

Summary
Parkinson’s disease (PD) is a progressive neurodegenerative disorder associated with Lewy body disease (LBD) pathology in central and peripheral nervous system structures. While the etiology of PD is not fully understood, recent clinicopathologic analyses by Braak and colleagues have led to the development of a staging system of LBD pathology in the evolution of prototypical PD. This system posits a relatively predictable topography of progression of LBD pathology in the central nervous system, from olfactory structures and the medulla, which then progresses rostrally from the medulla to the pons, then midbrain/substantia nigra, then limbic, and then neocortical structures. If this topography and temporal evolution of LBD pathology indeed occur, one could hypothesize that other manifestations of LBD which reflect degeneration of olfactory and pontomedullary structures may begin many years prior to the development of prominent nigral degeneration and the associated parkinsonian features of classic PD. One such manifestation of prodromal PD is rapid eye movement (REM) sleep behavior disorder (RBD), which is a parasomnia manifested by vivid dreams associated with dream enactment behavior during REM sleep. Animal and human studies have implicated lesions or dysfunction in REM sleep and motor control circuitry in the pontomedullary structures cause RBD phenomenology, and degeneration of these structures could explain the presence of RBD years or decades prior to the onset of parkinsonism in those who develop PD. This review incorporates the rapidly growing literature on RBD and other prodromal features of PD as it pertains to the Braak staging system, and presents a framework from which many hypotheses can be (and already are being) tested. An important outcome of this framework will be to determine the natural history of RBD and associated features in the evolution to PD in the current era of no disease-modifying therapies – these natural history data will permit the development of clinical trail methodology with key measures and adequate power to detect if such therapies delay the onset or prevent the development of PD and associated morbidity.
Keywords: REM sleep behavior disorder, parasomnia, Lewy body disease, Parkinson disease, Lewy body dementia, synucleinopathy, neurodegenerative disease
Overview
The days of viewing Parkinson’s disease (PD) as a primary motor/extrapyramidal syndrome associated primarily with dopaminergic deficiency are long gone. The underlying substrate for PD – Lewy body disease (LBD) – is a complex neurodegenerative disorder with the histopathologic hallmarks – the Lewy body and Lewy neurite – being comprised of abnormal accumulations of α-synuclein protein in neurons. While it is still debated whether the Lewy bodies and Lewy neurites are neurotoxic or bystanders of another primary pathophysiologic process, their presence marks the presence of the disease. LBD is not only a brain disorder – it is a systemic disorder that involves several key structures in the peripheral and central nervous systems, and is manifested by a spectrum of clinical features which can include the cognitive, neuropsychiatric, motor, sleep, autonomic and sensory domains. Dopamine is one of many altered neurochemical systems in LBD. LBD can manifest as three primary clinical syndromes – PD +/− dementia, dementia with Lewy bodies, and pure autonomic failure. Yet with all of its complexity, in the majority of affected individuals, LBD tends to affect some neuronal structures and systems and spare others (ie, selective vulnerability). Therefore, most patients with underlying LBD who develop the phenotype of prototypical PD appear to evolve in manner that is selective, sequential, and relatively predictable.
The key structures pertinent to PD phenomenology are shown in Figure 1. Obvious parkinsonism is present when sufficient nigral degeneration has occurred. Of particular interest is the identification of patients who are experiencing symptoms or exhibiting signs of very early or “prodromal” PD – meaning that such individuals are not showing the motor signs of PD as yet. In the current era of no disease-modifying therapies being available for PD and other neurodegenerative disorders, identifying those with prodromal PD may not seem all that important. Yet early identification is important for many reasons, particularly when potential disease-modifying therapies become available in the future, as these therapies are far more likely to alter the progression of neurodegeneration, and hence evolution of symptoms, if they are commenced as early in the disease course as possible. Furthermore, despite the absence of disease-modifying therapies currently, it is critical to identify individuals during the prodromal phase and study the natural history of their progression to plan for future intervention studies.
Figure 1. Structures pertinent to Parkinson’s disease phenomenology.
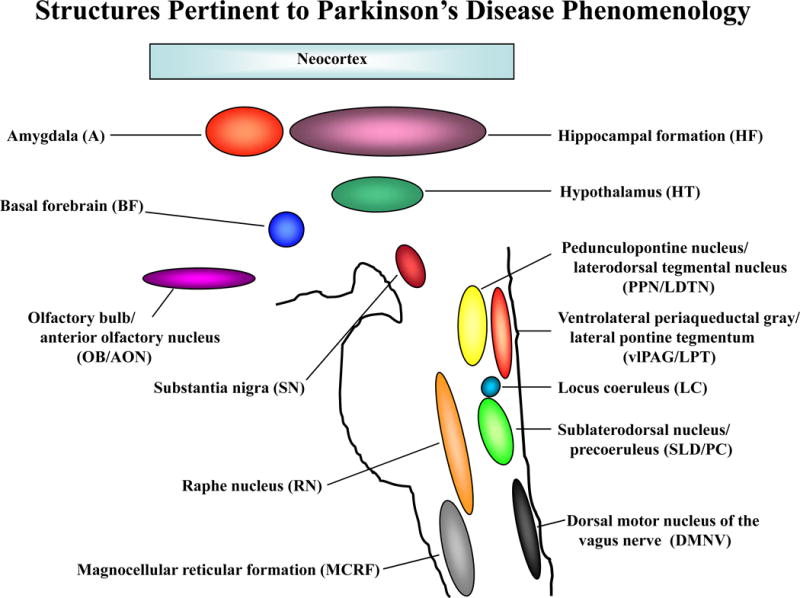
Figure depicting the key brain structures typically affected by Lewy bodies, Lewy neurites and neuronal loss in the Parkinson’s disease phenotype of Lewy body disease.
Note: the nuclei in the brainstem are positioned in this figure as approximations but their relative sizes are not drawn to scale.
There is no consensus on the term to apply to the early phase of evolving LBD. The term “premotor” is often used to describe the phase prior to the onset of parkinsonism, but as will be emphasized in detail below, REM sleep behavior disorder (RBD) is a motor manifestation of evolving LBD and hence “premotor” is not entirely fitting.1 The term “presymptomatic” is also not applicable, as many features and symptoms preceding parkinsonism reflect “symptomatic” LBD. The term “prodromal” will therefore be used when referring to the phase of LBD preceding overt parkinsonism.2, 3
The work of Braak and colleagues has led to the development of a staging scheme for the pathoanatomy of LBD as it relates to the phenotype of prototypical PD.4, 5 While still a matter of debate,6 this staging scheme has several important implications for clinicians and researchers. This review will present data on the known and suspected prodromal features of evolving LBD as they relate to the prototypical PD phenotype, and then view these features in the context of the Braak staging scheme. Many of these features are common in the normal population and may be unrelated to underlying LBD, suggesting that they may not be good markers (at least not in isolation) for early identification of LBD. However, the parasomnia of RBD is striking in its clinical phenomenology, relatively easy to recognize and diagnose [particularly when collateral history from a bedpartner is obtained and polysomnography (PSG) is performed with synchronous video-PSG monitoring and additional EMG leads on the upper and lower limbs are used], and relatively common and reasonably specific for LBD,7 leading many groups of investigators to focus on RBD as an early manifestation of evolving PD.2, 8–23 The primary goals of this review are to synthesize that the rapidly growing data on RBD as a distinct early manifestation of PD and related disorders, and provide a framework to foster increased interest and research in this fascinating parasomnia, particularly as it relates to neurodegenerative disease.
Importantly, the Braak staging scheme for PD was developed based on autopsied case material from subjects without any apparent neurologic symptoms or findings, and from patients who had exhibited features of typical PD during life. As such, the applicability to patients with the other LBD phenotypes – namely dementia with Lewy bodies and pure autonomic failure – may or may not be appropriate. Furthermore, a significant minority of patients with Parkinson’s disease do not exhibit all of the prodromal features of typical PD, or the various features manifest over a time course that “does not fit” into the Braak staging scheme. Also, this staging scheme emphasizes the topography and evolution of Lewy body and Lewy neurite accumulation, which may or may not correlate with neuronal dysfunction with or without neuronal loss which is responsible for clinical manifestions to be apparent. These issues will be discussed in more detail below. Therfore, this review focuses on RBD and other clinical features of prototypical prodromal and symptomatic PD, realizing that determining who is prototypical vs atypical during life is a challenge for all clinicians. These and other important issues are discussed in more detail in the section below on Controversies and Uncertainties.
Clinical Features of REM Sleep Behavior Disorder
The clinical features of RBD are distinctive, and the manifestations are often colorfully described by spouses.24–26 RBD tends to affect middle-aged to older males. Abnormal vocalizations, abnormal motor behavior, and abnormal dream mentation form the core clinical manifestations, in which patients appear to “act out their dreams” by yelling, screaming, flailing limbs, punching, kicking, etc., usually at a perceived attacker in their dreams (the attacker is usually a human, animal, or insect). Since RBD reflects, at least in part, the loss of the usual active paralysis of skeletal muscles during REM sleep [note: the updated nomenclature pertaining to sleep stages refers to REM sleep as “stage R,”27 but for historical reasons the term REM sleep is used throughout this review], and most REM sleep transpires over the second half of the sleep period, the abnormal behaviors are typically manifested well after midnight and particularly during the terminal third of the sleep period. Increased electromyographic (EMG) tone during REM sleep with or without abnormal behaviors during REM sleep are the defining PSG features.28 Management is directed at minimizing the potential for injury to the patient and bedpartner, and decreasing the unpleasant nature of the dream content. Approaches include making adjustments to the bedroom environment to move sharp or injurious objects out of harms way, and using nightly therapy with melatonin and/or clonazepam.13, 25, 29
Pathoanatomy of REM Sleep Behavior Disorder
Based on work in the animal models,30–36 the sublaterodorsal nucleus and precoeruleus complex (SLD/PC) and magnocellular reticular formation (MCRF) have been implicated in the pathophysiology of human RBD (Figure 2).12, 13 These nuclei send projections either directly or indirectly to the anterior horn cells of the bulbar, trunk and limb skeletal musculature in the brainstem and spinal cord. During normal REM sleep, the descending influences from the SLD/PC and MCRF effectively inhibit the anterior horn cells such that most of the cranial muscles and essentially all of the skeletal muscles are paralyzed. This is manifested by electromyographic atonia on polysomnography during essentially all of REM sleep and the absence of elaborate vocalizations and complex motor behavior (Figure 2). While muscle twitches and brief vocalizations can occur during normal REM sleep, the characteristic features of REM sleep without atonia (RSWA) and RBD are not present. There is still debate on what constitutes normal and mildly increased EMG tone during REM sleep,37 and PSG interpretation can be challenging to interpret in those with coexisting parkinsonism and/or dementia or in those using antidepressant agents, but in the majority of individuals who undergo PSG, the distinctions between normal REM sleep, RSWA, and clinical RBD are obvious.
Figure 2. Proposed structures involved in normal human REM sleep.
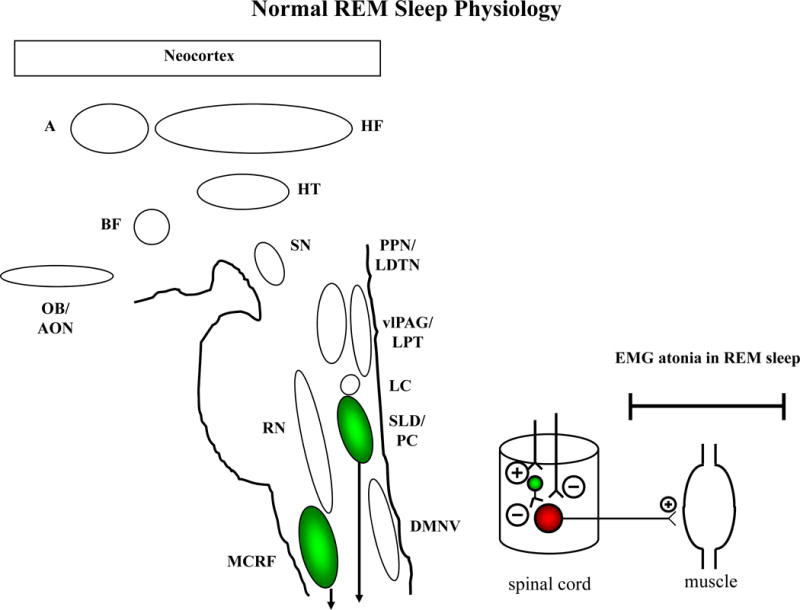
The REM-off region is represented by the vlPAG and LPT, and the REM-on region is represented by the PC and SLD. The SLD (or analogous nucleus in humans) +/− PC projects to spinal interneurons (“direct route,” denoted by the line from SLD/PC to spinal interneurons) and likely represents the final common pathway that causes active inhibition of skeletal muscle activity in REM sleep. The “indirect route,” denoted by the line from the MCRF to the spinal interneurons, may also contribute to EMG atonia. Excitatory projections represented by ⊕, inhibitory projections represented by ⊝, with the size of these symbols representing the relative effect of each projection on the synapsing nuclei. The net effect of these pontomedullary influences on bulbar and spinal motor neurons is to actively inhibit neuronal firing during REM sleep, which is reflected by the absence of electromyographic (EMG) activity on polysomnography (PSG).
Note: the abbreviations for each structure are spelled out in Figure 1.
Dysfunction in the SLD +/− MCRF and peri-LC structures (Stage 2 – see below) is suspected to lead to RSWA, and further degeneration of these structures, and possibly alterations in other structures such as the locomotor generators, lead to overt RBD (Stage 3 – see below). This temporal sequence of pathology likely explains why RBD precedes parkinsonism and cognitive decline (Stages 3 and 4) and dementia (Stages 4–6) in most patients who develop the PD phenotype associated with Lewy body pathology, as detailed below.
Clinical Features of Parkinson’s Disease
The defining clinical features for the diagnosis of PD include limb rigidity, resting limb tremor, bradykinesia, and postural instability – these represent the core “motor” features of the PD phenotype. The rigidity, tremor, and bradykinesia tend to be asymmetric. Masked facies, reduced armswing while walking, shuffling gait, micrographia and poor fine motor dexterity are other typical motor features. These features largely relate to the well-known dopamine deficiency in the disorder due to marked degeneration and neuronal loss in the substantia nigra. In those with PD, many will develop other clinical manifestations of the widespread LBD pathology, including cognitive features (particularly with reduced psychomotor speed and impairment in attention/concentration, executive functioning, learning and memory, and visuospatial functioning); neuropsychiatric features such as depression, anxiety, apathy, visual hallucinations and delusions; progression of motor features with gradual diminished response to dopaminergic therapy; sleep disorders such as RBD, periodic limb movements during sleep, hypersomnia and insomnia; autonomic dysfunction such as erectile dysfunction, orthostatic hypotension, constipation and urinary incontinence; and sensory changes such as reduced olfaction and color vision. The presence of many of these features, the timing of their onset, and their frequency and severity tend to be variable across individuals.38, 39 However, many of these features develop around the same time as the motor features, or years later – all during the “motor” or “symptomatic” phase of PD. Dementia that evolves in PD likely reflects involvement of many neurochemical systems as well as degeneration in limbic and neocortical structures.40
Pathoanatomy of Parkinson’s Disease Features
Referring again to the clinical phenomenology of LBD, and to the schematic representation of the key structures in the brain involved in PD phenomenology, the cognitive/neuropsychologic features likely relate to changes in the cholinergic, dopaminergic, serotonergic, and other neurotransmitter systems, and their associated nuclei (i.e., basal forebrain, pedunculopontine nucleus/laterodorsal tegmental nucleus, ventral tegmental area, lower raphe nuclei, etc.) and neuronal networks, which clearly involve networks in the limbic system and neocortex.40 The behavioral/neuropsychiatric features likely relate to many of these same neurochemical systems and networks. The motor/extrapyramidal features of PD surely relate, at least in part, to degeneration of the nigrostriatal system and the associated dopaminergic deficiency. Regarding the sleep disorder features associated with PD, while the key neuronal networks involved in human RBD pathophysiology are not yet known with certainty, degeneration in the subcoeruleus/precoeruleus and/or magnocellular reticular formation and dysfunction in their afferent/efferent connections are likely at play, as noted above. Clinicopathologic studies in humans have not clarified the precise anatomic structures involved in those who had RBD antemortem compared to those who did not.41, 42 Hypersomnia and/or insomnia can be caused by factors relating the primary brain dysfunction, non-neurologic factors (eg, restless legs syndrome, periodic limb movements during sleep, obstructive or central sleep apnea, medications), or a combination of both. Primary brain factors could include changes in the pedunculopontine nucleus/laterodorsal tegmental nucleus, ventral tegmental area, lower raphe nuclei, hypocretinergic neurons in the lateral hypothalamus, or histaminergic neurons in the hypothalamic tuberomamillary nucleus, as well as coexisting depression or dementia. Furthermore, alterations in the “flip-flop switch” in the brainstem and/or hypothalamic influences on this switch36 could account for the frequent arousals for no apparent reason during sleep, and also for the elements of state dissociation that is characteristic of narcolepsy and for some of the sleep related features in PD – muscle tone and vocalizations invading into REM sleep (RBD), sleep invading into wakefulness (hypersomnia and sleep attacks), dream imagery of REM sleep invading into wakefulness (visual hallucinations), etc. The autonomic features likely relate to degeneration of the dorsal motor nucleus of the vagus nerve, the autonomic centers of the spinal cord, and the autonomic circuitry in and around the heart, abdominal/pelvic/lower limb venous system, gut, sex organs, etc. For the sensory features, degeneration of the olfactory bulb and anterior olfactory nucleus likely explains changes in odor appreciation, identification and discrimination. Color vision dysfunction may relate to degeneration in the retina and/or visual cortex. Electrophysiologic and imaging studies have also shown abnormalities in those with PD.
“Prodromal” Features of Parkinson’s Disease
Retrospective and prospective studies have suggested or substantiated many prodromal features and findings in those with PD38, 39, 43–45 – a list of a number of these are shown in Table 1. While these features are rather common (and sometimes pervasive) in those diagnosed with PD, each may be an early feature of prodromal PD in isolation or in combination.
Table.
Features/Symptoms, Associated Clinical and Ancillary Test Findings, and Their Interpretation in Patients with “Idiopathic” REM Sleep Behavior Disorder
| Clinical Feature or Symptom | Findings Detectable on Measurement Techniques/Biomarkers | Interpretation |
|---|---|---|
| Dysnosmia/anosmia | Smell testing – impaired odor identification and discrimination | Degeneration of olfactory structures (begin in Stage 1) |
| Orthostatic hypotension Constipation Erectile dysfunction Urinary incontinence Decreased sweating |
Peripheral and central autonomic function testing Cardiac MIBG imaging – reduced cardiac sympathetic activity |
Degeneration of structures in the brainstem, intermediolateral cell column, and peripheral ganglia involved in autonomic functioning (? begin in Stage 1) |
| REM sleep behavior disorder | Polysomnography – degree of REM sleep without atonia | Degeneration of the brainstem structures (sublaterodorsal nucleus, precoeruleus, and magnocellular reticular formation) involved in REM sleep circuitry (begins in Stage 2) |
| Depression | Various measures for assessing mood | Degeneration of raphe nucleus and/or locus coeruleus (begins in Stage 2) |
| Apathy | Various measures for assessing apathy | ? degeneration of raphe nucleus and/or locus coeruleus (begins in Stage 2) |
| Anxiety | Various measures for assessing apathy | ? degeneration of raphe nucleus and/or locus coeruleus (begins in Stage 2) |
| Sleep fragmentation with insomnia | Polysomnography – frequent arousals for no apparent reason | ? degeneration of raphe nucleus, locus coeruleus, sublaterodorsal nucleus, precoeruleus, pedunculopontine nucleus, laterodorsal nucleus, tuberomamillary nucleus (begins in Stage 2/3) |
| Hypersomnia | Epworth Sleepiness Scale – increased tendency to doze Multiple Sleep Latency Test or Maintenance of Wakefulness Test – reduced initial sleep latency |
? degeneration of brainstem nuclei (begins in Stage 2/3) Degeneration of hypocretin-1 cells in the lateral hypothalamus (begins in Stage 3) |
| Parkinsonism | Timed up and go Gait testing – increased variability in gait measures Clinical examination/Unified Parkinson’s Disease Rating Scale – rest tremor, bradykinesia, rigidity, postural instability, reduced armswing, masked facies Dopamine transporter scan (DaTscan) – reduced striatonigral uptake of dopamine transporter on SPECT imaging Fluorodopa positron emission tomography (FD-PET) – reduced fluorodopa metabolism in nigrostriatal system Transcranial ultrasonography (TCS) – midbrain echogeneity |
Degeneration of substantia nigra (begins in Stage 3) |
| Cognitive impairment | Neuropsychological testing – impairment in one or more cognitive domains | Degeneration of the cholinergic basal forebrain and limbic/neocortical structures and their efferent/afferent connections (begins in Stage 4) |
| Color vision | Farnsworth-Munsell-100-Hue test (FM-100) – impaired color vision testing | Degeneration of retinal and/or visual cortex structures (begins in Stage 6) |
| Asymptomatic – electrophysiologic and imaging measures | Electroencephalography (EEG) – diffuse slowing, increased theta/delta power Magnetic resonance spectroscopy (MRS) – altered metabolic ratios in key regions of interest Magnetic resonance imaging (MRI) diffusion tensor imaging (DTI) – altered fractional inosotropy and mean diffusivity in key brain regions MRI voxel based morphometry (VBM) – altered gray matter density of hippocampus MRI based corticometery – decreased neocortical thickness in frontal and/or parietal regions Brain single photon emission computed tomorgraphy (SPECT) – hypoperfusion of cortical structures, particularly occipital region Fluorodeoxyglucose positron emission tomography (FDG-PET) – hypometabolism of cerebral cortex, particularly occipital region |
Degeneration of various brain structures (begins in Stages 2–4) |
The Braak Staging Scheme of Parkinson’s Disease Pathoanatomy and Associated Clinical Manifestations
This staging system proposes a temporal sequence of α-synuclein pathology – Lewy bodies and Lewy neurites – in the brain and spinal cord beginning in the medulla (and olfactory structures) and gradually ascending to more rostral structures.4, 5, 46 This system was developed based on autopsy material, and any neuropathologic analyses are inherently cross-sectional. However, this system provides a framework to potentially explain the sequence of clinical manifestations which tend to evolve in patients with PD, including in the prodromal phase. Furthermore, this permits hypotheses to be generated and tested.47
There are some key points in interpreting the potential clinical associations of the Braak staging scheme. Each stage represents an expanding constellation of structures which has demonstrable Lewy bodies and Lewy neurites, with each successive stage having a greater burden of Lewy body/Lewy neurite pathology in previously affected structures and some degree of burden in newly affected structures.4, 5 Recent evidence suggests that a prion-like propagation occurs whereby those neurons with LB/LN may affect other neurons synapsing on them.48–52 Except for the olfactory structures (Stage 1), the propagation of α-synuclein positive pathology ascends from the medulla and up to the neocortex. However, some structures are invariably hit, while others are almost never hit. LBD and its propagation therefore involves concepts such as selective vulnerability, slow rate of progression (likely over many years or decades), and the structures involved in the early stages may be so decimated from neurodegeneration that no neurons are remaining, and hence no LB nor LN can be found. However, a critical point in LBD phenomenology reflects cellular and neurochemical alterations – neurodegenerative symptomatology must reflect neurons +/− glia becoming dysfunctional or expiring, neurochemical systems being altered (usually decreasing due to degeneration of neurotransmitter-producing nuclei), or some combination of these.53
While it makes intuitive sense that cells with LB and LN are dysfunctional and will likely die, the degree to which neurons with LB and LN can still function normally is not well-understood. Therefore, for example, if the substantia nigra is invariably affected early in those with PD, do such patients invariably develop symptoms reflecting damage to this nucleus, or is some level of LB/LN burden “tolerable” and hence asymptomatic. In other words, can some degree of measurable change in functioning be detected with 20%, 50%, or 80% of a nuclear group or network degenerated? And there may be a difference between the degree of degeneration necessary to be detected by some measure compared to the degree needed to be manifested as a clinical symptom or finding. For example, perhaps nigral degeneration can be detected using a neuroimaging modality when 50% of the nigral population has died, whereas symptomatic parkinsonism may not become manifest until 80% of nigral neurons are lost. For purposes of this review, it will be assumed that if a structure is invariably hit with LB/LN pathology at a certain stage (e.g, substantia nigra in Stage 3), then the functions carried out by that structure may become detectable at that stage, at the earliest, by some measure (e.g. reduced nigrostriatal uptake on dopamine transporter scanning), and symptoms evolve at some point thereafter (e.g. overt parkinsonism in Stage 4) and the measure that detects that abnormality shows greater dysfunction with advancing disease (e.g. increasingly greater reduction in nigrostriatal uptake in Stages 4 to 5 to 6).
With these speculations in mind, the schematic representations of each Braak stage are shown in Supplemental Figures 1–6 with the presumed or substantiated clinical manifestations associated with degeneration of those structures. While not specifically stated in the Braak staging scheme, if one proposes Stages 1–6, one can infer there must be a “Stage 0,” in which all structures are not affected by the disease process and thus are functioning normally.
Recent evidence suggests that at least in some patients, LB/LN changes occur in the peripheral nervous system (particularly autonomic nervous system)54, but the temporal sequence of peripheral nervous system involvement and brain involvement is still being studied. This review will focus on the Braak staging scheme as it pertains to LB/LN changes in the brain with the presumption that clinical manifestations of LBD reflecting peripheral/autonomic nervous system involvement (eg, constipation, orthostatic hypotension, erectile dysfunction) may be present to some degree in those with early stage LBD in the brain.
In Stage 1, the dorsal motor nucleus of the vagus nerve (DMNVN) and olfactory bulb and anterior olfactory nucleus (OB/AON) complex are affected (Supplemental Figure 1). Once a threshold of degeneration is reached, the likely clinical manifestations of autonomic dysfunction and smell dysfunction become manifest, which could be detectable by measures such as orthostatic blood pressure and pulse testing, cardiac beat-to-beat variability, gut transit time, erectile and urinary functioning, cardiac MIBG imaging, and formal smell appreciation, identification, and discrimination. Presumably, detectable changes on these ancillary tests will precede the development of overt symptoms.
Stage 2 of this scheme involves further LB/LN formation in the structures involved in Stage 1, and the beginning of LB/LN formation in the SLD/PC and MCRF as well as raphe nucleus (RN) and locus coeruleus (LC) (Supplemental Figure 2). Once a threshold is reached, changes in mood, behavior and sleep likely develop. Detectable alterations in serotonin and noradrenalin may be present at this stage. The degeneration in the SLD/PC and/or MCRF could lessen the inhibitory influences on the caudal anterior horn cells, thereby potentially leading to the polysomnographic finding of REM sleep without atonia (RSWA). It is presumed, but certainly not proven, that additional changes in these and likely other networks are necessary for the full expression of RBD to be manifested. Hence, RSWA – beginning in Stage 2 at the earliest – may reflect the electrophysiologic precursor of evolving RBD – which may manifest in Stage 2 or 3.
Most of the prodromal features of evolving PD (Supplemental eTable) become evident by Stage 3 (Supplemental Figure 3). Overt RBD is now present, and abnormalities are detectable or overtly symptomatic on measures assessing smell, autonomic functioning, motor functioning, and neuropsychological functioning. Apathy, anxiety, depression, or some combination of these may be present. Hypersomnia and/or insomnia may occur in some. Subtle changes in hypothalamic-mediated functions may also be evident.
Overt parkinsonism becomes evident in Stage 4 (Supplemental Figure 4), and along with the features from Stage 3 progressing, clinically-relevant features also evolve in some patients – more measurable changes in neuropsychological functioning, more obvious emotional/behavioral manifestations, and more obvious hypothalamic-mediated changes.
Cognitive decline to the point of a formal diagnosis of mild cognitive impairment (MCI) or dementia associated with PD (Parkinson’s disease with dementia or PDD), motor fluctuations, visual hallucinations, sleep fragmentation and/or hypersomnia, are common manifestations of Stage 5 (Supplemental Figure 5). Overt PDD with all of the aforementioned features are present by Stage 6 (Supplemental Figure 6).
An important caveat on the cognitive aspects of PD must be emphasized – the neuropathologic substrate for MCI and PDD associated with LBD pathology continues to be studied.55–59 The presence of limbic +/− neocortical LBD pathology is associated with cognitive impairment, recurrent visual hallucinations, and delusions, but LBs in the cerebral cortex have been found in patients with PD who have no obvious cognitive impairment or hallucinations/delusions.60 Surely overt neocortical +/− limbic degeneration contributes to cognitive impairment. Frontostriatal neural network dysfunction likely contributes to cognitive impairment as well, particularly aspects of attention, concentration and executive functioning.61 However, to what degrees 1) LBD pathology in the limbic and neocortical structures, 2) alterations in neurochemical systems, 3) alterations in wake-sleep mechanisms, or 4) some combination of these, contributes to the cognitive and neuropsychiatric manifestations of PD with MCI and PDD are not clear. While not proven, a reasonable conclusion from clinicopathologic studies is that LBD pathology in limbic +/− neocortical structures (i.e., Braak Stage 5 or 6) is often associated with, but not sufficient, to cause cognitive impairment/dementia in those with PD.
In Search of a Window into the Evolution and Progression of LBD Pathology in Prodromal PD
The evolution of LBD pathology and associated clinical manifestations of the prodromal phase, and parkinsonism phase, and parkinsonism plus cognitive impairment phase, in the prototypical PD phenotype is becoming increasingly understood.14, 17, 21, 22, 38, 39, 62, 63 One can theorize on the progression of LB/LN deposition and/or neuronal degeneration over time being linear, sinusoidal, or curvilinear, and perhaps it differs between patients (a theoretical example is shown in Figure 3). Also, even if the rate of progression is relatively linear over time, as discussed previously, symptoms and features likely require some threshold to be reached before they are detectable using biomarker measures and also clinically manifested. A reasonable assumption is that biomarker measures are more sensitive to neuronal dysfunction and death compared to clinical features being manifested. For example, a biomarker measure may detect a 30% decrease in a neuronal network (eg, detection of nigral degeneration), but an 80% decrease in neuronal network functioning is required before a clinical manifestation (eg, parkinsonism) becomes apparent. The challenge for investigators is to identify persons who are in the midst of Stage 1 to Stage 3 LBD, and then to use measures which reliably reflect degenerative changes in the nervous system, and perform them longitudinally, to determine the natural history of dynamic biomarker changes in LBD progression as the typical features of PD evolve. Identifying and monitoring RBD subjects is a reasonable approach to investigate this issue.
Figure 3. Theoretical evolution of clinical manifestations according to Braak stage in the Parkinson’s disease phenotype of Lewy body disease.

This figure depicts the theoretical evolution of manifestations beginning with changes in smell functioning, then autonomic functioning, then onset of REM sleep behavior disorder, then changes in motor functioning, then changes in cognition, and then prominent neuropsychiatric changes (particularly visual hallucinations). There is wide variability within and across individuals, with some experiencing minimal changes in some domains and some experiencing changes in a sequence that is different from what is shown. Furthermore, the degree of any abnormality is likely variable in terms of when it can be detected by some measure and when it becomes symptomatic. Whether the changes evolve in a linear, curvilinear, sinusoidal, or some other pattern for each domain will require further study.
Longitudinal Assessment of iRBD Subjects – Testing the Braak Staging System in vivo
Without a mechanism to easily identify individuals with prodromal LBD, it will be impossible to confirm or refute elements of the Braak staging system. The early identification of prodromal LBD will also be important for future treatment trials, assuming that the earlier the intervention is initiated in the neurodegenerative process, the more likely such intervention will modify the rate of progression. The identification of patients with idiopathic RBD (iRBD), and their longitudinal assessment, may be reasonable mechanisms to test this Braak staging system in vivo, and to prepare for future therapeutic trials.
As summarized in the Table, cross-sectional studies in patients with iRBD have shown that a significant proportion of them have detectable abnormalities on measures of smell testing,64–69 color vision and discrimination,17, 69 cardiac autonomic activity,17, 70, 71 cardiac (123)I-metaiodobenzylguanidine (MIBG) imaging,72–74 motor and gait functioning,17 neuropsychological testing,75–78 electroencephalography (EEG),79–81 transcranial sonography (TCS),15, 22, 66, 68, 82 magnetic resonance imaging (MRI)/magnetic resonance spectroscopy (MRS),83–86 single photon emission computed tomography (SPECT),87, 88 dopamine transporter imaging using SPECT,15, 16, 22, 64, 66, 89–91 fluoro-deoxyglucose positron emission tomography (FDG-PET),92, 93 and dihydrotetrabenazine (DTBZ) PET.94. Therefore, one could surmise that most patients with iRBD who have abnormalities on one or more of these measures, if they indeed have underlying LBD, represent Stage 2 or Stage 3 disease. The profile of some of these key abnormalities is shown in Figure 4.
Figure 4. Hypothesized associations between detectable features/findings and Braak stage of Lewy body pathology in idiopathic RBD patients evolving into the Parkinson’s disease phenotype.
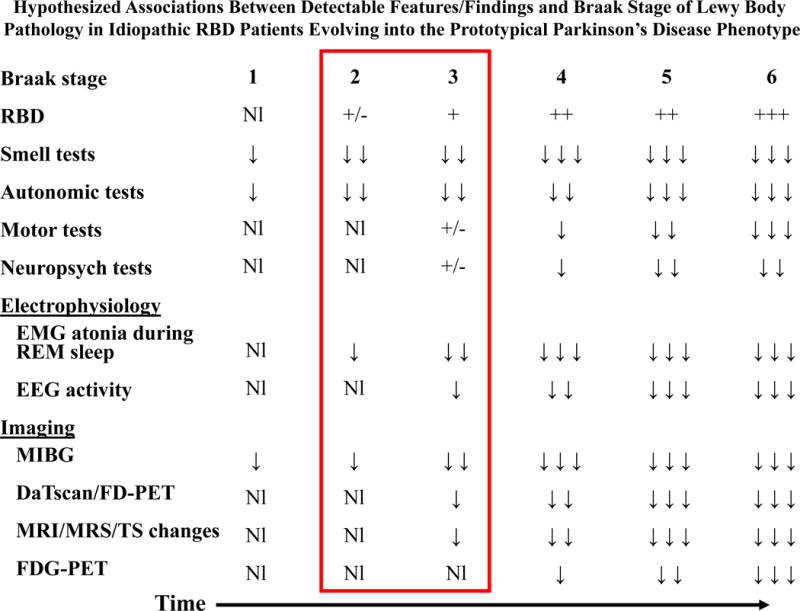
In Braak Stage 1, since the peripheral autonomic system and olfactory structures are affected, one would predict this stage to be the earliest period when detectable abnormalities on measures of smell and autonomic functioning would occur. In Braak Stage 2, the pontomedullary nuclei involved in REM sleep control are initially affected, suggesting that this stage would be the earliest period when electromyographic evidence of reduced atonia would be detectable on polysomnography. Recurrent dream enactment behavior could begin at this stage. Abnormalities on measures of smell and autonomic functioning would be more prominent. Overt RBD should be present by Braak Stage 3, and changes on measures assessing motor functioning, neuropsychological functioning, background electroencephalographic activity and several neuroimaging studies may first be detected during this stage. The window of testing the Braak staging system during the preparkinsonian phase of Lewy body disease is during Stages 2 and 3 (reflected by the red rectangle encircling these stages and associated findings). Parkinsonism (bradykinesia, rest tremor, limb rigidity, postural instability, masked facies, stooped posture, reduced armswing, or some combination of these findings) should be apparent clinically during Stage 4, and detectable changes should be apparent in most or all measures shown during this stage. Abnormalities on these measures become increasingly prominent during Stages 5 and 6. Inherent in this framework is that individual variability in the degree of degenerative changes in each affected structure will result in variability in the degrees of clinical manifestations and detectable changes on measures.
There are many questions that arise in those who have iRBD.13, 95 Question 1 may be “in whom with iRBD is a neurodegenerative disorder underlying their symptoms?” Not all patients with iRBD have an underlying neurodegenerative disorder, and if many of these measures are performed and all findings are normal, then either the underlying substrate for RBD is due to a non-degenerative process, or the degenerative process is so early and mild that no other correlate can be found. If one or more of these measures are abnormal, the suspicion of an underlying neurodegenerative disorder is heightened. The next question involves “among those with a suspected underlying neurodegenerative disorder, is a synucleinopathy or a disorder within the spectrum of tauopathies or trinucleotide repeated disorders at play?” This is where the Braak staging system provides the theoretical framework to test hypotheses. The profile of abnormalities on the measures reviewed in the Table, and particularly the findings on other biomarkers that may be sensitive and specific for evolving LBD (such biomarkers are still being studied, but none are considered sufficient for widespread clinical or research testing yet), will likely provide differential findings. Another challenging two questions in those with iRBD who undoubtedly have an underlying synucleinopathy is 1) “which phenotype will evolve – PD, MCI to DLB, MSA, PAF, or some overlap syndrome?” and 2) “when will the more definitive features of a neurodegenerative syndrome manifest?”13, 95
Current data suggests that the risk of developing cognitive impairment and/or parkinsonism is in the 50–73% range within 12–14 years after iRBD onset.18, 22 Since MSA is far less common in the population than LBD-associated neurodegenerative syndromes, one can presume that the majority of patients with iRBD due to an underlying neurodegenerative disease have LBD. The relatively few iRBD patients with abnormalities on one or more of these measures, who have been followed prospectively, have developed MCI, PD, dementia (usually DLB), or multiple system atrophy (MSA) years after the measures were performed.14, 15, 17, 18, 22, 69, 96, 97 The admittedly limited data thus far suggests 1) the degree of increased EMG tone during REM sleep may be predictive of future PD risk,97 and 2) abnormalities on olfaction, color vision, quantitative motor testing, and DaTscan and/or transcranial ultrasonography may be at increased short term risk for development of cognitive impairment and/or parkinsonism.15, 16, 21, 22, 69 Therefore, several hypotheses could be examined using a series of clinical, smell, and autonomic tests performed at periodic intervals (such as every 1–5 years) in those with iRBD, with these cases followed prospectively to determine the predictive value of various measures. Many groups of investigators are doing just that, and these should continue particularly in view of the relatively inexpensive measures being used in these studies.
A potentially even more informative approach, although clearly more rigorous and expensive, is to complete a battery with measures described above assessing clinical, smell, and autonomic functioning, and add additional electrophysiologic and imaging tests, with the plan to perform these at periodic intervals (perhaps every 1–5 years) to better answer the four questions posed above. Model programs applicable to this concept include the Alzheimer’s Disease Neuroimaging Initiative (ADNI) [http://www.adni-info.org/] and particularly the Prodromal Parkinson’s Progression Markers Initiative (PPPMI) [http://www.michaeljfox.org/living_PPMI.cfm]. To elaborate further, what change on serial testing is most predictive of a “conversion” from iRBD to a defined neurodegenerative syndrome – PD, MCI, DLB, MSA, or PAF? What profile of changes on this battery of tests best predicts each syndrome? Also, which threshold of changes best predicts short term risk of “conversion” (i.e., within five years). The battery of measures being utilized in the PPPMI continues to expand, and those with iRBD will be included in this protocol. Yet other groups are already performing longitudinal studies with similar or more expanded batteries of measures, the PPPMI protocol will not involve large numbers of subjects, and the findings from the PPPMI won’t be available for several years, suggesting that these and additional longitudinal studies in subjects with iRBD are warranted.
The theoretical considerations as shown in Figures 3 and 4 are for illustrative purposes, and clinical experience and several published series indicate that significant variability exists in the clinical manifestations of iRBD evolving into PD +/− cognitive impairment/dementia, and in the timing of when specific features evolve, if at all. The observation that the manifestations of LBD can evolve over a remarkably long period of time in some patients, as many have had iRBD features for 10–50 years prior to the onset of overt parkinsonism,14, 18, 98, 99 bodes well for many patients. However, this slow rate of progression complicates the development of clinical trials as pharmaceutical companies and funding agencies are understandably weary of supporting longitudinal studies when endpoints may be difficult to identify or are so prolonged in their evolution.
Controversies and Uncertainties
In the discussion that follows, the terms “nucleus” or “nuclei” will be used when considering a nuclear system that synthesizes and secretes one or more neurotransmitters or peptides (eg, substantia nigra), and the term neuronal network will refer to those neurons and their afferent/efferent projections which work as a functional unit (eg, olfactory network).
Clinical correlations of LB/LN vs neuronal dysfunction vs neurotransmitter dysfunction vs overt neurodegeneration in the Braak staging scheme
One criticism of the Braak staging scheme relates to the clinical correlates of intraneuronal LB/LN in key nuclei and neuronal networks, with proponents of this view arguing that the Braak staging scheme is based on the distribution pattern of the pathology alone and not on cell loss, which in and of itself should not cause any symptoms if cell loss is negligible or absent.6, 100 Another criticism is that, “the Braak staging scheme is primarily based on the expanding constellation of structures involved and not on the severity of Lewy pathologies and definitely not on the degree of cell loss. It would be misleading to suggest that Braak staging relies on increasing severity of lesions in all vulnerable structures with consequent progressive neuronal loss.”101 Also, some argue that many patients do not follow the staged model as proposed.102–105
As emphasized by Braak et al, there is evidence of dysfunction in those neurons with LB/LN inclusions.4, 106], and the severity of pathology within key structures increases with advancing stages.4, 5, 107 Some of the findings that have been discrepant from those of Braak et al might have been due to the use of variable laboratory protocols for selecting tissue blocks from the brain, processing and analyzing tissue.108–110 Furthermore, neurotransmitter dysfunction could potentially cause biomarker changes and symptoms in the absence of overt neuronal loss.106 Plus, recent evidence indicates that alterations in synaptic functioning are key aspects of neuronal dysfunction,111 which stands to reason considering that α-synuclein is a synaptic protein and the pathophysiologic processes involved in LBD may alter the synapse with the formation of LB/LN inclusions being a more downstream result. An in-depth interpretation of the data for and against these hypotheses is beyond the scope of this review, but considering that the clinical phenomenology of prodromal and symptomatic PD corresponds reasonably well with the nuclei and networks affected by LB/LN inclusions as per the Braak model, the model has solid face value and is worthy of continued research into its clinical correlates.47, 109, 112
Thresholds and variabilities for biomarker detection and clinical feature expression
A related issue pertains to what threshold of dysfunction in a nucleus and/or neuronal network permits 1) detection by a biomarker and 2) expression of a clinical feature. Again, one would expect that biomarker detection of dysfunctional a nucleus or neuronal network will be evident prior to overt clinical expression of symptoms or findings, and the time difference between biomarker detection and clinical expression likely spans years or decades for many networks. Using the association of nigral degeneration with parkinsonism example again, nigral changes may begin in Stage 3 (each stage presumably evolves over years), and biomarker measures such as DaTscan or TCS may detect abnormalities compared to aged matched norms when (for example) a 30% decrease in functional activity is present (perhaps reflecting early to mid Stage 3), subtle but asymptomatic motor changes based on gait assessment and timed motor activities may not be evident until (for example) a 50% decrement has occurred (perhaps reflecting mid to late Stage 3 or early Stage 4), and overt symptomatic parkinsonism may not become evident until 80% of the substantia nigra has degenerated, which according to the Braak staging scheme would represent Stage 4 at the earliest. Assessment of longitudinal motor changes using the neuroimaging and clinical measures described above in prodromal PD would be very important to plan for disease-modifying therapies, and with a clinical feature such as RBD potentially revealing those who may be in the midst of Stage 2 or 3 disease, identification of iRBD could permit clarification of measureable changes in dopaminergic functioning and their corresponding clinical correlates.
Another important concept regarding thresholds involves variability in nuclear structures (eg, brainstem nuclei) and functional networks (eg, striatonigral system). While LB/LN involvement in several vulnerable nuclei may consistently occur within and across patients, is it realistic to expect that the same degree of neuronal dysfunction or neuronal loss will occur in all affected structures, even in those structures which develop LB/LN at the same stage in the Braak model? Maybe or maybe not. Without this knowledge, it is challenging to know how to interpret biomarkers which measure functional activity of nuclei or networks which are considered within the same stage of the Braak model. For example, one nucleus or network undergoing a functional change may be detectable by a biomarker when a 30% decrease in functional activity has occurred, whereas another structure or network undergoing a change may be detectable by a biomarker measure only when a 70% decrease has occurred. Are these differences due to the different sensitivities of the biomarker measures themselves, or do they represent true differences in the degrees of dysfunction within the nuclei or networks being measured? Plus, a biomarker measure may be markedly abnormal when 30%, or 50%, of functional activity of that nucleus or network has become dysfunctional. For example, if markedly impaired olfaction on formal smell testing might be present with a 50% reduction in functional activity in the olfactory structures (this is also speculative and only used as an example), then such a biomarker may be a sensitive marker of prodromal LBD, but that measure may not be useful as a longitudinal marker of progression if the degree of abnormality is marked early in the course (ie, floor effect). These uncertainties represent challenges to the field as comprehensive batteries of measures are performed in iRBD patients, particularly as the findings are analyzed longitudinally. Yet they must be done in order to develop protocols so that potential disease-modifying therapies can be carried out with appropriate measures and endpoints, and be adequately powered to detect changes compared to natural history data or (more ideally) to placebo-controlled subjects.
One other issue to consider is the potential impact of multiple factors contributing to a biomarker finding or clinical feature. The most obvious example of a complex construct is cognition, in which multiple neurotransmitter systems and several neuronal networks subserving the different cognitive domains surely impact on cognitive functioning. Does one consider cognition as abnormal when a global measure is impaired? Or only when neuropsychologic impairment is associated with functional decline? Or does one consider impairment compared to aged-matched norms on single neuropsychological measures, or sets of measures that assess the same cognitive domain, as the best markers for determining impairment and then tracking longitudinal change? While impairment on certain neuropsychological measures have been documented in those with iRBD,75–78 there is clearly variability in the degree of impairment within and across measures. Plus, there is inherent variability for each measure depending on the state of the individual at the time of testing (ie, adequacy of recent sleep, mood state, concurrent stressors and medications, etc.). These questions are complex to address,113 but clearly warrant further study.
These issues underscore the need to better determine 1) what each biomarker is measuring at the nuclear and network levels, 2) what threshold must be reached to view each biomarker as abnormal, 3) how to consider the inherent variability of functional decreases in the different biomarker measures and clinical features within and across subjects, and 4) whether the various possible biomarkers will track with progression of the disease.
Prototypical versus atypical PD phenotypes in the context of the Braak staging scheme
The Braak staging scheme explains the temporal sequence of clinical feature evolution of prototypical PD reasonably well, which is remarkable considering that the stages were based on pathologic material (which “by definition” is cross-sectional) from individuals who presumably had died at some point along the continuum of prodromal to symptomatic PD.109 Seasoned neurologists who have evaluated patients with very early PD and followed them over many years, as well as the many sleep clinicians who have followed iRBD patients over years through their development of PD, can in hindsight view the temporal sequence of what they have witnessed in their clinical practice to mirror the temporal sequence of feature evolution that is predicted by the Braak model. Recent studies using more rigorous methodology have substantiated what clinicians have been observing.38, 39 Yet these same clinicians will comment on the minority of patients who “haven’t followed the model,” and some authors have used the findings in case reports or series of cases in these “atypical PD” cases as arguments suggesting that the Braak staging model is not accurate.6, 100, 102–105 Perhaps there is another perspective to view all PD patients within this context.
It is very difficult to determine, particularly early in the course of disease when features are mild or not clinically expressed yet, whether any individual PD patient is following a prototypical versus an atypical course. For example, it is well-established that not near every PD patient will ever exhibit RBD, or they might start exhibiting RBD years after the onset of otherwise typical PD symptomatology.114 Plus, there are examples of patients who experience typical RBD features over ten or more years and never exhibit any other neurologic sign or symptom of a Lewy body disease-spectrum disorder, yet have pathologically-proven LBD.115, 116 Also, a significant proportion of PD patients do not experience anosmia, or obvious autonomic dysfunction, or ever develop dementia despite otherwise typical PD features over decades.38, 39, 117
The bottom line is that individual variability can never be fully captured in any model of human neurodegenerative disease, as there will always be exceptions to any model. If one considers Alzheimer’s disease (AD), patients with prototypical AD experience features initially most consistent with amnestic mild cognitive impairment, and subsequently develop impairment in other cognitive domains associated with functional impairment in everyday activities, and thus meet criteria for clinically probable AD.118 The Braak staging scheme for the topography and evolution of neurofibrillary tangle development, in which NFTs are initially present in the transentorrhinal regions, then spread to the limbic regions, and then to isocortical regions,119, 120 correlates quite well with the clinical, neuropsychological and neuroimaging findings from amnestic MCI to mild then moderate then severe AD.118, 121 With the focus over recent years on identifying and characterizing subjects with presymptomatic as well as symptomatic Alzheimer’s disease and following them longitudinally with a battery of measures,122 the initial findings fostered the development of a hypothetical model of dynamic changes in evolving Alzheimer’s disease123. Continued longitudinal assessments along with the expansion of additional subjects are already leading to refinements in the model,124–126 which will impact many aspects of AD research and therapeutic trials for years to come. The PD field is not at this degree of prodromal characterization yet, although this not due to lack of effort, insufficient patients, or inadequate investigator motivation. Rather, characterization of prodromal PD – particularly involving iRBD subjects – has not received the needed attention, high prioritization and necessary funding across centers of excellence globally to prepare for disease-modifying trials once promising agents are identified. This will hopefully change in the near future.
Also, there are numerous examples of patients with ultimately autopsy-proven AD that have minimal to no significant memory impairment and minimal to no mesial temporal lobe pathology (so-called “hippocampal-sparing AD”) who present in life as frontotemporal dementia, primary progressive aphasia, corticobasal syndrome or posterior cortical atrophy depending on which focal/asymmetric cortical regions become dysfunctional early in the course.127 Do these “atypical AD” cases “fit” the Braak model of NFT topography and progression? No. Yet do they negate the utility of the Braak model for prototypical AD? Also no. The model has proven very useful, and as noted above, the dynamic model of biomarker changes as it applies to beta-amyloid and hyperphosphorylated tau deposition in the brain is being revised based on ongoing longitudinal studies. The revised model incorporates some of the atypical observations that previously didn’t “fit” the model.
One would likely expect that a similar appreciation will occur in the years to come as the biomarker correlates in prototypical and atypical PD will offer insights on an updated model provided that an adequate battery of measures are utilized longitudinally in a large number of subjects which surely will include prototypical and atypical cases.
The Braak staging scheme as it pertains to the non-PD phenotypes of LBD
Another criticism of the Braak staging scheme is that it does not appear to apply to the non-PD phenotypes of LBD such as PAF and particularly DLB.6, 100, 102, 104, 105 The authors of some of these papers seem to neglect the point that has always been emphasized by Braak and his colleagues – their staging scheme was designed to apply to prototypical PD.4, 106 They have never claimed that their model also applied to DLB or PAF, although many investigators still find it attractive to do so, including this author.9, 11, 12, 128 Again, due to the cross-sectional nature of any autopsy study which attempts to characterize a disease process that may have clinical applicability, it is impossible to know which cases studied in a prodromal state (for example, Stages 1–3 LBD) would have evolved to a PD versus DLB phenotype had those individuals lived long enough to develop any symptoms. Furthermore, neuropathologists have found it very difficult to hypothesize which clinical phenotype was exhibited in patients who have neuropathologic evidence of brainstem, limbic and neocortical LB/LN pathology as the syndromic phenotypes of PD, PDD and DLB can appear indistinguishable based on histologic examination.57 As noted above, these and other observations suggest that alterations beyond the presence of LB/LN in neurons and their processes are likely contributing to the prodromal and symptomatic aspects of LBD.
On a related note, in contrast to the caudal to rostral or “bottom-up” evolution of LBD topography that applies aptly to prototypical PD, perhaps prototypical DLB is best considered as LBD progressing in a rostral to caudal or “top-down” evolution of topography. This latter view appears inconsistent with most of the published data on DLB, in which RBD and other typical features of prodromal PD begin years or decades prior to the onset of cognitive decline.38, 39, 55, 129 A more parsamonious view would be that most patients with PD evolve in a relatively prototypical manner, and the same occurs in most DLB patients, with a “bottom-up” sequence of evolution, and there are atypical cases who evolve in a more patchy or diffuse manner with some vulnerable networks affected relatively consistently and profoundly while others are affected more minimally such that symptoms never become manifest (and perhaps biomarkers cannot detect anything is abnormal).130 Furthermore, some features of PD are qualitatively different than in DLB, such as elements of parkinsonism (DLB patients have more symmetric bradykinesia/rigidity/tremor and the tremor is more obvious with posture/action than at rest than in PD), and cognitive impairment (the neuropsychological deficits in DLB are more prominent in the visuospatial domain than in similarly functionally impaired PDD patients), and the biologic reasons for these differences remain unclear.131 An ample number of additional cases who are well-characterized (using many of the biomarker measures discussed in this review) during life and expire in the midst of the various phenotypes that span the spectrum of LBD – prodromal LBD, MCI, PD plus MCI, PDD, and DLB (particularly DLB with minimal to no parkinsonism prior to death) – will be needed to better determine how the Braak model might be reconsidered or adapted to apply to prototypical DLB.
Conclusions
There are few other disorders as distinctive as RBD in its clinical features and as early a manifestation in the course of a neurodegenerative disease. Staging schemes based on autopsied material designed to posit the evolution of neurodegenerative disease need reliable markers of disease during life to verify its utility, and when inconsistencies are found, then modifications should be made as the nuances of disease evolution are appreciated. The Braak staging scheme provides a welcome framework from which to test hypotheses in the second most common neurodegenerative disorder affecting humans – LBD – particularly for the prototypical PD phenotype, and iRBD is a relatively consistent early clinical manifestation which provides a window for gaining further insights into LBD evolution in vivo.
Supplementary Material
Search Strategy and Selection Criteria Statement.
Medline (www.pubmed.gov) was used to search for articles published in the English language between January, 1986, and February, 2013, with the search terms “REM sleep”, “REM sleep behavior disorder”, “Parkinson disease”, “Parkinson’s disease”, “Dementia with Lewy bodies”, “Lewy body”, “Lewy body disease”, and “Braak staging,” and the papers and additional referenced papers were reviewed as they pertained to prodromal Parkinson’s disease. Those papers which included concepts and/or data on prodromal Parkinson’s disease, REM sleep behavior disorder, underlying Lewy body disease and the Braak staging scheme for Parkinson’s disease, were included in this review and referenced accordingly.
Acknowledgments
Supported by grants AG015866, AG016574, AG006786, NS040256, the Mangurian Foundation for Lewy Body Dementia Research, and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program of the Mayo Foundation
Disclosures : Dr. Boeve has served as an investigator for clinical trials sponsored by Cephalon, Inc., Allon Pharmaceuticals and GE Healthcare. He receives royalties from the publication of a book entitled Behavioral Neurology Of Dementia (Cambridge Medicine, 2009). He has received honoraria from the American Academy of Neurology. He serves on the Scientific Advisory Board of the Tau Consortium. He receives research support from the National Institute on Aging (P50 AG016574, U01 AG006786, RO1 AG032306, RO1 AG041797 and the Mangurian Foundation.
Abbreviations
- DaTscan
ioflupane dopamine transporter scanning
- EEG
electroencephalographic
- EMG
electromyographic
- FD-PET
fluorodopa positron emission tomography
- FDG-PET
fluorodeoxyglucose positron emission tomography
- MIBG
Cardiac (123)I-metaiodobenzylguanidine imaging
- MRI
magnetic resonance imaging
- MRS
magnetic resonance spectroscopy
- RBD
REM sleep behavior disorder
- REM
rapid eye movement
- TS
transcranial sonography
- Notations
Nl=normal
- +/−
equivocal abnormalities
- +
mild degree of frequency/severity
- ++
moderate degree of frequency/severity
- +++
marked degree of frequency/severity
- ↓
mildly abnormal
- ↓↓
moderately abnormal
- ↓↓↓
severely abnormal
References
- 1.Schenck C, Boeve B. The strong presence of REM sleep behavior disorder in PD: Clinical and research implications. Neurology. 2011;77:1030–1032. doi: 10.1212/WNL.0b013e31822e14d7. [DOI] [PubMed] [Google Scholar]
- 2.Postuma RB, Aarsland D, Barone P, Burn DJ, Hawkes CH, Oertel W, et al. Identifying prodromal Parkinson’s disease: pre-motor disorders in Parkinson’s disease. Mov Disord. 2012;27(5):617–26. doi: 10.1002/mds.24996. [DOI] [PubMed] [Google Scholar]
- 3.Olanow C, Obeso J. The significance of defining preclinical or prodromal Parkinson’s disease. Mov Disord. 2012;27:666–9. doi: 10.1002/mds.25019. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Del Tredici K, Rub U, de Vos R, Jansen Steur E, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–34. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 6.Jellinger K. A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol. 2008;116:1–16. doi: 10.1007/s00401-008-0406-y. [DOI] [PubMed] [Google Scholar]
- 7.Boeve B, Silber M, Ferman T, Lin S, Benarroch E, Schmeichel A, et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med. 2013 doi: 10.1016/j.sleep.2012.10.015. Available online 7 March 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boeve B, Silber M, Ferman T, Lucas J, Parisi J. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16:622–30. doi: 10.1002/mds.1120. [DOI] [PubMed] [Google Scholar]
- 9.Boeve B, Silber M, Parisi J, Dickson D, Ferman T, Benarroch E, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61:40–5. doi: 10.1212/01.wnl.0000073619.94467.b0. [DOI] [PubMed] [Google Scholar]
- 10.Boeve B, Silber M, Ferman T. REM sleep behavior disorder in Parkinson’s disease and dementia with Lewy bodies. J Ger Psychiatry Neurol. 2004;17:146–57. doi: 10.1177/0891988704267465. [DOI] [PubMed] [Google Scholar]
- 11.Boeve B, Saper C. REM sleep behavior disorder: A possible early marker for synucleinopathies. Neurology. 2006;66:796–7. doi: 10.1212/01.wnl.0000209264.61035.bb. [DOI] [PubMed] [Google Scholar]
- 12.Boeve B, Silber M, Saper C, Ferman T, Dickson D, Parisi J, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain. 2007;130:2770–88. doi: 10.1093/brain/awm056. [DOI] [PubMed] [Google Scholar]
- 13.Boeve B. REM sleep behavior disorder: Updated review of the core features, the REM sleep behavior disorder-neurodegenerative disease association, evolving concepts, controversies, and future directions. Ann NY Acad Sci. 2010;1184:17–56. doi: 10.1111/j.1749-6632.2009.05115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iranzo A, Molinuevo J, Santamaría J, Serradell M, Martí M, Valldeoriola F, et al. Rapid-eye-movement sleep behaviour disorder as an early marker for a neurodegenerative disorder: a descriptive study. Lancet Neurol. 2006;5:572–7. doi: 10.1016/S1474-4422(06)70476-8. [DOI] [PubMed] [Google Scholar]
- 15.Iranzo A, Lomeña F, Stockner H, Valldeoriola F, Vilaseca I, Salamero M, et al. Decreased striatal dopamine transporter uptake and substantia nigra hyperechogenicity as risk markers of synucleinopathy in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study. Lancet Neurol. 2010;9(11):1070–7. doi: 10.1016/S1474-4422(10)70216-7. [DOI] [PubMed] [Google Scholar]
- 16.Iranzo A, Valldeoriola F, Lomeña F, Molinuevo J, Serradell M, Salamero M, et al. Serial dopamine transporter imaging of nigrostriatal function in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study. Lancet Neurol. 2011;10:797–805. doi: 10.1016/S1474-4422(11)70152-1. [DOI] [PubMed] [Google Scholar]
- 17.Postuma R, Lang A, Massicotte-Marquez J, Montplaisir J. Potential early markers of Parkinson disease in idiopathic REM sleep behavior disorder. Neurology. 2006;66:845–51. doi: 10.1212/01.wnl.0000203648.80727.5b. [DOI] [PubMed] [Google Scholar]
- 18.Postuma R, Gagnon J, Vendette M, Fantini M, Massicotte-Marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology. 2009;72:1296–300. doi: 10.1212/01.wnl.0000340980.19702.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Postuma RB, Gagnon JF, Montplaisir JY. REM sleep behavior disorder: From dreams to neurodegeneration. Neurobiol Dis. 2012;46(3):553–8. doi: 10.1016/j.nbd.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Postuma RB, Bertrand JA, Montplaisir J, Desjardins C, Vendette M, Rios Romenets S, et al. Rapid eye movement sleep behavior disorder and risk of dementia in Parkinson’s disease: A prospective study. Mov Disord. 2012;27(6):720–6. doi: 10.1002/mds.24939. [DOI] [PubMed] [Google Scholar]
- 21.Postuma R, Lang A, Gagnon J, Pelletier A, Montplaisir J. How does parkinsonism start? Prodromal parkinsonism motor changes in idiopathic REM sleep behaviour disorder. Brain. 2012;135:1860–70. doi: 10.1093/brain/aws093. [DOI] [PubMed] [Google Scholar]
- 22.Iranzo A, Tolosa E, Gelpi E, Molinuevo J, Valldeoriola F, Serradell M, et al. Rapid eye movement sleep behaviour disorder in the prodromal phase of Lewy body disorders: An observational study. Lancet Neurol. 2013;XX:XXX–XXX. [Google Scholar]
- 23.Schenck C, Boeve B, Mahowald M. Delayed emergence of a parkinsonian disorder or dementia in 81% of older males initially diagnosed with idiopathic REM sleep behavior disorder (RBD): 16year update on a previously reported series. Sleep Med. 2013 doi: 10.1016/j.sleep.2012.10.009. Available online March 2013. [DOI] [PubMed] [Google Scholar]
- 24.Schenck CH, Bundlie SR, Patterson AL, Mahowald MW. Rapid eye movement sleep behavior disorder. A treatable parasomnia affecting older adults. Jama. 1987;257(13):1786–9. [PubMed] [Google Scholar]
- 25.Schenck C, Mahowald M. REM sleep behavior disorder: Clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep. 2002;25:120–38. doi: 10.1093/sleep/25.2.120. [DOI] [PubMed] [Google Scholar]
- 26.Olson E, Boeve B, Silber M. Rapid eye movement sleep behavior disorder: demographic, clinical, and laboratory findings in 93 cases. Brain. 2000;123:331–9. doi: 10.1093/brain/123.2.331. [DOI] [PubMed] [Google Scholar]
- 27.Iber C, Ancoli-Israel S, Chesson A, Quan S, Medicine ftAAoS . The AASM manual for the scoring of sleep and associated events: rules, terminology and technical specifications. Westchester, IL: American Academy of Sleep Medicine; 2007. [Google Scholar]
- 28.International Classification of Sleep Disorders: Diagnostic and Coding Manual. 2nd. Westchester, IL: American Academy of Sleep Medicine; 2005. [Google Scholar]
- 29.McCarter S, St Louis E, Boeve B. REM sleep behavior disorder and REM sleep without atonia as an early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep. 2012;12:182–92. doi: 10.1007/s11910-012-0253-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai Y, Siegel J. Medullary regions mediating atonia. J Neurosci. 1988;8:4790–6. doi: 10.1523/JNEUROSCI.08-12-04790.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai Y, Siegel J. Brainstem-mediated locomtion and myoclonic jerks. II. Pharmacological effects. Brain Res. 1997;745:265–70. doi: 10.1016/s0006-8993(96)01180-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siegel J. REM Sleep Behavior Disorder. In: Schenck C, editor. Associated Professional Sleep Societies. Vol. 2001. Chicago, IL: 2001. [Google Scholar]
- 33.Boissard R, Gervasoni D, Schmidt M, Barbagli B, Fort P, Luppi P. The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci. 2002;16:1959–73. doi: 10.1046/j.1460-9568.2002.02257.x. [DOI] [PubMed] [Google Scholar]
- 34.Boissard E, Fort P, Gervasoni D, Barbagli B, Luppi P. Localization of the GABAergic and non-GABAergic neurons projecting to the sublaterodorsal nucleus and potentially gating paradoxical sleep onset. Eur J Neurosci. 2003;18:1627–39. doi: 10.1046/j.1460-9568.2003.02861.x. [DOI] [PubMed] [Google Scholar]
- 35.Siegel J. The stuff dreams are made of: anatomical substrates of REM sleep. Nature Neurosci. 2006;9:721–2. doi: 10.1038/nn0606-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu J, Sherman D, Devor M, Saper C. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–94. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- 37.Frauscher B, Iranzo A, Högl B, Casanova-Molla J, Salamero M, Gschliesser V, et al. Quantification of electromyographic activity during REM sleep in multiple muscles in REM sleep behavior disorder. Sleep. 2008;31:724–31. doi: 10.1093/sleep/31.5.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaenslen A, Swid I, Liepelt-Scarfone I, Godau J, Berg D. The patients’ perception of prodromal symptoms before the initial diagnosis of Parkinson’s disease. Mov Disord. 2011;26:653–8. doi: 10.1002/mds.23499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ross G, Abbott R, Petrovitcha H, Tanner C, White L. Pre-motor features of Parkinson’s disease: the Honolulu-Asia Aging Study experience. Parkinsonism Relat Disord. 2012;18S1:S199–S202. doi: 10.1016/S1353-8020(11)70062-1. [DOI] [PubMed] [Google Scholar]
- 40.Emre M, Aarsland D, Brown R, Burn D, Duyckaerts C, Mizuno Y, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord. 2007;22:1689–707. doi: 10.1002/mds.21507. [DOI] [PubMed] [Google Scholar]
- 41.Dugger B, Boeve B, Murray M, Parisi J, Fujishiro H, Dickson D, et al. Rapid eye movement sleep behavior disorder and subtypes in autopsy-confirmed dementia with Lewy bodies. Mov Disord. 2012;27(1):72–8. doi: 10.1002/mds.24003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dugger B, Murray M, Boeve B, Parisi J, Benarroch E, Ferman T, et al. Neuropathological analysis of brainstem cholinergic and catecholaminergic nuclei in relation to rapid eye movement (REM) sleep behaviour disorder. Neuropathol Appl Neurobiol. 2012;38(2):142–52. doi: 10.1111/j.1365-2990.2011.01203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savica R, Rocca W, Ahlskog J. When does Parkinson disease start? Arch Neurol. 2010;67:798–801. doi: 10.1001/archneurol.2010.135. [DOI] [PubMed] [Google Scholar]
- 44.Lang A. A critical appraisal of the premotor symptoms of Parkinson’s disease: potential usefulness in early diagnosis and design of neuroprotective trials. Mov Disord. 2011;26:775–83. doi: 10.1002/mds.23609. [DOI] [PubMed] [Google Scholar]
- 45.Siderowf A, Lang A. Premotor Parkinson’s disease: concepts and definitions. Mov Disord. 2012;27:608–16. doi: 10.1002/mds.24954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Tredici K, Braak H. Spinal cord lesions in sporadic Parkinson’s disease. Acta Neuropathol. 2012;124(5):643–64. doi: 10.1007/s00401-012-1028-y. [DOI] [PubMed] [Google Scholar]
- 47.Halliday G, McCann H, Shepherd C. Evaluation of the Braak hypothesis: how far can it explain the pathogenesis of Parkinson’s disease? Expert Rev Neurotherap. 2012;12:673–86. doi: 10.1586/ern.12.47. [DOI] [PubMed] [Google Scholar]
- 48.Desplats P, Lee H, Bae E, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. PNAS. 2009;106:13010–5. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freundt E, Maynard N, Clancy E, Roy S, Bousset L, Sourigues Y, et al. Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann Neurol. 2012;72:517–24. doi: 10.1002/ana.23747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Polymenidou M, Cleveland D. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med. 2012;209:889–93. doi: 10.1084/jem.20120741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hansen C, Li J. Beyond alpha-synuclein transfer: pathology propagation in Parkinson’s disease. Trends Mol Med. 2012;18:248–55. doi: 10.1016/j.molmed.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 52.Luk K, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski J, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Volpicelli-Daley L, Luk K, Patel T, Tanik S, Riddle D, Stieber A, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Braak H, Sastre M, Bohl J, de Vos R, Del Tredici K. Parkinson’s disease: lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol. 2007;113:421–9. doi: 10.1007/s00401-007-0193-x. [DOI] [PubMed] [Google Scholar]
- 55.Molano J, Boeve B, Ferman T, Smith G, Parisi J, Dickson D, et al. Mild cognitive impairment associated with limbic and neocortical Lewy body disease: A clinicopathological study. Brain. 2009;133:540–56. doi: 10.1093/brain/awp280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Apaydin H, Ahlskog J, Parisi J, Boeve B, Dickson D. Parkinson’s disease neuropathology: Later-developing dementia and loss of the levodopa response. Arch Neurol. 2002;59:102–12. doi: 10.1001/archneur.59.1.102. [DOI] [PubMed] [Google Scholar]
- 57.Tsuboi Y, Dickson D. Dementia with Lewy bodies and Parkinson’s disease with dementia: are they different? Parkinsonism Relat Disord. 2005;11:S47–S51. doi: 10.1016/j.parkreldis.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 58.Weintraub D, Doshi J, Koka D, Davatzikos C, Siderowf A, Duda J, et al. Neurodegeneration across stages of cognitive decline in Parkinson disease. Arch Neurol. 2011;68:1562–8. doi: 10.1001/archneurol.2011.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Irwin D, White M, Toledo J, Xie S, Robinson J, Van Deerlin V, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72:587–98. doi: 10.1002/ana.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson’s disease. Arch Neurol. 1993;50(2):140–8. doi: 10.1001/archneur.1993.00540020018011. [DOI] [PubMed] [Google Scholar]
- 61.Williams-Gray C, Evans J, Goris A, Foltynie T, Ban M, Robbins T, et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain. 2009;132:2958–69. doi: 10.1093/brain/awp245. [DOI] [PubMed] [Google Scholar]
- 62.Frigerio R, Fujishiro H, Maraganore D, Klos K, DelleDonne A, Heckman M, et al. Comparison of risk factor profiles in incidental Lewy body disease and Parkinson disease. Arch Neurol. 2009;66:1114–9. doi: 10.1001/archneurol.2009.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DelleDonne A, Klos K, Fujishiro H, Ahmed Z, Parisi J, Josephs K, et al. Incidental Lewy body disease and preclinical Parkinson disease. Arch Neurol. 2008;65(8):1074–180. doi: 10.1001/archneur.65.8.1074. [DOI] [PubMed] [Google Scholar]
- 64.Stiasny-Kolster K, Doerr Y, Möller J, Höffken H, Behr T, Oertel W, et al. Combination of ‘idiopathic’ REM sleep behaviour disorder and olfactory dysfunction as possible indicator for -synucleinopathy demonstrated by dopamine transporter FP-CIT-SPECT. Brain. 2005;128:126–37. doi: 10.1093/brain/awh322. [DOI] [PubMed] [Google Scholar]
- 65.Fantini M, Postuma R, Montplaisir J, Ferini-Strambi L. Olfactory deficit in idiopathic rapid eye movements sleep behavior disorder. Brain Res Bull. 2006;70:386–90. doi: 10.1016/j.brainresbull.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 66.Unger M, Möller J, Stiasny-Kolster K, Mankel K, Berg D, Walter U, et al. Assessment of idiopathic rapid-eye-movement sleep behavior disorder by transcranial sonography, olfactory function test, and FP-CIT-SPECT. Mov Disord. 2008;23:596–9. doi: 10.1002/mds.21908. [DOI] [PubMed] [Google Scholar]
- 67.Miyamoto T, Miyamoto M, Iwanami M, Hirata K, Kobayashi M, Nakamura M, et al. Olfactory dysfunction in idiopathic REM sleep behavior disorder. Sleep Med. 2010;11:458–61. doi: 10.1016/j.sleep.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 68.Iwanami M, Miyamoto T, Miyamoto M, Hirata K, Takada E. Relevance of substantia nigra hyperechogenicity and reduced odor identification in idiopathic REM sleep behavior disorder. Sleep Med. 2010;11:361–5. doi: 10.1016/j.sleep.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 69.Postuma R, Gagnon J, Vendette M, Desjardins C, Montplaisir J. Olfaction and color vision identify impending neurodegeneration in rapid eye movement sleep behavior disorder. Ann Neurol. 2011;69:811–8. doi: 10.1002/ana.22282. [DOI] [PubMed] [Google Scholar]
- 70.Ferini-Strambi L, Oldani A, Zucconi M, Smirne S. Cardiac autonomic activity during wakefulness and sleep in REM sleep behavior disorder. Sleep. 1996;19:367–9. doi: 10.1093/sleep/19.5.367. [DOI] [PubMed] [Google Scholar]
- 71.Postuma R, Lanfranchi P, Blais H, Gagnon J, Montplaisir J. Cardiac autonomic dysfunction in idiopathic REM sleep behavior disorder. Mov Disord. 2010;25:2304–10. doi: 10.1002/mds.23347. [DOI] [PubMed] [Google Scholar]
- 72.Miyamoto T, Miyamoto M, Inoue Y, Usui Y, Suzuki K, Hirata K. Reduced cardiac 123I-MIBG scintigraphy in idiopathic REM sleep behavior disorder. Neurology. 2006;67:2236–8. doi: 10.1212/01.wnl.0000249313.25627.2e. [DOI] [PubMed] [Google Scholar]
- 73.Miyamoto T, Miyamoto M, Suzuki K, Nishibayashi M, Iwanami M, Hirata K. 123I-MIBG cardiac scintigraphy provides clues to the underlying neurodegenerative disorder in idiopathic REM sleep behavior disorder. Sleep. 2008;31:717–23. doi: 10.1093/sleep/31.5.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oguri T, Tachibana N, Mitake S, Kawanishi T, Fukuyama H. Decrease in myocardial 123I-MIBG radioactivity in REM sleep behavior disorder: two patients with different clinical progression. Sleep Med. 2008;9:583–5. doi: 10.1016/j.sleep.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 75.Ferini-Strambi L, Di Gioia M, Castronovo V, Oldani A, Zucconi M, Cappa S. Neuropsychological assessment in idiopathic REM sleep behavior disorder (RBD): Does the idiopathic form of RBD really exist? Neurology. 2004;62:41–5. doi: 10.1212/01.wnl.0000101726.69701.fa. [DOI] [PubMed] [Google Scholar]
- 76.Massicotte-Marquez J, Décary A, Gagnon J, Vendette M, Mathieu A, Postuma R, et al. Executive dysfunction and memory impairment in idiopathic REM sleep behavior disorder. Neurology. 2008;70:1250–7. doi: 10.1212/01.wnl.0000286943.79593.a6. [DOI] [PubMed] [Google Scholar]
- 77.Terzaghi M, Sinforiani E, Zucchella C, Zambrelli E, Pasotti C, Rustioni V, et al. Cognitive performance in REM sleep behaviour disorder: a possible early marker of neurodegenerative disease? Sleep Med. 2008;9:343–51. doi: 10.1016/j.sleep.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 78.Fantini ML, Farini E, Ortelli P, Zucconi M, Manconi M, Cappa S, et al. Longitudinal study of cognitive function in idiopathic REM sleep behavior disorder. SLEEP. 2011;34:619–25. [PMC free article] [PubMed] [Google Scholar]
- 79.Fantini ML, Gagnon JF, Petit D, Rompre S, Decary A, Carrier J, et al. Slowing of electroencephalogram in rapid eye movement sleep behavior disorder. Ann Neurol. 2003;53(6):774–80. doi: 10.1002/ana.10547. [DOI] [PubMed] [Google Scholar]
- 80.Massicotte-Marquez J, Carrier J, Decary A, Mathieu A, Vendette M, Petit D, et al. Slow-wave sleep and delta power in rapid eye movement sleep behavior disorder. Ann Neurol. 2005;57:277–82. doi: 10.1002/ana.20373. [DOI] [PubMed] [Google Scholar]
- 81.Iranzo A, Isetta V, Molinuevo J, Serradell M, Navajas D, Farre R, et al. Electroencephalographic slowing heralds mild cognitive impairment in idiopathic REM sleep behavior disorder. Sleep Med. 2010;11:534–9. doi: 10.1016/j.sleep.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 82.Stockner H, Iranzo A, Seppi K, Serradell M, Gschliesser V, Sojer M, et al. Midbrain hyperechogenicity in idiopathic REM sleep behavior disorder. Mov Disord. 2009;24:1906–9. doi: 10.1002/mds.22483. [DOI] [PubMed] [Google Scholar]
- 83.Iranzo A, Santamaria J, Pujol J, Moreno A, Deus J, Tolosa E. Brainstem proton magnetic resonance spectroscopy in idopathic REM sleep behavior disorder. Sleep. 2002;25(8):867–70. [PubMed] [Google Scholar]
- 84.Unger M, Belke M, Menzler K, Heverhagen J, Keil B, Stiasny-Kolster K, et al. Diffusion tensor imaging in idiopathic REM sleep behavior disorder reveals microstructural changes in the brainstem, substantia nigra, olfactory region, and other brain regions. SLEEP. 2010;33:767–73. doi: 10.1093/sleep/33.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ellmore T, Hood A, Castriotta R, Stimming E, Bick R, Schiess M. Reduced volume of the putamen in REM sleep behavior disorder patients. Parkinsonism Relat Disord. 2010;16:645–9. doi: 10.1016/j.parkreldis.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 86.Scherfler C, Frauscher B, Schocke M, Iranzo A, Gschliesser V, Seppi K, et al. White and gray matter abnormalities in idiopathic rapid eye movement sleep behavior disorder: a diffusion-tensor imaging and voxel-based morphometry study. Ann Neurol. 2011;69:400–7. doi: 10.1002/ana.22245. [DOI] [PubMed] [Google Scholar]
- 87.Mazza S, Soucy J, Gravel P, Michaud M, Postuma R, Massicotte J, et al. Assessing whole brain perfusion changes in REM sleep behavior disorder. Neurology. 2006;67:1618–22. doi: 10.1212/01.wnl.0000242879.39415.49. [DOI] [PubMed] [Google Scholar]
- 88.Hanyu H, Inoue Y, Sakurai H, Kanetaka H, Nakamura M, Miyamoto T, et al. Regional cerebral blood flow changes in patients with idiopathic REM sleep behavior disorder. Eur J Neurol. 2011;18:784–8. doi: 10.1111/j.1468-1331.2010.03283.x. [DOI] [PubMed] [Google Scholar]
- 89.Eisensehr I, Linke R, Noachtar S, Schwarz J, Gildehaus F, Tatsch K. Reduced striatal dopamine transporters in idiopathic rapid eye movement sleep behaviour disorder: Comparison with Parkinson’s disease and controls. Brain. 2000;123:1155–60. doi: 10.1093/brain/123.6.1155. [DOI] [PubMed] [Google Scholar]
- 90.Eisensehr I, Linke R, Tatsch K, Kharraz B, Gildehaus JF, Wetter CT, et al. Increased muscle activity during rapid eye movement sleep correlates with decrease of striatal presynaptic dopamine transporters. IPT and IBZM SPECT imaging in subclinical and clinically manifest idiopathic REM sleep behavior disorder, Parkinson’s disease, and controls. Sleep. 2003;26(5):507–12. doi: 10.1093/sleep/26.5.507. [DOI] [PubMed] [Google Scholar]
- 91.Kim Y, Yoon I, Kim J, Jeong S, Kim K, Shin Y, et al. The implication of nigrostriatal dopaminergic degeneration in the pathogenesis of REM sleep behavior disorder. Eur J Neurol. 2010;17:487–92. doi: 10.1111/j.1468-1331.2009.02854.x. [DOI] [PubMed] [Google Scholar]
- 92.Caselli R, Chen K, Bandy D, Smilovici O, Boeve B, Osborne D, et al. A preliminary fluorodeoxyglucose positron emission tomography study in healthy adults reporting dream-enactment behavior. Sleep. 2006;29:927–33. doi: 10.1093/sleep/29.7.927. [DOI] [PubMed] [Google Scholar]
- 93.Fujishiro H, Iseki E, Murayama N, Yamamoto R, Higashi S, Kasanuki K, et al. Diffuse occipital hypometabolism on [18 F]-FDG PET scans in patients with idiopathic REM sleep behavior disorder: prodromal dementia with Lewy bodies? Psychogeriatrics. 2010;10:144–52. doi: 10.1111/j.1479-8301.2010.00325.x. [DOI] [PubMed] [Google Scholar]
- 94.Albin R, Koeppe R, Chervin R, Consens F, Wernette K, Frey K, et al. Decreased striatal dopaminergic innervation in REM sleep behavior disorder. Neurology. 2000;55:1410–2. doi: 10.1212/wnl.55.9.1410. [DOI] [PubMed] [Google Scholar]
- 95.Boeve B. Predicting the future in idiopathic rapid-eye movement sleep behaviour disorder. Lancet Neurol. 2010;9:1040–2. doi: 10.1016/S1474-4422(10)70221-0. [DOI] [PubMed] [Google Scholar]
- 96.Gagnon J-F, Postuma R, Mazza S, Doyon J, Montplaisir J. Rapid-eye-movement sleep behaviour disorder and neurodegenerative diseases. Lancet Neurol. 2006;5:424–32. doi: 10.1016/S1474-4422(06)70441-0. [DOI] [PubMed] [Google Scholar]
- 97.Postuma R, Gagnon J, Rompré S, Montplaisir J. Severity of REM atonia loss in idiopathic REM sleep behavior disorder predicts Parkinson disease. Neurology. 2010;74:239–44. doi: 10.1212/WNL.0b013e3181ca0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology. 1996;46(2):388–93. doi: 10.1212/wnl.46.2.388. [DOI] [PubMed] [Google Scholar]
- 99.Claassen D, Josephs K, Ahlskog J, Smith G, Silber M, Tippmann-Peikert M, et al. REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology. 2010;75:494–9. doi: 10.1212/WNL.0b013e3181ec7fac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jellinger K. A critical evaluation of current staging of a-synuclein pathology in Lewy body disorders. Biochim Biophys Acta. 2009;1792:730–40. doi: 10.1016/j.bbadis.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 101.Kingsbury A, Bandopadhyay R, Silveira-Moriyama L, Ayling H, Kallis C, Sterlacci W, et al. Brain stem pathology in Parkinson’s disease: An evaluation of the Braak staging model. Mov Disord. 2010;25:2508–15. doi: 10.1002/mds.23305. [DOI] [PubMed] [Google Scholar]
- 102.Parkkinen L, Kauppinen T, Pirttila T, Autere J, Alafuzoff I. Alpha synuclein pathology does not predict extra-pyramidal symptoms or dementia. Ann Neurol. 2005;57:81–91. doi: 10.1002/ana.20321. [DOI] [PubMed] [Google Scholar]
- 103.Attems J, Jellinger K. The dorsal motor nucleus of the vagus is not an obligatory trigger site in Parkinson’s disease. Neuropathol Appl Neurobiol. 2008;34:466–7. doi: 10.1111/j.1365-2990.2008.00937.x. [DOI] [PubMed] [Google Scholar]
- 104.Kalaitzakis M, Graeber M, Gentleman S, Pearce R. The dorsal motor nucleus of the vagus is not an obligatory trigger of Parkinson’s disease: a critical analysis of a2synuclein staging. Neuropathol Appl Neurobiol. 2008;34:284–95. doi: 10.1111/j.1365-2990.2007.00923.x. [DOI] [PubMed] [Google Scholar]
- 105.Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta neuropathologica. 2009;117(6):613–34. doi: 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Del Tredici K, Braak H. Lewy pathology and neurodegeneration in premotor Parkinson’s disease. Mov Disord. 2012;27(5):597–607. doi: 10.1002/mds.24921. [DOI] [PubMed] [Google Scholar]
- 107.Braak H, Del Tredici K. Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv Anat Embryol Cell Biol. 2009;201:1–119. [PubMed] [Google Scholar]
- 108.Dickson D, Braak H, Duda J, Duyckaerts C, Gasser T, Halliday G, et al. Neuropathological assessment of Parkinson’s disease: Refining the diagnostic criteria. Lancet Neurol. 2009;8(12):1150–7. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 109.Dickson D, Uchikado H, Fujishiro H, Tsuboi Y. Evidence in favor of Braak staging of Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S78–82. doi: 10.1002/mds.22637. [DOI] [PubMed] [Google Scholar]
- 110.Beach T, Adler C, Sue L, Vedders L, Lue L, White C, et al. Multi-organ distribution of phophorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119:689–702. doi: 10.1007/s00401-010-0664-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schulz-Schaeffer W. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010;120:131–43. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Halliday G, Del Tredici K, Braak H. Critical appraisal of brain pathology staging related to presymptomatic and symptomatic cases of sporadic Parkinson’s disease. J Neural Trans. 2006;(Suppl):99–103. doi: 10.1007/978-3-211-45295-0_16. [DOI] [PubMed] [Google Scholar]
- 113.Boeve B, Ferman T. Neuropsychological characterization of evolving cognitive decline in idiopathic REM sleep behavior disorder is important, but not easy. Sleep. 2011;34:619–25. doi: 10.1093/sleep/34.5.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gagnon J-F, Medard M-A, Fantini M, Petit D, Panisset M, Rompre S, et al. REM sleep behavior disorder and REM sleep without atonia in Parkinson’s disease. Neurology. 2002;59:585–9. doi: 10.1212/wnl.59.4.585. [DOI] [PubMed] [Google Scholar]
- 115.Uchiyama M, Isse K, Tanaka K, Yokota N, Hamamoto H, Aida S, et al. Incidental Lewy body disease in a patient with REM sleep behavior disorder. Neurology. 1995;45:709–12. doi: 10.1212/wnl.45.4.709. [DOI] [PubMed] [Google Scholar]
- 116.Boeve B, Dickson D, Olson E, Shepard J, Silber M, Ferman T, et al. Insights into REM sleep behavior disorder pathophysiology in brainstem-predominant Lewy body disease. Sleep Med. 2007;8:60–4. doi: 10.1016/j.sleep.2006.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Aarsland D, Zaccai J, Brayne C. A systematic review of prevalence studies of dementia in Parkinson’s disease. Mov Disord. 2005;20:1255–63. doi: 10.1002/mds.20527. [DOI] [PubMed] [Google Scholar]
- 118.McKhann G, Knopman D, Chertkow H, Hyman B, Jack CJ, Kawas C, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 120.Braak H, Braak E, Bohl J, Bratzke H. Evolution of Alzheimer’s disease related cortical lesions. J Neural Transm Suppl. 1998;54:97–106. doi: 10.1007/978-3-7091-7508-8_9. [DOI] [PubMed] [Google Scholar]
- 121.Albert M, DeKosky S, Dickson D, Dubois B, Feldman H, Fox N, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, et al. The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 2012;8(Suppl 1):S1–68. doi: 10.1016/j.jalz.2011.09.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jack CJ, Knopman D, Jagust W, Shaw L, Aisen P, Weiner M, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jack CR, Jr, Vemuri P, Wiste HJ, Weigand SD, Aisen PS, Trojanowski JQ, et al. Evidence for ordering of Alzheimer disease biomarkers. Arch Neurol. 2011;68(12):1526–35. doi: 10.1001/archneurol.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jack CR, Jr, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Lowe V, et al. Shapes of the Trajectories of 5 Major Biomarkers of Alzheimer Disease. Arch Neurol. 2012;69(7):856–67. doi: 10.1001/archneurol.2011.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jack C, Jr, Knopman D, Jagust W, Petersen R, Weiner M, Aisen P, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Murray M, Graff-Radford N, Ross O, Petersen R, Duara R, Dickson D. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10(9):785–96. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Boeve B. Update on the Diagnosis and Management of Sleep Disturbances in Dementia. In: MS, editor. Sleep Medicine Clinics. Philadelphia: Saunders/Elsevior; 2008. pp. 347–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jicha G, Schmitt F, Abner E, Nelson P, Cooper G, Smith C, et al. Prodromal clinical manifestations of neuropathologically confirmed Lewy body disease. Neurobiol Aging. 2010;31:1805–13. doi: 10.1016/j.neurobiolaging.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Frigerio R, Fujishiro H, Ahn T, Josephs K, Maraganore D, DelleDonne A, et al. Incidental Lewy body disease: do some cases represent a preclinical stage of dementia with Lewy bodies? Neurobiol Aging. 2011;32(5):857–63. doi: 10.1016/j.neurobiolaging.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lippa C, Duda J, Grossman M, Hurtig H, Aarsland D, Boeve B, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68(11):812–9. doi: 10.1212/01.wnl.0000256715.13907.d3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.