosis but synergistically enhanced by IFN-γ. Such synergistic induction of hepatocyte apoptosis may be due to synergistic induction of STAT1 protein by IFN-γ/STZ (S2). Our data show that STZ-induced hyperglycemia is also associated with the induction of diabetes and hepatic injury, but highly attenuated by STAT1 or IFN-γ depletion using STAT1−/− or IFN-γ−/− mice (Figs. 2 and 3). Also, STZ did not directly induce STAT1 phosphorylation and its downstream IRF-1 induction in hepatocytes, but STZ alone increased STAT1 and ATF3 protein (Fig. 3B, C). So, it is likely that IFN-γ produced in STZ-treated mice directly involved in STAT1 activation and STAT1-mediated liver injury rather than STZ direct response to hepatocyte. In fact, Fig. 3D–G using IFN-γ−/− mice show that STZ-induced diabetic liver injury might be mediated by IFN-γ produced in vivo, thereby upregulate STAT1 and ATF3, results in liver injury. Therefore, ATF3 induced by IFN-γ and/or STZ may contribute to the synergistic induction of STAT1 protein by IFN-γ/STZ (see below).
4.2. ATF3 plays an important role in stabilizing STAT1 protein and contributes to STZ-induced diabetic liver injury
Although it was well-established that the mechanism by which STAT1 mediates cell death appears to involve either transcription-dependent or -independent process [37], the relevant upstream regulators or downstream target molecules of STAT1 remain poorly understood. In this paper, we identified that ATF3 is an important factor to stabilize STAT1 protein and plays an important role in induction of hepatocyte apoptosis. Previous reports showed that IFN-γ induction of ATF3 in monocytes and macrophages is STAT1-dependent [32]. However, our findings here suggest that STAT1 is not required for ATF3 induction in hepatocytes as STZ induction of ATF3 in the liver was not abolished in STAT1−/− mice and its hepatocytes, suggesting that STAT1-mediated induction of ATF3 is cell type dependent. In contrast, we provided evidence suggesting that ATF3 may act as a novel upstream regulator to stabilize STAT1 protein in hepatocytes. First, STAT1 protein was significantly increased in ATF3-transfected cells. Second, IFN-γ induction of STAT1 protein was almost completely abolished by transfection with inactive ATF3(ΔC) (Fig. 5) or ATF3 siRNA (S3). Furthermore, IFN-γ produced by STZ-injected mice may play an upstream regulator of ATF3 in liver tissues since STZ-induced ATF3 protein was completely abolished in IFN-γ−/− mice or anti-IFN-γ-injected mice. These results indicated that STZ produced IFN-γ, which was associated with the induction of ATF3, and IFN-γ-induced ATF3 may be involved in STAT1-mediated liver injury via STAT1 activation and its stabilization.
It has been shown that after activation, STAT1 signaling is tightly controlled by several inhibitory factors [33] including (1) suppressor of cytokine signaling (SOCS) that inhibits cytokine-associated JAKs, (2) protein inhibitor of STAT (PIAS) families of proteins that inhibit activated STAT molecule, (3) tyrosine phosphatases that dephosphorylate JAKs or STATs. However, these inhibitory pathways only negatively regulate the phosphorylation of JAKs or STATs but do not affect STAT1 protein. Increasing evidence suggests that posttranslational modifications of STAT1 proteins play an important role in regulating STAT1 expression. Among them, polyubiquitination of STAT1 protein and subsequent degradation by the 26S proteasome have been shown to be an important factor to degrade STAT1 protein [33]. Here we showed that ATF3 inhibited the proteolytic activities of 20S proteasome and inhibited the ubiquitination of STAT1 protein in vivo and in vitro, strongly suggesting that ATF3 stabilizes STAT1 protein via inhibiting STAT1 ubiquitination and subsequent degradation. Further studies indicate that a C-terminal domain of ATF3 was required for the interaction with STAT1 and inhibition of STAT1 ubiquitination and proteasome activity induced by ATF3 (S3). At present, although the precise molecular mechanism underlying the ATF3 inhibition of STAT1 ubiquitination remains unknown, it is likely that ATF3 may inhibit STAT1 ubiquitination through binding to the STAT1 domain harboring most of the lysine residues targeted for ubiquitination [38].
In summary, we demonstrated that during STZ-mediated diabetic liver injury, elevated ATF3 interacted directly with STAT1 and enhanced its stability, and therefore it may serve as a liver injury-inducing factor via stimulation of the apoptotic functions of STAT1. Also, STAT1 is a common signaling pathway contributing to STZ-induced diabetes and diabetic liver injury, which may provide a novel therapeutic target to treat type-I diabetes and its associated liver injury.
Supplementary Material
Acknowledgments
We thank Dr. Bin Gao for planning and peer reviewing this paper.
Abbreviations
- STAT1
signal transducer and activator of transcription factor 1
- STZ
streptozotocine
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
Footnotes
Grants: This study was supported by research grants from the Korean National Institutes of Health (4845-300-210-13).
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.cellsig.2009.07.011.
References
- 1.El-Serag HB, Tran T, Everhart JE. Gastroenterology. 2004;126:460. doi: 10.1053/j.gastro.2003.10.065. [DOI] [PubMed] [Google Scholar]
- 2.Davila JA, Morgan RO, Shaib Y, McGlynn KA, El-Serag HB. Gut. 2005;54:533. doi: 10.1136/gut.2004.052167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fracanzani AL, Valenti L, Bugianesi E, Andreoletti M, Colli A, Vanni E, Bertelli C, Fatta E, Bignamini D, Marchesini G, Fragion S. Hepatology. 2008;48:792. doi: 10.1002/hep.22429. [DOI] [PubMed] [Google Scholar]
- 4.Veldt BJ, Chen W, Heathcote EJ, Wedemeyer H, Reichen J, Hofmann WP, de knegt RJ, Zeuzem S, Manns MP, Hansen BE, Schalm SW, Janssen HL. Hepatology. 2008;47:1856. doi: 10.1002/hep.22251. [DOI] [PubMed] [Google Scholar]
- 5.Vuppalanchi R, Chalasani N. Hepatology. 2009;49:306. doi: 10.1002/hep.22603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ota T, Takamura T, Kurita S, Matsuzawa N, Kita Y, Uno M, Akahori H, Misu H, Sakurai M, Zen Y, Nakanuma Y, Kaneko S. Gastroenterology. 2007;132:282. doi: 10.1053/j.gastro.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 7.Campos GM, Bambha K, Vittinghoff E, Rabl C, Posselt AM, Ciovica R, Tiwari U, Ferrel L, Pabst M, Bass NM, Merriman RB. Hepatology. 2008;47:1916. doi: 10.1002/hep.22241. [DOI] [PubMed] [Google Scholar]
- 8.Mofrad P, Contos MJ, Haque M, Sargeant C, Fisher RA, Luketic VA, Sterling RK, Shiffman ML, Stravitz RT, Sanyal AJ. Hepatology. 2003;37:1286. doi: 10.1053/jhep.2003.50229. [DOI] [PubMed] [Google Scholar]
- 9.Shoelson SE, Herrero L, Naaz A. Gastroenterology. 2007;132:2169. doi: 10.1053/j.gastro.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 10.Maher JJ, Leon P, Ryan JC. Hepatology. 2008;48:670. doi: 10.1002/hep.22399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olsson R, Wesslau C, William-Olsson T, Zettergren L. J Clin Gastroenterol. 1989;11:541. doi: 10.1097/00004836-198910000-00010. [DOI] [PubMed] [Google Scholar]
- 12.Torbenson M, Chen YY, Brunt E, Cummings OW, Gottfried M, Jakate S, Liu YC, Yeh MM, Ferrell L. Am J Surg Pathol. 2006;30:508. doi: 10.1097/00000478-200604000-00012. [DOI] [PubMed] [Google Scholar]
- 13.Like AA, Rossini AA. Science. 1976;193:415. doi: 10.1126/science.180605. [DOI] [PubMed] [Google Scholar]
- 14.Flodstrom M, Tyrberg B, Eizirik DL, Sandler S. Diabetes. 1999;48:706. doi: 10.2337/diabetes.48.4.706. [DOI] [PubMed] [Google Scholar]
- 15.Guven A, Yavuz O, Cam M, Ercan F, Bukan N, Comunoglu C, Gokce F. Acta Histochem. 2006;108:85. doi: 10.1016/j.acthis.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Gezginci-Oktayoglu S, Basaraner H, Yanardag R, Bolkent S. Dig Dis Sci. 2009;54:538. doi: 10.1007/s10620-008-0381-0. [DOI] [PubMed] [Google Scholar]
- 17.Yanardag R, Ozsoy-Sacan O, Bolkent S, Orak H, Karabulut-Bulan O. Hum Exp Toxicol. 2005;24:129. doi: 10.1191/0960327104ht507oa. [DOI] [PubMed] [Google Scholar]
- 18.Gysemans CA, Ladriere L, Callewaert H, Rasschaert J, Flamez D, Levy DE, Matthys P, Eizirik DL, Mathieu C. Diabetes. 2005;54:2396. doi: 10.2337/diabetes.54.8.2396. [DOI] [PubMed] [Google Scholar]
- 19.Callewaert HI, Gysemans CA, Ladriere L, D'Hertog W, Hagenbrock J, Overbergh L, Eizirik DL, Mathieu C. Diabetes. 2007;56:2169. doi: 10.2337/db07-0052. [DOI] [PubMed] [Google Scholar]
- 20.Hong F, Jaruga B, Kim WH, Radaeva S, El-Assal ON, Tian Z, Nguyen VA, Gao B. J Clin Invest. 2002;110:1503. doi: 10.1172/JCI15841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim WH, Hong F, Radaeva S, Jaruga B, Fan S, Gao B. Am J Physiol: Gasterointest Liver Physiol. 2003;285:G761. doi: 10.1152/ajpgi.00224.2003. [DOI] [PubMed] [Google Scholar]
- 22.Lee HJ, Oh YK, Rhee M, Lim JY, Hwang JY, Park YS, Kwon Y, Choi KH, Jo I, Park SI, Gao B, Kim WH. J Mol Biol. 2007;369:967. doi: 10.1016/j.jmb.2007.03.072. [DOI] [PubMed] [Google Scholar]
- 23.Sun R, Park O, Horiguchi N, Kulkarni S, Jeong WI, Sun HY, Radaeva S, Gao B. Hepatology. 2006;44:955. doi: 10.1002/hep.21344. [DOI] [PubMed] [Google Scholar]
- 24.Liang G, Wolfgang CD, Chen BP, Chen TH, Hai T. J Biol Chem. 1996;271:1695. doi: 10.1074/jbc.271.3.1695. [DOI] [PubMed] [Google Scholar]
- 25.Yan C, Lu D, Hai T, D D. EMBO J. 2005;24:2425. doi: 10.1038/sj.emboj.7600712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nobori K, Ito H, Tamamori-Adachi M, Adachi S, Ono Y, Kawauchi J, Kitajima S, Marumo F, Isobe M. J Mol Cell Cardiol. 2002;34:1387. doi: 10.1006/jmcc.2002.2091. [DOI] [PubMed] [Google Scholar]
- 27.Chen BP, Liang G, Whelan J, Hai T. J Biol Chem. 1994;269:15819. [PubMed] [Google Scholar]
- 28.Wolfgang CD, Chen BP, Martindale JL, Holbrook NJ, Hai T. Mol Cell Biol. 1997;17:6700. doi: 10.1128/mcb.17.11.6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim HB, Kong M, Kim TM, Suh YH, Kim WH, Lim JH, Song JH, Jung MH. Diabetes. 2006;55:1342. doi: 10.2337/db05-1507. [DOI] [PubMed] [Google Scholar]
- 30.Lee JW, Kim WH, Lim JH, Song EH, Song J, Choi KY, H M. Cell Signal. 2009;21:69. doi: 10.1016/j.cellsig.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 31.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Proc Natl Acad Sci U S A. 1999;96(18):10403. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ho HH, Antoniv TT, Ji JD, Ivashkiv LB. J Immunol. 2008;181:5089. doi: 10.4049/jimmunol.181.7.5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shuai K, Liu B. Nat Rev Immunol. 2003;3:900. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 34.Wong FS, Siew LK, Wen L. Biochem Soc Trans. 2008;36:316. doi: 10.1042/BST0360316. [DOI] [PubMed] [Google Scholar]
- 35.Kay TW, Thomas HE, Harrison LC, Allison J. Trends Endocrinol Metab. 2000;11:11. doi: 10.1016/s1043-2760(99)00210-6. [DOI] [PubMed] [Google Scholar]
- 36.Cockfield SM, Ramassar V, Urmson J, Halloran PF. J Immunol. 1989;142:1120. [PubMed] [Google Scholar]
- 37.Kim HS, Lee MS. Cell Signal. 2007;19:454. doi: 10.1016/j.cellsig.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Ungureanu D, Silvennoinen O. Sci STKE. 2005;304:49. doi: 10.1126/stke.3042005pe49. [DOI] [PubMed] [Google Scholar]
- 39.Kim WH, Lee JW, Gao B, Jung MH. Cell Signal. 2005;17:1516. doi: 10.1016/j.cellsig.2005.03.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.