

Abstract
The mitochondrial genomes of flowering plants experience frequent insertions of foreign sequences, including linear plasmids that also exist in standalone forms within mitochondria, but the history and phylogenetic distribution of plasmid insertions is not well known. Taking advantage of the increased availability of plant mitochondrial genome sequences, we performed phylogenetic analyses to reconstruct the evolutionary history of these plasmids and plasmid-derived insertions. Mitochondrial genomes from multiple land plant lineages (including liverworts, lycophytes, ferns, and gymnosperms) include fragmented remnants from ancient plasmid insertions. Such insertions are much more recent and widespread in angiosperms, in which approximately 75% of sequenced mitochondrial genomes contain identifiable plasmid insertions. Although conflicts between plasmid and angiosperm phylogenies provide clear evidence of repeated horizontal transfers, we were still able to detect significant phylogenetic concordance, indicating that mitochondrial plasmids have also experienced sustained periods of (effectively) vertical transmission in angiosperms. The observed levels of sequence divergence in plasmid-derived genes suggest that nucleotide substitution rates in these plasmids, which often encode their own viral-like DNA polymerases, are orders of magnitude higher than in mitochondrial chromosomes. Based on these results, we hypothesize that the periodic incorporation of mitochondrial genes into plasmids contributes to the remarkable heterogeneity in substitution rates among genes that has recently been discovered in some angiosperm mitochondrial genomes. In support of this hypothesis, we show that the recently acquired ψtrnP-trnW gene region in a maize linear plasmid is evolving significantly faster than homologous sequences that have been retained in the mitochondrial chromosome in closely related grasses.
Keywords: angiosperms, DNA polymerase, mtDNA, mitochondrial plasmids, mutation rate
Introduction
Identifying the factors that determine rates of DNA sequence evolution remains a fundamental challenge in the field of molecular evolution. Land plant mitochondrial genomes offer valuable opportunities for pursuing this challenge because they are some of the slowest evolving eukaryotic genomes ever identified (Wolfe et al. 1987; Drouin et al. 2008; Richardson et al. 2013). Rates of nucleotide substitution in plant mitochondrial DNA (mtDNA) are generally slower than in the plastid and nuclear genomes. The relatively low mitochondrial substitution rates are a derived state in land plants (Smith et al. 2014) and contrast with the rapid sequence evolution in mitochondrial genomes in many other eukaryotes, including bilaterian animals and yeast (Brown et al. 1979; Lynch et al. 2008).
Plants also exhibit remarkable heterogeneity in mitochondrial substitution rates. In several angiosperm lineages, there have been mysterious genome-wide increases in rates of mtDNA evolution (Cho et al. 2004; Parkinson et al. 2005; Sloan, Alverson, et al. 2012; Skippington et al. 2015). In even more puzzling cases, some species have experienced massive gene-specific accelerations, while the rest of the mitochondrial genes maintain typically slow rates of nucleotide substitution (Mower et al. 2007; Sloan et al. 2009). For example, protein-coding genes within the Ajuga reptans mitochondrial genome differ by 340-fold in synonymous (i.e., “silent”) substitution rates (Zhu et al. 2014). These increases in nucleotide substitution rate have been interpreted as resulting from changes in underlying mutation rates, but the specific mechanisms remain elusive.
Another important feature of many plant mitochondrial genomes is their propensity to acquire foreign or “promiscuous” DNA, which comes from diverse sources including the nucleus, plastids, bacteria, viruses, and mitochondria from other plant species (Ellis 1982; Knoop et al. 2011; Mower et al. 2012). In addition, plant mitochondria contain linear plasmids that are similar to mitochondrial plasmids found in other eukaryotic lineages and were likely acquired by horizontal transmission from fungi (Handa 2008). The genealogical relationships among angiosperm plasmid and plasmid-derived genes conflict with established angiosperm phylogenetic relationships (Robison and Wolyn 2005; McDermott et al. 2008), indicating a possible history of horizontal transfer among flowering plants, which has also been observed for plant mitochondrial genes, introns, and even entire genomes (Bergthorsson et al. 2003; Sanchez-Puerta et al. 2008; Rice et al. 2013; Park et al. 2015).
Linear plasmids can exist as standalone extrachromosomal elements, but their sequences can also be physically integrated into the mitochondrial genome. They are not known to encode an integrase function but instead undergo recombination involving repeated sequences that are shared between plasmids and the mitochondrial chromosome (Brown and Zhang 1995). Plant mitochondrial plasmids often contain DNA polymerase (DPO) and RNA polymerase (RPO) genes, suggesting that they are capable of autonomous replication and transcription (Kuzmin and Levchenko 1987). The plasmid-encoded DPO genes are related to family B DNA polymerases found in some viruses (Knopf 1998; Filee et al. 2002) and are clearly distinct from the nuclear-encoded Pol I-like polymerases that are responsible for replication of plant mitochondrial and plastid genomes (Cupp and Nielsen 2014). Mitochondrial plasmids also exhibit lower guanine-cytosine (GC) content (Handa 2008) than the rest of the mitochondrial genome (Sloan and Taylor 2010), further supporting the interpretation that they are replicated independently.
Analysis of complete mitochondrial genomes in Zea mays has found that integrated copies of linear plasmids have a disproportionately large number of single nucleotide polymorphisms (SNPs; Allen et al. 2007), which raises the possibility that these plasmids may contribute to elevated and variable rates of sequence evolution. Plasmids are capable of taking up genes from the mitochondrial genome (Leon et al. 1989), and plasmid-derived sequences have been transferred to the mitochondrial chromosome in both angiosperms (McDermott et al. 2008) and liverworts (Weber et al. 1995). However, our understanding of the phylogenetic distribution and evolutionary history of plasmid-derived sequences remains limited. Here, we take advantage of the large number of green plant mitochondrial genomes that are now available to address the following questions: 1) How widely distributed are mitochondrial plasmid sequences among the major lineages of green plants? 2) To what extent is the diversity of plasmid-derived sequences consistent with a history of vertical versus horizontal transmission? and 3) Do the rates of sequence evolution in plasmid-derived sequences differ from those of typical plant mitochondrial genes?
The Phylogenetic Distribution and Evolutionary History of Linear Plasmids in Plant Mitochondria
Plasmid-derived sequences are widespread in land plant mitochondrial genomes. We performed a BLAST-based search for DPO and/or RPO genes in all sequenced green plant mitochondrial genomes and found hits below an e-value threshold of 1 × 10−6 in all major embryophyte lineages except hornworts and mosses (fig. 1 and supplementary table S1, Supplementary Material online). No green algae matches were found that met this significance threshold, but a TBLASTN search returned a weak hit (e-value of 3 × 10−5) aligning a small portion of an RPO open reading frame (ORF) from the angiosperm Lolium perenne to the mtDNA of the chlorophyte Pseudendoclonium akinetum (56 amino acids with 46% identity and 1 indel). This region (nucleotide position 10,486–10,653) is not found within any ORF or annotated gene within the P. akinetum mitochondrial genome (GenBank accession NC_005926). Therefore, although it is possible that the hit represents a small fragment of an ancient linear plasmid insertion in this chlorophyte lineage, we are not able draw any definitive conclusions about the origins of this short sequence or about the history, if any, of linear mitochondrial plasmids in P. akinetum.
Fig. 1.—

Summary of the distribution of plasmid-derived insertions of DPO and RPO genes in mitochondrial genomes across the green plant phylogeny as detected by BLAST searches (supplementary table S1, Supplementary Material online). The phylogenetic relationships follow the reconstruction in figure 2 from Wickett et al. (2014).
Plasmid-derived sequence was most abundant in angiosperm mtDNA (fig. 1 and supplementary table S1, Supplementary Material online), with 74.5% of surveyed angiosperm mitochondrial genomes having significant similarity to DPO and/or RPO sequences. Identified sequences within angiosperms were also much more intact than those in other land plants, with many ORFs >3 kb in length. In contrast, the longest sequence fragments detected outside angiosperms were <600 bp, which suggest that these other land plant lineages may have had ancient associations with mitochondrial plasmids that are no longer active. In contrast, free plasmids still exist in the mitochondria of many angiosperms (Handa 2008), so it is not surprising that integrated plasmid gene sequences are much more common and intact in flowering plant mtDNA (fig. 1 and supplementary table S1, Supplementary Material online).
For multiple reasons, it is likely that we are underestimating the prevalence of plasmid-derived insertions and the distribution of linear mitochondrial plasmids across the plant phylogeny. First, our search was based on only two genes (DPO and RPO), but plasmids sometimes lack one or both of these polymerase genes and usually contain additional genes (predominantly uncharacterized ORFs, which are difficult to detect or compare across species because of their lack of sequence conservation). Therefore, some insertions would be undetectable based on our methods. Second, the extreme level of sequence divergence in plasmid genes and the fact that copies inserted into mitochondrial chromosomes generally appear to degenerate as pseudogenes make it difficult to detect significant similarity between plasmid-derived sequences that are truly homologous. Finally, linear plasmids may be present in mitochondria without leaving any inserted fragments in the mitochondrial chromosome. For example, the sequenced mitochondrial genomes of Daucus carota (Iorizzo et al. 2012) and Brassica napus (Handa 2003) lacked any detectable insertions (supplementary table S1, Supplementary Material online) even though these species are known to have free plasmids and integrated plasmid sequence have been documented in other D. carota cytotypes (Robison and Wolyn 2005).
We performed more detailed phylogenetic and cophylogenetic analyses to infer the transmission history of linear mitochondrial plasmids. These analyses were restricted to angiosperms because the identified sequences outside of flowering plants were too short and fragmented to provide a robust phylogenetic signal. We identified numerous well-supported conflicts between plasmid(-derived) and angiosperm phylogenies (figs. 2 and 3 and supplementary figs. S1–S6 and file S7, Supplementary Material online), which is consistent with previous findings rejecting a single plasmid origin and strict vertical inheritance (Robison and Wolyn 2005; McDermott et al. 2008). In addition, there were instances where multiple copies of DPO and RPO sequences from the same species failed to form monophyletic clades. For example, two divergent copies of the DPO sequence present in the same Ferrocalamus rimosivaginus mitochondrial genome were clearly resolved into two different clades (supplementary figs. S1 and S2, Supplementary Material online). Therefore, the history of linear mitochondrial plasmids must involve horizontal transfer among angiosperms and/or multiple independent acquisitions from fungi or other taxa. The latter scenario would mean that the closest extant relatives of angiosperm mitochondrial plasmids have yet to be identified because known plasmids in flowering plants appear to form a monophyletic group to the exclusion of fungal sequences (McDermott et al. 2008).
Fig. 2.—

Partial concatenation tree with reduced terminal sampling (27 terminals) that is arbitrarily rooted using midpoint rooting for consistency with the cophylogenetic analyses. The most likely topology identified by PhyML is shown with likelihood bootstrap (left) and SH-like aLRT (right) values above branches and parsimony bootstrap values (when applicable) below branches. The three pairs of cases wherein the DPO and RPO sequences from the same genome assembly that were not concatenated because of clear topological conflict between the gene trees are labeled, with the exception that the RPO sequence from Rhazya stricta has been excluded. Branches that are contradicted on the strict consensus of most parsimonious trees are indicated by asterisks flanking the highest contradictory jackknife value. The phylogram of this tree is presented in supplementary fig. S7, Supplementary Material online.
Fig. 3.—

"Tanglegram" illustrating the similarities and conflicts between the phylogenies of linear mitochondrial plasmids and angiosperms, based on the inferred maximum-likelihood topology (fig. 2) with branches with <50% bootstrap support collapsed into polytomies. This figure was generated with TreeMap v3 build 1243 (https://sites.google.com/site/cophylogeny/home, last accessed December 21, 2015).
Despite the evidence for horizontal transfer, cophylogenetic analyses identified a nonrandom level of topological similarity between plasmid gene trees and the angiosperm phylogeny (fig. 3 and table 1). For all plasmid data sets (DPO, RPO, and a partial concatenation of both genes at different levels of taxon sampling; see Materials and Methods), we found more topological congruence with the angiosperm phylogeny than expected based on random tip-mapping (table 1). Nonrandom congruence between gene trees is typically taken as evidence of cotransmission or cospeciation (Brooks and McLennan 1993; Moran and Baumann 1994), although other mechanisms can potentially create this pattern (de Vienne et al. 2007; Andam et al. 2010). In this case, regions of similarity between plasmid and angiosperm phylogenies most likely reflect sustained periods of vertical transmission or mechanisms of horizontal transmission that favor transfers among very close relatives.
Table 1.
Summary of Cophylogenetic Analyses
| Gene | Species | Sequences | Observed Cost | Random Cost (mean) | P Value |
|---|---|---|---|---|---|
| DPO | 20 | 24 | 27 | 31.34 | 0.017 |
| DPO | 23 | 28 | 32 | 37.84 | 0.002 |
| RPO | 19 | 24 | 26 | 31.84 | 0.008 |
| RPO | 23 | 28 | 32 | 38.42 | 0.004 |
| Concatenated | 20 | 27 | 27 | 39.85 | <0.001 |
| Concatenated | 34 | 39 | 48 | 56.38 | <0.001 |
Note.—For each data set, the observed cost is the minimum total event costs identified as being needed to reconcile the plasmid gene tree with the angiosperm phylogeny. Lower costs are indicative of more congruent trees. The random cost is derived from the mean of 1,000 permutations of the data set (random tip mappings), and the P value indicates where the observed cost falls within that random distribution. Two different analyses are reported for each gene/concatenation, corresponding to the full and reduced taxon samplings described in the Materials and Methods.
Rapid Evolution of Plasmid and Plasmid-Derived Sequences in Angiosperms
The levels of sequence divergence between plasmid-derived sequences in angiosperm mitochondrial genomes greatly exceed what is typically observed for mitochondrial genes (supplementary fig. S6 and tables S2 and S3, Supplementary Material online). Even after extensive trimming to remove the most variable positions within the DPO and RPO alignments, plasmid-derived ORFs in angiosperms share as little as 52% amino acid identity (supplementary tables S2 and S3, Supplementary Material online). The divergence is even more striking when considered across the entire length of the untrimmed sequences. For example, two DPO sequences obtained from different populations of Silene vulgaris were identified as each other’s closest relatives in the dataset (fig. 2 and supplementary figs. S1 and S2, Supplementary Material online) and yet shared only 51% amino acid identity across their entire lengths. Similarly high levels of divergence were observed between each of these S. vulgaris DPO sequences and the copy found in the mitochondrial genome of its congener Silene latifolia, which diverged ∼5 Ma (Rautenberg et al. 2012). For comparison, there is only 0.2% amino acid sequence polymorphism between the two S. vulgaris populations and only 0.4% fixed divergence with S. latifolia for the set of eight complex I proteins encoded by the mitochondrial genome (Sloan, Muller, et al. 2012). In cases such as this where the plasmid gene trees reflect expected phylogenetic relationships (i.e., all the Silene samples cluster together), the extreme levels of sequence divergence between plasmid-derived genes are likely a result of high rates of sequence evolution rather than ancient divergence times that greatly exceed the divergence times between their angiosperm host species.
There is one known case in which a sequence from a plant mitochondrial genome has been transferred to a linear plasmid, which occurred recently in the Z. mays lineage. A 474-bp region containing the functional transfer RNA (tRNA) gene trnW and the pseudogene ψtrnP is found in a 2.3-kb linear mitochondrial plasmid in Z. mays (Leon et al. 1989) but located in the mitochondrial chromosome in other grasses, including other Zea species (supplementary fig. S7, Supplementary Material online). This recent transfer event creates an opportunity to directly compare rates of evolution for sequences located on plasmids versus the mitochondrial chromosome. Maximum likelihood analysis of the ψtrnP-trnW region resulted in a longer branch length for Z. mays than in related grasses (fig. 4), indicating an accelerated rate of sequence evolution for the copy in the Z. mays plasmid. Relative rate tests confirmed that there was a statistically significant difference in nucleotide substitution rates between the plasmid-encoded ψtrnP-trnW region in Z. mays and the homologous region in four close relatives: Zea perennis (P = 0.011), Zea luxurians (P = 0.011), Tripsacum dactyloides (P = 0.005), and Sorghum bicolor (P = 0.035). None of the observed substitutions in Z. mays occurred within the functional trnW gene, which is completely identical to the inferred ancestral sequence for grasses (supplementary fig. S7, Supplementary Material online).
Fig. 4.—
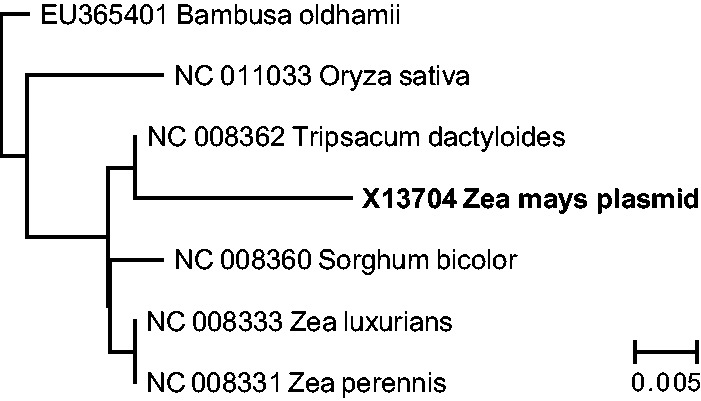
Maximum-likelihood phylogram based on the ψtrnP-trnW sequence region that is present in a small linear plasmid in Zea mays but located in the mitochondrial chromosome in related grasses. The branch lengths are scaled to number of nucleotide substitutions per site, illustrating the accelerated rate of sequence evolution in the plasmid copy. The placement of the root of the tree was determined using the outgroup Phoenix dactylifera.
One potential nonbiological explanation for the higher observed levels of sequence divergence in Z. mays is that there were errors that occurred in the original sequencing of the plasmid (Leon et al. 1989). However, resequencing the ψtrnP-trnW region from 15 accessions of Z. mays (including B37, which was used for the original sequencing study) consistently produced a sequence that was almost identical to the previously published sequence except that it differed by a single SNP (supplementary fig. S7 and table S4, Supplementary Material online; GenBank accession KT444594). Repeating the relative rate tests with this new sequence produced qualitatively similar results (data not shown). Although we did not find SNPs within our chosen set of Z. mays samples, there was evidence of length polymorphism within individuals associated with a homopolymer region (positions 116–123 in GenBank accession KT444594). We consistently observed stuttering in sequencing reads after this region, indicating the presence of multiple competing products with varying homopolymer lengths.
Linear Plasmids as Causes of Heterogeneous Substitution Rates in Plant Mitochondrial Genomes
Based on three key observations, we hypothesize that linear plasmids are partially responsible for variation in rates of molecular evolution among genes in angiosperm mitochondrial genomes. First, there is a history of bidirectional transfer of DNA sequence between mitochondrial chromosomes and plasmids. The presence of the trnW gene in the small linear plasmid of Z. mays demonstrates that functional mitochondrial genes can be moved to plasmids (Leon et al. 1989), and whole-genome sequencing has revealed that mitochondrial chromosomes are littered with plasmid-derived insertions (fig. 1 and supplementary table S1, Supplementary Material online). Second, linear plasmids replicate independently of the mitochondrial chromosome and often encode their own viral-like DNA polymerases. Therefore, plasmids may experience more error-prone replication and/or fail to utilize the recombinational repair machinery that is likely responsible for low rates of nucleotide substitutions in plant mtDNA (Christensen 2013, 2014). Third, rates of sequence evolution for plasmid genes appear to be dramatically higher than for the mitochondrial chromosome. Based on these three observations, we propose a simple model in which mitochondrial genes are occasionally transferred to extrachromosomal plasmids, resulting in episodes of accelerated sequence evolution before being reincorporated into the mitochondrial chromosome.
Our hypothesized model is supported by the observation that the chromosomally derived ψtrnP-trnW region in the small maize linear plasmid is evolving significantly faster than homologous sequences that are retained in the mitochondrial genome in closely related species (fig. 4). This model could explain the recent finding that some angiosperm mitochondrial genomes have experienced major gene-specific accelerations in synonymous substitution rates. The clearest examples of this phenomenon have been described in Ajuga (Zhu et al. 2014) and Silene (Sloan et al. 2009). Notably, we found relatively full-length insertions of plasmid polymerase genes in the mitochondrial genomes from species in each of these genera (supplementary table S1 and files S1 and S2, Supplementary Material online), suggesting especially recent interactions with linear plasmids. Furthermore, free linear mitochondrial plasmids have been identified (but not yet characterized with respect to sequence content) in some Swedish populations of S. vulgaris (Andersson-Ceplitis and Bengtsson 2002). Under a slight variant of this proposed model, it is also possible that plasmids and mitochondrial chromosomes could have duplicate copies of the same gene and that recombination (gene conversion) between the two copies could periodically introduce plasmid mutations into the mitochondrial genome. This mechanism could explain why the atp9 gene, which is unusually fast evolving throughout the tribe Sileneae, was found to exist in multiple copies in many Sileneae species (Sloan et al. 2009).
Based on the hypothesis that mitochondrial linear plasmids are responsible for gene-specific accelerations in some angiosperm mitochondrial genomes, we would predict that further identification and sequencing of free linear plasmids in plant mitochondria will reveal additional examples of mitochondrial genes that have been acquired by plasmids and undergone accelerated rates of sequence evolution. To date, free mitochondrial plasmids have only been sequenced in four angiosperm species (Handa 2008), and the small linear plasmid in Z. mays is the only documented case in plants of a functional mitochondrial gene being transferred to a plasmid (Leon et al. 1989). Examining additional free linear plasmids in angiosperm mitochondria would be particularly valuable because there are some important uncertainties related to the accelerated rate of sequence evolution in the ψtrnP-trnW region in Z. mays. In particular, unlike many other plasmids, the small linear plasmid in Z. mays does not encode its own DNA polymerase gene, so it is not clear if and how it replicates autonomously. Nevertheless, the plasmid’s low GC content (36.5%) indicates that it is subject to different mutation pressures than the mitochondrial chromosome. Also, although the ψtrnP-trnW region on the Z. mays plasmid was subject to a significant rate acceleration (fig. 4), its overall level of sequence divergence is still low (>97% nucleotide identity with other Zea species), and we did not find evidence of SNPs in the plasmid-borne ψtrnP-trnW region among different Z. mays accessions (supplementary table S4, Supplementary Material online). Therefore, the extent to which the transfer of mitochondrial genes to linear plasmids could be responsible for much larger observed levels of sequence divergence remains unclear.
The distribution and evolutionary history of linear mitochondrial plasmids in plants and their potential role in altering rates of sequence evolution have a number of parallels in mitochondrial evolution throughout the eukaryotic phylogeny. For example, the spread of linear plasmids bears many similarities to the distribution of mitoviruses (Bruenn et al. 2015). In addition, a similar hypothesis to what we present here regarding the effect of linear mitochondrial plasmids on rates of mitochondrial genome evolution has been proposed for the ciliate Oxytricha trifallax (Swart et al. 2012). It is also noteworthy that Pol γ, which is encoded in the nucleus but responsible for replication of the rapidly evolving mitochondrial genomes in fungi and metazoans, appears to be phage derived (Shutt and Gray 2006). Therefore, the invasion of selfish genetic elements with error-prone, viral-like replication machinery may be a recurring process that has shaped the dramatic variation in rates of mitochondrial sequence evolution across eukaryotes.
Materials and Methods
Green Plant Mitochondrial Genome Data Set
We obtained the complete nucleotide sequences of all published green plant mitochondrial genomes in the National Center for Biotechnology Information (NCBI) Genome website as of March 10, 2015 (supplementary table S1, Supplementary Material online). In addition, we were provided access to unpublished mitochondrial genome assemblies from the gymnosperms Ginkgo biloba and Welwitschia mirabilis and the ferns Equisetum hyemale and Ophioglossum californicum (Mower JP, personal communication). All genomes were analyzed based on their reported sequence on GenBank. Therefore, we cannot rule out the possibility that misassemblies may have occurred in the original studies if both integrated and free plasmids were present in the same mtDNA samples.
BLAST Searches to Identify Plasmid and Plasmid-Derived Sequences and to Determine Presence/Absence in Green Plant Mitochondrial Genomes
To identify published DNA sequences related to plant linear plasmids, DPO and RPO gene sequences from the B. napus mitochondrial linear plasmid (GenBank accession AB073400) were first searched against the entire NCBI nucleotide collection (nr/nt) database with NCBI-TBLASTN. Predicted DNA and RNA polymerase ORFs were extracted from identified BLAST hits using the program ORF Finder at the NCBI website (http://www.ncbi.nlm.nih.gov/gorf/gorf.html, last accessed December 21, 2015). To perform a more thorough search specifically in our set of green plant mitochondrial genomes, we used all identified plant ORFs longer than 1,500 bp as queries for NCBI-TBLASTN and NCBI-BLASTN version 2.2.30+ searches against the mitochondrial genomes. The TBLASTN searches were run with default parameters, and the BLASTN searches were run with the “-task BLASTN” option. BLAST hits were parsed and filtered based on an e-value threshold of 1 × 10−6 with a custom Perl script utilizing BioPerl modules (Stajich et al. 2002).
Alignment of Angiosperm DPO and RPO Sequences
To infer the evolutionary history of plasmid-derived DPO and RPO sequences found in angiosperms, we performed multiple sequence alignments followed by parsimony- and maximum-likelihood-based phylogenetic inference methods. Our BLAST searches against the NCBI nr/nt databases resulted in numerous hits outside of land plants, including fungal mitochondrial plasmids, bacteria, and viruses. However, our exploratory analyses indicated that these hits were highly divergent and could not be reliably aligned along most of their length. With one exception, the only hits to ORFs that had a minimum length of 500 bp and enough sequence similarity to be confidently aligned were found in angiosperms. The exception was a whole-genome assembly for the nematode Brugia timori (GenBank assembly accession GCA_000950975.1), which contained short contigs (<2 kb) that were highly similar to plant mitochondrial plasmid sequences. Given that these hits were only found on short contigs from an unfiltered genome assembly, we considered it likely that they were the result of contamination rather than true nematode sequence, and they were not included in subsequent alignments and phylogenetic analyses.
Angiosperm DPO and RPO ORF sequences longer than 500 bp were translated into amino acids using the standard genetic code in MacClade version 4.08 (Maddison DR and Maddison WP 2001). To implement a form of the heads-or-tails alignment check (Landan and Graur 2007), the amino acid sequences were reversed using a custom Perl script. DPO and RPO sequences were aligned independently of each other using MAFFT version 7 online (Katoh and Standley 2013; supplementary files S1 and S2, Supplementary Material online). Nondefault options implemented in MAFFT were as follows: Iterative refinement method E-INS-i, amino acid scoring matrix BLOSUM45, and “leave gappy regions.”
Numerous DPO and RPO sequences were identical or nearly identical to each other and could have biased our alignment trimming step (see below) by inflating estimated sequence similarity, thereby favoring inclusion of regions with several such sequences. The forward alignments were uploaded into MEGA version 6.06 (Tamura et al. 2013), which was used to calculate pairwise p distances between all sequences, with pairwise deletion for nonoverlapping sequences. Sequences with a pairwise distance of ≤ 0.06 were identified and the single longest sequence was maintained while the others were deleted. In cases with two or more sequences of identical length, one sequence was selected at random. Six sets of (near) identical DPO sequences were merged (from Beta, Cucumis, Ferrocalamus, Lolium, and Zea [two sets]), with a total of 24 sequences deleted (supplementary table S5, Supplementary Material online). Nine sets of (near) identical RPO sequences were merged (from Beta, Cucumis, Ferrocalamus, Lolium, Silene, Triticum, Vitis, and Zea [2 sets]), with a total of 21 sequences deleted (supplemented table S6, Supplementary Material online).
Because of the high sequence divergence and the confounding effect caused by numerous indels, many regions appeared arbitrarily aligned in MAFFT’s global alignment. Sequences were trimmed using trimAl version 1.2 (Capella-Gutierrez et al. 2009). The first trimming step was used for the heads-and-tails alignments of DPO and RPO using a consistency score of 0.5, thereby decreasing the DPO (forward) alignment from 1,378 to 933 positions and the RPO (forward) alignment from 1,606 to 1,194 positions. The second trimming step applied a similarity score of 0.001, thereby decreasing the DPO alignment from 933 to 444 positions and the RPO alignment from 1,194 to 351 positions. Taken together, the two trimming steps reduced the average DPO sequence length from 626 to 309 amino acids and the average RPO sequence length from 732 to 252 amino acids. The resulting alignments were manually examined in MEGA and regions of individual sequences that were adjacent to gapped positions and appeared arbitrarily aligned were rescored as missing data (a total of 72 cells from 4 DPO sequences and a total of 55 cells from 4 RPO sequences). The final data matrices are provided as supplementary files S3–S5, Supplementary Material online.
Phylogenetic Analysis of Angiosperm DPO, RPO, and Partial Concatenation Sequences
Many of the DPO and RPO sequences are fragments rather than the entire gene (missing and inapplicable data represent 31% of the DPO data matrix and 29% of the RPO data matrix), and several sequences have zero sequence overlap. Therefore, it is important that our gene tree analysis methods be robust to cases wherein clades can only be ambiguously supported because of the distribution of missing data. Rigorous parsimony analyses followed by calculating the strict consensus of all most parsimonious trees (for the entire matrix as well as within each resampling pseudoreplicate) are highly robust to these cases (Goloboff and Pol 2005; Simmons and Goloboff 2014).
Parsimony-based gene tree analyses were conducted using TNT version 1.1 May 2014 (Goloboff et al. 2008), with branch support calculated using the strict consensus jackknife (Farris et al. 1996; Davis et al. 1998). Ten thousand tree bisection reconnection (TBR) tree searches with up to 1,000 trees held per search were conducted to search for the most parsimonious trees with TBR collapsing implemented (Goloboff and Farris 2001), followed by calculation of the strict consensus (Schuh and Polhemus 1980). Jackknife analyses were conducted using 1,000 pseudoreplicates and a deletion probability of 0.37. Each pseudoreplicate consisted of 100 TBR searches with up to 1,000 trees held per search and TBR collapsing implemented. Jackknife values were then mapped onto the strict consensus of most parsimonious trees using TreeGraph2 version 2.2.0 (Stöver and Müller 2010), following Simmons and Freudenstein (2011).
Different implementations of maximum likelihood, including different programs, models, and search settings, can produce divergent topologies and branch support values when applied to data matrices with high amounts of nonrandomly distributed missing data (Simmons and Norton 2013; Simmons and Randle 2014). PhyML (Guindon et al. 2010) and the Shimodaira-Hasegawa-like approximate likelihood ratio test (SH-like aLRT; Anisimova and Gascuel 2006; Guindon et al. 2010) have been identified as relatively robust to the artifact of providing high support for clades that can only be ambiguously supported because of the distribution of missing data.
Likelihood-based gene tree analyses were conducted using PhyML version. 20120412, with branch support calculated using the bootstrap (Felsenstein 1985) and SH-like aLRT. The best-fit model for the complete sequence sampling version of each matrix was selected using the Akaike Information Criterion (AIC; Akaike 1974) in ProtTest version 3.2 (Abascal et al. 2005). In all cases the LG model (Le and Gascuel 2008) with the gamma distribution (Yang 1993) and estimated amino acid frequencies was chosen by the AIC and implemented in PhyML. One thousand subtree pruning regrafting (SPR) searches were conducted to search for the most likely tree (PhyML only ever outputs a single fully resolved optimal tree) and 1,000 bootstrap pseudoreplicates, with a single SPR search per pseudoreplicate, were conducted. Bootstrap and SH-like aLRT branch support values were then mapped onto the most likely tree using TreeGraph 2 version 2.2.0.
Gene tree analyses that included potential outgroup DPO and RPO sequences from fungi were attempted in exploratory analyses (data not shown), but the likelihood-estimated branch lengths connecting these outgroup(s) to the plant-sourced ingroup sequences were >1 for DPO and >2.4 for RPO. Furthermore, the alignments were dubious in multiple regions and these outgroup(s) connected to the ingroup at very weakly supported internal branches. Therefore, no outgroups were included in our analyses and the trees are considered unrooted.
Many clades in the DPO and RPO gene trees received very low branch support values (<50% by both the likelihood bootstrap and parsimony jackknife), even after exclusion of three very short and problematic sequences from each of the analyses (supplementary figs. S1–S4, Supplementary Material online). Therefore, in an attempt to increase branch support values and confidence in our trees, we performed a partial concatenation-based analysis (Kluge 1989; Lecointre and Deleporte 2005). DPO and RPO sequences that were obtained from the same plant genome assembly were concatenated, with three exceptions wherein strong topological conflict was identified between the DPO and RPO gene trees. First, the RPO sequence from L. perenne (JX999996) was resolved in a clade with the other two RPO sequences from this taxon in the main grass clade (supplementary fig. S4, Supplementary Material online), whereas the DPO sequence was resolved as sister to that from F. rimosivaginus JQ235168 well outside the main grass clade (supplementary fig. S2, Supplementary Material online). Second, the DPO sequence from Rhazya stricta was resolved as sister to that from Vaccinium macrocarpon (supplementary fig. S2, Supplementary Material online), whereas the RPO sequence was resolved as distantly related to that from Vaccinium (supplementary fig. S4, Supplementary Material online). Exploratory analyses (data not shown) confirmed that the resolution of the DPO and RPO sequences from Vaccinium were largely consistent with each other, unlike those from Rhazya. Third, all DPO sequences from members of Poaceae tribe Andropogoneae (i.e., Tripsacum, Zea; Grass Phylogeny Working Group II 2012) were resolved in a clade (supplementary fig. S2, Supplementary Material online), whereas the RPO sequence from T. dactyloides DQ984517 was resolved in a clade with those from Ferrocalamus and Lolium (supplementary fig. S4, Supplementary Material online). For these three cases the DPO sequence was treated as a different terminal from the associated RPO sequence in the partial concatenation analyses. The partial concatenation data matrix is provided as supplementary file S5, Supplementary Material online.
In addition to the complete sequence sampling DPO, RPO, and partial concatenation analyses, another set of analyses was conducted using sequence subsampling. These sequence subsampling analyses were performed to help increase branch support and our confidence for resolution of the remaining terminals. In all three cases, a subset of the shortest sequences was excluded because these sequences were not resolved in well-supported clades in exploratory analyses (data not shown). Four sequences were removed for the DPO and RPO gene tree analyses (of 52–135 positions vs. the average of 307 positions for DPO; 81–103 positions vs. the average of 250 positions for RPO), and 12 sequences were removed from the partial concatenation analyses (48–250 positions vs. the average of 400 positions).
Cophylogenetic Analysis
To make inferences about the mode of linear plasmid transmission in plant mitochondrial genomes, the cophylogeny program Jane version 4 (Conow et al. 2010) was used to compare DPO and RPO gene trees with established species relationships among angiosperms. Jane is an event-cost–based method to quantify cophylogenetic signal. Event-cost methods aim to reconcile pairs of tree topologies by assigning costs to biologically plausible events, and finding the best reconstructions by minimizing global cost (de Vienne et al. 2013; Bellec et al. 2014). The event-cost parameters were set to default values of cospeciation = 0, duplication = 1, host switch = 2, sorting = 1, and failure to diverge = 1. For the genetic algorithm parameters, the population size and number of generations were set to 200 and 100, respectively. Otherwise, default genetic parameters were used, with the “Prevent Mid-Polytomy Events” option. All models were tested against a null distribution generated with 1,000 random tip mappings. The “host” tree was constructed based on the Angiosperm Phylogeny website version 13 (Stevens 2015) and individual phylogenetic studies for finer-scale relationships among grasses (Grass Phylogeny Working Group II 2012) and Zea species (Doebley 1990). Analyses were performed on plasmid gene tree topologies that were identified in the maximum-likelihood searches described above with branches with <50% bootstrap support collapsed into polytomies. Jane requires rooted trees as inputs, so each plasmid tree was midpoint rooted for these analyses. Separate tests were run for gene trees inferred from DPO, RPO, and partial concatenations.
Maize 2.3-kb Plasmid Comparison, Relative Rates Test, and Resequencing
We analyzed rates of sequence evolution in the region containing the tRNA genes ψtrnP and trnW that are normally found in angiosperm mitochondrial genomes but have recently been transferred to the 2.3-kb linear plasmid in Z. mays (Leon et al. 1989). Branch lengths were estimated by maximum likelihood for this region in Z. mays and in a sample of other monocots where it is still located in the mitochondrial chromosome in related monocots. Nucleotide sequences were aligned using MAFFT (supplementary file S6, Supplementary Material online). The scoring matrix was set to 1PAM, the leave gappy regions option was selected, and the remaining settings were left as default. The TVM+G model was selected as the best fitting based on AIC using jModelTest2 version 2.1.7 (Guindon and Gascuel 2003; Darriba et al. 2012). A maximum-likelihood tree search was performed with PhyML as described above. The resulting tree was visualized using MEGA.
Relative rate tests (Tajima 1993) were conducted with MEGA to compare rates of nucleotide substitution in the ψtrnP-trnW gene region of the 2.3-kb linear plasmid in Z. mays to the homologous region in four close relatives (Z. perennis, Z. luxurians, T. dactyloides, and S. bicolor), using Bambusa oldhamii as the outgroup.
To verify the accuracy of the originally reported Z. mays ψtrnP-trnW sequence (Leon et al. 1989), we performed polymerase chain reactions (PCR) and Sanger sequencing for multiple Z. mays accessions, including a representative of the B37 line that was used in the original study (supplementary table S4, Supplementary Material online). DNA was extracted from approximately 200 mg of leaf tissue collected within 30 days of germination, using a Qiagen DNeasy Plant Mini kit. The ψtrnP-trnW region was amplified using standard PCR protocols with the following primers: 5′-ATTATCCCTGTCCTGGGAAC-3′ and 5′-CCAACCGATACACAATTACGA-3′. The resulting PCR products were used as templates for Sanger sequencing with internal primers 5′-GGGAACAGATGGGAGACATA-3′ and 5′-TACGACATTGGGTTTTGGAG-3′ performed at the University of Chicago CCC DNA Sequencing and Genotyping Facility.
Supplementary Material
Supplementary figures S1–S7, tables S1–S6, and files S1–S7 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
We thank Jeff Mower for performing BLAST searches against his unpublished mitochondrial genome assemblies from ferns and gymnosperms. We also thank the USDA’s National Center for Genetic Resources Preservation for providing Z. mays seed and Cody Kalous for his assistance in amplifying and resequencing portions of the small linear plasmid. Rachel Mueller, members of the Sloan laboratory, and two anonymous reviewers provided insightful comments on an earlier version of the manuscript. This research was supported by Colorado State University (CSU) and the National Science Foundation (NSF MCB-1412260). J.M.W. is a participant in the NSF-funded GAUSSI graduate training program at CSU (DGE-1450032).
Literature Cited
- Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105. [DOI] [PubMed] [Google Scholar]
- Akaike H. 1974. A new look at the statistical model identification. IEEE Trans Autom Control 19:716–723. [Google Scholar]
- Allen JO, et al. 2007. Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genetics 177:1173–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andam CP, Williams D, Gogarten JP. 2010. Biased gene transfer mimics patterns created through shared ancestry. Proc Natl Acad Sci U S A. 107:10679–10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson-Ceplitis H, Bengtsson BO. 2002. Transmission rates and phenotypic effects of mitochondrial plasmids and cytotypes in Silene vulgaris. Evolution 56:1586–1591. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Gascuel O. 2006. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 55:539–552. [DOI] [PubMed] [Google Scholar]
- Bellec L, et al. 2014. Cophylogenetic interactions between marine viruses and eukaryotic picophytoplankton. BMC Evol Biol. 14:59–2148. 14-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergthorsson U, Adams KL, Thomason B. 2003. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 424:197–201. [DOI] [PubMed] [Google Scholar]
- Brooks DR, McLennan DA. 1993. Parascript: Parasites and the Language of Evolution. Washington (DC): Smithsonian Institution Press. [Google Scholar]
- Brown GG, Zhang M. 1995. Mitochondrial plasmids: DNA and RNA In: Levings CS, Vasil IK, editors. The Molecular Biology of Plant Mitochondria. Dordrecht (The Netherlands): Kluwer Academic Publishers; p. 61–91. [Google Scholar]
- Brown WM, George M, Wilson AC. 1979. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A. 76:1967–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruenn JA, Warner BE, Yerramsetty P. 2015. Widespread mitovirus sequences in plant genomes. PeerJ. 3:e876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Mower JP, Qiu YL, Palmer JD. 2004. Mitochondrial substitution rates are extraordinarily elevated and variable in a genus of flowering plants. Proc Natl Acad Sci U S A. 101:17741–17746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AC. 2013. Plant mitochondrial genome evolution can be explained by DNA repair mechanisms. Genome Biol Evol. 5:1079–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AC. 2014. Genes and junk in plant mitochondria-repair mechanisms and selection. Genome Biol Evol. 6:1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conow C, Fielder D, Ovadia Y, Libeskind-Hadas R. 2010. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms Mol Biol. 5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupp JD, Nielsen BL. 2014. DNA replication in plant mitochondria. Mitochondrion 19:231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JI, Simmons MP, Stevenson DW, Wendel JF. 1998. Data decisiveness, data quality, and incongruence in phylogenetic analysis: an example from the monocotyledons using mitochondrial atp A sequences. Syst Biol. 47:282–310. [DOI] [PubMed] [Google Scholar]
- de Vienne DM, Giraud T, Shykoff JA. 2007. When can host shifts produce congruent host and parasite phylogenies? A simulation approach. J Evol Biol. 20:1428–1438. [DOI] [PubMed] [Google Scholar]
- de Vienne DM, et al. 2013. Cospeciation vs host-shift speciation: methods for testing, evidence from natural associations and relation to coevolution. New Phytol. 198:347–385. [DOI] [PubMed] [Google Scholar]
- Doebley J. 1990. Molecular evidence and the evolution of maize. Econ Bot. 44:6–27. [Google Scholar]
- Drouin G, Daoud H, Xia J. 2008. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol Phylogenet Evol. 49:827–831. [DOI] [PubMed] [Google Scholar]
- Ellis J. 1982. Promiscuous DNA—chloroplast genes inside plant mitochondria. Nature 299:678–679. [DOI] [PubMed] [Google Scholar]
- Farris JS, Albert VA, Källersjö M, Lipscomb D, Kluge AG. 1996. Parsimony jackknifing outperforms neighbor-joining. Cladistics 12:99–124. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. [DOI] [PubMed] [Google Scholar]
- Filee J, Forterre P, Sen-Lin T, Laurent J. 2002. Evolution of DNA polymerase families: evidences for multiple gene exchange between cellular and viral proteins. J Mol Evol. 54:763–773. [DOI] [PubMed] [Google Scholar]
- Goloboff PA, Farris JS. 2001. Methods for quick consensus estimation. Cladistics 17:S26–S34. [Google Scholar]
- Goloboff PA, Farris JS, Nixon KC. 2008. TNT, a free program for phylogenetic analysis. Cladistics 24:774–786. [Google Scholar]
- Goloboff PA, Pol D. 2005. Parsimony and bayesian phylogenetics In: Albert VA, editor. Parsimony, phylogeny, and genomics. New York: Oxford University Press; p. 148–159. [Google Scholar]
- Grass Phylogeny Working Group II. 2012. New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol 193:304–312. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52:696–704. [DOI] [PubMed] [Google Scholar]
- Guindon S, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59:307–321. [DOI] [PubMed] [Google Scholar]
- Handa H. 2003. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 31:5907–5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa H. 2008. Linear plasmids in plant mitochondria: peaceful coexistences or malicious invasions? Mitochondrion 8:15–25. [DOI] [PubMed] [Google Scholar]
- Iorizzo M, et al. 2012. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluge AG. 1989. A concern for evidence and a phylogenetic hypothesis of relationships among Epicrates (boidae, serpentes). Syst Zool. 38:7–25. [Google Scholar]
- Knoop V, Volkmar U, Hecht J, Grewe F. 2011. Mitochondrial genome evolution in the plant lineage In: Kempken F, editor. Plant mitochondria. New York: Springer; p. 3–29. [Google Scholar]
- Knopf CW. 1998. Evolution of viral DNA-dependent DNA polymerases. Virus Genes 16:47–58. [DOI] [PubMed] [Google Scholar]
- Kuzmin EV, Levchenko IV. 1987. S1 plasmid from cms-S-maize mitochondria encodes a viral type DNA-polymerase. Nucleic Acids Res. 15:6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landan G, Graur D. 2007. Heads or tails: a simple reliability check for multiple sequence alignments. Mol Biol Evol. 24:1380–1383. [DOI] [PubMed] [Google Scholar]
- Le SQ, Gascuel O. 2008. An improved general amino acid replacement matrix. Mol Biol Evol. 25:1307–1320. [DOI] [PubMed] [Google Scholar]
- Lecointre G, Deleporte P. 2005. Total evidence requires exclusion of phylogenetically misleading data. Zool Scr. 34:101–117. [Google Scholar]
- Leon P, Walbot V, Bedinger P. 1989. Molecular analysis of the linear 2.3 kb plasmid of maize mitochondria: apparent capture of tRNA genes. Nucleic Acids Res. 17:4089–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A. 105:9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison DR, Maddison WP. 2001. MacClade: analysis of phylogeny and character evolution version 4.03. Sunderland (MA): Sinauer Associates, Inc. [Google Scholar]
- McDermott P, Connolly V, Kavanagh TA. 2008. The mitochondrial genome of a cytoplasmic male sterile line of perennial ryegrass (Lolium perenne L.) contains an integrated linear plasmid-like element. Theor Appl Genet. 117:459–470. [DOI] [PubMed] [Google Scholar]
- Moran N, Baumann P. 1994. Phylogenetics of cytoplasmically inherited microorganisms of arthropods. Trends Ecol Evol. 9:15–20. [DOI] [PubMed] [Google Scholar]
- Mower JP, Sloan DB, Alverson AJ. 2012. Plant mitochondrial diversity—the genomics revolution In: Wendel JF, editor. Plant genome diversity. Vienna (Austria): Springer; p. 123–144. [Google Scholar]
- Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD. 2007. Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol Biol. 7:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, et al. 2015. Dynamic evolution of geranium mitochondrial genomes through multiple horizontal and intracellular gene transfers. New Phytol. 208:570–583. [DOI] [PubMed] [Google Scholar]
- Parkinson CL, et al. 2005. Multiple major increases and decreases in mitochondrial substitution rates in the plant family geraniaceae. BMC Evol Biol. 5:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautenberg A, Sloan DB, Aldén V, Oxelman B. 2012. Phylogenetic relationships of Silene multinervia and Silene section Conoimorpha (caryophyllaceae). Syst Bot 37:226–237. [Google Scholar]
- Rice DW, et al. 2013. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342:1468–1473. [DOI] [PubMed] [Google Scholar]
- Richardson AO, Rice DW, Young GJ, Alverson AJ, Palmer JD. 2013. The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison MM, Wolyn DJ. 2005. A mitochondrial plasmid and plasmid-like RNA and DNA polymerases encoded within the mitochondrial genome of carrot (Daucus carota L.). Curr Genet. 47:57–66. [DOI] [PubMed] [Google Scholar]
- Sanchez-Puerta MV, Cho Y, Mower JP, Alverson AJ, Palmer JD. 2008. Frequent, phylogenetically local horizontal transfer of the cox1 group I intron in flowering plant mitochondria. Mol Biol Evol. 25:1762–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh RT, Polhemus JT. 1980. Analysis of taxonomic congruence among morphological, ecological, and biogeographic data sets for the Leptopodomorpha (Hemiptera). Syst Biol. 29:1–26. [Google Scholar]
- Shutt TE, Gray MW. 2006. Bacteriophage origins of mitochondrial replication and transcription proteins. Trends Genet. 22:90–95. [DOI] [PubMed] [Google Scholar]
- Simmons MP, Freudenstein JV. 2011. Spurious 99% bootstrap and jackknife support for unsupported clades. Mol Phylogenet Evol. 61:177–191. [DOI] [PubMed] [Google Scholar]
- Simmons MP, Goloboff PA. 2014. Dubious resolution and support from published sparse supermatrices: the importance of thorough tree searches. Mol Phylogenet Evol. 78:334–348. [DOI] [PubMed] [Google Scholar]
- Simmons MP, Norton AP. 2013. Quantification and relative severity of inflated branch-support values generated by alternative methods: an empirical example. Mol Phylogenet Evol. 67:277–296. [DOI] [PubMed] [Google Scholar]
- Simmons MP, Randle CP. 2014. Disparate parametric branch-support values from ambiguous characters. Mol Phylogenet Evol. 78:66–86. [DOI] [PubMed] [Google Scholar]
- Skippington E, Barkman TJ, Rice DW, Palmer JD. 2015. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112:E3515–E3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Muller K, McCauley DE, Taylor DR, Storchova H. 2012. Intraspecific variation in mitochondrial genome sequence, structure, and gene content in Silene vulgaris, an angiosperm with pervasive cytoplasmic male sterility. New Phytol. 196:1228–1239. [DOI] [PubMed] [Google Scholar]
- Sloan DB, Oxelman B, Rautenberg A, Taylor DR. 2009. Phylogenetic analysis of mitochondrial substitution rate variation in the angiosperm tribe Sileneae (Caryophyllaceae). BMC Evol Biol. 9:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Taylor DR. 2010. Testing for selection on synonymous sites in plant mitochondrial DNA: the role of codon bias and RNA editing. J Mol Evol. 70:479–491. [DOI] [PubMed] [Google Scholar]
- Sloan DB, Alverson AJ, et al. 2012. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10:e1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Jackson CJ, Reyes-Prieto A. 2014. Nucleotide substitution analyses of the glaucophyte Cyanophora suggest an ancestrally lower mutation rate in plastid vs mitochondrial DNA for the archaeplastida. Mol Phylogenet Evol. 79:380–384. [DOI] [PubMed] [Google Scholar]
- Stajich JE, et al. 2002. The bioperl toolkit: perl modules for the life sciences. Genome Res. 12:1611–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens PF. 2015. Angiosperm phylogeny website. Version 13, December 2015. Available from: http://www.mobot.org/MOBOT/research/APweb/
- Stöver BC, Müller KF. 2010. TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinformatics 11:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart EC, et al. 2012. The Oxytricha trifallax mitochondrial genome. Genome Biol Evol. 4:136–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. 1993. Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 135:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30:2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber B, Borner T, Weihe A. 1995. Remnants of a DNA polymerase gene in the mitochondrial DNA of Marchantia polymorpha. Curr Genet. 27:488–490. [DOI] [PubMed] [Google Scholar]
- Wickett NJ, et al. 2014. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc Natl Acad Sci U S A. 111:E4859–E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Li WH, Sharp PM. 1987. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci U S A. 84:9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 1993. Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol Biol Evol. 10:1396–1401. [DOI] [PubMed] [Google Scholar]
- Zhu A, Guo W, Jain K, Mower JP. 2014. Unprecedented heterogeneity in the synonymous substitution rate within a plant genome. Mol Biol Evol. 31:1228–1236 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.