

Abstract
The natural biology of Saccharomyces cerevisiae, the best known unicellular model eukaryote, remains poorly documented and understood although recent progress has started to change this situation. Studies carried out recently in the Northern Hemisphere revealed the existence of wild populations associated with oak trees in North America, Asia, and in the Mediterranean region. However, in spite of these advances, the global distribution of natural populations of S. cerevisiae, especially in regions were oaks and other members of the Fagaceae are absent, is not well understood. Here we investigate the occurrence of S. cerevisiae in Brazil, a tropical region where oaks and other Fagaceae are absent. We report a candidate natural habitat of S. cerevisiae in South America and, using whole-genome data, we uncover new lineages that appear to have as closest relatives the wild populations found in North America and Japan. A population structure analysis revealed the penetration of the wine genotype into the wild Brazilian population, a first observation of the impact of domesticated microbe lineages on the genetic structure of wild populations. Unexpectedly, the Brazilian population shows conspicuous evidence of hybridization with an American population of Saccharomyces paradoxus. Introgressions from S. paradoxus were significantly enriched in genes encoding secondary active transmembrane transporters. We hypothesize that hybridization in tropical wild lineages may have facilitated the habitat transition accompanying the colonization of the tropical ecosystem.
Keywords: microbe population genomics, yeast molecular ecology, introgression, genome evolution, Saccharomyces paradoxus
Introduction
The wealth of knowledge on the cellular biology, physiology, genetics, and genomics of Saccharomyces cerevisiae makes it the quintessential unicellular model eukaryote and prime biotechnology workhorse. In spite of this, the natural biology of S. cerevisiae remains poorly known. Since the onset of cultivation methods more than 180 years ago, the recurrent isolation of S. cerevisiae from fermented beverages and foods, and the difficulty in finding it in habitats with limited or no human influence, has supported the common view that this species is only prevalent in anthropic habitats associated to wine, beer, bread, and related products (Martini 1993; Vaughan-Martini and Martini 1995; Ciani et al. 2004). Recent studies have challenged the classical ecological model and have provided compelling evidence linking the natural ecology of the genus Saccharomyces, with the oak habitat in the Northern Hemisphere and with the Nothofagus (Southern beech) system in the Southern Hemisphere (Naumov et al. 1998; Sniegowski et al. 2002; Johnson et al. 2004; Sampaio and Gonçalves 2008; Libkind et al. 2011; Wang et al. 2012; Almeida et al. 2014, 2015; Bing et al. 2014; Charron et al. 2014; Sylvester et al. 2015). In addition, other studies have shown, first using multilocus sequencing (Fay and Benavides 2005), and afterwards employing whole-genome data (Liti et al. 2009; Cromie et al. 2013; Hyma and Fay 2013; Almeida et al. 2015) that S. cerevisiae is composed of both domesticated and wild populations.
Data currently available are not sufficient to determine the natural distribution of S. cerevisiae at a global scale. Besides an apparent overlap with the distribution of oaks and other Fagaceae in the northern hemisphere, the distribution of S. cerevisiae can be confounded by its association with anthropic niches. Therefore, although the ubiquitous distribution of S. cerevisiae has been abundantly described (Goddard and Greig 2015; Liti 2015), its main underlying causes and mechanisms are presently unknown.
The isolation of strains of this species from artisanal fermented foods and beverages and from fruits of cultivated plants in Africa, Europe, Asia, Central, and South America allows the formulation of two distinct hypotheses: 1) local natural S. cerevisiae populations were already present in these different locations when humans unwittingly promoted yeast fermentations, and were therefore independently coopted in domestication or 2) after a single and ancestral domestication event, starter S. cerevisiae cultures were geographically disseminated with human migrations. These two scenarios are not mutually exclusive and an intermediate one is also likely. In this case, foreign domesticated lineages were recurrently used in new fermentations but contact and recombination with local autochthonous populations also occurred. To disentangle the natural history of S. cerevisiae from the likely multiple domestication events promoted by humans, it is important to obtain a comprehensive map of natural populations and of their genetic attributes. A detailed characterization of such populations will be critical for a proper understanding of the ecology of S. cerevisiae in nature and for a better knowledge of the genetic underpinnings of domestication.
In this study, we hypothesize that the oak niche (Almeida et al. 2015) is not the sole natural habitat of S. cerevisiae and investigate the natural occurrence of S. cerevisiae in a tropical region in which oaks or other members of the Fagaceae are absent. Here, we report for the first time a candidate natural habitat of S. cerevisiae in South America. Using whole-genome data, we analyze the genetic structure of a wild collection of South American isolates of S. cerevisiae found in different bioclimatic regions of Brazil and uncover new lineages that appear to be more phylogenetically related to the wild populations found in North America and Japan than any other populations. Moreover, a survey of potential traces of hybridization with other Saccharomyces species reveals widespread introgression from the American population of Saccharomyces paradoxus into the newly uncovered Brazilian population.
Materials and Methods
Strain Isolation, Identification, and Typing
Isolation of Saccharomyces strains was based on the selective enrichment protocol previously described (Sampaio and Gonçalves 2008). Putative Saccharomyces isolates were confirmed by the observation of Saccharomyces-type ascospores and species identifications were done by sequencing of the ITS and D1/D2 regions of the rDNA.
Genome Sequencing, Read Alignment, and Genotype Calling
For sequencing purposes, strains were selected to maximize the variety of sources and locations of isolation. DNA was extracted from overnight cultures of monosporic derivatives and sequenced either for 100 cycles (single-end) and 2 × 100 cycles or 2 × 300 (paired-end) cycles using the Illumina HiSeq2000 and MiSeq systems, respectively.
Genomic information for other isolates was obtained from the NCBI-SRA collection and from Saccharomyces Genome Resequencing Project v2 (SGRP2) (Bergström et al. 2014). Where only finished genome sequences were available in public databases (NCBI), the corresponding error-free Illumina reads were simulated using dwgsim (http://sourceforge.net/apps/mediawiki/dnaa/).
Reads for each isolate were mapped to S. cerevisiae reference genome (UCSC version sacCer3) using SMALT v0.7.5 aligner (http://www.sanger.ac.uk/resources/software/smalt/). The reference index was built with a word length of 13 and a sampling step size of 2 (-k 13 –s 2). An exhaustive search for alignments (-x) was performed during the mapping step with the random assignment of ambiguous alignments switched off (-r -1) and the base quality threshold for the look-up of the hash index set to 10 (-q 10). With these settings, SMALT v0.7.5 only reports the best unique gapped alignment for each read. Whenever paired-end information was available, the insert size distribution was inferred with the “sample” command of SMALT prior to mapping. Conversion of SAM format to BAM, sorting, indexing, several mapping statistics, and consensus genotype calling were performed using the tools available in the SAMtools package v1.18 (Li et al. 2009) as described previously (Almeida et al. 2014). Multiple sequence alignments for each reference chromosome were generated from the resulting fasta files. For downstream analysis, all bases with Phred quality score below Q40 (equivalent to a 99.99% base call accuracy) or ambiguous base calls were converted to an “N”.
Phylogeny and Population Structure
Chromosomal single nucleotide polymorphisms (SNPs) were extracted from multiple sequence alignments only if the evaluated site was represented by unambiguous high-confidence alleles in all isolates. SNPs were then concatenated to generate a whole-genome SNP alignment.
Unrooted maximum likelihood phylogeny was estimated using the rapid bootstrap algorithm as implemented in RAxML v7.3.5 (Stamatakis 2006) with GTRGAMMA model of sequence evolution. Population structure of S. cerevisiae was explored using the model-based Bayesian clustering method implemented in STRUCTURE v2.3.4 (Falush et al. 2003). STRUCTURE was run with a subset of 9,860 equally spaced parsimony informative sites. The number of Markov chain Monte Carlo iterations was set to an initial burn-in period of 100,000 iterations, followed by 100,000 iterations of sampling. The ancestry model allowed for admixture and allele frequencies were assumed to be correlated among populations. Five independent simulations were run for each value of K, varying from K = 1 to K = 15, and stability was assessed by monitoring the standard deviation between simulations. The run with the highest estimated log probability of the data was chosen to represent each value of K.
Multilocus Sequence Analysis
Thirteen loci previously used to characterize Chinese isolates (Wang et al. 2012) were retrieved from the available de novo genome sequences using BLASTN (see above) and aligned with FSA v1.15 (Bradley et al. 2009). After alignment, loci were concatenated and sequences with less than 80% of the total length were removed. The phylogenetic history was inferred from the concatenated alignment using the Neighbor-Joining method in MEGA 5 v6.06 (Tamura et al. 2011). Evolutionary distances were computed with the Kimura two-parameter model of sequence evolution and are in units of the number of base substitutions per site. All positions with less than 95% site coverage were eliminated, that is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position. There were a total of 13,830 positions in the final data set. Branch support was estimated from 1,000 nonparametric bootstrap replicates.
Polymorphism and Divergence Analyses
Whole-genome levels of polymorphism and divergence were estimated using Variscan v2.0 (Hutter et al. 2006). Only sites with valid high quality alleles (Q > 40) in at least 75% of ingroup sequences were used in calculations. Sites with more than this threshold were randomly subsampled to 75% of the total number of sequences (defined with the NumNuc parameter together with CompleteDeletion = 0 and FixNum = 1). For divergence estimates between populations, only positions with valid alleles in at least four ingroup individuals were used for calculations (defined with the NumNuc parameter together with CompleteDeletion = 0 and FixNum = 0).
For these analyses, whenever the wine group was compared it was represented only by strains isolated from wine environments (i.e., commercial and wine must strains but not vineyard strains). Strains ZP 530, ZP 1050, and UWOPS 83-787.3 were excluded from the North America group because they were isolated in regions outside North America and strain YPS 1000 was excluded from the Brazilian B1 group for similar reasons.
Screening for Introgressions from Other Saccharomyces Species
We searched for evidence of introgressions from other Saccharomyces species by mapping the reads to a combined reference that includes all the available annotated coding sequences of six Saccharomyces species (S. arboricola, S. cerevisiae, S. kudriavzevii, S. mikatae, S. paradoxus, and S. uvarum) (Scannell et al. 2011; Liti et al. 2013). Reads were mapped to this combined reference using BWA v0.6.2 (Li and Durbin 2009) with default parameters but setting the quality threshold to 10 (-q 10). SAMtools v1.1852 (Li et al. 2009) was used for the manipulation of the resulting BAM files. Only genes with orthologs unambiguously annotated in all six species were analyzed. An ORF was considered to have a foreign origin to S. cerevisiae if its coverage was at least higher than one-fourth of the median whole-genome coverage for the analyzed strain. The ORF coverage was defined as the product of the total number of mapped reads to the orthologous ORFs by the read size, dividing by the sum of the length of each ORF, considering only the ones with more than 25% of reads mapped (relative to the orthologous ORF with the highest number of reads) to control for spurious alignment counts. This coverage threshold allowed for some heterogeneity in the read counts and for the eventual presence of a foreign ORF together with the native S. cerevisiae ORF.
Pairwise divergence between the S. paradoxus (strain YPS 138) and S. cerevisiae (strain S288c) was used as a proxy to search for evidence of DNA segments of S. paradoxus in the genomes of S. cerevisiae strains. Divergence per site, k, (with Jukes–Cantor correction) was calculated using a nonoverlapping sliding window of 10,000 sites, using Variscan v2.0 (Hutter et al. 2006).
Inference of Orthologous Gene Pairs between S. cerevisiae and the North America Population C of S. paradoxus
We predicted coding regions in 24 published de novo assemblies of genomes from North American strains of S. paradoxus using AUGUSTUS, with the built-in set of parameters resulting from training on S. cerevisiae S288C (Stanke and Morgenstern 2005). All predicted ORFs were then translated into protein sequences. Duplicated sequences were eliminated within each putative proteome and a clustering by Reciprocal Best Hits (cRBH) was conducted. The cRBH consisted in making every possible pairwise comparison of proteomes with BLASTP (Camacho et al. 2009), finding all interproteome pairs of proteins which are each other’s best BLASTP hit (on the basis of their e-values), building a network out of those pairs and searching for clusters containing one protein per proteome. We used the BLASTP option for soft filtering of low information segments, since it is known to improve orthology detection (Moreno-Hagelsieb and Latimer 2008). In total, 5,887 curated protein sequences of S. cerevisiae from the Saccharomyces Genome Database—SGD (Cherry et al. 2011) were also included in the cRBH. In order to eliminate weakly connected clusters, the pairs of RBH and the associated alignment scores were passed to the MCL software with an inflation parameter of 3 (Van Dongen 2000). This parameter controls the sizes of the resulting clusters, but testing across the range of typically used values (1.2–5.0) (Van Dongen 2000) showed negligible variation in the output. We then selected the clusters that contained exactly one protein from each of the 24 S. paradoxus proteomes. In order to avoid errors associated with the misprediction of intron boundaries, we also excluded clusters where at least one intron was predicted in any strain or identified in S. cerevisiae. Among the 4,555 remaining clusters, 4,521 contained exactly one curated S. cerevisiae protein sequence and were considered reliable orthogroups. The ORFs of the strain LL2012_027 that were associated with proteins from those orthogroups were used in subsequent analyses as representative of the North America population C of S. paradoxus. The orthogroup inference pipeline described here corresponds to a more stringent version of a cRBH method shown to be more accurate than several other popular methods of orthogroup inference (Salichos and Rokas 2011).
De Novo Assemblies for a More Detailed Analysis of the Introgressed Genes
In order to further explore the origin of the introgressed genes, which are not present in the reference genome, we performed de novo genome assemblies for all the strains included in this study, using SPAdes v.3.1.0 (Bankevich et al. 2012). Prior to assembly, reads were processed with Trimmomatic (Bolger et al. 2014) based on a quality score threshold of 20 for windowed trimming, discarding reads with length less than 30 for single-end and 100 bp for paired-end reads, respectively, or with any “Ns” on them. To retrieve the introgressed genes, a local BLAST database was set up for each genome. The introgressed ORFS were searched by BLASTN, using the correspondent S. cerevisiae ORF sequences available at SGD as queries. Evolutionary distances were computed with the Kimura two-parameter model of sequence evolution and are in units of the number of base substitutions per site. Branch support was estimated from 1,000 nonparametric bootstrap replicates. Homologous sequences retrieved from previously described S. paradoxus populations (European, NRRL Y-17217; Far East, N-44; North America population B, YPS 138; and North America population C, LL2012_027) were used for comparison purposes. Whenever available, homologous sequences from other Saccharomyces species were used as outgroups.
Gene Ontology Analysis
The Standard GO (gene ontology) term discovery was performed with the GO Term Finder tool, available at SGD.
Results
Isolation of S. cerevisiae from Natural Habitats
Using a selective isolation protocol that previously allowed the consistent isolation of Saccharomyces spp. in different natural ecosystems (e.g., Sampaio and Gonçalves 2008; Libkind et al. 2011), we carried out a survey of wild Saccharomyces populations in the Brazilian states of Bahia, Minas Gerais, Paraná, Tocantins, and Roraima, between 2010 and 2013. Given the previously documented association of wild Saccharomyces with the bark of Fagaceae in the Northern Hemisphere (Sniegowski et al. 2002; Sampaio and Gonçalves 2008; Wang et al. 2012) and Nothofagus in Patagonia and Australasia (Almeida et al. 2014) and the absence of such trees in Brazil, we collected preferentially tree bark of various native trees. In total, we analyzed 545 samples, 71% of which from tree bark and the remaining from soil, moss, mushrooms, and plant inflorescences and fruits (table 1). Due to the vast geographic and floristic dimensions of Brazil, our sampling effort must necessarily be viewed as partial. In Bahia, Minas Gerais, and Paraná, in northeastern, southeastern, and south Brazil, respectively, sampling was conducted in mountain regions with a predominant Atlantic rainforest ecosystem, sometimes including contact zones with “Cerrado” (savannah) and “Caatinga” ecosystems. The Atlantic rainforest features a humid tropical climate with mild temperatures. In Tocantins (north Brazil) the predominant biome is Cerrado, whereas in Roraima, the predominant vegetation corresponds to the Amazonian Forest biome and the climate is hot and humid.
Table 1.
Number and Type of samples Used for Saccharomyces Isolations and Respective Results
| No. of Samples | No. of Isolates | Success Rate (%) | |
|---|---|---|---|
| Bark from miscellaneous trees | 251 | 10 | 4 |
| Bark from Tapirira guianensis | 131 | 17 | 13 |
| Bark from cultivated Quercus rubra | 7 | 5 | 71 |
| Other (nonbark) | 156 | 2 | 1 |
Since after the initial round of isolation the most consistent positive results were obtained for bark samples of Tapirira guianensis (Anacardiaceae), a common tree in Brazil especially in riparian areas, we concentrated our efforts on obtaining additional samples from this tree, originating from different regions. Globally the success rate for Saccharomyces isolations from T. guianensis was 13% (table 1). In one of the sampling sites (Santuário do Caraça, Minas Gerais) a few ornamental oak trees (Quercus rubra) imported from North America and planted in the first half of the 1900’s were sampled with a high success rate of isolation (table 1). The other substrates (other trees and other natural substrates) provided lower isolation frequencies that ranged from 1 to 4% (table 1).
In spite of the limited scope of our survey, we obtained 34 Saccharomyces isolates (table 1), which after molecular identification (sequencing of the D1/D2 domains of the 26S rDNA subunit) were all found to belong to S. cerevisiae, even if all samples were simultaneously incubated at high (30 °C) and low (10 °C) temperatures, shown before to allow efficient isolation of both thermotolerant and cryotolerant species, respectively. In this study, all Saccharomyces strains were obtained after incubation at 30 °C.
Phylogeny of Wild Brazilian Isolates
A subset of 28 wild Brazilian S. cerevisiae isolates were selected for whole-genome sequencing and their phylogenetic relationships were analyzed based on 69,321 high-quality polymorphic sites, using for comparison representatives of all the known S. cerevisiae populations for which genomic data is available. This broad phylogenetic analysis included the previously known wild lineages associated with Mediterranean oaks, multiple lineages associated with North American and Japanese oaks, and a Malaysian lineage associated with the inflorescences of Bertram palms as well as a few groups associated with specific fermentation products (supplementary table S1, Supplementary Material online). Whenever possible we avoided the use of strains with admixed (mosaic) genomes since they hamper the reconstruction of phylogenetic relationships and the assessment of population structure. Therefore, we use the same restricted data set of strains as in Almeida et al. (2015) to select for representatives of the known lineages. As shown in figure 1, most of the new Brazilian isolates selected for whole-genome sequencing (68%) were resolved in one new clade (B1), composed almost entirely of Brazilian strains except one North American isolate (YPS 1000). Curiously, two Brazilian isolates were placed in a clade composed mostly of North American isolates and the remaining strains were placed in separate lineages (B2–B4) positioned between the Brazilian main clade and the Mediterranean oak/Wine clades (fig. 1). Therefore, we may conclude that the vast majority of Brazilian wild isolates belong to new, genetically distinct lineages. Within the B1 clade, strains UFMG-CM-Y651 and UFMG-CM-Y652, the only isolates from the state of Paraná, had exceptionally long branches. The genomes of these two strains had a hybrid composition, with conspicuous contributions from S. paradoxus (fig. 2). Not surprisingly the S. paradoxus progenitor could be assigned to the North American population of this species—population B sensu Kuehne et al. (2007) and Leducq et al. (2016) rather than to the European or Far Eastern populations. Therefore these two strains should be viewed as natural hybrids of S. cerevisiae and S. paradoxus. We also assessed the phylogenetic relationships of the remaining Brazilian isolates with Chinese lineages, some of which represent the most divergent S. cerevisiae populations known to date, and for which whole-genome data are not available (Wang et al. 2012). To this purpose we used the available partial sequences of nine genes and four intergenic regions from these isolates (Wang et al. 2012) and the corresponding sequences of representatives of the populations depicted in figure 1 and of our Brazilian isolates to construct an extended multilocus phylogeny (fig. 3). This analysis revealed that the Brazilian isolates are unique and do not belong to any of the Chinese lineages to a close extent.
Fig. 1.—

Whole-genome phylogeny of S. cerevisiae. Phylogenetic tree of 143 strains, inferred from 69,321 SNPs, using the maximum likelihood method as implemented in RAxML with the GTRGAMMA model of sequence evolution. Branch lengths correspond to the expected number of substitutions per site. Support values from bootstrap replicates above 75% are shown. Representatives of previously described populations (Wine, Sake, West Africa, Malaysia, Wild Mediterranean, and Japan/North America) are included, as well as representatives of the new wild lineages from Brazil indicated as B1, B2, B3, and B4. Branch colors highlight the phylogenetic groups and “color-coded circles” highlight individual strains. The five collecting regions in Brazil are indicated in the insert map and in the phylogeny (1-Minas Gerais; 2-Tocantins; 3-Roraima; 4-Paraná; 5-Bahia, no isolates were obtained).
Fig. 2.—
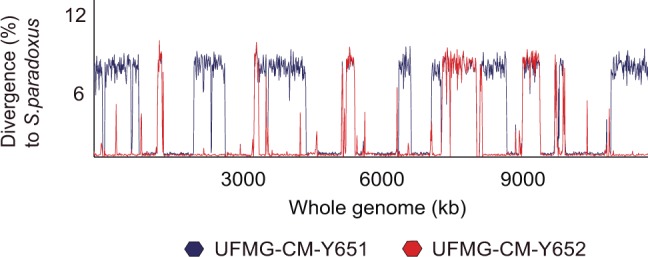
Saccharomyces cerevisiae X S. paradoxus hybrid genomes. Sliding window analysis of strains UFMG-CM-Y651 (“blue”) and UFMG-CM-Y652 (“red”) depicting whole genome % divergence relative to S. paradoxus YPS 138 (North America—population B).
Fig. 3.—
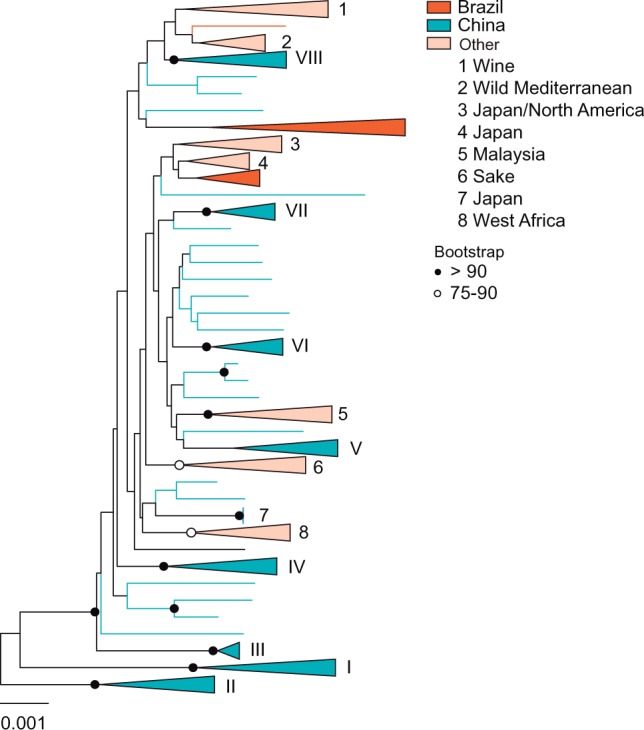
Multilocus phylogeny of Brazilian and Chinese lineages. Neighbor-joining tree inferred from the concatenated alignment of nine genes and four intergenic loci using the Kimura two-parameter model of sequence evolution, depicting the phylogenetic relationships of Brazilian and Chinese (represented by numbers I–VIII) lineages. Some branches are collapsed to indicate major phylogenetic groups. Bootstrap values above 75% (1,000 replicates) are indicated. The “scale bar” represents 0.001 substitutions per nucleotide position.
Population Structure
The impact of the newly uncovered South American lineages on the population structure of S. cerevisiae was analyzed using STRUCTURE (Falush et al. 2003) and testing 2–15 ancestral (K) clusters. The more comprehensive representation of sequence ancestry was achieved with K = 10 (fig. 4) since analyses using higher K values did not reveal new meaningful clusters. Similarly to other recent studies (e.g., Almeida et al. 2015) our analysis recovered the main groups of industrial variants or geographically delimited populations such as Wine (1), Mediterranean oak (2), Sake (3), Philippines (4), Malaysia (5), West Africa (6), North American and/or Japanese populations (7–8) and, in addition, two new Brazilian genetic clusters (9–10). The largest group of Brazilian isolates was grouped in a single cluster (cluster 9—Brazil I) that included almost equal proportions of strains with “pure” genomes and strains with minor contributions of the wine genotype (cluster 1). In the phylogeny of figure 1, all strains with a major contribution of cluster 9 formed the main Brazilian clade (B1). Cluster 10 (Brazil II) was rarely found, being only detected in four strains. Two strains possessed only this genotype and two additional strains had also minor contributions from cluster 9 (Brazil I) and larger contributions from cluster 1 (wine). Together they formed the B3 clade in the phylogeny (fig. 1). The last three Brazilian strains on the right end side of figure 4 had complex mosaic genomes dominated by wine (cluster 1) and sake (cluster 3) ancestry and with minor contributions of Brazilian genotypes. They formed the B2 and B4 clades in the phylogeny of figure 1. Finally, two additional Brazilian strains were placed in the Japan–North America clade in figure 1 and we believe they might represent North American migrants. Whereas strain UFMG-CM-Y255 from Minas Gerais had a pure North American ancestry (cluster 7), strain UFMG-CM-Y640, isolated in Roraima, had a complex structure dominated by cluster 7 but with a relevant component of Brazilian ancestry (cluster 9). Taking into consideration all the Brazilian genotypes that were analyzed with STRUCTURE, the two local genetic clusters represented 75% of the global genetic ancestry (Brazil I: 67%; Brazil II: 8%) whereas the Wine cluster represented 14% and the Japan–North America cluster represented 7% (fig. 5).
Fig. 4.—

—Population structure of S. cerevisiae. STRUCTURE plot based on a subset of 9,860 parsimony informative sites for K = 10. Numbers from 1 to 10 represent the different clusters that capture the maximum representation of population ancestry. The phylogenetic groups inferred in figure 1 are “color-coded” at the bottom of the plot. Migrant strains are marked with “colored diamonds”: “red” (isolated in Brazil), “green” (isolated in Europe), and “blue” (isolated in North America). “Yellow circles” depict non-Brazilian strains with partial Brazilian ancestry.
Fig. 5.—
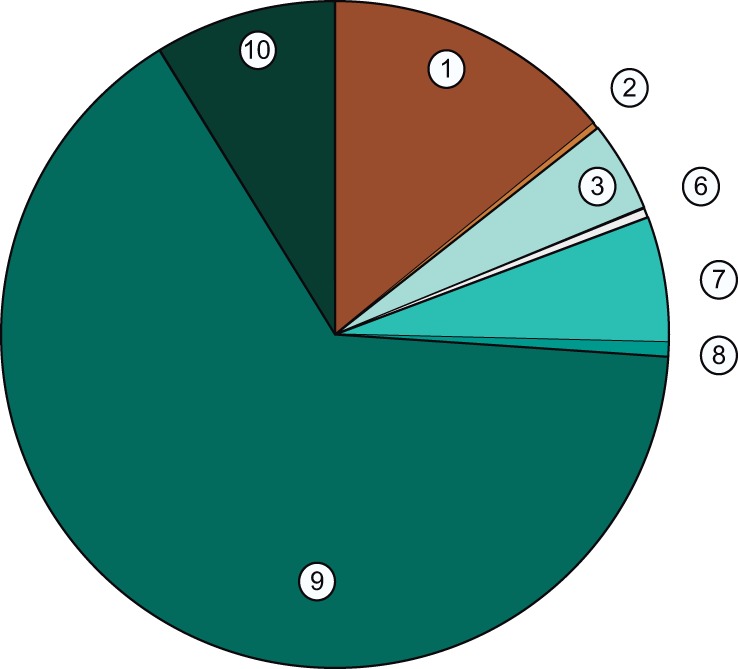
Global genetic ancestry of wild Brazilian strains. Combined STRUCTURE data of the analysed Brazilian strains showing the relative weight of the genetic clusters depicted in figure 4.
Diversity and Divergence
Nucleotide diversity (pairwise differences, π*100) of the Brazilian population for which a more representative number of isolates is available (B1) was 0.0022%. This value is similar to that of the Japan–North America wild population (0.0026%) and higher than the diversity of the Mediterranean oak population (0.0010%) (table 2). The genome-wide Tajima’s D values of the Brazilian and Japan–North America populations were positive, contrary to what was found for the wine and Mediterranean oak populations (table 2). The causes of such differences are at this stage unclear but the negative values could represent population expansion, an expected situation for wine yeasts due to their widespread use in wine fermentation, whereas the positive values could be due to population structure. Nucleotide divergence between Brazil B1 and the wild populations found in Japan and in North America was lower that the divergence from the other populations considered (supplementary table S2, Supplementary Material online).
Table 2.
Whole-Genome Diversity within Populations of Saccharomyces cerevisiae
| No. of Strains | Analyzed Sites | Segregating Sites | Π | θw | Tajima’s D | |
|---|---|---|---|---|---|---|
| Brazil (B1) | 17 | 11,075,830 | 70,292 | 0.002169 | 0.001882 | 0.664367 |
| North America–Japana | 42 | 11,348,218 | 119,184 | 0.002560 | 0.002448 | 0.171544 |
| Mediterranean oaka | 31 | 11,286,153 | 56,053 | 0.000991 | 0.001250 | −0.810951 |
| Winea | 19 | 11,216,288 | 56,367 | 0.001116 | 0.001447 | −0.973222 |
Note.—Diversity values are per site estimates calculated for the total length of the genome
aTaken from Almeida et al. (2015).
Introgressions from S. paradoxus
Since various cases of introgressions involving S. cerevisiae and other species of the genus Saccharomyces (S. arboricola, S. kudriavzevii, S. mikatae, S. paradoxus, and S. uvarum) have been detected (e.g., Liti et al. 2006; Almeida et al. 2014), we investigated the presence of introgressions in the Brazilian populations. Our genome screening of foreign genomic DNA was negative for all species tested except S. paradoxus. Besides the two hybrid genomes already discussed (fig. 2), we detected introgressions from S. paradoxus in 21 out of the remaining 26 Brazilian strains that were investigated. A total of 62 ORFs in 13 chromosomes were implicated in such events and are listed in supplementary table S3, Supplementary Material online. No instance of introgression was found to be fixed in the population (fig. 6a). In most cases the introgression involved a single foreign ORF but in some occasions larger introgressed regions encompassing up to five ORFs were detected (fig. 6a). Although their pattern of distribution among the Brazilian strains was not uniform, two major configurations generally corresponding to the phylogenetic groups B1 and B3 could be recognized (fig. 6a). Moreover, the introgressions appear to be more prevalent in clade B3 that corresponds to cluster 10 (Brazil II) in the STRUCTURE analysis, but are also conspicuous in clade B1. Not surprisingly, most of the introgressions could be assigned to the North American S. paradoxus, as shown in comparisons involving orthologs of the main populations of S. paradoxus (European, Far Eastern, and North American B and C) (fig. 6b and c). More specifically, most of the introgressed ORFs (47 out of 62) were identical to those of the most widespread genetic group in North America, the population B (fig. 6b and c). In seven cases we detected recombinant introgressed sequences involving North American S. paradoxus and S. cerevisiae (fig. 6b and c). In other situations (nine cases) we could not determine with confidence the donor of the introgression that nevertheless appears to belong to an unknown S. paradoxus population that shows more sequence identity to the North American populations than to the European or Far Eastern populations (fig. 6b and c). Interestingly, a GO analysis of the complete set of introgressed genes revealed that it was significantly enriched in genes encoding secondary active transmembrane transporters (supplementary table S4, Supplementary Material online).
Fig. 6.—

Introgressions of Saccharomyces paradoxus into S. cerevisiae. (a) Profiles of presence/absence of introgressions are numbered from 1 to 28 (profiles 1, 19, 24, and 25 depict absence of introgressions) and “color coded” according to the phylogenetic group (B—Brazil; N/J—North America/Japan; MO—Mediterranean oaks). The number of strains in each profile is depicted in the “gray bar” (numerals in “black boxes” refer to the number of strains without introgressions). The introgressed ORFS are listed on the “left side” and the four classes of introgressions are “color coded” (“dark blue”: S. paradoxus North American population B; “light blue”: recombinant S. cerevisiae × S. paradoxus North American population B; “gray”: S. paradoxus—unknown population; and “green”: S. paradoxus—European population). (b) Divergence plots of exemplary sequences showing cases of ORFs introgressed from S. paradoxus North American population B (I); recombinant S. cerevisiae X S. paradoxus North American population B (II); and unknown population of S. paradoxus (III). (c) Phylogeny of introgressed ORFs (same ORFs as in b), using for comparison the available sequences from the different populations of S. paradoxus, and the orthologs of S. cerevisiae, S. mikatae, and S. uvarum.
Using the same approach we searched for introgressions from other Saccharomyces species in the other wild populations of S. cerevisiae for which a relevant number of strains was available. Although for the Japanese and North American wild S. cerevisiae populations no introgressions were detected, we detected introgressions from S. paradoxus in the wild population associated with Mediterranean Oaks (fig. 6a). In this case, introgressed regions originated in the European population of S. paradoxus and involve only three ORFs, one of which is also introgressed in the Brazilian population B3. Taking into consideration that the number of Mediterranean strains investigated is comparable to the number of Brazilian strains studied (24 and 28, respectively), a more regular pattern of distribution is observed among the Mediterranean strains although the number of introgressed ORFs is smaller (fig. 6a).
Discussion
New Tropical Isolates and a Candidate Natural Habitat
Saccharomyces cerevisiae is one of the best understood model organisms but it still has an enigmatic natural ecology (Liti 2015). Besides its association with oak trees in the Northern Hemisphere, natural habitats in other latitudes have not been systematically investigated, suggesting that our knowledge of the natural ecology of this species is far from complete. Moreover, domestication associated with a myriad of foods and beverages like wine, beer, bread, and various traditional and/or local fermented products likely promoted the emergence of domesticated lineages, adding another layer of complexity to the identification and characterization of wild populations. Here we investigated if natural populations of S. cerevisiae are present in Brazil, a country of tropical climate and where oaks and other members of the Fagales do not occur naturally. We surveyed more than 500 natural samples, mainly the bark of native trees. T. guianensis, a tree whose distribution spans a large area from Mexico and Central America to tropical South America, emerged as a possible tropical habitat of S. cerevisiae with an isolation success rate of 13%. Although these values are lower than those obtained for some oak trees in the Northern Hemisphere—for example Sampaio and Gonçalves (2008) reported a success rate of 40% for Quercus pyrenaica—they are comparable with the figures reported by Wang et al. (2012) regarding the isolation of S. cerevisiae in natural environments in China. These findings have to be corroborated by additional field work in order to clarify whether this tree is a consistent tropical S. cerevisiae habitat. It is also noteworthy that the few oaks sampled during the present study yielded a high frequency of S. cerevisiae isolates (71%).
Another important observation of our survey is that unlike in other regions of the globe (e.g., North America, Southern Europe, China, and Japan), S. cerevisiae was the only species of the genus retrieved. We speculate that the cryotolerant species S. eubayanus and S. uvarum, although present in South America (Patagonia) (Libkind et al. 2011), are restricted to regions with cooler climates in the same way that S. cerevisiae (thermotolerant) was not found in Patagonia, or in Quebec (Canada) (Charron et al. 2014), another region with a cool climate. Since both S. cerevisiae and S. paradoxus are thermotolerant it is intriguing why during our field surveys not a single strain of S. paradoxus was isolated. A tentative explanation is that in Brazil the habitats of S. paradoxus and S. cerevisiae overlap less than in other regions, thus reducing the density of S. paradoxus in the type of substrates we surveyed.
New Wild Populations
A phylogenomic analysis of representatives of the Brazilian isolates showed that almost 70% of them clustered in a new clade (B1), the remaining strains being grouped in three additional clades (B2–B4). These groups did not overlap with the previously known main populations of S. cerevisiae that were also used in the analysis. Therefore, the new isolates appear to represent new lineages of S. cerevisiae. Our population analysis carried out in STRUCTURE recovered ten ancestral genetic clusters, eight representing populations already described (Liti et al. 2009; Schacherer et al. 2009; Wang et al. 2012; Cromie et al. 2013; Almeida et al. 2015), and two new clusters associated with the Brazilian isolates. Brazil I (cluster 9) was dominant in most of the isolates, whereas Brazil II (cluster 10) was rare. The conspicuous presence of the Wine genotype in the wild Brazilian populations is noteworthy, since 54% of the strains had some proportion of ancestry in the Wine group. The penetration of the Wine genotype, that represents a notable case of microbe domestication whose origins have been traced to the Mediterranean region (Almeida et al. 2015), in Brazilian wild populations is a probable consequence of human activities that promoted the transatlantic transport of wine strains. These observations provide palpable evidence of the impact of a domesticated lineage in the natural history of S. cerevisiae. Moreover, the detection of domesticated (wine yeast) ancestry in wild Brazilian populations parallels for the first time in microbes observations made earlier in domesticated plants (e.g., Papa and Gepts 2003; Hufford et al. 2013) and animals (e.g., Marshall et al. 2014) documenting gene flow from domesticated to wild populations. Interestingly, the degree of dissemination of the wine genotype in Brazilian natural isolates is not observed for the North American lineages, where only 13% of the strains had traces of this genotype (fig. 4). The causes for such discrepancy are presently unknown and we speculate that they might reside on fundamental ecological differences that the two populations face. The locally dominant Brazil I cluster was rarely found outside Brazil being detected in three non-Brazilian strains (fig. 4).
Migrant Strains
We detected what appear to be transcontinental migrant strains. YPS 1000 represents a likely Brazilian migrant having a pure Brazil I genotype but isolated in North America (figs. 1 and 4). On the other hand, UFMG-CM-Y255 and UFMG-CM-Y640 have a North American genotype and were found in Brazil. Finally, two of the European oak isolates included in this study have also a North American genotype (figs. 1 and 4). Therefore, although the number of candidate migrant strains is low, we document cases of probable transcontinental migration North–South and South–North in the Americas and also from North America to Europe. While cases of transoceanic migration from Europe to North America (Kuehne et al. 2007) and to New Zealand (Zhang et al. 2010) have been documented in S. paradoxus, the transcontinental movement of wild strains of S. cerevisiae has not yet been studied. Some of the migrant strains detected in this investigation revealed mosaic genomes in STRUCTURE, a likely consequence of their more complex history.
A possible source of migrant strains from North America is exotic trees like the red oaks (Q. rubra) that we sampled in Minas Gerais, imported more than seven decades ago from North America. Previous studies have documented the transport to New Zealand of European S. paradoxus probably associated with European oak acorns (Zhang et al. 2010). Although we did not obtain North American migrant strains from oak samples, we detected two representatives of the B1 clade. Nevertheless, we isolated from the same collecting locality, from an unidentified tree, one of the two North American migrant strains (UFMG-CM-Y255) found in this study (supplementary table S1, Supplementary Material online). Interestingly, in the largest phylogenetic group (B1), the only strain that did not show any introgression was YPS 1000, a likely migrant strain that has a Brazilian genotype but was found in North America. With respect to other candidate migrant strains, neither of the two North American genotypes found in Brazil (UFMG-CM-Y255 and UFMG-CM-Y640) nor the two North American strains found in Europe have introgressions.
Hybridization
We obtained evidence for the presence of multiple introgressions, indicative of hybridization of wild Brazilian S. cerevisiae strains with strains closely related to the North American population of S. paradoxus. Introgressions are prevalent in the Brazilian strains as they were detected in 21 of the 26 strains whose genomes were analyzed. Currently three S. paradoxus genetic groups are known in North America (Kuehne et al. 2007). Group A comprises probable European migrants, group B is apparently the most widespread and group C appears limited to cooler climates since in Northeastern North America groups B and C were found to be distributed according to a North–South gradient with C strains more frequent in the North (Leducq et al. 2016). Interestingly, the reports of the presence of S. paradoxus in South America are very scarce and are limited to two isolates of Saccharomyces cariocanus (Naumov et al. 2000), currently considered to be a synonym of S. paradoxus group B (Boynton and Greig 2014). Since we could confirm through sequence comparisons that the observed introgressions into the Brazilian population originate in S. paradoxus group B and since the most logical explanation for the geographic origin of the introgressions is Brazil, because equivalent introgressions have not been found in the Northern Hemisphere, we suggest that the current or historical range of S. paradoxus group B extends to South America.
Overall, the pattern of distribution of the introgressions is compatible with a limited number of original hybridizations between S cerevisiae and S. paradoxus followed by independent losses of S. paradoxus genes. The two strains that belong to clade B1 and have a S. paradoxus subgenome (fig. 2) could represent the initial stage which, after repeated backcrosses with S. cerevisiae, leads to the observed patterns of introgressions in the various strains. Taking into consideration the phylogenetic placement of the introgressed strains in groups B1 and B3, the hypothesis of a single hybridization per lineage followed by independent losses of S. paradoxus regions cannot be discarded. Since the introgressions are not fixed in the Brazilian population and their pattern of occurrence is variable (fig. 6a), they appear to be relatively recent. Even if originating in rare events, the prevalence of introgressed genes in the vast majority of Brazilian strains is in sharp contrast with their absence in the North American and Japanese wild isolates. This disparity does not appear to be caused by lack of contact between S. cerevisiae and S. paradoxus in the Northern hemisphere since it is well-documented that the two species are sympatric in North America (Sniegowski et al. 2002; Leducq et al. 2014), Asia (Wang et al. 2012), and Europe (Sampaio and Gonçalves 2008). In a recent study, Strope et al. (2015) searched for highly diverged syntenic (putatively introgressed) genes in 93 S. cerevisiae genomes corresponding mostly to wine and clinical strains and including a considerable frequency of mosaic genomes. Interestingly, among the 381 protein-coding gene sequences with high similarity with S. paradoxus (>96% identity) identified by Strope et al. (2015), 30 were also found by us. Although the introgression patterns that we found are clearly distinct from those of Strope et al. (2015) and the frequency of introgressions tends to be higher in the Brazilian strains, further studies should aim at clarifying if the introgressions found in the two studies are related.
Hybridization and subsequent introgression are well-known adaptive mechanisms (Arnold and Martin 2010) and can occur as a consequence of environmental changes (Grant and Grant 2002). These and other processes, including the horizontal acquisition of genes seem to be correlated with the need for swift adaptation to new niches or life styles (Wisecaver and Rokas 2015). In line with this, horizontally acquired genes were often found to be involved in interactions with the cell environment (Wisecaver and Rokas 2015), which seems also to be the case for the introgressions identified in the present study, since analysis of the complete set of introgressed genes was found to be significantly enriched in active transporter proteins and transcriptional activators of RNA Polymerase II-transcribed genes (supplementary table S4, Supplementary Material online). Transporters have been found to be among the most frequent functional categories involved in horizontal transfers, possibly due to their preponderant role in the interaction of the cell with the environment (Wisecaver and Rokas 2015) and to their relatively low connectivity that facilitates successful integration of laterally acquired genes in the host. Also, changes in transcriptional regulation have been shown to explain most intraspecific phenotypic differences in a set of S. cerevisiae strains (Treu et al. 2014), mainly due to promoter variability and differentially expressed transcription factors. In face of these findings, it seems plausible that the acquisition of transcription factor genes from a sibling species, which may be differently regulated or show slight but relevant functional differences, may provide an expeditious means to readjust expression of a large number of genes, thereby favoring swift adaptation to a new environment.
We could hypothesize that adaptation of S. cerevisiae to the Brazilian ecosystem, but not to the North American/Asian ecosystem, could select for the retention of introgressed regions. Therefore we tentatively propose that wild S. cerevisiae are originally best adapted to the oak environments of China and Japan, where genetic diversity of this species is the highest (Wang et al. 2012; Almeida et al. 2015), which fits to the model that posits that Asia is the ancestral radiation center of S. cerevisiae wild lineages. One possible scenario may have involved migration to North America, proceeding probably through the Bering Strait, as observed for various animal species, including humans (Hewitt 2004). This migration resulted in the introduction of some of the Asian lineages in the New World. The South American populations could derive from their North American counterparts as the genetic divergence between the Brazilian B1 population and the Japanese and North American populations is lower than the calculated divergence from other populations, which supports a more recent population split from the Asian–North American population complex. In this scenario, adaptation to the tropical ecosystem was facilitated by the access to the S. paradoxus gene pool provided by hybridization events, as a source of rapid diversification. This type of acquisition of genes previously adapted to the environment is probably a much swifter mode of adaptation to a new environment than evolving new functions from preexisting genes, even when compared with the relatively rapid neofunctionalization that often follows gene duplication. A somewhat parallel situation might have occurred at the origin of the Mediterranean wild population of S. cerevisiae. It was recently observed that this population has approximately half of the genetic diversity of other wild populations (Almeida et al. 2015), which is compatible with a relatively recent origin and a founder effect. Hybridization and introgression has been shown to be associated with the formation of phenotypically and genetically distinct lineage of S. paradoxus in North America (Leducq et al. 2016), again supporting the role of these processes in generating diversification. We speculate that the adaptation to the specific conditions of oak forests of Southern Europe may also have been facilitated by the horizontal acquisition of S. paradoxus genes via hybridization. In this hypothetical scenario, both the Brazilian and Mediterranean wild ecosystems appear as secondarily colonized habitats that impose a new threshold of selective pressures as compared with the North American and Asian oak forests that would constitute the primary habitat of wild S. cerevisiae populations. Therefore, the presence or absence of introgressions from S. paradoxus could reveal the strength of the environmental challenges acting on a given S. cerevisiae population.
Data Accessibility
Genome sequencing data have been deposited in EBI’s ENA (https://www.ebi.ac.uk/ena) as PRJEB11698.
Supplementary Material
Supplementary tables S1–S4 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by Fundação para a Ciência e a Tecnologia, Portugal, grants PTDC/BIA-EVF/118618/2010 (J.P.S., P.A., P.G.), PTDC/AGR-ALI/118590/2010 (J.P.S., P.A., P.G., R.B.), UID/Multi/04378/2013 (J.P.S., P.G.), and SFRH/BD/77390/2011 (P.A.), by Conselho Nacional de Desenvolvimento Científico e Tecnologico-CNPq (CAR, process numbers 560715/2010-2 and 457499/2014-1, PBM process number 457443/2012-0) and Fundação de Amparo a Pesquisa de Minas Gerais FAPEMIG and VALE S.A (CAR, process number RCP-00094-10). Work of C.R.L. on this project was supported by a NSERC Discovery grant. C.R.L. holds the Canada Research Chair in Evolutionary Cell and Systems Biology. The authors thank Dr. Siu Mui Tsai, Universidade de São Paulo, Brazil, for making available strain UFMG-CM-Y640.
Literature Cited
- Almeida P, et al. 2014. A Gondwanan imprint on global diversity and domestication of wine and cider yeast Saccharomyces uvarum. Nat Commun. 5:4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida P, et al. 2015. A population genomics insight into the Mediterranean origins of wine yeast domestication. Mol Ecol. 24:5412–5427. [DOI] [PubMed] [Google Scholar]
- Arnold ML, Martin NH. 2010. Hybrid fitness across time and habitats. Trends Ecol Evol. 25:530–536. [DOI] [PubMed] [Google Scholar]
- Bankevich A, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergström A, et al. 2014. A high-definition view of functional genetic variation from natural yeast genomes. Mol Biol Evol. 31:872–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing J, Han PJ, Liu WQ, Wang QM, Bai FY. 2014. Evidence for a Far East Asian origin of lager beer yeast. Curr Biol. 24:380–381. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boynton PJ, Greig D. 2014. The ecology and evolution of non-domesticated Saccharomyces species. Yeast 31:449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley RK, et al. 2009. Fast statistical alignment. PLoS Comput Biol. 5:e1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron G, Leducq JB, Bertin C, Dubé AK, Landry CR. 2014. Exploring the northern limit of the distribution of Saccharomyces cerevisiae and Saccharomyces paradoxus in North America. FEMS Yeast Res. 14:281–288. [DOI] [PubMed] [Google Scholar]
- Cherry JM, et al. 2011. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 40:D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani M, Mannazzu I, Marinangeli P, Clementi F, Martini A. 2004. Contribution of winery-resident Saccharomyces cerevisiae strains to spontaneous grape must fermentation. Antonie Van Leeuwenhoek 85:159–164. [DOI] [PubMed] [Google Scholar]
- Cromie GA, et al. 2013. Genomic sequence diversity and population structure of Saccharomyces cerevisiae assessed by RAD-seq. G3 3:2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Benavides JA. 2005. Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLoS Genet. 1:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard MR, Greig D. 2015. Saccharomyces cerevisiae: a nomadic yeast with no niche? FEMS Yeast Res. 15: fov009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant PR, Grant BR. 2002. Unpredictable evolution in a 30-year study of Darwin’s finches. Science. 296:707–711. [DOI] [PubMed] [Google Scholar]
- Hewitt GM. 2004. The structure of biodiversity—insights from molecular phylogeography. Front Zool. 1:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufford MB, et al. 2013. The genomic signature of crop-wild introgression in maize. PLoS Genet. 9:e1003477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter S, Vilella AJ, Rozas J. 2006. Genome-wide DNA polymorphism analyses using VariScan. BMC Bioinformatics 7:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyma KE, Fay JC. 2013. Mixing of vineyard and oak-tree ecotypes of Saccharomyces cerevisiae in North American vineyards. Mol Ecol. 22:2917–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LJ, et al. 2004. Population genetics of the wild yeast Saccharomyces paradoxus. Genetics 166:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehne HA, Murphy HA, Francis CA, Sniegowski PD. 2007. Allopatric divergence, secondary contact, and genetic isolation in wild yeast populations. Curr Biol. 17:407–411. [DOI] [PubMed] [Google Scholar]
- Leducq JB, et al. 2014. Local climatic adaptation in a widespread microorganism. Proc Biol Sci. 281:20132472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leducq JB, et al. 2016. Speciation driven by hybridization and chromosomal plasticity in a wild yeast. Nat Microbiol. 1:1–10 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libkind D, et al. 2011. Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proc Natl Acad Sci U S A. 108:14539–14544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G. 2015. The fascinating and secret wild life of the budding yeast S. cerevisiae. eLife 4:e05835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Barton DBH, Louis EJ. 2006. Sequence diversity, reproductive isolation and species concepts in Saccharomyces. Genetics 174:839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, et al. 2009. Population genomics of domestic and wild yeasts. Nature 458:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, et al. 2013. High quality de novo sequencing and assembly of the Saccharomyces arboricolus genome. BCM Genomics 14:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall FB, Dobney K, Denham T, Capriles JM. 2014. Evaluating the roles of directed breeding and gene flow in animal domestication. Proc Natl Acad Sci U S A. 111:6153–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini A. 1993. Origin and domestication of the wine yeast Saccharomyces cerevisiae. J Wine Res. 4:165–176. [Google Scholar]
- Moreno-Hagelsieb G, Latimer K. 2008. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 24:319–324. [DOI] [PubMed] [Google Scholar]
- Naumov GI, James SA, Naumova ES, Louis EJ, Roberts IN. 2000. Three new species in the Saccharomyces sensu stricto complex: Saccharomyces cariocanus, Saccharomyces kudriavzevii and Saccharomyces mikatae. Int J Syst Evol Microbiol. 50:1931–1942. [DOI] [PubMed] [Google Scholar]
- Naumov GI, Naumova ES, Sniegowski PD. 1998. Saccharomyces paradoxus and Saccharomyces cerevisiae are associated with exudates of North American oaks. Can J Microbiol. 44:1045–1050. [PubMed] [Google Scholar]
- Papa R, Gepts P. 2003. Asymmetry of gene flow and differential geographical structure of molecular diversity in wild and domesticated common bean (Phaseolus vulgaris L.) from Mesoamerica. Theor Appl Genet. 106:239–250. [DOI] [PubMed] [Google Scholar]
- Salichos L, Rokas A. 2011. Evaluating ortholog prediction algorithms in a yeast model clade. PLoS One 6:e18755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio JP, Gonçalves P. 2008. Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl Environ Microbiol. 74:2144–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell DR, et al. 2011. The awesome power of yeast evolutionary genetics: new genome sequences and strain resources for the Saccharomyces sensu stricto genus. G3 1:11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacherer J, Shapiro JA, Ruderfer DM, Kruglyak L. 2009. Comprehensive polymorphism survey elucidates population structure of S. cerevisiae. Nature 458:342–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski PD, Dombrowski PG, Fingerman E. 2002. Saccharomyces cerevisiae and Saccharomyces paradoxus coexist in a natural woodland site in North America and display different levels of reproductive isolation from European conspecifics. FEMS Yeast Res. 1:299–306. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Stanke M, Morgenstern B. 2005. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33:W465–W467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strope P, et al. 2015. The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res. 25:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvester K, et al. 2015. Temperature and host preferences drive the diversification of Saccharomyces and other yeasts: a survey and the discovery of eight new yeast species. FEMS Yeast Res. 15: fov002. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treu L, et al. 2014. The impact of genomic variability on gene expression in environmental Saccharomyces cerevisiae strains. Environ Microbiol. 16:1378–1397. [DOI] [PubMed] [Google Scholar]
- Van Dongen SM. 2000. Graph clustering by flow simulation [PhD thesis]. The Netherlands: University of Utrecht.
- Vaughan-Martini A, Martini A. 1995. Facts, myths and legends on the prime industrial microorganism. J Ind Microbiol. 14:514–522. [DOI] [PubMed] [Google Scholar]
- Wang QM, Liu WQ, Liti G, Wang SA, Bai FY. 2012. Surprisingly diverged populations of Saccharomyces cerevisiae in natural environments remote from human activity. Mol Ecol. 21:5404–5417. [DOI] [PubMed] [Google Scholar]
- Wisecaver JH, Rokas A. 2015. Fungal metabolic gene clusters—caravans traveling across genomes and environments. Microb Physiol Metab. 6:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Skelton A, Gardner RC, Goddard MR. 2010. Saccharomyces paradoxus and Saccharomyces cerevisiae reside on oak trees in New Zealand: evidence for migration from Europe and interspecies hybrids. FEMS Yeast Res. 10:941–947 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome sequencing data have been deposited in EBI’s ENA (https://www.ebi.ac.uk/ena) as PRJEB11698.