

Abstract
Background
Sugarcane (Saccharum officinarum L.) is an important sugar crop which belongs to the grass family and can be used for fuel ethanol production. The growing demands for sugar and biofuel is asking for breeding a sugarcane variety that can shed their leaves during the maturity time due to the increasing cost on sugarcane harvest.
Results
To determine leaf abscission related genes in sugarcane, we generated 524,328,950 paired reads with RNA-Seq and profiled the transcriptome of new born leaves of leaf abscission sugarcane varieties (Q1 and T) and leaf packaging sugarcane varieties (Q2 and B). Initially, 275,018 transcripts were assembled with N50 of 1,177 bp. Next, the transcriptome was annotated by mapping them to NR, UniProtKB/Swiss-Prot, Gene Ontology and KEGG pathway databases. Further, we used TransDecoder and Trinotate to obtain the likely proteins and annotate them in terms of known proteins, protein domains, signal peptides, transmembrane regions and rRNA transcripts. Different expression analysis showed 1,202 transcripts were up regulated in leaf abscission sugarcane varieties, relatively to the leaf packaging sugarcane varieties. Functional analysis told us 62, 38 and 10 upregulated transcripts were involved in plant-pathogen interaction, response to stress and abscisic acid associated pathways, respectively. The upregulation of transcripts encoding 4 disease resistance proteins (RPM1, RPP13, RGA2, and RGA4), 6 ABC transporter G family members and 16 transcription factors including WRK33 and heat stress transcription factors indicate they may be used as candidate genes for sugarcane breeding. The expression levels of transcripts were validated by qRT-PCR. In addition, we characterized 3,722 SNPs between leaf abscission and leaf packaging sugarcane plants.
Conclusion
Our results showed leaf abscission associated genes in sugarcane during the maturity period. The output of this study provides a valuable resource for future genetic and genomic studies in sugarcane.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2552-2) contains supplementary material, which is available to authorized users.
Keywords: Leaf abscission, Leaf shedding, Sugarcane, Plant-pathogen interaction, Abscisic acid, ABA, Saccharum officinarum, Transcriptome
Background
Sugarcane (Saccharum officinarum L.) is an important sugar crop, which is widely grown in the tropical and subtropical areas [1]. The total sugarcane acreage all over the world is more than 200 million hectares, yielding over 13 billion tons of sugar per year [2]. Apart of this, sugarcane is an ideal non-food biomass crop which can produce two kinds of biofuel: ethanol and high biomass raw sugar [3]. In China, sugarcane is mainly distributed in the provinces of Guangxi, Yunnan, Guangdong and Hainan, of them Guangxi province produces more than 60 % of the total sugar per year. The sugarcane harvest mainly relies on hand-cut in several countries like China, large-scale mechanized harvest is less than 15 % of the total amount, and the cost of sugarcane is increasing. Hence, it is important to study the mechanism of leaf abscission in sugarcane and breed sugarcane varieties which can shed their leaves during the maturity time.
Abscission is the programmed developmental process by which some of the organs such as leaves, flowers, or fruits are shed during the life of a plant [4]. It occurs within a specific tissue, called abscission zone (AZ), which is formed at the base of the petiole [5]. The abscission process can be divided into four major steps [6]: development of the AZ tissue (step 1), acquisition of competence to respond to abscission-promoting signaling (step 2), activation of abscission (step 3), and post abscission trans-differentiation (step 4). Steps 2 and 3 have been extensively investigated, by transcriptome analyses, in the gene expression changes during the shedding of AZs tissues, such as leaves, flowers and fruits [7–12]. It is interesting that different plant species and different organs share a majority of genes involved in steps 2 and 3, including genes involved in ethylene and auxin biosynthesis and signal transduction, cell wall modification and various stress responses [12].
Although some genes have been found to regulate the development of AZs tissues (step 1), they are different across species and organs. In Arabidopsis, the formation of seed AZs is regulated by the MADS-box transcription factor (TF) gene SEEDSTICK (STK) and the bHLH transcription factor gene HECATE3 (HEC3) [13, 14], but the formation of Arabidopsis floral organ AZs is regulated by BLADE-ON-PETIOLE1 (BOP1) and BOP2 which encode BTB/POZ domain and Ankyrin repeat containing NPR1-like proteins [5]. In tomato, MACROCALYX and JOINTLESS containing a MADS-box controls the fruit and flower AZ development [12, 15]. In rice, several genes and their products have been reported to regulate the pedicel AZ formation for seed shattering, including qSH1 (a major chromosome 1 quantitative trait locus for seed shattering, encoding a BELL-type homeobox TF) [16], SH4 (a major chromosome 4 seed shattering quantitative trait locus, encoding a MYB3 DNA-binding domain containing protein) [17], HATTERING ABORTION1 (SHAT1, encoding an AP2 family TF) [18], the rice SHATTERING1 homologue (OsSH1, encoding a YAB family TF) [19], and CTD phosphatase-like protein1 (OsCPL1) [20]. Overall, AZ development associated genes vary from one to another plant species. In sugarcane, genes controlling leaf AZ development and leaf abscission are still unknown.
RNA-Seq, a next generation sequencing technology, is used for profiling gene expression and plant breeding programs in many plants including rice [21], maize [22] and millet [23]. The genome sequence of sugarcane is not available currently, so transcriptome studies in sugarcane have been proposed, in progress or accomplished in different countries like South Africa [24, 25], Australia2 [26, 27] and USA [28]. Here, we performed an RNA-Seq study to profile the gene expression of new born leaf tissues using the HiSeq 2000 platform. Differential expression analysis revealed 1,202 transcripts upregulated in leaf abscission sugarcane plants (LASP) which can shed their leaves during the maturity time, compared to the leaf packaging sugarcane plants (LPSP) which are packed by the leaves during the maturity time. Functional analysis showed the upregulated transcripts in LASP were enriched in “plant-pathogen interaction”, “response to stress” and abscisic acid (ABA) associated pathways. Due to their up regulation, we assumed these transcripts may involve in the processes of AZ development and leaf abscission in sugarcane. This is the first time to study the genes associated with AZ development and leaf abscission in sugarcane. Our results will provide a valuable resource for understanding the mechanism of leaf abscission in sugarcane and will contribute to the researchers in the field of sugarcane breeding.
Results and discussion
Deep sequencing and de novo assembly
In order to understand the mechanism of leaf abscission in sugarcane, six transcriptome libraries for two parent sugarcane varieties (Q1 and Q2), two F1 generation sugarcane varieties (T1 and T2), which can shed their leaves during the maturity time, and another two F1 generation sugarcanes (B1 and B2), which are packed by the leaves during the maturity, were constructed and sequenced. As shown in Table 1, by using the HiSeq 2000 platform we generated a total of 524,328,950 paired raw reads for six libraries. The Q20 values of six libraries were from 97.58 % to 97.96 %. After removing low quality reads and reads with adaptors, we obtained ~76.9 M, ~83.2 M, ~81.4 M, ~77.2 M, ~78.6 M and ~80.9 M clean reads for B1, B2, Q1, Q2, T1 and T2, respectively. As the estimated genome sizes of S. officinarum accessions ranged from 7.50 to 8.55 Gb with an average size of 7.88 Gb [29], our data equivalent to ~10-fold coverage of the sugarcane genome sequences. The clean reads (478.1 M) were then used for de novo assembly analysis by Trinity software [30], resulting a total of 275,018 unique transcripts corresponding to 164,803 genes. The GC percentage of the assembled transcripts was 48.23 %, the N50 statistic was 1,177 which represented at least 50 % of the sum of the lengths of all contigs include contigs that were at least 1,177 bp long, and the distribution of contig length can be seen in Fig. 1. Approximately 40 % of the assembled contigs were 200 ~ 400 bp in length, however, we obtained 67,238 contigs longer than 1,000 bp. The longest contig was 15,553 bp in length and a total of 6,234 contigs were longer than 3,000 bp. The number of total assembled bases was 215,019,578, which meant about 215 M size of mRNA sequences were characterized in this study.
Table 1.
Overview of transcriptome sequencing and de novo assembl results
| B1 | B2 | Q1 | Q2 | T1 | T2 | |
|---|---|---|---|---|---|---|
| Raw reads | 83,879,424 | 92,222,556 | 89,370,426 | 89,370,426 | 85,447,812 | 88,421,042 |
| Q20 percentage | 97.96 % | 97.58 % | 97.77 % | 97.66 % | 97.94 % | 97.86 % |
| Clean reads | 76,869,838 | 83,161,522 | 81,439,136 | 77,208,082 | 78,574,934 | 80,853,072 |
| Total reads | 478,106,584 | |||||
| Total trinity ‘genes’ | 164,803 | |||||
| Total trinity transcripts | 275,018 | |||||
| Percent GC | 48.23 % | |||||
| ContigN10 | 3,155 | |||||
| ContigN50 | 1,177 | |||||
| Total assembled bases | 215,019,578 | |||||
Fig. 1.
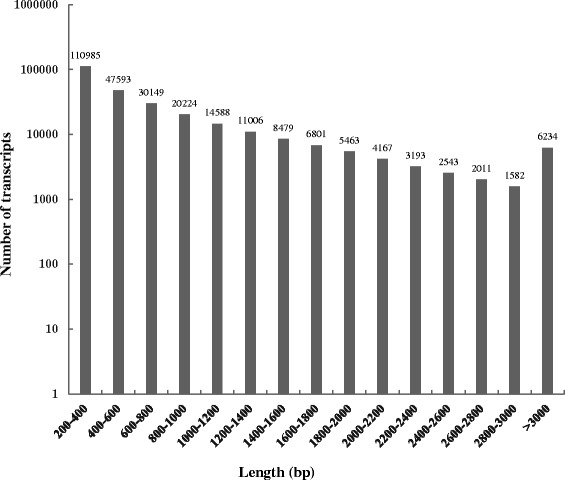
Length distribution of sugarcane leaf transcriptome assembled by Trinity
Functional annotation of assembled transcripts
To understand the features and functions of the assembled transcripts, we annotated the assembled transcripts by mapping them to several public databases, like NCBI non-redundant (NR), UniProtKB/Swiss-Prot, GO and KEGG pathway. The numbers of transcripts aligned to each database can be seen in Fig. 2a. A total of 110,486 (40.17 %) transcripts were annotated, of which 110,039 (40.01 %) were mapping to NR database under the e-value of 1 × 10−5. As expected, in the NR mapping results we found 64,902 (58.98 %), 25,410 (23.09 %), 9,433 (8.57 %) and 2,606 (2.37 %) assembled transcripts were aligned to Sorghum bicolor, Zea mays, Oryza sativa Japonica Group and Oryza sativa Indica Group, respectively (Fig. 2b, Additional file 1). Due to the unavailability of sugarcane genome and gene sequences in public databases, we found only 550 transcripts were mapping to Saccharum species. GO analysis is an international standardized gene function classification system that provides a controlled vocabulary to facilitate high-quality functional gene annotation [31, 32]. GO term distribution (Fig. 2c) of the assembled transcripts showed the “cellular process” and “metabolic process” were two most abundantly represented with 24,907 (53.32 %) and 27,955 (59.85 %) transcripts, respectively. In the “cellular components” ontology transcripts were mainly distributed in “cell” (36,954, 79.12 %), “cell part” (36,954, 79.12 %) and “organelle” (30,693, 65.71 %). GO analysis also showed 33,849 (72.47 %) transcripts had “binding” function and 30,060 (64.36 %) with “catalytic activity” function in “molecular function” ontology. Detailed information can be found in Additional file 2. In addition, some transcripts (59.83 %) showed no similarity to any known protein database, there were probabilities that they were putative long noncoding RNAs or novel genes in sugarcane [33]. More experiments are required to characterize them [34, 35].
Fig. 2.
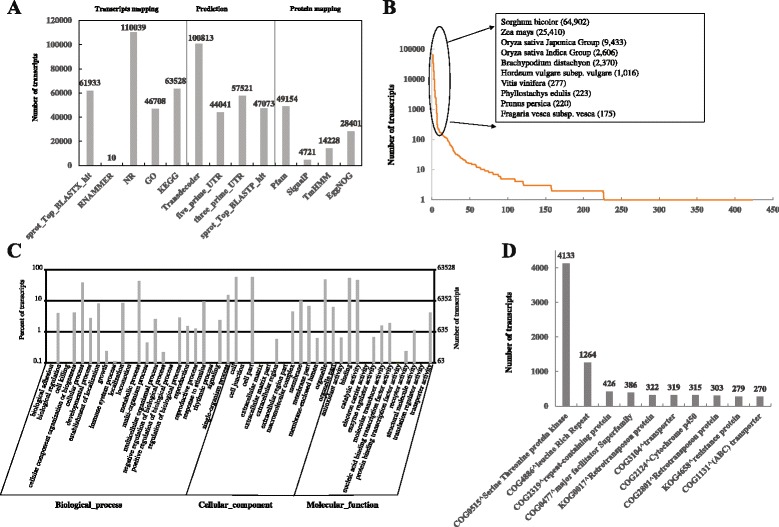
Functional annotation for the assembled transcripts. a The numbers of transcripts or putative proteins mapping to public databases or annotated based on the conservation. b Distribution of species aligned by the assembled transcripts. c Gene Ontology annotation for the assembled transcriptome. d Top 10 COGs annotations
Then, we extracted likely coding sequences from the assembled transcripts using TransDecoder. In total, 100,813 likely protein sequences, 44,041 5’-prime-UTRs and 57,521 3’-prime-UTRs (Fig. 2a) were obtained. By using the Trinotate pipeline likely protein sequences were annotated in terms of known proteins, protein domains, signal peptides, transmembrane regions and rRNA transcripts (Fig. 2a). It showed 47,073 (46.69 %) of total likely protein sequences were aligned to UniProtKB/Swiss-Prot under e-values of 1e-5 by BLASTX, 49,154 (48.76 %) protein sequences were characterized to have protein domains of Pfam, 4,721 (4.68 %) potential signal proteins were identified by SignalP [36] and 14,228 (14.11 %) proteins were found with high similarity to membrane proteins by TMHMM Sever v2.0 [37]. In addition, 10 transcripts were identified as ribosomal rRNAs, 28,401 protein sequences were aligned to EggNOG database (v4.1) [38] resulting 1,363 COGs, 43 KOGs, 21 euNOGs and 333 NOGs. According to the numbers of transcripts mapping to the EggNOG groups, top 10 were shown in Fig. 2d. Likely protein sequences of sugarcane leaves were annotated with “threonine protein kinase” (4,133 transcripts) which had more than three times of the second “leucine rich repeat” (1264 transcripts). The threonine protein kinase has been reported to play a key role in the regulation of cell proliferation, cell differentiation and cell death [39, 40]. These annotations for the assembled transcripts including gene and protein description, putative conserved domains and potential biological pathways provided a valuable resource for subsequent investigation of specific biological processes, functions and pathways in cell death and sugarcane leaf shedding.
Transcriptome profile
We next aligned the clean reads of all six samples to the assembled transcripts using Bowtie2 [41] and estimated the abundance of each transcript using RSEM (RNA-Seq by Expectation-Maximization) [42]. According to the phenotypes of F1 generation sugarcane varieties, B1 and B2 were considered as B, T1 and T2 samples were considered as T in this study. As shown in Fig. 3a, we detected a total of 198,816, 189,635, 229,117 and 227,900 transcripts in Q1, Q2, T and B, respectively. There were 143,021 transcripts common to all the samples. It is notable that 11,988, 3955, 10,035 and 8400 transcripts were exclusively detected in Q1, Q2, T and B, respectively. KEGG pathway analysis showed specifically expressed transcripts were enriched in different biological pathways. For example, transcripts unique to B were mainly enriched in “ribosome” (ko03011, 34 transcripts), “polycyclic aromatic hydrocarbon degradation” (ko00624, 12 transcripts), and “stilbenoid, diarylheptanoid and gingerol biosynthesis” (ko00945, 15 transcripts). Transcripts exclusively detected in T might involve in “plant-pathogen interaction” (ko04626, 94 transcripts), “NOD-like receptor signaling pathway” (ko04621, 12 transcripts), and “antigen processing and presentation” (ko04612, 16 transcripts). Different KEGG pathways for transcripts detected only in B and T indicated they may have differences on their genetic information and phenotypes.
Fig. 3.
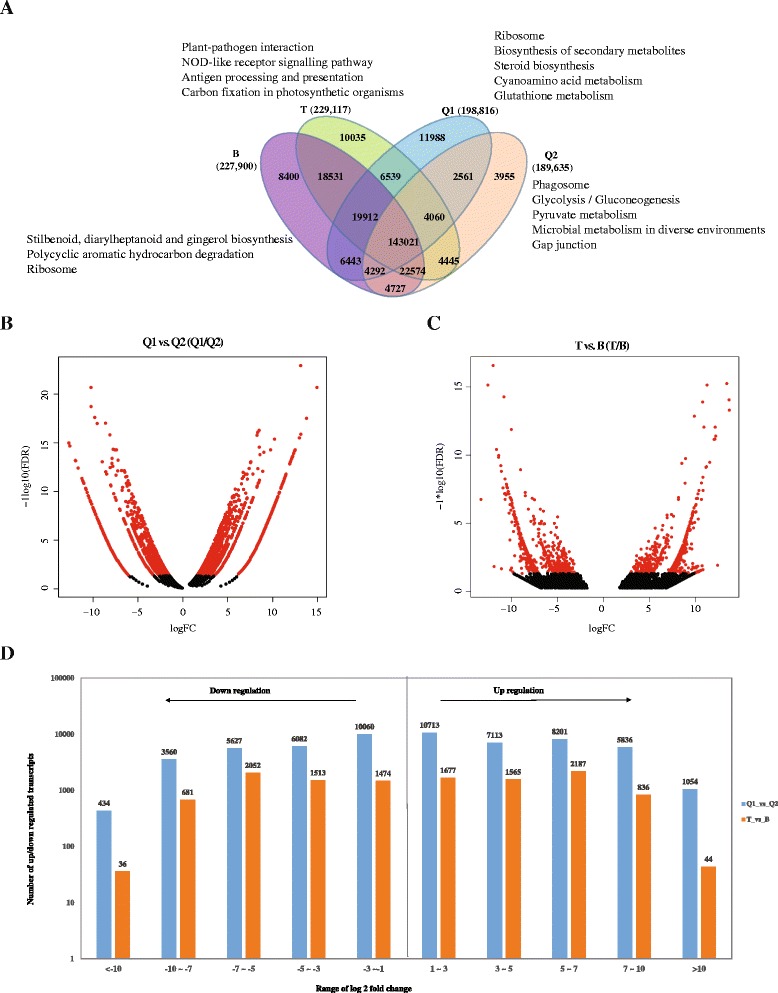
Transcriptome profile and different expression. a Venn diagram of transcripts detected in Q1, Q2, T and B. b Volcano plot of differentially expressed transcripts between Q1 and Q2. c Volcano plot of differentially expressed transcripts between T and B. d Numbers of up- and down-regulated transcripts identified in Q1 vs. Q2 and T vs. B
Differentially expressed transcripts
We next used edgeR [43] to identify differentially expressed transcripts in LASP (Q1 and T), compared to LPSP (Q2 and B). As described in Material and Methods, we filtered the transcripts by their fold changes (>2) and p-values (<0.05). By using this critical, differentially expressed transcripts were shown in red in the volcano plots of Fig. 3b and c. In total, we detected 32,917 transcripts upregulated and 25,764 transcripts down regulated in Q1 sugarcane in comparison of Q2 sugarcane (Fig. 3b), and 6,309 upregulated and 5,757 down regulated transcripts in T sugarcane relatively to B sugarcane (Fig. 3c). The numbers of up and down regulated transcripts in different ranges can be found in Fig. 3d. Notably, 1,054 and 44 transcripts were upregulated very significantly (log2FC >10) in Q1 and T in comparison of Q1 and B, respectively. In addition, we found Q1 and T shared a total of 1,202 upregulated and 953 down regulated transcripts (Additional file 3). GO analysis of the commonly upregulated transcripts (Table 2) showed 38, 35 and 15 transcripts were enriched in “response to stress”, “transition metal ion binding” and “cellular protein modification process”, respectively. KEGG pathway analysis (Table 3) showed commonly upregulated transcripts were enriched in the pathways of “plant-pathogen interaction”, “one carbon pool by folate” and “diterpenoid biosynthesis”.
Table 2.
GO analysis for the commonly up regulated transcripts
| Gene Ontology | GO_item | Transcript_number | P-value | Q-value |
|---|---|---|---|---|
| Cellular component | intracellular part | 9 | 2.43E-02 | 4.38E-01 |
| Molecular function | transition metal ion binding | 35 | 1.02E-05 | 8.43E-04 |
| iron ion binding | 9 | 1.41E-05 | 1.17E-03 | |
| amine transmembrane transporter activity | 1 | 1.08E-04 | 8.96E-03 | |
| nucleic acid binding transcription factor activity | 6 | 6.72E-04 | 5.58E-02 | |
| Biological process | cellular homeostasis | 3 | 5.30E-13 | 4.19E-11 |
| organic substance transport | 5 | 1.05E-09 | 8.32E-08 | |
| establishment of localization | 2 | 3.04E-07 | 2.40E-05 | |
| response to stress | 38 | 1.08E-06 | 8.50E-05 | |
| amino acid transport | 1 | 7.57E-06 | 5.98E-04 | |
| DNA metabolic process | 5 | 1.21E-05 | 9.53E-04 | |
| translation | 2 | 1.16E-04 | 9.14E-03 | |
| purine nucleoside monophosphate biosynthetic process | 1 | 3.29E-04 | 2.60E-02 | |
| cell morphogenesis | 1 | 3.62E-04 | 2.86E-02 | |
| cellular protein modification process | 15 | 7.43E-04 | 5.87E-02 | |
| transport | 3 | 1.00E-03 | 7.91E-02 | |
| proteasomal ubiquitin-dependent protein catabolic process | 2 | 1.59E-03 | 1.26E-01 | |
| cell wall biogenesis | 2 | 1.94E-03 | 1.54E-01 | |
| vitamin K biosynthetic process | 1 | 4.69E-03 | 3.71E-01 | |
| primary metabolic process | 4 | 5.85E-03 | 4.62E-01 | |
| one-carbon metabolic process | 7 | 6.35E-03 | 5.02E-01 | |
| branched-chain amino acid metabolic process | 2 | 6.99E-03 | 5.52E-01 | |
| response to metal ion | 4 | 7.13E-03 | 5.63E-01 | |
| polysaccharide metabolic process | 1 | 7.30E-03 | 5.76E-01 | |
| glucose catabolic process | 3 | 1.08E-02 | 8.55E-01 |
Table 3.
Significant KEGG pathways of commonly up-regulated transcripts in Q1 vs. Q2 and T vs. B
| Pathway | ID | Transcritps_number | P-value | Q-value |
|---|---|---|---|---|
| Plant-pathogen interaction | ko04626 | 62 | 1.49E-09 | 2.68E-07 |
| One carbon pool by folate | ko00670 | 20 | 5.73E-06 | 1.03E-03 |
| Diterpenoid biosynthesis | ko00904 | 8 | 4.39E-05 | 7.90E-03 |
| Homologous recombination | ko03440 | 20 | 1.18E-04 | 2.13E-02 |
| Epstein-Barr virus infection | ko05169 | 24 | 3.37E-04 | 6.07E-02 |
| Cytosolic DNA-sensing pathway | ko04623 | 17 | 4.01E-04 | 7.22E-02 |
| Two-component system | ko02020 | 18 | 5.82E-04 | 1.05E-01 |
| Nitrogen metabolism | ko00910 | 10 | 9.44E-04 | 1.70E-01 |
| Drug metabolism - cytochrome P450 | ko00982 | 10 | 1.21E-03 | 2.17E-01 |
| Histidine metabolism | ko00340 | 6 | 1.38E-03 | 2.49E-01 |
| Chloroalkane and chloroalkene degradation | ko00625 | 9 | 1.99E-03 | 3.58E-01 |
| RNA polymerase | ko03020 | 17 | 2.59E-03 | 4.65E-01 |
| Metabolism of xenobiotics by cytochrome P450 | ko00980 | 9 | 2.76E-03 | 4.97E-01 |
| Naphthalene degradation | ko00626 | 7 | 4.81E-03 | 8.66E-01 |
We were surprised that Q1-up-regulated transcripts and T-up-regulated transcripts were enriched in different KEGG pathways (Additional file 4 and Additional file 5), indicating they might have different regulatory pathways of leaf abscission during the maturity time. Transcripts upregulated in Q1 (compared to Q2) were enriched in the auxin related pathways, like “flavonoid biosynthesis” (151 transcripts, p-value: 2.20E-16), “limonene and pinene degradation” (427 transcripts, p-value: 2.20E-16), “plant hormone signal transduction” (606 transcripts, p-value: 2.20E-16) and “phenylpropanoid biosynthesis” (343 transcripts, p-value: 2.20E-16). However, transcripts upregulated in T (compared to B) were enriched in the pathways of “plant-pathogen interaction” (210 transcripts, p-value: 2.20E-16), “one carbon pool by folate” (59 transcripts, p-value: 1.96E-11) and “homologous recombination” (64 transcripts, p-value: 7.24E-10). As we know, internal and environmental signals can influence and proceed the leaf senescence and death, including abiotic factors like drought, nutrient limitation, extreme temperature, and oxidative stress by UV-B (ultraviolet B) irradiation and ozone [44, 45]. It is inferred that leaf abscission of sugarcane is linked to at least one of these pathways.
Transcripts associated with leaf abscission
The leaf abscission mechanism in sugarcane is still unknown, however, genes associated with hormonal regulation, stress and diseases has been studied to regulate the process of leaf abscission in other plants [46–49]. In this study, we analyzed the commonly upregulated transcripts in LASP in comparison of LPSP and found genes related to leaf abscission in sugarcane, which involved in plant-pathogen interaction, responses to stress and ABA-associated pathways.
Plant-pathogen interactions
First, we analyzed the transcripts of “plant-pathogen interaction” because it was the most significant KEGG pathway of commonly upregulated transcripts. Unlike in animals, pathogen interactions often trigger programmed cell death in plants [50–52]. Among the commonly upregulated transcripts, 62 were annotated to regulate the pathway of plant-pathogen interaction. Table 4 showed their log 2 values of fold changes, ranging from 1.45 to 11.45 in Q1/Q2 and 1.35 to 11.12 in T/B. Of these 62 transcripts, 36 (58.06 %) transcripts were annotated to encode disease resistance proteins, including 7 RPM1 (resistance to Pseudomonas syringae pv. Maculicola 1), transcripts, 7 RPP13 (recognition of Peronospora parasitica 13) transcripts and 6 RPP13-like transcripts. Both RPM1 and RPP13 can trigger the plant defense process [53, 54]. In Arabidopsis, RPM1 acts through its interaction with RIN4 (RPM1-interacting protein 4), an essential regulator of plant defense, and triggers plant disease resistance when RIN4 is phosphorylated by AvrRpm1 [55, 56]. After infection of tomato yellow leaf curl virus, RPP13 is upregulated in a resistant tomato line [57]. In addition, a WRKY transcription factor called WRK33 was identified to regulate the plant-pathogen interaction. WRK33 can interact with the W box (5’-TTGAC[CT]-3’, an elicitor-responsive cis-acting element) [58] and function in ABA signaling [59]. In Arabidopsis, WRK33 is involved in regulation of the antagonistic relationship between defense pathways mediating responses to the bacterial pathogen P. syringae and the necrotrophic pathogen B. cinerea promoters [60]. The up regulation of transcripts encoding plant disease resistance proteins suggested that the defense system of sugarcane was activated by some reason and might contribute on shedding sugarcane leaves during the maturity time.
Table 4.
Transcripts involved in the pathway of plant-pathogene interaction
| Gene family | gene_id | log2FC(Q1/Q2) | PValue | log2FC(T/B) | PValue | Description |
|---|---|---|---|---|---|---|
| RX24L | c97780_g1_i3 | 3.228 | ####### | 6.523 | ####### | Probable disease resistance protein RXW24L |
| c105254_g1_i2 | 1.990 | ####### | 2.181 | ####### | Calcium-dependent protein kinase isoform 11 | |
| DRL | c101464_g2_i5 | 2.348 | ####### | 2.145 | ####### | Putative disease resistance protein At1g50180 |
| c77488_g1_i1 | 2.807 | ####### | 6.112 | ####### | ||
| c93620_g2_i1 | 5.716 | ####### | 5.264 | ####### | ||
| c97780_g3_i1 | 6.722 | ####### | 5.438 | ####### | ||
| c142188_g1_i1 | 3.527 | ####### | 4.753 | ####### | Probable disease resistance protein RDL6 | |
| ERECT | c105005_g2_i1 | 3.139 | ####### | 1.797 | ####### | LRR receptor-like serine/threonine-protein kinase ERECTA |
| FB95 | c82142_g1_i1 | 2.097 | ####### | 7.978 | ####### | F-box protein At2g02240 |
| FLS2 | c104719_g1_i2 | 6.103 | ####### | 2.347 | ####### | LRR receptor-like serine/threonine-protein kinase FLS2 |
| HSP82 | c108102_g3_i1 | 3.494 | ####### | 2.543 | ####### | Heat shock protein 82 |
| c76241_g1_i1 | 2.373 | ####### | 2.374 | ####### | ||
| LRC40 | c15793_g1_i1 | 5.998 | ####### | 5.839 | ####### | Leucine-rich repeat-containing protein 40 |
| LRK91 | c99649_g1_i3 | 5.808 | ####### | 5.880 | ####### | L-type lectin-domain containing receptor kinase IX.1 |
| MPK5 | c83394_g1_i1 | 4.061 | ####### | 1.615 | ####### | Mitogen-activated protein kinase 5 |
| R13L family | c105793_g1_i13 | 2.664 | ####### | 2.311 | ####### | Putative disease resistance RPP13-like protein 2 |
| c105793_g1_i8 | 8.150 | ####### | 4.113 | ####### | ||
| c105793_g1_i9 | 1.563 | ####### | 7.504 | ####### | ||
| c67555_g1_i1 | 3.908 | ####### | 2.860 | ####### | Putative disease resistance RPP13-like protein 3 | |
| c101028_g2_i1 | 9.403 | ####### | 8.190 | ####### | Disease resistance RPP13-like protein 4 | |
| c101028_g2_i5 | 8.861 | ####### | 6.967 | ####### | ||
| R1B | c104414_g3_i1 | 4.078 | ####### | 4.774 | ####### | Putative late blight resistance protein homolog R1B-12 |
| c82142_g2_i1 | 2.352 | ####### | 6.412 | ####### | ||
| c97780_g2_i3 | 2.354 | ####### | 6.259 | ####### | ||
| c105793_g1_i1 | 3.268 | ####### | 1.792 | ####### | Putative late blight resistance protein homolog R1B-16 | |
| c68709_g1_i1 | 3.122 | ####### | 3.125 | ####### | ||
| RGA | c91884_g5_i3 | 1.449 | ####### | 2.760 | ####### | Putative disease resistance protein RGA3 |
| c102886_g1_i4 | 5.643 | ####### | 2.903 | ####### | Putative disease resistance protein RGA4 | |
| c91884_g5_i4 | 2.167 | ####### | 2.247 | ####### | ||
| RP8L3 | c97780_g1_i1 | 7.405 | ####### | 6.223 | ####### | Disease resistance RPP8-like protein 3 |
| c97780_g1_i5 | 5.565 | ####### | 6.223 | ####### | ||
| RPM1 | c107814_g1_i1 | 2.025 | ####### | 1.350 | ####### | Disease resistance protein RPM1 |
| c108644_g1_i2 | 2.759 | ####### | 2.277 | ####### | ||
| c66665_g1_i1 | 9.403 | ####### | 7.371 | ####### | ||
| c68216_g1_i1 | 9.485 | ####### | 5.894 | ####### | ||
| c95380_g2_i1 | 8.265 | ####### | 9.699 | ####### | ||
| c95380_g2_i2 | 8.438 | ####### | 9.519 | ####### | ||
| c99461_g1_i2 | 2.318 | ####### | 3.144 | ####### | ||
| RPP | c104393_g1_i1 | 3.763 | ####### | 3.659 | ####### | Disease resistance protein RPP13 |
| c104393_g1_i3 | 3.376 | ####### | 5.385 | ####### | ||
| c104393_g1_i8 | 2.979 | ####### | 3.708 | ####### | ||
| c104393_g1_i9 | 2.827 | ####### | 2.926 | ####### | ||
| c107654_g2_i3 | 2.151 | ####### | 2.678 | ####### | ||
| c89572_g4_i3 | 1.611 | ####### | 3.859 | ####### | ||
| c97780_g5_i1 | 2.086 | ####### | 5.392 | ####### | ||
| RSLE2 | c99739_g2_i1 | 2.603 | ####### | 1.697 | ####### | Zinc finger BED domain-containing protein RICESLEEPER 2 |
| c99739_g2_i3 | 8.733 | ####### | 3.065 | ####### | ||
| c99739_g2_i7 | 9.022 | ####### | 2.909 | ####### | ||
| SD25 | c99065_g1_i9 | 6.315 | ####### | 5.297 | ####### | G-type lectin S-receptor-like serine/threonine-protein kinase SD2-5 |
| WAK1 | c48856_g1_i1 | 11.447 | ####### | 11.123 | ####### | Wall-associated receptor kinase 1 |
| WRK33 | c99739_g2_i8 | 6.689 | ####### | 3.657 | ####### | Probable WRKY transcription factor 33 |
| Y1571 | c103804_g1_i1 | 6.863 | ####### | 2.780 | ####### | Probable leucine-rich repeat receptor-like protein kinase At1g35710 |
| c103804_g1_i2 | 6.400 | ####### | 2.506 | ####### | ||
| Y3475 | c103043_g1_i3 | 4.158 | ####### | 3.636 | ####### | Probable LRR receptor-like serine/threonine-protein kinase At3g47570 |
| c104719_g1_i1 | 6.051 | ####### | 2.048 | ####### | ||
| Y4885 | c103804_g1_i3 | 6.967 | ####### | 3.236 | ####### | Probable LRR receptor-like serine/threonine-protein kinase At4g08850 |
| c105253_g3_i2 | 4.866 | ####### | 2.290 | ####### | ||
| gi|414591554|tpg|DAA42125.1| | c101028_g1_i1 | 7.789 | ####### | 6.349 | ####### | TPA: hypothetical protein ZEAMMB73_852544 [Zea mays] |
| gi|242077232|ref|XP_002448552.1| | c65099_g1_i1 | 3.245 | ####### | 2.407 | ####### | hypothetical protein SORBIDRAFT_06g028920 [Sorghum bicolor] |
| c65099_g1_i2 | 2.978 | ####### | 1.949 | ####### | ||
| gi|242047620|ref|XP_002461556.1| | c75847_g1_i2 | 5.808 | ####### | 6.270 | ####### | hypothetical protein SORBIDRAFT_02g004690 [Sorghum bicolor] |
| gi|115489000|ref|NP_001066987.1| | c91026_g3_i1 | 1.813 | ####### | 5.859 | ####### | Os12g0553200 [Oryza sativa Japonica Group] |
Stress responses
Several stresses can lead cell death and leaf abscission in plants, like drought [61, 62] and salt [63] stresses. Hence, we analyzed the transcripts involved in the response to stresses. Compared to LPSP, 38 transcripts were upregulated in LASP and annotated to respond the stresses (Table 5). Notably, 18 (47.37 %) transcripts of them had the ability of encoding disease resistance proteins, including 4 RGA2 (resistance gene analog 2) transcripts, 3 At1g50180 homolog transcripts and 2 RGA4 (resistance gene analog 4) transcripts. In addition, we identified 6 upregulated transcripts encoding the hypothetical protein (SORBIDRAFT_08g002290) in response to stresses. Although the function of SORBIDRAFT_08g002290 is not clear, several important motifs like leucine-rich repeats, AAA ATPase and NB-ARC domains are found in SORBIDRAFT_08g002290 [64]. It is said that proteins containing the short motif of leucine-rich repeats can regulate the interactions between proteins [65] and NB-ARC domain has been found in many resistance proteins [66]. These evidence told us that transcripts encoding proteins in response of stresses can act in the pathway of plant-pathogen interactions, and their over-expression indicated some relationship with leaf abscission in sugarcane.
Table 5.
Transcripts involved in the response of stress
| Gene family | gene_id | logFC(Q1/Q2) | PValue | logFC(T/B) | PValue | Description |
|---|---|---|---|---|---|---|
| DRL4 | c101464_g2_i5 | 2.348 | 2.01E-03 | 2.145 | 2.57E-02 | Putative disease resistance protein At1g50180 |
| c77488_g1_i1 | 2.807 | 2.91E-03 | 6.112 | 3.75E-03 | ||
| c97780_g3_i1 | 6.722 | 5.41E-11 | 5.438 | 4.32E-02 | ||
| DRL45 | c142188_g1_i1 | 3.527 | 6.40E-05 | 4.753 | 1.21E-02 | Probable disease resistance protein RDL6 |
| ESAG8 | c53232_g2_i1 | 1.943 | 1.03E-02 | 5.900 | 3.71E-02 | Putative adenylate cyclase regulatory protein |
| PER1 | c97626_g3_i1 | 5.639 | 2.59E-11 | 2.054 | 4.31E-02 | Cationic peroxidase SPC4 |
| c98359_g1_i4 | 9.281 | 1.47E-08 | 4.586 | 1.56E-02 | ||
| PPD4 | c108711_g1_i2 | 3.677 | 4.47E-07 | 2.058 | 4.54E-03 | PsbP domain-containing protein 4, chloroplastic |
| R1B12 | c97780_g2_i3 | 2.354 | 1.35E-02 | 6.259 | 2.52E-03 | Putative late blight resistance protein homolog R1B-12 |
| RGA2 | c108711_g2_i1 | 4.177 | 2.28E-08 | 2.425 | 1.46E-03 | Disease resistance protein RGA2 |
| c108711_g2_i2 | 3.511 | 1.23E-06 | 2.322 | 2.15E-03 | ||
| c71260_g1_i1 | 12.593 | 7.40E-18 | 2.771 | 4.90E-02 | ||
| c89143_g1_i1 | 8.593 | 7.45E-07 | 2.820 | 4.35E-02 | ||
| RGA4 | c102886_g1_i4 | 5.643 | 1.63E-06 | 2.903 | 2.82E-02 | Putative disease resistance protein RGA4 |
| c91884_g5_i4 | 2.167 | 1.72E-02 | 2.247 | 6.76E-03 | ||
| RPM1 | c107814_g1_i1 | 2.025 | 2.98E-03 | 1.350 | 4.78E-02 | Disease resistance protein RPM1 |
| c108644_g1_i2 | 2.759 | 2.86E-04 | 2.277 | 9.81E-03 | ||
| RPP13 | c107654_g2_i3 | 2.151 | 2.26E-03 | 2.678 | 3.36E-03 | Disease resistance protein RPP13 |
| c89572_g4_i3 | 1.611 | 3.32E-02 | 3.859 | 2.17E-03 | ||
| c97780_g5_i1 | 2.086 | 1.55E-02 | 5.392 | 2.94E-02 | ||
| RPP8 | c107654_g1_i3 | 2.435 | 1.44E-03 | 2.793 | 1.78E-02 | Disease resistance protein RPP8 |
| RSH3C | c74407_g1_i1 | 9.516 | 3.94E-09 | 3.288 | 1.93E-02 | Probable GTP diphosphokinase RSH3, chloroplastic |
| gi|218187380|gb|EEC69807.1| | c83558_g1_i2 | 2.199 | 1.68E-02 | 4.774 | 4.36E-06 | hypothetical protein OsI_00114 [Oryza sativa Indica Group] |
| gi|242061230|ref|XP_002451904.1| | c86140_g4_i1 | 6.894 | 1.41E-03 | 2.842 | 1.89E-02 | hypothetical protein SORBIDRAFT_04g009750 [Sorghum bicolor] |
| c86140_g4_i4 | 3.382 | 1.98E-02 | 5.202 | 3.32E-04 | ||
| gi|242082690|ref|XP_002441770.1| | c108670_g3_i2 | 8.228 | 5.19E-06 | 4.179 | 7.33E-03 | hypothetical protein SORBIDRAFT_08g002056 [Sorghum bicolor] |
| c87617_g7_i1 | 7.443 | 1.65E-04 | 7.436 | 1.33E-04 | ||
| gi|242082722|ref|XP_002441786.1| | c104391_g1_i1 | 3.805 | 3.77E-07 | 2.579 | 6.18E-04 | hypothetical protein SORBIDRAFT_08g002290 [Sorghum bicolor] |
| c104391_g1_i3 | 4.440 | 7.67E-08 | 2.081 | 5.48E-03 | ||
| c108711_g1_i1 | 3.783 | 9.38E-07 | 2.252 | 3.43E-03 | ||
| c81573_g3_i1 | 8.593 | 7.45E-07 | 4.211 | 5.15E-03 | ||
| c81573_g4_i1 | 6.315 | 9.22E-03 | 2.583 | 4.09E-02 | ||
| c88513_g2_i1 | 2.802 | 6.63E-04 | 4.141 | 1.07E-05 | ||
| gi|242082800|ref|XP_002441825.1| | c86447_g1_i2 | 5.341 | 8.23E-06 | 3.133 | 1.34E-02 | hypothetical protein SORBIDRAFT_08g002950 [Sorghum bicolor] |
| gi|242084342|ref|XP_002442596.1| | c96572_g2_i5 | 1.844 | 9.69E-03 | 2.067 | 9.74E-03 | hypothetical protein SORBIDRAFT_08g022670 [Sorghum bicolor] |
| gi|27542778|gb|AAO16711.1| | c104391_g1_i4 | 10.834 | 9.54E-13 | 5.610 | 1.23E-08 | truncated Xa1-like protein [Sorghum bicolor] |
| gi|357144043|ref|XP_003573148.1| | c83558_g1_i1 | 3.287 | 1.02E-04 | 1.832 | 3.90E-02 | PREDICTED: putative disease resistance protein RGA4-like [Brachypodium distachyon] |
| gi|414591554|tpg|DAA42125.1| | c101028_g1_i1 | 7.789 | 3.57E-05 | 6.349 | 2.15E-02 | TPA: hypothetical protein ZEAMMB73_852544 [Zea mays] |
ABA-associated transcripts
In the abscission process, ABA plays a critical role and involves in different pathways [67]. In citrus, leaf abscission induced by rehydration after a period of water stress requires ABA accumulation [62]. In our study, we found different upregulated transcripts involved in ABA signaling pathways in the two sugarcane generations. Compared to Q2 and B, a total of 341 and 26 upregulated ABA-associated transcripts were identified Q1 (Additional file 6) and T (Table 6), respectively. Q1 and T shared 10 upregulated transcripts involved in ABA signaling pathways. GO annotation showed commonly upregulated transcripts were involved in ABA D-glucopyranosyl ester transmembrane transport, ABA catabolic process, ABA-activated signaling pathway, response to ABA and ABA binding. Compared to T sugarcane, Q1 had more upregulated ABA-associated transcripts, like ABC (ATP-binding cassette) transporter G family members (5, 11, 15, 25, 36, and 40), ABA receptor (PYL2 and PYL8) and zeaxanthin epoxidase (ZEP). In Arabidopsis, ABA transport can be mediated by PDR-type ABC transporter, ABG25 and AB40G [68, 69]. ZEP also plays an important role in the xanthophyll cycle and ABA biosynthesis, can convert zeaxanthin into antheraxanthin and subsequently violaxanthin, and is required for resistance to osmotic and drought stresses, seed development and dormancy [70].
Table 6.
ABA associated transcripts up regulated in T sugarcane plants, compared to B
| Gene family | gene_id | logFC(Q1/Q2) | PValue | logFC(T/B) | PValue | Description | Relationship with abscisic acid |
|---|---|---|---|---|---|---|---|
| AB2C | c53535_g1_i1 | 0.000 | 1.00E + 00 | 5.712 | 4.91E-02 | ABC transporter C family member 2 | abscisic acid D-glucopyranosyl ester transmembrane transport |
| ABAH2 | c60338_g1_i1 | −0.050 | 1.00E + 00 | 2.701 | 3.56E-02 | Abscisic acid 8′-hydroxylase 2 | abscisic acid catabolic process |
| ABAH3 | c78622_g1_i1 | 0.307 | 8.54E-01 | 2.924 | 9.01E-03 | Abscisic acid 8′-hydroxylase 3 | |
| c78622_g2_i1 | −0.440 | 7.00E-01 | 2.794 | 1.07E-02 | |||
| AI5L3 | c84393_g3_i1 | −1.790 | 9.68E-02 | 3.982 | 5.51E-04 | ABSCISIC ACID-INSENSITIVE 5-like protein 3 | abscisic acid-activated signaling pathway |
| AMO | c101773_g1_i1 | 8.258 | 2.79E-19 | 3.002 | 3.94E-02 | Primary amine oxidase | |
| CDPKU | c64532_g1_i2 | 0.000 | 1.00E + 00 | 10.382 | 1.56E-04 | Calcium-dependent protein kinase 30 | |
| CRK29 | c100815_g2_i1 | 5.974 | 2.54E-07 | 4.499 | 1.04E-02 | Cysteine-rich receptor-like protein kinase 29 | response to abscisic acid |
| c80509_g3_i1 | 0.000 | 1.00E + 00 | 7.704 | 3.33E-03 | |||
| FERON | c86070_g1_i3 | 5.144 | 2.25E-05 | 2.851 | 3.28E-02 | Receptor-like protein kinase FERONIA | abscisic acid-activated signaling pathway |
| c89220_g1_i1 | −0.241 | 9.09E-01 | 2.685 | 2.75E-02 | |||
| GG3 | c67489_g1_i3 | 0.000 | 1.00E + 00 | 6.142 | 2.91E-02 | Guanine nucleotide-binding protein subunit gamma 3 | response to abscisic acid |
| IP5P3 | c103894_g2_i1 | −7.938 | 2.25E-05 | 2.181 | 4.56E-02 | Type I inositol 1,4,5-trisphosphate 5-phosphatase CVP2 | abscisic acid-activated signaling pathway; response to abscisic acid |
| c91503_g1_i4 | −0.239 | 1.00E + 00 | 1.926 | 2.67E-02 | |||
| IP5PB | c95707_g1_i3 | 2.637 | 1.03E-03 | 2.479 | 1.55E-02 | Type I inositol 1,4,5-trisphosphate 5-phosphatase 11 | response to abscisic acid |
| LPP2 | c101924_g4_i1 | 2.513 | 4.64E-03 | 1.874 | 2.85E-02 | Lipid phosphate phosphatase 2 | abscisic acid-activated signaling pathway |
| LTI65 | c97091_g1_i2 | 12.717 | 3.19E-18 | 4.848 | 1.35E-03 | Low-temperature-induced 65 kDa protein | abscisic acid-activated signaling pathway; |
| MFT | c87994_g2_i6 | 9.022 | 6.69E-08 | 5.653 | 2.18E-02 | Protein MOTHER of FT and TF 1 | response to abscisic acid |
| MPK5 | c83394_g1_i1 | 4.061 | 5.22E-08 | 1.615 | 2.92E-02 | Mitogen-activated protein kinase 5 | abscisic acid-activated signaling pathway |
| NCED1 | c102008_g2_i1 | 2.225 | 4.93E-02 | 3.563 | 2.23E-02 | 9-cis-epoxycarotenoid dioxygenase 1, chloroplastic | abscisic acid biosynthetic process |
| c73153_g1_i1 | 4.064 | 5.24E-01 | 5.300 | 2.15E-03 | |||
| PXG4 | c60264_g1_i1 | 5.998 | 2.19E-02 | 2.708 | 3.13E-02 | Probable peroxygenase 4 | response to abscisic acid |
| PYL2 | c100077_g1_i1 | −9.503 | 3.94E-09 | 6.381 | 1.09E-05 | Abscisic acid receptor PYL2 | abscisic acid binding; |
| VIV1 | c127665_g1_i1 | 0.000 | 1.00E + 00 | 5.318 | 2.48E-02 | Regulatory protein viviparous-1 | abscisic acid-activated signaling pathway |
| Y1571 | c97220_g1_i1 | −0.460 | 7.64E-01 | 3.447 | 5.68E-03 | Probable leucine-rich repeat receptor-like protein kinase At1g35710 | response to abscisic acid |
| c97220_g1_i2 | −1.118 | 2.71E-01 | 3.092 | 2.27E-02 |
Transcription factors
Further, 2,031 of the assembled transcripts were annotated to encode TFs in this study. There were 603 and 58 transcripts were upregulated in Q1 and T relatively to Q2 and B, respectively. Of the upregulated transcripts encoding TFs, 16 were in common, including two transcripts encoding MADS-box transcription factors, six transcripts encoding heat stress transcription factors, two transcripts encoding transcription factor DIVARICATA, one WRKY33 transcript, two transcripts encoding NAC transcription factor 29, and three transcripts encoding ethylene-responsive transcription factor. Although the function of these TFs in sugarcane is not clear, they have been reported to regulate the leaf senescence and abscission in Arabidopsis [71–73].
Overall, pathways of plant-pathogen interaction, response to stresses and ABA signaling are important pathways in plants and involve in several bio-activities including cell development and death [50, 74]. Although it is hard to tell which pathways are involved in the regulation of leaf abscission in sugarcane, the up regulation of transcripts involved in these pathways strongly supports that they may play an important role in the AZ development and leaf abscission in sugarcane. Q1 and T upregulated transcripts were enriched in different pathways, we can infer that they may promote the shedding of their leaves in different ways during the maturity time. Transcripts identified here showed their potential availability which can be used in sugarcane breeding programs.
qRT-PCR validation
Differentially expressed transcripts between LASP and LPSP identified by RNA-Seq were confirmed by quantitative real-time PCR (qRT-PCR) experiment. A total of 20 transcripts were randomly selected as candidates. We used RT2 First Strand Kit (QIAGEN) and PCR mix (QIAGEN) to perform the cDNA synthesis and real-time PCR experiment. Forward and reverse primers for these 20 candidate transcripts were designed by Primer Express Software (v2.0, ABI) and actin gene was used as the reference gene. The information of primers used in this study can be found in Additional file 7. As shown in Fig. 4, we used relatively normalized expression (RNE) and log 2 fold change (log2FC) to show the expression changes of candidate transcripts in Q1 vs. Q2 and T vs. B identified by qRT-PCR and RNA-Seq, respectively. The dysregulation of 19 transcripts (90.48 %) were consistent between qRT-PCR and RNA-Seq. Especially for those transcripts absent in several samples, they were not detected by both qRT-PCR and RNA-Seq. The Pearson correlations showed high relevance between RNA-Seq and qRT-PCR (0.911 for Q1/Q2 and 0.971 for T/B).
Fig. 4.
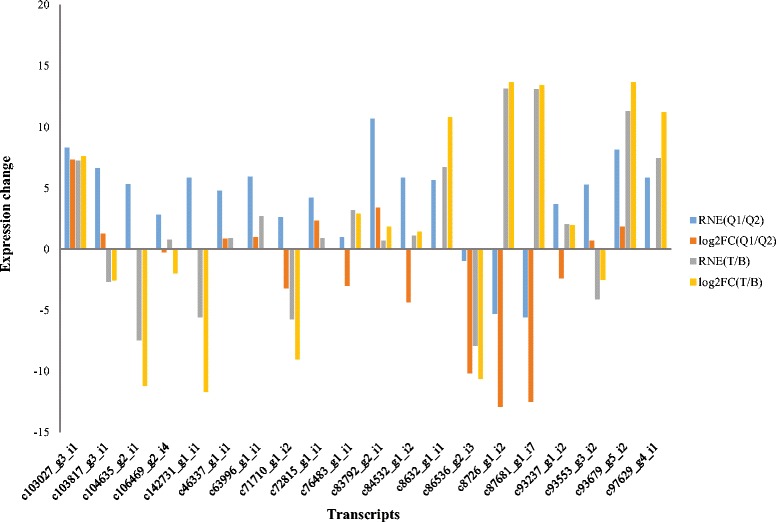
qRT-PCR validation for candidate transcripts
SNP discovery
Specificity and high abundance of genes support we can call single nucleotide polymorphisms (SNPs) using RNA-Seq [75]. RNA-Seq has been used for SNP discovery in non-model plants such as Eucalyptus grandis [76], Brassica napus [77], and Medicago sativa [78]. In this study, we used samtools [79] and bcftools to call SNPs, and found a total of 1,544,787 SNPs in the transcriptome alignment files for six libraries. The numbers of different SNP types were shown in Fig. 5. Because multiple SNP types could happen at one site, the total number of SNP types is a little greater than the total SNP site number (1,544,787). A total of 285,080 SNPs (18.45 %) were annotated to occur in the potential protein coding regions. Four SNP types (A- > G, C- > T, G- > A and T- > C) were significant because they took 61.50 % of the total SNP types in this study. Finally, we identified 3,722 SNPs, which were different between LASP and LPSP but same in LASP or LPSP. As shown in Additional file 8, these 3,722 SNPs were divided into 1,134 nonsynonymous SNPs and 2,588 synonymous SNPs. KEGG pathway analysis showed the transcripts containing nonsynonymous SNPs were enriched in the pathways of cytosolic DNA-sensing pathway, ribosome biogenesis in eukaryotes and inositol phosphate metabolism. Due to limit sugarcane gene annotation, SNP results seemed not relevant to our aim, which is simply to identify leaf abscission associated genes in sugarcane, more experiments and data are required to investigate the functions of these SNPs in leaf abscission.
Fig. 5.
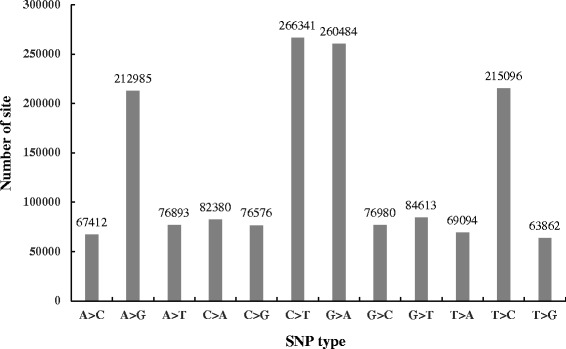
Different types of SNPs identified in this study
Conclusions
In current study we employed next generation sequencing method RNA-Seq to analyze the transcriptome of sugarcane leaves and characterize candidate genes related to the leaf abscission in sugarcane during the maturity time. A total of 215,019,578 transcripts were initially assembled with N50 of 1,177 bp in length. We annotated them by mapping them to several known databases such as NR, UniProtKB/Swiss-Prot, GO and KEGG pathway databases. Further annotations including signal protein, rRNAs, protein domains and membrane proteins were performed by Transdecoder and Trinotate pipelines. Based on the assembled transcriptome, we identified several transcripts differentially expressed between LASP and LPSP, which were annotated to involve in the plant-pathogen interaction, stress response and ABA-associated pathways. qRT-PCR was used to validate the expression levels of 20 randomly selected transcripts. The results showed high consistency between qRT-PCR and RNA-Seq methods. Furthermore, a total of 1,544,787 SNPs were identified successfully, of them 1,134 were nonsynonymous SNPs and 2,588 were synonymous SNPs. The transcriptome produced by this study will provide a valuable resource of molecular information for future investigations and process understanding the roles of genes in the leaf abscission of sugarcane. Our study may also help develop sugarcane varieties which may shed their leaves during the maturity time and benefit the sugarcane farmers.
Methods
Plant material
The sugarcane plants were grown and maintained in the experimental filed of Sugarcane Research Institute in Nanning, Guangxi Province of China. ROC-26 (from Taiwan) is a precocious and productive sugarcane variety, but the sugar cane is wrapped very tight at the physiological maturity [80]. GT96-167 is a late-maturing and high-yield sugarcane variety which is bred by Guangxi Sugarcane Research Institute [81–83]. In contrast with ROC-26, GT96-197 can shed their leaves easily during the maturity time. By using 19 GT96-167 and 12 ROC-26 sugarcane plants, we obtained 83 pairs of sugarcane sexual hybridizations. For each hybridization, 3–6 g seeds (germination number: 30-260/g) were used to cultivate a total of 34,000 sugarcane seedlings. After 4 years, 49 hybridizations were proved to have distinct phenotypes on leaf shedding among their F1 generations. In this study, we chose two F1 generation sugarcane varieties 42–1 and 42–2 (named as T1 and T2) which can shed their leaves during the maturity time and two another F1 generation sugarcane varieties 42–6 and 42–16 (named as B1 and B2) which cannot shed their leaves during the maturity time, as well as their parents GT96-167 and ROC-26 (named as Q1 and Q2). According to the cultivation and sugar content test, sugarcane harvest time is always in the period of mid-November to the next mid-March in China. Six sugarcane plants (Q1, Q2, T1, T2, B1 and B2) were planted in January of 2014. And on 24th December of 2014 we performed the sugar content test for all six sugarcane plants and found the sugar content of each plant reached (or was close to) the peak. New born leaf tissues approximately 5 cm above the growing point were taken on 5th December of 2014. The leaf tissues were stored in the liquid nitrogen before RNA extraction. The F1 generation sugarcane varieties were proved to have stable agronomic characteristic on leaf shedding by 5 years of filed observation.
RNA extraction
Total RNA was isolated from sugarcane leaves by using TRIzol® reagent (Invitrogen) according to the manufacture’s protocol. Briefly, 1 ml of TRIzol® reagent was added into 100 mg of leaf sample, the sample was homogenized using power homogenizer and centrifuged at 12,000 × g for 10 min at 4 °C. After the fatty layer was removed and discarded, the cleared supernatant was transferred into a new tube and mixed with 0.2 ml of chloroform. The tube was shaken vigorously for 15 s, followed by an incubation for 3 min at room temperature. Next, the sample was centrifuged at 12,000 × g for 15 min at 4 °C and the aqueous phase was moved into a new tube for RNA precipitation. For precipitating RNA from each sample, 10 μg of RNase-free glycogen was added into the aqueous phase as a carrier, followed by 0.5 ml of 100 % isopropanol, then, samples were incubated at room temperature for 10 min and centrifuged at 12,000 × g for 10 min at 4 °C. To wash the RNA pellet, we added 1 ml of 75 % ethanol into the tube, vortexed the tube gently, centrifuged the tube 7,500 × g for 5 min at 4 °C and discarded the wash. The RNA pellet was air-dried, suspended in RNase-free water, water bathed at 60 °C for 10 min, quality controlled by Agilent 2100 Bioanalyzer and used for cDNA library construction and deep sequencing.
cDNA library construction and transcriptome sequencing
Equal amount of total RNA (20 μg) was used for cDNA library construction using TruSeq™ RNA Sample Preparation Kit v2 (Illumina) and for transcriptome sequencing on the Illumina HiSeq 2000 platform, according to protocols. Briefly, total RNA was used to isolate poly(A) mRNAs using Dynal Oligo(dT) beads (Invitrogen). Then, mRNAs were chemically fragmented to ~200 nt fragments by using divalent cations (Elute/Prime/Fragment Mix buffer, Illumina) under elevated temperature. Cleaved RNA fragments were then copied into first strand cDNA by using reverse transcriptase and random primers, followed by the second strand cDNA synthesis using DNA Polymerase I (Invitrogen) and RNase H (Invitrogen) treatment. The cDNA fragments were next end repaired by using End Repair Mix (Illumina) reagent, purified and enriched with PCR to create the final cDNA libraries. Six cDNA libraries were sequenced by using pair-end (2 × 90 bp) sequencing technology with an Illumina HiSeq™ 2000 sequencer. The sequencing raw data (FASTQ formatted files) for six samples can be accessed in the NCBI Sequence Read Archive (SRA) database (http://trace.ncbi.nlm.nih.gov/Traces/sra/) under the accession number of SRA291189.
De novo assembly of the transcriptome
Raw reads of six libraries were cleaned by removing adapter sequences and low-quality sequences (reads with ambiguous bases ‘N’ and reads with more than 10 % Q < 20 bases). The resulting high quality reads of each library were quality controlled by using the program FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) before assembly. De novo assembly of the transcriptome was performed by Trinity software (release 2014-07-17) [30], according to the protocol [84]. Initially, highly quality RNA-Seq reads were used to generate overlapping k-mers (25). Based on the (k-1)-mer overlaps, Inchworm was used to assemble sorted k-mers into transcript contigs. Next, Chrysalis was used to cluster related Inchworm contigs into components by using grouped raw reads and paired read links. Then, a de Bruijn graph for each cluster was built by Chrysalis and reads were partitioned among the clusters. Finally, Butterfly was used to process the individual graphs and ultimately report the full length transcripts. To ensure a uniform transcriptome reference across samples, all clean reads were pooled together for assembly, then clean reads of six samples were individually aligned back to the assembled transcriptome reference.
Extract likely coding sequences from Trinity transcripts
We used TransDecoder, which is included in the Trinity software distribution, to extract potential coding sequences from assembled transcripts. Briefly, the longest open reading frame (ORF) was first identified within the assembled transcripts. Using a Markov model based on hexamers, a subset corresponding to the very longest transcripts were identified from all the longest ORFs. Then, all the longest ORFs identified were scored according to the Markov Model (log likelihood ratio based on coding/noncoding potential) in each of the six possible reading frames. For a particular transcript, the ORF was reported when its proper coding frame score calculated by GeneID software [85] was positive and highest of the other presumed wrong reading frames. A high scored putative ORF was excluded when it was fully encapsulated by the coordinates of another candidate ORF. The identified ORFs were set to encode a protein at least 100 amino acids.
Functional annotation of assembled transcripts using Trinotate
After the ORFs were extracted from the assembled transcripts, the deduced proteins were annotated using Trinotate (r2014-07-08, available at http://trinotate.github.io/). Briefly, deduced proteins were used to search against UniProtKB/Swiss-Prot database (ftp://ftp.broadinstitute.org/pub/Trinity/Trinotate_v2.0_RESOURCES/uniprot_sprot.trinotate_v2.0.pep.gz) to identify known protein sequences. Functional domains were identified by mapping them to the PFAM domain database (ftp://ftp.broadinstitute.org/pub/Trinity/Trinotate_v2.0_RESOURCES/Pfam-A.hmm.gz) [86] using HMMER [87]. Potential signal peptides, transmembrane domains and rRNA transcripts were predicted by SignalP [36], TMHMM Sever v2.0 [37] and RNAMMER [88], respectively. Then, the likely protein sequences were used to search against EggNOG database (v4.1, http://eggnogdb.embl.de) [38] which enables to identify the proteins distributed in EuKaryotic Orthologous Groups (KOG), Clusters of Orthologous Groups (COGs), and non-supervised orthologous groups (NOGs). Finally, above annotations were loaded into a Trinotate SQLite database and a final annotation report was generated. The maximum e-values for reporting best hit and associated annotation were no more than 1e-5.
GO and KEGG pathway annotation
To identify the assembled transcripts related with Gene Ontology (GO) and biology pathways, they were annotated by comparing to previously annotated genes in three public databases, NCBI non-redundant (NR) database, Swiss-Prot database and Kyoto Encyclopedia of Genes and Genomes (KEGG) database. We used BLAST software [89] to map assembled transcripts against NR database and filtered the mapping hits using a cut-off of e-value (1 × 10−5). The resulting transcripts were then processed to retrieve associated Gene Ontology (GO) items describing biological processes (BPs), cellular components (CCs) and molecular functions (MFs) by BLAST2GO software [90]. By using unique gene accession numbers, BLAST2GO also produces corresponding enzyme commission (EC) numbers for sequences with an e-value ≤1e-5. Then, the transcripts with corresponding ECs were obtained and mapped to KEGG metabolic pathway database.
Transcriptome profile and different expression
The abundance of each transcript was evaluated by Bowtie2 and RSEM (RNA-Seq by Expectation-Maximization) tools in every sample. First, high quality reads of each library were mapped to Trinity assembled transcripts by Bowtie2 [41]. Then, an R package RSEM was used to evaluate the expression levels of each transcript in every library by estimating the abundance of reads that aligned to the transcript. Differential expression of transcripts across samples was identified by using an R package called edgeR [43]. edgeR can proceed differential expression of a transcript in two groups as we performed biological replicates for T and B sugarcane varieties in this study. The significance of differential expression was evaluated by the fold change (≥2) and p-value (<0.05).
SNP discovery
As the phenotypes of sugarcanes used in this study were different, we used samtools (v1.2, https://github.com/samtools/samtools/releases/download/1.2/samtools-1.2.tar.bz2) [79] and bcftools (v1.2, https://github.com/samtools/bcftools/releases/download/1.2/bcftools-1.2.tar.bz2) to find possible single nucleic polymorphisms (SNPs). In brief, clean reads of each sample were aligned against the assembled transcriptome reference by Bowtie2, generating BAM formatted files. The BAM files were then indexed and processed by the mplieup function of samtools to produce a BCF file that contains all the locations in the genome. The BCF file was then used to call genotypes and reduce the list of sites to those found to be variant by passing this file into bcftools call. Finally, after filtering low quality SNPs, reliable SNPs were left and stated by a self- developed Perl script.
GO and KEGG pathway enrichment analysis
Functional analysis was performed using the Gene Ontology and KEGG pathway annotations for the transcripts. To find enriched GO items and KEGG pathways, we used p-value (Fisher’s exact test) and q- value [91] to show the significance of enrichment and control the false discovery rate. Significant GO items and KEGG pathways should satisfy the critical of p-value < 0.05 and q-value < 0.9. Detected pathways only related to animal or human GO items and KEGG pathways were filtered.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) experiment was used to validate the expression patterns of RNA transcripts in different sugarcane varieties. Total RNA (4 μg) which was used for RNA-Seq previously described was used for cDNA synthesis by using RT2 First Strand Kit (QIAGEN) and qRT-PCR was performed by using PCR mix (QIAGEN), according to the manufactures’ protocols. Briefly, genomic DNA elimination mix (10 μl) was first made by using total RNA (4 μg), Buffer GE and RNase-free water. After incubated at 42 °C for 5 min and on ice for 5 min, the genomic DNA elimination mix was mixed with 5x Buffer BC3 (4 μl), control P2 (1 μl), RE3 Reverse Transcriptase Mix (2 μl) and RNase-free water (3 μl). The final reverse transcription mix (20 μl) was incubated at 42 °C for exactly 15 min and at 95 °C for 5 min to finish cDNA synthesis. Primers for qRT-PCR were designed by Primer Express Software (v2.0, ABI) and synthesized by BGI (Additional file 7). A total of 16 μl reaction mix was made by 2x PCR mix (8 μl), forward primer (0.2 μl, 50pM/ul), reverse primer (0.2 μl, 50pM/ul), cDNA template (1 μl) and RNase-free water (6.6 μl). The final cDNA concentration of each reaction was 12.5 ng/μl. Actin was used as control and three reactions were performed for each transcript in every sample. The PCR reaction was performed and analyzed by using ABI ViiA 7 Real Time PCR System. After the threshold cycle (CT) numbers of each transcript in every samples were evaluated, mean CT values were calculated for subsequent analysis. Base on the mean CT value, ΔCT was used to present and normalize the expression of a candidate transcript. Relatively normalized expression (RNE, −ΔΔCT method) was used to show the expression change of a transcript in two samples. CT values greater than 35 were set to 35.
Acknowledgements
This work was financially supported by the National Science Foundation of China (31460093, 31400281), Guangxi Scientific Research and Technology Development Plan (GuiKeZhong14121005-1-2,Guikegong1598006-1-1D), Nanning Scientific Research and Technology Development Plan (20131038) and the development foundation of Guangxi Academy of Agricultural Science (2015JZ15, 2015JZ91).
Abbreviations
- ABA
abscisic acid
- ABC
ATP-binding cassette
- AZ
abscission zone
- COG
Clusters of Orthologous Group
- EC
enzyme commission
- GO
gene ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KOG
EuKaryotic Orthologous Group
- LASP
leaf abscission sugarcane plants
- LPSP
leaf packaging sugarcane plants
- NOG
non-supervised orthologous group
- ORF
open reading frame
- qRT-PCR
quantitative real-time polymerase chain reaction
- RIN4
RPM1-interacting protein 4
- RPM1
resistance to Pseudomonas syringae pv. Maculicola 1
- RPP13
recognition of Peronospora parasitica 13
- RSEM
RNA-Seq by Expectation-Maximization
- SNP
single nucleotide polymorphism
- SRA
sequence read archive
- TF
transcription factor
- ZEP
zeaxanthin epoxidase
Additional files
Number of sugarcane transcripts aligned to different species. (XLSX 19 kb)
Gene Ontology annotation for the assembled transcriptome of sugarcane leaves. (TXT 3759 kb)
Commonly upregulated transcripts in Q1/Q2 and T/B. (XLSX 320 kb)
KEGG pathway analysis for Q1 sugarcane upregulated transcripts relatively to Q2 sugarcane. (XLSX 14 kb)
KEGG pathway analysis for T sugarcane upregulated transcripts relatively to B sugarcane. (XLSX 11 kb)
ABA-associated transcripts upregulated in Q2 in comparison of Q1. (XLSX 38 kb)
Primer sequences used in qRT-PCR experiment. (XLSX 10 kb)
1,134 nonsynonymous SNPs and 2,588 synonymous SNPs identified in this study. (XLSX 404 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
ML and ZL designed research; KW, YJ and GW performed sample preparation and total RNA extraction; SH, YZ, JL and ZM analyzed the RNA-Seq data; FT, LW and SL performed qRT-PCR experiment and new reagents; ML and ZL wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Ming Li, Email: mingli9606@163.com.
Zhaoxu Liang, Email: 33350797@qq.com.
Yuan Zeng, Email: 50506252@qq.com.
Yan Jing, Email: gxjingyan@163.com.
Kaichao Wu, Email: kaichaowu@126.com.
Jun Liang, Email: jliang@biotgr.com.
Shanshan He, Email: Shanbh613@126.com.
Guanyu Wang, Email: 2593998841@qq.com.
Zhanghong Mo, Email: mozhanghong@126.com.
Fang Tan, Email: tanfang64@163.com.
Song Li, Email: ls19009@163.com.
Lunwang Wang, Email: wlunwang@163.com.
References
- 1.Que Y, Pan Y, Lu Y, Yang C, Yang Y, Huang N, Xu L. Genetic analysis of diversity within a Chinese local sugarcane germplasm based on start codon targeted polymorphism. Biomed Res Int. 2014;2014:468375. doi: 10.1155/2014/468375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Menossi M, Silva-Filho MC, Vincentz M, Van-Sluys MA, Souza GM. Sugarcane functional genomics: gene discovery for agronomic trait development. Int J Plant Genomics. 2008;2008:458732. doi: 10.1155/2008/458732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lam E, Shine J, Da Silva J, Lawton M, Bonos S, Calvino M, Carrer H, SILVA‐FILHO MC, Glynn N, Helsel Z. Improving sugarcane for biofuel: engineering for an even better feedstock. Gcb Bioenergy. 2009;1(3):251–255. doi: 10.1111/j.1757-1707.2009.01016.x. [DOI] [Google Scholar]
- 4.Sexton R, Roberts JA. Cell biology of abscission. Annu Rev Plant Physiol. 1982;33(1):133–162. doi: 10.1146/annurev.pp.33.060182.001025. [DOI] [Google Scholar]
- 5.McKim SM, Stenvik GE, Butenko MA, Kristiansen W, Cho SK, Hepworth SR, Aalen RB, Haughn GW. The BLADE-ON-PETIOLE genes are essential for abscission zone formation in Arabidopsis. Development. 2008;135(8):1537–1546. doi: 10.1242/dev.012807. [DOI] [PubMed] [Google Scholar]
- 6.Patterson SE. Cutting loose. Abscission and dehiscence in Arabidopsis. Plant Physiol. 2001;126(2):494–500. doi: 10.1104/pp.126.2.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts JA, Elliott KA, Gonzalez-Carranza ZH. Abscission, dehiscence, and other cell separation processes. Annu Rev Plant Biol. 2002;53:131–158. doi: 10.1146/annurev.arplant.53.092701.180236. [DOI] [PubMed] [Google Scholar]
- 8.Agusti J, Merelo P, Cercos M, Tadeo FR, Talon M. Ethylene-induced differential gene expression during abscission of citrus leaves. J Exp Bot. 2008;59(10):2717–2733. doi: 10.1093/jxb/ern138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai S, Lashbrook CC. Stamen abscission zone transcriptome profiling reveals new candidates for abscission control: enhanced retention of floral organs in transgenic plants overexpressing Arabidopsis ZINC FINGER PROTEIN2. Plant Physiol. 2008;146(3):1305–1321. doi: 10.1104/pp.107.110908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meir S, Philosoph-Hadas S, Sundaresan S, Selvaraj KS, Burd S, Ophir R, Kochanek B, Reid MS, Jiang CZ, Lers A. Microarray analysis of the abscission-related transcriptome in the tomato flower abscission zone in response to auxin depletion. Plant Physiol. 2010;154(4):1929–1956. doi: 10.1104/pp.110.160697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Botton A, Eccher G, Forcato C, Ferrarini A, Begheldo M, Zermiani M, Moscatello S, Battistelli A, Velasco R, Ruperti B, et al. Signaling pathways mediating the induction of apple fruitlet abscission. Plant Physiol. 2011;155(1):185–208. doi: 10.1104/pp.110.165779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakano T, Kimbara J, Fujisawa M, Kitagawa M, Ihashi N, Maeda H, Kasumi T, Ito Y. MACROCALYX and JOINTLESS interact in the transcriptional regulation of tomato fruit abscission zone development. Plant Physiol. 2012;158(1):439–450. doi: 10.1104/pp.111.183731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogawa M, Kay P, Wilson S, Swain SM. ARABIDOPSIS DEHISCENCE ZONE POLYGALACTURONASE1 (ADPG1), ADPG2, and QUARTET2 are Polygalacturonases required for cell separation during reproductive development in Arabidopsis. Plant Cell. 2009;21(1):216–233. doi: 10.1105/tpc.108.063768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinyopich A, Ditta GS, Savidge B, Liljegren SJ, Baumann E, Wisman E, Yanofsky MF. Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature. 2003;424(6944):85–88. doi: 10.1038/nature01741. [DOI] [PubMed] [Google Scholar]
- 15.Mao L, Begum D, Chuang HW, Budiman MA, Szymkowiak EJ, Irish EE, Wing RA. JOINTLESS is a MADS-box gene controlling tomato flower abscission zone development. Nature. 2000;406(6798):910–913. doi: 10.1038/35022611. [DOI] [PubMed] [Google Scholar]
- 16.Konishi S, Izawa T, Lin SY, Ebana K, Fukuta Y, Sasaki T, Yano M. An SNP caused loss of seed shattering during rice domestication. Science. 2006;312(5778):1392–1396. doi: 10.1126/science.1126410. [DOI] [PubMed] [Google Scholar]
- 17.Li C, Zhou A, Sang T. Rice domestication by reducing shattering. Science. 2006;311(5769):1936–1939. doi: 10.1126/science.1123604. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Y, Lu D, Li C, Luo J, Zhu BF, Zhu J, Shangguan Y, Wang Z, Sang T, Zhou B, et al. Genetic control of seed shattering in rice by the APETALA2 transcription factor shattering abortion1. Plant Cell. 2012;24(3):1034–1048. doi: 10.1105/tpc.111.094383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin Z, Li X, Shannon LM, Yeh CT, Wang ML, Bai G, Peng Z, Li J, Trick HN, Clemente TE, et al. Parallel domestication of the Shattering1 genes in cereals. Nat Genet. 2012;44(6):720–724. doi: 10.1038/ng.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji H, Kim SR, Kim YH, Kim H, Eun MY, Jin ID, Cha YS, Yun DW, Ahn BO, Lee MC, et al. Inactivation of the CTD phosphatase-like gene OsCPL1 enhances the development of the abscission layer and seed shattering in rice. Plant J. 2010;61(1):96–106. doi: 10.1111/j.1365-313X.2009.04039.x. [DOI] [PubMed] [Google Scholar]
- 21.Venu R, Sreerekha M, Nobuta K, Belo A, Ning Y, An G, Meyers BC, Wang GL. Deep sequencing reveals the complex and coordinated transcriptional regulation of genes related to grain quality in rice cultivars. BMC Genomics. 2011;12:190. doi: 10.1186/1471-2164-12-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li D, Wang L, Liu X, Cui D, Chen T, Zhang H, Jiang C, Xu C, Li P, Li S, et al. Deep sequencing of maize small RNAs reveals a diverse set of microRNA in dry and imbibed seeds. PLoS One. 2013;8(1):e55107. doi: 10.1371/journal.pone.0055107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qi X, Xie S, Liu Y, Yi F, Yu J. Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol Biol. 2013;83(4–5):459–473. doi: 10.1007/s11103-013-0104-6. [DOI] [PubMed] [Google Scholar]
- 24.Carson DL, Botha FC. Preliminary analysis of expressed sequence tags for sugarcane. Crop Science. 2000;40(6):1769–1779. doi: 10.2135/cropsci2000.4061769x. [DOI] [Google Scholar]
- 25.Carson D, Botha F. Genes expressed in sugarcane maturing internodal tissue. Plant Cell Rep. 2002;20(11):1075–1081. doi: 10.1007/s00299-002-0444-1. [DOI] [Google Scholar]
- 26.Casu RE, Grof CP, Rae AL, McIntyre CL, Dimmock CM, Manners JM. Identification of a novel sugar transporter homologue strongly expressed in maturing stem vascular tissues of sugarcane by expressed sequence tag and microarray analysis. Plant Mol Biol. 2003;52(2):371–386. doi: 10.1023/A:1023957214644. [DOI] [PubMed] [Google Scholar]
- 27.Casu RE, Dimmock CM, Chapman SC, Grof CP, McIntyre CL, Bonnett GD, Manners JM. Identification of differentially expressed transcripts from maturing stem of sugarcane by in silico analysis of stem expressed sequence tags and gene expression profiling. Plant Mol Biol. 2004;54(4):503–517. doi: 10.1023/B:PLAN.0000038255.96128.41. [DOI] [PubMed] [Google Scholar]
- 28.Ma HM, Schulze S, Lee S, Yang M, Mirkov E, Irvine J, Moore P, Paterson A. An EST survey of the sugarcane transcriptome. Theor Appl Genet. 2004;108(5):851–863. doi: 10.1007/s00122-003-1510-y. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Nagai C, Yu Q, Pan Y-B, Ayala-Silva T, Schnell RJ, Comstock JC, Arumuganathan AK, Ming R. Genome size variation in three Saccharum species. Euphytica. 2012;185(3):511–519. doi: 10.1007/s10681-012-0664-6. [DOI] [Google Scholar]
- 30.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu T, Zhu S, Tang Q, Chen P, Yu Y, Tang S. De novo assembly and characterization of transcriptome using Illumina paired-end sequencing and identification of CesA gene in ramie (Boehmeria nivea L. Gaud) BMC Genomics. 2013;14:125. doi: 10.1186/1471-2164-14-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32(Database issue):D258–261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cardoso-Silva CB, Costa EA, Mancini MC, Balsalobre TW, Canesin LE, Pinto LR, Carneiro MS, Garcia AA, de Souza AP, Vicentini R. De novo assembly and transcriptome analysis of contrasting sugarcane varieties. PLoS One. 2014;9(2):e88462. doi: 10.1371/journal.pone.0088462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Xu L, Chen Y, Shen H, Gong Y, Limera C, Liu L. Transcriptome profiling of radish (Raphanus sativus L.) root and identification of genes involved in response to Lead (Pb) stress with next generation sequencing. PLoS One. 2013;8(6):e66539. doi: 10.1371/journal.pone.0066539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, Jiang CJ, Sun J, Li YY, Chen Q, Xia T, et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genomics. 2011;12:131. doi: 10.1186/1471-2164-12-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 37.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 38.Powell S, Forslund K, Szklarczyk D, Trachana K, Roth A, Huerta-Cepas J, Gabaldon T, Rattei T, Creevey C, Kuhn M, et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014;42(Database issue):D231–239. doi: 10.1093/nar/gkt1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cross TG, Scheel-Toellner D, Henriquez NV, Deacon E, Salmon M, Lord JM. Serine/threonine protein kinases and apoptosis. Exp Cell Res. 2000;256(1):34–41. doi: 10.1006/excr.2000.4836. [DOI] [PubMed] [Google Scholar]
- 40.Clark DE, Errington TM, Smith JA, Frierson HF, Jr, Weber MJ, Lannigan DA. The serine/threonine protein kinase, p90 ribosomal S6 kinase, is an important regulator of prostate cancer cell proliferation. Cancer Res. 2005;65(8):3108–3116. doi: 10.1158/0008-5472.CAN-04-3151. [DOI] [PubMed] [Google Scholar]
- 41.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim PO, Woo HR, Nam HG. Molecular genetics of leaf senescence in Arabidopsis. Trends Plant Sci. 2003;8(6):272–278. doi: 10.1016/S1360-1385(03)00103-1. [DOI] [PubMed] [Google Scholar]
- 45.Lim PO, Kim HJ, Nam HG. Leaf senescence. Annu Rev Plant Biol. 2007;58:115–136. doi: 10.1146/annurev.arplant.57.032905.105316. [DOI] [PubMed] [Google Scholar]
- 46.Jacobs WP. Hormonal regulation of leaf abscission. Plant Physiol. 1968;43(9 Pt B):1480–1495. [PMC free article] [PubMed] [Google Scholar]
- 47.Kozlowski T. Water supply and leaf shedding. Soil water measurements, plant responses, and breeding for drought resistance. 1976;4:191–231.
- 48.Vloutoglou I, Kalogerakis S. Effects of inoculum concentration, wetness duration and plant age on development of early blight (Alternaria solani) and on shedding of leaves in tomato plants. Plant Pathol. 2000;49(3):339–345. doi: 10.1046/j.1365-3059.2000.00462.x. [DOI] [Google Scholar]
- 49.Tyree M, Cochard H, Cruiziat P, Sinclair B, Ameglio T. Drought‐induced leaf shedding in walnut: evidence for vulnerability segmentation. Plant Cell Environ. 1993;16(7):879–882. doi: 10.1111/j.1365-3040.1993.tb00511.x. [DOI] [Google Scholar]
- 50.Greenberg JT. Programmed Cell Death in Plant-Pathogen Interactions. Annu Rev Plant Physiol Plant Mol Biol. 1997;48:525–545. doi: 10.1146/annurev.arplant.48.1.525. [DOI] [PubMed] [Google Scholar]
- 51.Greenberg JT, Yao N. The role and regulation of programmed cell death in plant-pathogen interactions. Cell Microbiol. 2004;6(3):201–211. doi: 10.1111/j.1462-5822.2004.00361.x. [DOI] [PubMed] [Google Scholar]
- 52.Peterhansel C, Freialdenhoven A, Kurth J, Kolsch R, Schulze-Lefert P. Interaction Analyses of Genes Required for Resistance Responses to Powdery Mildew in Barley Reveal Distinct Pathways Leading to Leaf Cell Death. Plant Cell. 1997;9(8):1397–1409. doi: 10.1105/tpc.9.8.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grant M, Brown I, Adams S, Knight M, Ainslie A, Mansfield J. The RPM1 plant disease resistance gene facilitates a rapid and sustained increase in cytosolic calcium that is necessary for the oxidative burst and hypersensitive cell death. Plant J. 2000;23(4):441–450. doi: 10.1046/j.1365-313x.2000.00804.x. [DOI] [PubMed] [Google Scholar]
- 54.Glazebrook J. Genes controlling expression of defense responses in Arabidopsis--2001 status. Curr Opin Plant Biol. 2001;4(4):301–308. doi: 10.1016/S1369-5266(00)00177-1. [DOI] [PubMed] [Google Scholar]
- 55.Mackey D, Holt BF, 3rd, Wiig A, Dangl JL. RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1-mediated resistance in Arabidopsis. Cell. 2002;108(6):743–754. doi: 10.1016/S0092-8674(02)00661-X. [DOI] [PubMed] [Google Scholar]
- 56.Mackey D, Belkhadir Y, Alonso JM, Ecker JR, Dangl JL. Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell. 2003;112(3):379–389. doi: 10.1016/S0092-8674(03)00040-0. [DOI] [PubMed] [Google Scholar]
- 57.Chen T, Lv Y, Zhao T, Li N, Yang Y, Yu W, He X, Liu T, Zhang B. Comparative transcriptome profiling of a resistant vs. susceptible tomato (Solanum lycopersicum) cultivar in response to infection by tomato yellow leaf curl virus. PLoS One. 2013;8(11):e80816. doi: 10.1371/journal.pone.0080816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buscaill P, Rivas S. Transcriptional control of plant defence responses. Curr Opin Plant Biol. 2014;20:35–46. doi: 10.1016/j.pbi.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 59.Rushton DL, Tripathi P, Rabara RC, Lin J, Ringler P, Boken AK, Langum TJ, Smidt L, Boomsma DD, Emme NJ, et al. WRKY transcription factors: key components in abscisic acid signalling. Plant Biotechnol J. 2012;10(1):2–11. doi: 10.1111/j.1467-7652.2011.00634.x. [DOI] [PubMed] [Google Scholar]
- 60.Zheng Z, Qamar SA, Chen Z, Mengiste T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 2006;48(4):592–605. doi: 10.1111/j.1365-313X.2006.02901.x. [DOI] [PubMed] [Google Scholar]
- 61.Xu D, Fang X, Su P, Wang G. Ecophysiological responses of Caragana korshinskii Kom. under extreme drought stress: Leaf abscission and stem survives. Photosynthetica. 2012;50(4):541–548. doi: 10.1007/s11099-012-0060-4. [DOI] [Google Scholar]
- 62.Agusti J, Gimeno J, Merelo P, Serrano R, Cercos M, Conesa A, Talon M, Tadeo FR. Early gene expression events in the laminar abscission zone of abscission-promoted citrus leaves after a cycle of water stress/rehydration: involvement of CitbHLH1. J Exp Bot. 2012;63(17):6079–6091. doi: 10.1093/jxb/ers270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tounekti T, Abreu ME, Khemira H, Munné-Bosch S. Canopy position determines the photoprotective demand and antioxidant protection of leaves in salt-stressed Salvia officinalis L. plants. Environ Exp Bot. 2012;78:146–156. doi: 10.1016/j.envexpbot.2011.12.037. [DOI] [Google Scholar]
- 64.Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457(7229):551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 65.Kobe B, Deisenhofer J. Proteins with leucine-rich repeats. Curr Opin Struct Biol. 1995;5(3):409–416. doi: 10.1016/0959-440X(95)80105-7. [DOI] [PubMed] [Google Scholar]
- 66.van Ooijen G, Mayr G, Kasiem MM, Albrecht M, Cornelissen BJ, Takken FL. Structure-function analysis of the NB-ARC domain of plant disease resistance proteins. J Exp Bot. 2008;59(6):1383–1397. doi: 10.1093/jxb/ern045. [DOI] [PubMed] [Google Scholar]
- 67.Gomez-Cadenas A, Tadeo FR, Talon M, Primo-Millo E. Leaf Abscission Induced by Ethylene in Water-Stressed Intact Seedlings of Cleopatra Mandarin Requires Previous Abscisic Acid Accumulation in Roots. Plant Physiol. 1996;112(1):401–408. doi: 10.1104/pp.112.1.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kang J, Hwang JU, Lee M, Kim YY, Assmann SM, Martinoia E, Lee Y. PDR-type ABC transporter mediates cellular uptake of the phytohormone abscisic acid. Proc Natl Acad Sci U S A. 2010;107(5):2355–2360. doi: 10.1073/pnas.0909222107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuromori T, Miyaji T, Yabuuchi H, Shimizu H, Sugimoto E, Kamiya A, Moriyama Y, Shinozaki K. ABC transporter AtABCG25 is involved in abscisic acid transport and responses. Proc Natl Acad Sci U S A. 2010;107(5):2361–2366. doi: 10.1073/pnas.0912516107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Agrawal GK, Yamazaki M, Kobayashi M, Hirochika R, Miyao A, Hirochika H. Screening of the rice viviparous mutants generated by endogenous retrotransposon Tos17 insertion. Tagging of a zeaxanthin epoxidase gene and a novel ostatc gene. Plant Physiol. 2001;125(3):1248–1257. doi: 10.1104/pp.125.3.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee S, Seo PJ, Lee HJ, Park CM. A NAC transcription factor NTL4 promotes reactive oxygen species production during drought-induced leaf senescence in Arabidopsis. Plant J. 2012;70(5):831–844. doi: 10.1111/j.1365-313X.2012.04932.x. [DOI] [PubMed] [Google Scholar]
- 72.Chen WH, Li PF, Chen MK, Lee YI, Yang CH. FOREVER YOUNG FLOWER Negatively Regulates Ethylene Response DNA-Binding Factors by Activating an Ethylene-Responsive Factor to Control Arabidopsis Floral Organ Senescence and Abscission. Plant Physiol. 2015;168(4):1666–1683. doi: 10.1104/pp.15.00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koyama T, Nii H, Mitsuda N, Ohta M, Kitajima S, Ohme-Takagi M, Sato F. A regulatory cascade involving class II ETHYLENE RESPONSE FACTOR transcriptional repressors operates in the progression of leaf senescence. Plant Physiol. 2013;162(2):991–1005. doi: 10.1104/pp.113.218115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ghassemian M, Nambara E, Cutler S, Kawaide H, Kamiya Y, McCourt P. Regulation of abscisic acid signaling by the ethylene response pathway in Arabidopsis. Plant Cell. 2000;12(7):1117–1126. doi: 10.1105/tpc.12.7.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumar S, Banks TW, Cloutier S. SNP Discovery through Next-Generation Sequencing and Its Applications. Int J Plant Genomics. 2012;2012:831460. doi: 10.1155/2012/831460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Novaes E, Drost DR, Farmerie WG, Pappas GJ, Jr, Grattapaglia D, Sederoff RR, Kirst M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics. 2008;9:312. doi: 10.1186/1471-2164-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Trick M, Long Y, Meng J, Bancroft I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol J. 2009;7(4):334–346. doi: 10.1111/j.1467-7652.2008.00396.x. [DOI] [PubMed] [Google Scholar]
- 78.Yang SS, Tu ZJ, Cheung F, Xu WW, Lamb JF, Jung HJ, Vance CP, Gronwald JW. Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genomics. 2011;12:199. doi: 10.1186/1471-2164-12-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.X-j LU, K-m LI, J-q YE, R-l XU, HUANG J. Comprehensive evaluation of 13 newly introduced sugarcane varieties by gray fuzzy analysis [J] Guangxi Agric Sci. 2008;6:009. [Google Scholar]
- 81.G-m ZHANG, H-b LIU, W-k FANG, Q-x CHEN. Analysis of Main Characteristics of New Sugarcane Parents and Their Potential Utilization in Breeding Program [J] Sugar Crops China. 2007;2:002. [Google Scholar]
- 82.ZHOU H. Evaluation on cold tolerance of sugarcane varieties under field conditions. J Plant Genetic Resour. 2012;13(6):968–973. [Google Scholar]
- 83.You Q, Pan Y-B, Xu L-P, Gao S-W, Wang Q-N, Su Y-C, Yang Y-Q, Wu Q-B, Zhou D-G, Que Y-X: Genetic diversity analysis of sugarcane germplasm based on fluorescence-labeled simple sequence repeat markers and a capillary electrophoresis-based genotyping platform. Sugar Tech. 2015:1–11.
- 84.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blanco E, Parra G, Guigo R. Using geneid to identify genes. Curr Protoc Bioinformatics / editoral board, Andreas D Baxevanis [et al.] 2007;Chapter 4:Unit 4 3. doi: 10.1002/0471250953.bi0403s18. [DOI] [PubMed] [Google Scholar]
- 86.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40(Database issue):D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29–37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 91.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B-Stat Methodol. 1995;57(1):289–300. [Google Scholar]