

Abstract
Almost 70% of breast cancers are estrogen receptor α (ERα) positive. Tamoxifen, a selective estrogen receptor modulator (SERM), represents the standard of care for many patients; however, 30-50% develop resistance, underlining the need for alternative therapeutics. Paradoxically, agonists at ERα such as estradiol (E2), have demonstrated clinical efficacy in patients with heavily-treated breast cancer, although side effects in gynecological tissues are unacceptable. A drug that selectively mimics the actions of E2 in breast cancer therapy, but minimizes estrogenic effects in other tissues is a novel, therapeutic alternative. We hypothesized that a selective human estrogen receptor partial agonist (ShERPA) at ERα would provide such an agent. Novel benzothiophene derivatives with nanomolar potency in breast cancer cell cultures were designed. Several showed partial agonist activity, with potency of 0.8-76 nM, mimicking E2 in inhibiting growth of tamoxifen-resistant breast cancer cell lines. Three ShERPAs were tested and validated in xenograft models of endocrine-independent and tamoxifen-resistant breast cancer, and, in contrast to E2, ShERPAs did not cause significant uterine growth.
Keywords: Breast cancer, tamoxifen resistance, estrogen receptor, partial agonist
Introduction
The estrogen receptors, ERα and ERβ, are transcription factors that regulate genes involved in cell proliferation, differentiation, and migration. Dysregulation of ERα signaling is associated with breast cancer development, which has made ERα an attractive drug target.1, 2 The archetype selective estrogen receptor modulator (SERM) tamoxifen is the standard of care for many patients with ER-positive (ER+) breast cancer and is intended to antagonize the actions of estrogens at ERα in the breast, although it poses an increased risk of endometrial cancer.3-5 Aromatase inhibitors that block estrogen biosynthesis are presently used as an alternative to tamoxifen; however, both tamoxifen and aromatase inhibitors cause compliance problems, due to side effects such as hot flashes, arising from antiestrogenic actions in non-gynecological tissues. Paradoxically, before the advent of tamoxifen as standard of care, estrogen and the full ERα agonist, diethylstilbesterol (DES), were used for the treatment of advanced ER+ breast cancer but were discontinued due to unacceptable, adverse effects.6, 7
Most importantly, in about 30-50% of women undergoing tamoxifen treatment, resistance occurs, leaving these women with no alternative but cytotoxic chemotherapy.4, 8 The cause of tamoxifen resistance and endocrine independence of ER+ breast cancer is not fully defined and may include remodeling of several cellular systems; for example, an overexpression of growth factor receptors, increased activity of downstream kinase pathways, phosphorylation of ER and its coregulators, and mutations in ER.9-11 The clinical benefits of estrogen or compounds with estrogenic activity have been largely forgotten or overlooked in the treatment of breast cancer,12, 13 despite demonstrated efficacy in recent clinical trials in patients with heavily-pretreated, metastatic ER+ breast cancer.14-16 In the present study, we sought to exploit the beneficial aspects of estrogenic compounds, whilst minimizing the adverse effects. Selective human Estrogen Receptor Partial Agonists (ShERPAs) stand out as a novel approach towards this goal. Ideally, these partial ERα agonists would mimic estradiol’s efficacy in treating tamoxifen-resistant breast cancer, while attenuating adverse effects that result from full estrogenicity.
SERMs are able to act as tissue-selective ER ligands, because of the ability of liganded-ER to cause gene activation or silencing when complexed with coactivators or corepressors.17-19 However, the extensive search for clinical SERMs has been focused on compounds that display ERα antagonist activity in breast tissues, and estrogenic, agonist activity in bone tissues in postmenopausal women.20 The SERM, raloxifene, in clinical use for postmenopausal osteoporosis for over a decade, appears safe and non-carcinogenic and is clinically indicated for chemoprevention of invasive breast cancer. Arzoxifene, an analogue of raloxifene and a “3rd generation” SERM, demonstrated a favorable profile in preclinical studies,21 and its active metabolite is more potent than raloxifene.22-26 The indole SERM, bazedoxifene, was recently approved in Europe for postmenopausal osteoporosis.27-29 We have focused upon a deeper understanding of ER ligands based upon the benzothiophene scaffold, common to raloxifene and arzoxifene.30-41 Recently, we demonstrated in two different animal models of tamoxifen-resistant breast cancer, that a prototype benzothiophene ShERPA caused regression of established xenografts, in the absence of the uterine weight gain shown by E2.42 Herein we report the design, synthesis, optimization, and characterization of potent ShERPAs with potential for treatment of endocrine-independent and tamoxifen-resistant breast cancer.
Structure design
The plasticity of the ER ligand-binding domain (LBD) is reflected by the structural diversity of ER ligand scaffolds (Fig. 1) and lends itself to design of novel pharmacological modulators.43, 44 The volume of the ER ligand-binding pocket is ~ 450 Å3 and can accommodate ligands as small as 250 Å3 (e.g. estradiol, E2; pdb: 1gwr) or as large as 380 Å3 (e.g. 17R-(2E-trifluoromethylphenylvinyl)-E2; pdb: 2p15).45 The ligand-binding domain of ER is comprised of twelve alpha helices and a beta sheet. These secondary structures create an interior hydrophobic cavity forming a ligand binding site and a hydrophobic, solvent-exposed surface to which transcriptional coregulators bind.46 In the “mousetrap” model of ER, helix 12 is closed, allowing coregulator binding (agonist mode), or open and hindering coregulator binding (antagonist mode).46 This binary model may have limited pursuit of partial agonists at ER; however, even in this mousetrap view, a ligand that can stabilize both agonist and antagonist conformations may act as a partial agonist.
Figure 1.

Structures of representative estrogen receptor ligands emphasizing the 2-4 carbon bond linking 2 phenol groups.
Partial estrogenic activity (defined by maximal activity significantly lower than E2) has been reported for genistein in cell cultures; however, this sub-maximal activity at micromolar concentrations may be associated with the many other known cellular targets of genistein.47-49 ERα partial agonist activity has also been reported for ligands in hepatocarcinoma cell lines transfected with ER or endometrial cell lines; however, these observations have not been followed up in mammary cells containing native, functional ER, nor to confirm pharmacological partial agonism.50-53 Relatively weak and partial agonist activity was reported for bisphenol AF (BPAF)54 and WAY-169916 congeners55, compatible with stabilization of sub-optimal conformations of ER, which might form weaker complexes with coregulators. The crystal structure with WAY-169916 (pdb: 2qzo)56 showed a conformation structurally different from the ERα/E2 complex due to a shift of helix 11 towards helix 12. A similar shift at helix 11 by placing a 4-methoxy phenyl group at the 11β position of E2 was also proposed as a potential mechanism for inducing ligand transition from an agonist to a partial agonist.53 In the case of ERα/BPAF crystal structures, two conformations were revealed in the same crystal structure, with one monomer of the homodimer occupying an “antagonist” conformation and the other an “agonist” conformation. This emphasizes that ligand:ER complexes can adopt multiple stable binding modes and that these might correspond to agonist, antagonist or partial agonist conformations (Fig. 2A).54
Figure 2.

Design of partial agonists. (A) Crystal structure of BPAF (6) (pdb ID: 3UUA) bound to ER LBD shows two binding modes with hydrogen bonding switch from His-524 to Thr-347, suggestive of alternate binding modes leading to agonist and antagonist conformations that contributes to partial agonist activity. (B) Overlay of E2 (1) and tamoxifen (2) crystal structures (pdb ID: 1GWR and 3ERT) shows the movement of helix 11 (His 524 and Leu 525) that can lead to strain of helix 12, and a partial agonist conformation. (C) Truncation of the side chain of raloxifene and substitution of phenolic OH leads to Design 1. Docking of ligand 19 shows two binding poses with alternate hydrogen bonding. (D) Insertion of a linker and diversifying substituents at C2 of the benzothiophene leads to Design 2. Docking of ligand 30a shows the effect of the elongation of the 2-substituent compatible with a shift of helix 11.
ER ligands commonly possess two phenol groups linked by two to four chemical bonds, with phenolic-OH separation of 11-12 Å. The central scaffold is usually planar and hydrophobic (Fig. 1).43 An overlay of the E2 and tamoxifen-bound ERα structures demonstrates the large shift in the terminal residues of helix 11, caused by the tamoxifen side chain, resulting in displacement of helix 12 and antagonist activity (Fig. 2B). A closer look at the structure of ERα complexed with WAY-169916 shows that the smaller shift of helix 11, repositioning His-524 and Leu-525, strains helix 12 without complete displacement. The ERα:BPAF structures show a switch from His-524 to Thr-347 as a key hydrogen bonding residue with the ligand, demonstrating a dynamic hydrogen bonding network that can accommodate diverse agonists or antagonists in various poses (Fig. 2A). We hypothesized that novel ligands could maintain high affinity and potency whilst acting as partial agonists by exerting small shifts in helix 11 and exploiting the dynamic hydrogen bonding network for stabilization.
To develop partial agonists, we chose the benzothiophene scaffold, common to the SERMs arzoxifene and raloxifene, the latter having a superior safety profile to tamoxifen. As depicted in Fig. 2C-D, the initial design step was to truncate the SERM side-chain to avert complete displacement of helix 12 and to avoid a pure antagonist. A small probe library was prepared using interchange and substitution of phenolic substituents to explore the effect on activity of truncation and replacing phenolic-OH. We hypothesized that partial agonist activity could result from: alternate binding modes, as seen in BPAF complexes, which might correspond to agonist and antagonist conformations (Design 1, Fig. 2C); or alternatively, from a shift in helix 11, causing strain in helix 12, and consequently sub-optimal coregulator binding (Design 2, Fig. 2D). We inserted an ether or ketone linker at C2 intended to displace the 2-benzothiophene substituent to interact directly with the terminal residues of helix 11, causing strain in helix 12 (Design 2).
Chemistry
2-(4-Fluorophenyl)-6-methoxybenzo[b]thiophene (9) was prepared by the cyclization rearrangement induced by polyphosphoric acid (PPA) as reported by Jones and colleagues.57 The regioisomer, 3-(4-fluorophenyl)-6-methoxybenzo[b]thiophene (10) was synthesized with BF3•OEt2 in dichloromethane (Scheme 1). Aroylation at C3 of benzothiophene was accomplished by direct Friedel-Crafts acylation. Coupling with the acid chloride bearing a strong electron withdrawing group, e.g. 14b, gave multiple isomers and minor products, and a C18 reverse-phase column was used to purify the final products (Scheme 2). A trans-halogenation minor product, 14c, was isolated from the preparation of 14b with 3% isolated yield, possibly from a SNAr displacement of fluoride by chloride.58, 59 C2 benzothiophene aroylation used a similar strategy with the exception of products containing strong electron withdrawing groups, which were obtained using n-butyllithiumto generate an organic lithium salt that reacts directly with the acid chloride (Scheme 3). The synthesis of ether-linked derivatives was accomplished adapting a reported procedure (Scheme 4).32 Bromination of the benzothiophene core with N-bromoacetamide provided exclusively the mono-brominated product in high yield, which was activated for SNAr reaction by oxidation to the sulfoxide under H2O2. Introduction of the substituted phenol was accomplished in the presence of NaH in DMF. Sulfoxide reduction was performed under LiAlH4 in THF. Deprotection of the methyl group was achieved by BBr3 or BF3•SMe2 to give the final product. Regioisomers were verified by 2D NMR, and one of the key intermediates was recrystallized for X-ray crystallography for structural confirmation (Supporting information_Fig. 1).
Scheme 1.

Synthesis of 2-(4-fluorophenyl) and 3-(4-fluorophenyl) benzothiophene coresa
aReagents and conditions: (a) KOH, EtOH/H2O, 3-methoxythiophenol, rt, 2 h; (b) PPA, 120 °C, 3 h, 55% over two steps; (c) BF3•OEt2, DCM, rt, overnight, 73% over two steps; (d) BBr3, DCM, −78 °C to rt, overnight.
Scheme 2.

Synthesis of 2-(4-fluorophenyl)-benzothiophenesa
aReagents and conditions: (a) N-bromoacetamide, DCM/EtOH, rt, 2 h, 78%; (b) H2O2, TFA, DCM, 0 °C, 2 h, 75%; (c) 4-methoxyphenol, NaH, DMF, rt, 3 h, 93%; (d) LiAlH4, THF, 0 °C, 2 h, 71%; (e) BBr3, DCM, −78 °C to rt, overnight; (f) AlCl3, R1COCl, DCM, rt, 2-48 h.
Scheme 3.

Synthesis of 3-(4-fluorophenyl)benzothiophene compounds with a ketone linkera
aReagents and conditions: (a) AlCl3, R1COCl, DCM, rt, 2-48 h; (b) BBr3, DCM, −78 °C to rt, 4-48 h for 21a-e and 26a-b; BF3•SMe2, DCM, 0 °C to rt, 24 h, for 21g and 23; (c) N-bromoacetamide, DCM/EtOH, rt, 89%, 2 h; (d) n-BuLi, R2COCl, THF, −78 °C to rt, 4 h; (e) Et3N, Pd(PPh3)2Cl2, DMF, CuI, ethynyltrimethylsilane, 4 h; (f)TBAF, THF, rt, 1 h, 50% over two steps.
Scheme 4.

Synthesis of 3-(4-fluorophenyl)benzothiophene compounds with an ether linkera
aReagents and conditions: (a) N-bromoacetamide, DCM/EtOH, rt, 89%, 2 h; (b) H2O2, TFA, DCM, 0 °C, 2 h, 84%; (c) R3OH, NaH, DMF, rt, 6 h, for 28a; Cs2CO3, DMF, 80 °C, overnight for 28b-d; (d) LiAlH4, THF, 0 °C, 2 h; (e) BBr3, DCM, −78 °C to rt, 4-48 h.
Biological testing
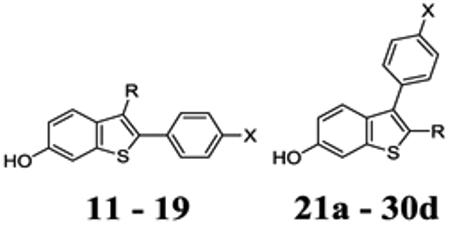
Ligand affinity and estrogenic potency were evaluated, respectively, in a fluorescence polarization (FP) binding assay using full-length ERα and a cell-based assay using an estrogen response element (ERE)-luciferase reporter (Table 1). Ligand affinity of selected ShERPAs was also studied by radioligand displacement (Table 2). Potency and efficacy (Emax) for ERE-luciferase activation were measured in endocrine-dependent ER+ MCF-7:ws8 cells, the response being normalized to control treated cells (0%) and E2 (1nM) treated cells (100 %).
Table 1.
Transcriptional activity and relative binding affinity of benzothiophene analogs.
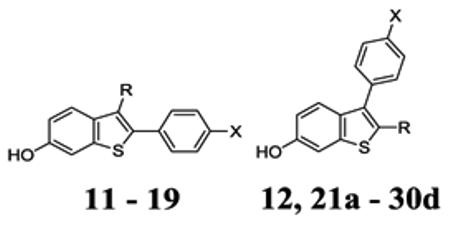
|
|||||
|---|---|---|---|---|---|
|
| |||||
| Compounds | X | R | ERE luciferasea
EC50 (nM) |
Efficacy(% E2) |
RBAral b |
| 3 | NA | 0 | 100 ± 14.4 | ||
| 11 | -OH | -H | NA | 0 | 0.6 ± 0.2 |
| 12 | -F | -H | NA | 0 | 0.2 ± 0.1 |
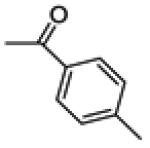
| 14a | -OH |
|
2.6 ± 0.4 | 100 | 3.3 ± 0.2 |
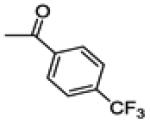
| 14b | -F |
|
61.5 ± 14.4 | 100 | 0.9 ± 0.4 |
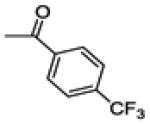
| 14c | -Cl |
|
121 ± 19.2 | 100 | 1.2 ± 0.2 |
| 19 | -F |
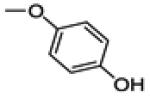
|
6.2 ± 2.8 | 100 | 32.6 ± 2.3 |
| 21a | -F |
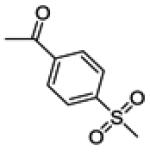
|
436.2 ± 112.1 | 100 | 4.4 ± 1.3 |
| 21b | -F |
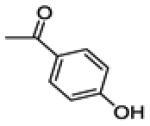
|
0.2 ± 0.1 | 100 | 13.8 ± 0.9 |
| 21c | -F |
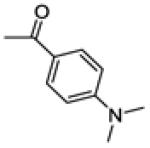
|
30.6 ± 14.4 | 51 | 0.4 ± 0.1 |
| 21d | -F |
|
717.1 ± 261.6 | 100 | 3.8 ± 1.0 |
| 21e | -F |
|
18.5 ± 9.5 | 100 | 5.1 ± 3.7 |
| 21f | -F |
|
13.9 ± 3.3 | 100 | 14.4 ± 5.7 |
| 21g | -F |
|
1.0 ± 0.1 | 100 | 1.9 ± 0.3 |
| 23 | -F |
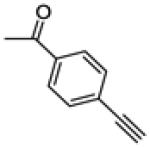
|
5.3 ± 1.7 | 65 | 2.1 ± 0.7 |
| 26a | -F |
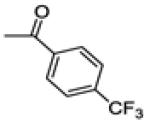
|
5.2 ± 1.4 | 85 | 0.5 ± 0.1 |
| 26b | -F |
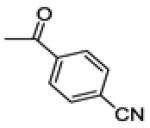
|
58.7 ± 8.6 | 100 | 0.9 ± 0.2 |
| 30a | -F |
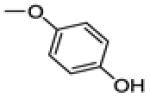
|
0.8 ± 0.3 | 60 | 14.2 ± 0.8 |
| 30b | -F |
|
76.1 ± 25.1 | 70 | 0.4 ± 0.1 |
| 30c | -F |
|
36.8 ± 4.4 | 100 | 0.4 ± 0.1 |
| 30d | -F |
|
33.4 ± 2.5 | 100 | 0.4 ± 0.1 |
In MCF-7:ws8 cells, estrogenic activity was measured using a luciferase reporter assay and normalized to Ctrl (0%) and E2-1nM (100%). Data show mean ± s.e.m. determined from an average of at least three passages.
RBAral measuring reduction in FP caused by displacement of fluorescein-E2 from ER, data are normalized to raloxifene binding at 100% ± s.e.m. NA = no activity.
Table 2.
Relative binding affinity of selected ShERPAs in radioligand displacement assay
| Compounds | RBAa |
|---|---|
| E2 | 100 |
| 26a | 1.3 ± 0.3 |
| 23 | 7.9 ± 0.3 |
| 30a | 92.4 ± 21 |
Relative binding affinity (RBA) values, determined by radiometric assays, are expressed as IC50 estradiol/IC50 compound × 100 ± the range or standard deviation (RBA, estradiol = 100%).
Pharmacological partial agonists are defined by the sub-maximal efficacy compared to the preferred, endogenous ligand and by antagonism of the activity of the endogenous agonist. Antagonism has rarely been tested in the literature for putative partial agonists at ER. By definition, a ShERPA must antagonize the actions of E2 in breast cancer cell cultures in which it exerts partial agonist activity. The therapeutic rationale for exploring ShERPAs is based upon the need for an ER ligand that mimics the actions of estrogen in killing tamoxifen-resistant breast cancer cells. Therefore, the ability of these mimics to inhibit growth of endocrine-independent, tamoxifen-resistant MCF-7:5C cells, on the 9th day after treatment, was assessed by MTS assay (confirmed by DNA assay). Results were reported as cell viability relative to vehicle (100%) (Fig. 3), or inhibition of cell growth relative to vehicle (0%) and E2 (100%) (Table 3).
Figure 3.

Profiling ERα ligands: endogenous agonist E2 (1); SERM antagonist raloxifene (3); benzothiophene partial agonist (ShERPA) (30a). (A) Activation of ERE (estrogen response element) in MCF-7:ws8 cells. (B) RBAral from FP assay of displacement of fluorescent-E2 from ERα. (C) Cell viability of tamoxifen-resistant MCF-7:5C breast cancer cells 9 days after drug treatment, normalized to vehicle (100%). Data show mean and s.e.m.
Table 3.
Cell viability of MCF-7:5C cells treated with E2 and test compounds
|
|||||
|---|---|---|---|---|---|
|
| |||||
| Compounds | X | R | Inhibition of cell growth relative to E2 %a |
||
| 1 nM | 10 nM | 100 nM | |||
| 3 | NA | NA | NA | ||
| 14a | -OH |
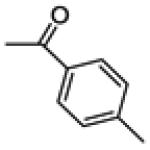
|
11.5 ± 4.0 | 60.2 ± 2.7 | 109.0 ± 1.4 |
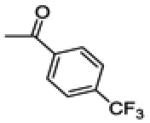
| 14b | -F |
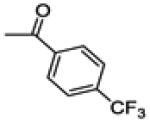
|
55.6 ± 3.5 | 69.7 ± 3.6 | 108.0 ± 2.6 |
| 14c | -Cl |
|
NA | NA | NA |
| 19 | -F |
|
NA | NA | NA |
| 21a | -F |
|
NA | NA | 18.9 ± 2.8 |
| 21b | -F |
|
95.4 ± 2.7 | 99.8 ± 2.8 | 100.0 ± 2.1 |
| 21c | -F |
|
4.1 ± 3.6 | 15.1 ± 3.1 | 110.9 ± 4.6 |
| 21d | -F |
|
13.3 ± 3.8 | 7.4 ± 4.8 | 110.0 ± 4.6 |
| 21e | -F |
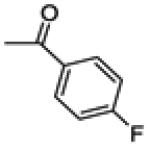
|
NA | 32.4 ± 5.9 | 131.0 ± 2.3 |
| 21f | -F |
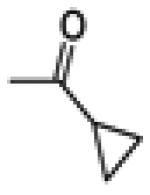
|
NA | 79.6 ± 3.9 | 146.0 ± 4.5 |
| 21g | -F |
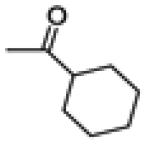
|
21.4 ± 3.7 | 134.0 ± 4.2 | 147 ± 5.4 |
| 23 | -F |
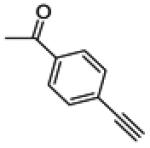
|
31.4 ± 2.9 | 79.9 ± 3.1 | 85.6 ± 1.9 |
| 26a | -F |
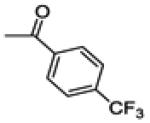
|
NA | 15.4 ± 1.7 | 88.6 ± 2.1 |
| 26b | -F |
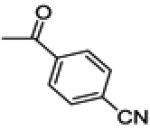
|
2.0 ± 2.3 | 0.9 ± 2.1 | 64.2 ± 5.0 |
| 30a | -F |
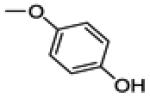
|
50.6 ± 3.3 | 104.0 ± 1.0 | 123.0 ± 1.7 |
| 30b | -F |
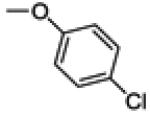
|
NA | NA | 30.7 ± 4.9 |
| 30c | -F |
|
NA | NA | 86.6 ± 8.7 |
| 30d | -F |
|
NA | NA | 84.6 ± 5.3 |
Cell death percentage was normalized to control (0 %) and E2 (100 %) assessed in a 9 day cell viability assay with drug treatments renewed every 3 days. Data represented as mean ± s.e.m. is an average of at least three cell passages (triplicates in each passage).
The three ERα ligands for which data are depicted in Fig. 3 represent examples of a full agonist (1), SERM antagonist (3), and a ShERPA (30a): Panel A shows ERE-luciferase response in MCF-7:ws8 cells; Panel B shows FP measured by displacement of fluorescent E2 from ERα; and, Panel C shows viability of treated tamoxifen-resistant MCF-7:5C cells.
ERE-luciferase guided structure optimization
A small probe library was prepared to explore side chain truncation and substitution of phenolic OH in derivatives containing the 2-phenyl-benzothiophene core common to arzoxifene and raloxifene. Truncation and substitution of a methyl group at the 4’-benzoyl position switched an antagonist (raloxifene) to a potent agonist (14a): EC50 = 2.6 nM, efficacy (Emax) ~ 100% (Table 1). Substitution of the phenolic OH by F in 14b reduced potency and affinity, but retained full agonist activity. Similarly, the chloro congener (14c) was a full agonist; and 19, containing an ether linkage and 4’-OH replaced by F was again a potent full agonist in MCF-7 cells: EC50 = 6.2 nM. Side chain truncation was confirmed as a route to ERα agonists and replacement of phenolic OH groups was tolerated while retaining nanomolar potency. In the FP assay using ligand displacement from full length ERα, all truncations and substitutions of raloxifene reduced relative binding affinity compared to raloxifene itself (RBAral). Even in this small conserved library of structures, correlation of affinity with potency in cell cultures was poor, driving use of the luciferase reporter assay for optimization.
Having demonstrated the simple hypothesis that side chain truncation would produce ER agonists, we proceeded to test the proposal that insertion and elongation of the 2-benzothiophene substituent would lead to partial agonist activity. The 3-fluorophenyl-benzothiophene series (21), yielded agonists that as in the probe library showed reduced RBAral, but nanomolar, and in one case picomolar potency for ERE activation. For example, compound 21b with 2 phenolic substituents was a potent full agonist (EC50 = 0.2 nM), but with RBAral only 14% that of raloxifene. One derivative in this series, 21c, proved to be a potent partial agonist (EC50 = 30 nM). Moreover, we were able to replace the 2-benzoyl group with sp3 alkanoyl substituents whilst maintaining potency (EC50 = 1-14 nM). Within the 2-benzoyl and 2-alkanoyl benzothiophene series, 3 of 10 compounds (21c, 23, 26a) demonstrated potent partial agonist activity, but with relatively low RBAral: for example, the trifluoromethyl derivative (26a) Emax = 80%, EC50 = 5 nM; and the alkynyl derivative (23) Emax = 65%, EC50 = 5 nM. A further series of 3-phenyl benzothiophenes with a 2-ether linkage (30) showed a similar pattern of activity, with relatively low RBAral, contrasting with nanomolar potency as agonists (0.8 – 76 nM). Directly comparing the ether- and ketone-linked series (21b vs 30a and 21e vs 30c) revealed bis-phenolic derivatives to have relatively high potency (<1 nM) and RBAral, whereas both bis-fluorophenyl derivatives had lower RBAral and 100-fold reduced potency (Table 1). We used G15, the inhibitor of GPER/GPR30, to test for any involvement of this extranuclear receptor in ShERPA activity. Co-treatment of cells with G15 and ShERPAs did not significantly reduce observed luciferase reporter activity (Supporting information_Fig. 2).
The relatively low RBAral values obtained from the FP assay accompanied by nanomolar potency in the reporter assay led to the testing of the three ShERPAs, 23, 26a and 30a in the classical radioligand displacement assay (Table 2). It was seen that, the potent ShERPA, 30a, exhibited 92% RBA relative to estradiol (RBA = 100%) while ShERPAs, 23 and 26a, exhibited 8% and 1% RBA respectively. While our compounds exhibit strong affinity in this assay, a lack of correlation between biochemical data and cellular response remains, confirming the use of the reporter assay as the optimal route for structure optimization.
Characterization of ShERPA partial agonism
All 2-phenyl and 3-phenyl benzothiophene derivatives obtained according to the design principles described in Fig. 2 acted as ERα agonists. As defined by ERE-luciferase activation in ER+ breast cancer cell lines, five of these were ShERPA candidates: 21c, 23, 26a, 30a and 30b. When compared to full agonists, these partial agonists did not uniformly display weaker affinity (RBAral) or reduced potency for ERE activation; for example, 30a is a partial agonist with picomolar potency, and affinity for isolated ERα (RBA) not significantly different from E2 itself. In addition to sub-maximal efficacy, a partial agonist should by definition antagonize the action of the endogenous agonist. The partial agonists, 30a, 23 and 30b, showing a range of potency (EC50 = 0.8 nM, 5.3 nM and 76.1 nM, respectively), were tested as antagonists in MCF-7:ws8 cells. Pharmacological partial agonist activity was validated by the antagonism of E2 induced ERE activation (Fig. 4).
Figure 4.

Partial agonist activity of three ShERPAs (30a, 23 and 30b) assessed by activation of ERE (estrogen response element) in MCF-7:ws8 cells (normalized to 0.1 nM E2 = 100%) using a luciferase assay after treatment for 18h. The antagonist action of these ShERPAs was assessed in the presence of 0.1 nM E2 and a dose dependent reduction in E2 luciferase activity is seen with increasing concentrations of ShERPAs (100-500 nM). Data show mean and s.e.m from at least three cell passages.
The mechanics preceding ERE activation by liganded ER include dimerization, nuclear translocation/accumulation, and binding to DNA at the ERE of a multi-protein complex containing ER in an agonist conformation. The recruitment, assembly, and disassembly of protein complexes at DNA controls transcriptional events triggered by liganded-ER.60-62 Recruitment of steroid receptor coregulator-3 (SRC3; or AIB1, Amplified in Breast Cancer-1) is a key component of ERα-mediated transcription. The interaction of ERα-LBD with SRC3 co-activator is therefore a minimal predictor of phenotype, as a necessary step in the canonical nuclear ER-transactivation pathway.63, 64 Time-resolved fluorescence (or Förster) resonance energy transfer (TR-FRET) assay of the interaction of ERα-LBD with an LxxLL-containing recognition sequence of SRC3, in the presence of E2, has been used to identify small molecules that directly inhibit protein-protein binding.65, 66 Ligand stabilization of the ERα/SRC3 complex is an initial and essential step in transmitting a functional, transcriptional signal and, unlike RBA measurements, will define agonist and antagonist activity. A full agonist is predicted to stabilize the ERα/SRC3 complex in a similar conformation to the E2-bound complex, yielding a similar TR-FRET signal; whereas, an antagonist in the presence of E2 will disrupt the ERα/SRC3 complex destroying the FRET signal.
In the TR-FRET assay, the increase of FRET signal on titration of SRC3 into solutions of E2, 23, or 30a was indicative of formation and stabilization of the ERα/SRC3 complex (Fig. 5A). In the presence of 23 and 30a, the maximal FRET signal was less than 50% of that generated by the E2-liganded ERα-LBD/SRC3 complex, compatible with 23 and 30a acting as partial agonists. In a second TR-FRET assay, conducted in the presence of a sub-maximal concentration of E2 (10 nM), titration of E2, 23, or 30a led to different outcomes. While E2 enhanced the FRET signal by increasing complexation, both 23 and 30a reduced FRET signal, compatible with weak antagonist activity. Observations in TR-FRET, although replicating only a minimal portion of the ERE-bound ERα complex, are compatible with the designation of ShERPA activity.
Figure 5.

(A) Agonist mediated recruitment of ERα coactivator (SRC3): Estradiol (1) induces an increase in FRET readout with increasing concentrations of SRC3. ShERPAs, 23 and 30a, also show an increase in FRET signal with increasing concentration of SRC3, but maximal activation is less than 50% of E2. (B) Increasing concentrations of ShERPAs, 23 and 30a, decrease the FRET signal induced by 10 nM E2, attributed to the antagonist action of these molecules. The FRET signal is amplified in the presence of increasing concentrations of added E2 (1) as is expected of an agonist. Data show mean and s.e.m.
Inhibition of growth of tamoxifen-resistant breast cancer cells
To further test the candidate ShERPAs, it was necessary to confirm their ability to mimic the actions of E2 in inhibiting growth of tamoxifen-resistant MCF-7 cells. A number of tamoxifen-resistant cell lines have been created by long-term exposure to tamoxifen (T47D:A18-TAM1), long-term estrogen deprivation (MCF-7:5C), or by ectopic upregulation of PKCα (T47D:A18/PKCα).42, 67, 68 These stable cell lines are characterized by endocrine independence (growth independent of estrogens) and resistance to any growth inhibitory effects of tamoxifen. Many of these lines are also characterized by a paradoxical inhibition of cell proliferation on treatment by E2 at nanomolar concentrations. In the case of MCF-7:5C cell cultures, treatment with E2 causes a significant reduction in cell viability measured at 9 days. E2 reduced cell viability by approximately 60% compared to vehicle control (Fig. 3C). Inhibition of cell growth relative to vehicle (0%) and to E2 (100%) was measured for the candidate selective estrogen mimics (SEMs) and ShERPAs (Table 3). For compounds 14a-b, 21b-g, and 30a at ≤ 100 nM, the percent of growth inhibition was equivalent or superior to the maximum inhibition observed after treatment with E2. Compound 21b gave 100% inhibition at 1 nM and both 21g and 30a, gave 100% inhibition at 10 nM. Of the candidate ShERPAs, only one did not show efficacy comparable to E2 in inhibition of MCF-7:5C cell growth. It is important to emphasize that inhibition of growth of MCF-7:5C cells is not caused by any general cytotoxicity of SEMs and ShERPAs. This was clearly demonstrated in the parent estrogen-responsive MCF-7:ws8 cells, in which all compounds (100 nM & 1 μM) supported growth at a level higher than DMSO control (data not shown).
Regression of tamoxifen-resistant xenografts uncoupled from uterine growth
We have previously reported that treatment with ShERPA, 30a, resulted in dramatic regression in the two tamoxifen-resistant breast cancer xenograft models, T47D:A18/PKCα and T47D:A18-TAM1, comparable to that observed on treatment with E2.42 The efficacy of ShERPAs, 23 and 26a, was evaluated in the T47D:A18/PKCα model of tamoxifen resistance. The tumor xenografts were allowed to establish for 12 weeks prior to treatment. Although dose optimization was not carried out, at the single dose studied, the tumor size was shown to reduce rapidly on ShERPA treatment and within two weeks to shrink to half the size of control tumors (Fig. 6A). Adverse estrogenic effects in gynecological tissues provided the rationale for development of ShERPAs. Estrogens are known uterotrophic agents, and even tamoxifen shows tissue-selective agonist activity in the uterus. In the T47D:A18/PKCα xenograft study, tumor regression induced by 30a was not accompanied by a significant increase in uterine weight, in contrast to E2.42 Uterine weight was measured in the xenograft study of 26a, presented herein, again showing no significant increase in uterine weight compared to vehicle control mice (Fig. 6B). It is tempting to link the lack of uterotrophic activity of ShERPAs in these studies with partial agonist, sub-maximal efficacy at ERα in uterine tissues. This hypothesis cannot be confirmed in vitro, because there is no cell-based model that reliably reproduces the uterotrophic actions of ER ligands. However, these observations do predict a reduced side effect profile for ShERPAs relative to full estrogens.
Figure 6.

(A) T47D:A18/PKCα tumors were established as described in the experimental section. Tumors were grown to an average size of 0.3 cm2. Mice were then randomized into three treatment groups: Ctrl (n = 4), 23 and 26a (n = 6). Administration of drug was by oral gavage daily (100 mg/kg). Within two weeks of treatment, a regression of established tumors was observed, while control tumors continued to grow. (B) Measured uterine weight (g) of Ctrl (n = 4) and 26a (n = 6) treatment groups showed no significant difference, suggesting an improved side effect profile. Data show mean and s.e.m.
Discussion
The “classic” genomic actions of ERα as a nuclear receptor and transcription factor have dominated thinking on the role of estrogen in cancer. ERα is thought to mediate differentiation, apoptosis, and proliferation in normal mammary epithelial cells; however, in malignant tissues, a shift to proliferation contributes to progression. Paradoxically, in a variety of mammary cell lines that have developed tamoxifen-resistance, the proliferative phenotype of estrogen switches to an antiproliferative and apoptotic phenotype, compatible with the clinical efficacy of E2 in advanced ER+ breast cancer.14-16, 42
Array and ChIP technologies have allowed mapping of ER to regulatory regions of activated and repressed target genes, showing that liganded-ER may cause gene activation or silencing, dependent on ligand stabilization of multi-protein transcriptional complexes, containing either coactivators or corepressors.17-19 ERα ligands may therefore act in a cell and tissue selective manner as exemplified by the SERMs raloxifene and tamoxifen: both SERMS act as estrogen antagonists in breast tissue; but as estrogen agonists in the bone tissue of postmenopausal women. In contrast to SERM antagonists, the goal of this study was to design agonists at ERα, selectively to mimic the actions of estrogen in ER+ tamoxifen-resistant breast cancer.69, 70 This goal was founded on the clinical efficacy of E2 and the ER agonist, DES, in advanced ER+ breast cancer.6, 7 Cognizant of the unacceptable side effects of these estrogens, it was hypothesized that ShERPAs, as partial agonists, would minimize excessive estrogenic activity in other gynecological tissues, for example, the uterus.
The design of ShERPAs, utilizing the benzothiophene core of raloxifene, was guided by structural information from published ERα-LBD crystal structures. In tamoxifen-resistant breast cancer cells, a ShERPA must stabilize ERα as part of a multi-protein complex at ERE, mimicking E2; however, partial agonism is desired to minimize full estrogenic side effects. Hypothetically, an ERα ligand that yields a less stable multi-protein complex than the E2-bound complex, or a ligand that stabilizes both activated and silenced complexes would produce partial agonist activity. Such ligands are predicted to have lower potency than E2 itself; however, potency similar to raloxifene should be achievable. The ShERPA ligands discovered in this study were profiled as pharmacological partial agonists with potency for ERE activation of 0.8-76 nM. Neither RBAral nor RBA for isolated ERα correlated with potency in cell cultures. Various studies have made similar observations on the discordance between biochemical affinity and potency in cell cultures,66, 71-73 and alternate methodologies have been proposed.74 Poor correlation was observed between RBA and growth inhibition in MCF-7 cells for a series of raloxifene analogues with similar RBAs, but up to 100-fold differences in potency in cell cultures.71 A lack of correlation between binding affinities (RBA), and both potency for coactivator recruitment, and cellular ERE-reporter activation was also apparent from a study of ERα ligands commonly used as chemical probes.66 This is compatible with the thermodynamics of ligand binding to ERα in a multi-protein complex being influenced by other binding partners in that complex.
ShERPAs were effective in killing tamoxifen-resistant breast cancer cells in culture. Three ShERPAs were further validated in tamoxifen-resistant xenograft models; these mice show no significant increase in uterine weight, a key side effect of estrogen treatment.42 It would be unsafe without further research to assign these observations entirely to the partial agonist activity of ShERPAs; for example, the involvement of extranuclear ER and ERβ needs to be considered. Nevertheless, we have demonstrated that potent, partial agonists at ERα can be created, and that these ShERPAs have activity in vivo that is of potential therapeutic benefit in treatment of a subset of ER+ breast cancers.
Experimental
Cell lines and culture conditions
MCF-7:ws8 cells were obtained from American Type Culture Collection (Manassas, VA). MCF-7:ws8 were cultured in phenol red containing RPMI 1640 media supplemented with 10% fetal bovine serum, 1% glutamax, 1% non-essential amino acids, insulin (10 µg/ml), 1% antibiotic-antimycotic and 5% CO2 at 37 °C. Treatment media was prepared by supplementing phenol red free RPMI 1640 media with charcoal-dextran treated fetal bovine serum while other supplements remained the same. MCF-7:5C cells were a gift from Dr. Tonetti’s lab. These cells served as a tamoxifen resistance model and were obtained by long term estrogen deprivation of MCF-7:ws8 cells. These cells were maintained in phenol-red free RPMI 1640 media supplemented with 10% charcoal-dextran treated fetal bovine serum, 1% glutamax, 1% non-essential amino acids, insulin (10 μg/ml), 1% antibiotic-antimycotic and 5% CO2 at 37 °C.
ER binding studies
The ligand binding studies for test compounds to full length ER were performed using a PolarScreenTM Nuclear Receptor Competitor Assay (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. Briefly, serial dilutions of test compounds were performed using a Biomek 3000 Laboratory Automation Workstation in a 384 deep well polypropylene block. This served as the 100X serial dilution plate of all test compounds in DMSO where dilutions of test compounds were 100X the final concentration. The 2X dilution plate was prepared by adding 2μl of the stocks from the 100X plate to 98μl of assay buffer and mixed well. The final assay plate was prepared by adding 10μl of 2X stocks to 10μl of ER + fluorescein-labeled E2 (final concentration of ER = 25 nM, fluorescein-labeled E2 = 4.5 nM) and mixing well. The plate was then incubated for 2 hrs in the dark and fluorescence polarization was measured on a SynergyH4 Hybrid Multi-Mode Microplate reader (Biotek). The fluorescence polarization was computed by measuring parallel and perpendicular polarizations at an excitation wavelength of 485 nm and the emission wavelength of 535 nm. IC50 values were computed for unlabeled E2, raloxifene and test compounds, and the relative binding affinities were computed using the following formula RBAral = [(IC50 of raloxifene)/(IC50 of test compounds)] *100. Relative binding affinities of 23, 26a, 30a were also determined by a competitive radiometric binding assay with 2 nM [3H]estradiol as tracer (PerkinElmer, Waltham, MA) and full-length purified human ERα (Pan Vera/Invitrogen, Carlsbad, CA), as described previously.75, 76 The RBA values were determined using the following equation: IC50 estradiol/IC50 compound × 100.
Induction of estrogen response elements in MCF-7 cells
Activation of ERα signaling by test compounds was measured using MCF-7:ws8 cells which were kept in stripped media three days prior to treatment. Cells were plated at a density of 6 × 10^5 cells/well in 12 well plates and were transfected with 3 µg of the pERE-luciferase plasmid, which contains three copies of the Xenopus laevis vitellogenin A2 ERE upstream of firefly luciferase. To normalize for cell viability and transfection efficiency, 1 µg pRL-TK plasmid (Promega, Madison, WI) containing a cDNA encoding Renilla luciferase was co-transfected along with ERE plasmid. Transfection was performed for 6 h using the Lipofectamine 2000 transfection reagent (Invitrogen), in Opti-MEM media according to the manufacturer’s instructions. Cells were treated with test compounds after 6 h and the luciferase activity was measured in cell lysates after 18h of treatment using the Dual Luciferase assay system (Promega) with FLUOstar OPTIMA (BMG LABTECH, Durham, NC). Antiestrogenic activity of ShERPAs was studied by cotreating with 0.1 nM E2 to observe inhibition of E2 response. Data is represented as an average of three passages reported as the relative luciferase percentage with 1 nM E2 treatment set as 100% and control treated cells as 0%. The initial luciferase activity values were calculated by dividing the firefly luciferase (ERE) reading by the renilla luciferase (pRL-TK) reading.
TR-FRET
FRET assay buffers (A & B) were prepared as previously reported.66 In the SRC3 titration assay, the 3X concentrations of Fluorescein-labeled SRC3 (5 μl) prepared in buffer A was added to a 96-well black microplate containing premixed streptavidin-terbium (SA-Tb) and biotinylated ERα LBD (5 μL). This was followed by the addition of 3X concentration of ligands 1, 23 and 30a (5 μL) prepared in buffer B. The final 15 μL assay volume contained 1nM ERα LBD, 20 μM ligand and 0.25 nM SA-Tb. Assay solutions were mixed and incubated for 1h at room temperature in the dark before being measured for TR-FRET. Parallel incubations without the biotinylated ERα LBD were performed to correct for diffusion-enhanced FRET and to correct for nonspecific binding. The background was subtracted from the total FRET values obtained from the test samples and plotted against log SRC3 concentrations. In the ligand titration assay, serial dilutions of 1, 23 and 30a were added to premixed 0.25 nM streptavidin-terbium, 1nM ERα LBD, 100 nM Fluorescein-labeled SRC3, and 10 nM E2. The plate was mixed and incubated in the dark for 1h before measurement of the FRET signal. The donor, SA-Tb was excited at 340/80 nm and emissions from the donor and the acceptor fluorescein were monitored at 495/20 and 520/25 nm, respectively, with a 100-μs delay. TR-FRET was measured on a Wallac Victor II plate reader (Molecular Devices, Sunnyvale, CA).
Cell Viability assays
MCF-7:5C tamoxifen resistant cells were plated overnight at a density of 5000 cells/well. Cells were then treated with either E2 or test compounds at 1, 10 and 100 nM. Treatments were renewed every 3 days and the cell viability was tested at day 9 using a CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS). The absorbance was measured at 490nm using a SynergyH4 Hybrid Multi-Mode Microplate reader (Biotek) and cell viability was normalized to control (DMSO) treated cells at 100%. Data is an average of at least triplicates from three different passages. The cell toxicity in MCF-7:5C was corroborated with a second cell viability DNA content assay. Briefly, cells were plated in 96 well plates at a density of 5000 cells/well and treated with test compounds for 6 days and then frozen in dH2O for 24h. Cells were then lysed with TNE buffer (100 μl/well) containing 50mL TE buffer, 1M NaCl, and 100μl Hoescht DNA stain. The fluorescence was then measured at an excitation wavelength of 355 nm and emission wavelength of 460 nm using the SynergyH4 Hybrid Mode Microplate Reader (Biotek). MCF-7:ws8 cells were stripped three days prior to plating a density of 5000 cells/well in 96 well plates and allowed to attach overnight. Cells were then treated with DMSO, E2 (1nM) and test compounds (100nM) for 3 days. Cell viability was measured using an MTT assay. Briefly, MTT powder was dissolved in PBS (5mg/ml stock solution) and was added to cell media to a final concentration of 0.5mg/mL. The media was removed after incubation and 100μL of DMSO was added to each well. After shaking, the absorbance of the plate was measured at 570nm using the SynergyH4 Hybrid Mode Microplate reader (Biotek) and cell viability was normalized to control at 100%.
Animal experiments
T47D:A18/PKCa tumors were established as previously described.77 23 and 26a were administered per os at a dose of 100 mg/kg daily for 2 weeks in a formulation of 0.1% Tween 80, 10% PEG400, and 0.5% CMC solution. Drinking water was replaced with a hydrogel suspension of drugs at a concentration of 0.25 mg/mL to maintain continuous drug exposure. Tumor cross-sectional area was determined weekly using Vernier calipers and calculated using the formula: length/2 × width/2 × π. Mean tumor area was plotted against time (in weeks) to monitor tumor growth. The mice were sacrificed by CO2 inhalation and cervical dislocation, and tumors and uteri were excised, cleaned of connective tissue, and immediately weighed. The Animal Care and Use Committee of UIC approved all of the procedures involving animals.
General
All chemicals and solvents were purchased from Sigma Aldrich, Fisher Scientific or Matrix Scientific and were used without further purification. Synthetic intermediates were purified by Biotage flash chromatography on 230−400 mesh silica gel. 1H and 13C NMR spectra were recorded on Bruker DPX-400 or AVANCE-400 spectrometer at 400 and 100 MHz, respectively. NMR chemical shifts were reported in δ (ppm) using residual solvent peaks as standard (CDCl3, 7.26 ppm (1H), 77.16 ppm (13C); CD3OD, 3.31 ppm (1H), 49.00 ppm (13C); DMSO-d6, 2.50 ppm (1H), 39.52 ppm (13C); Acetone-d6, 2.05 ppm (1H), 29.84 ppm (13C)). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, br = broad, m = multiplet, abq = ab quartet), number of protons, and coupling constants. Low-resolution (LR) mass spectra were acquired on an Agilent 6300 ion-trap LC−MS instrument. High resolution mass spectral data were collected in-house using a Shimadzu IT-TOF. All compounds submitted for biological testing were confirmed to be ≥ 95% pure by analytical HPLC. Synthetic procedures, spectral data, and HRMS for final compounds and novel intermediates are described below.
3-(4-fluorophenyl)-6-methoxybenzo[b]thiophene (10)
Methoxybenzenethiol (65 g, 7.1mmol) was added to a round-bottom flask charged with a freshly prepared solution containing 300 mL of ethanol, 100 mL of water, and 30 g of KOH (538 mmol). The solution was cooled in an ice water bath and a solution of 2-bromo-1-(4-fluorophenyl)ethanone (100 g, 461 mmol) in 100 mL of ethyl acetate was slowly added. The reaction mixture was monitored by TLC until finish. The solvents were evaporated under reduced pressure and the residue mixture was partitioned between water and ethyl acetate. The combined organic phase was washed with brine and dried by Na2SO4 to give 120 g of 1-(4-fluorophenyl)-2-(3-methoxyphenylsulfanyl)ethanone, which was used in the next step without further purification. BF3•OEt2 was slowly added to a flask charged with 1-(4-fluorophenyl)-2-(3-methoxyphenylsulfanyl)ethanone (120 g) under argon atmosphere in an ice bath. The reaction mixture was stirred until starting material was consumed as monitored by TLC. The reaction mixture was poured into saturated NaHCO3/ice water, stirred 30 min and extracted with dichloromethane. The crude product was purified by silica gel chromatography (5% dichloromethane in hexane). The combined fractions from the column were concentrated and recrystallized to give 75 g pale yellow solid with a 54% yield over the two steps. 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 8.9 Hz, 1H), 7.56 – 7.48 (m, 2H), 7.38 (d, J = 2.2 Hz, 1H), 7.21 – 7.12 (m, 3H), 7.02 (dd, J = 8.9, 2.2 Hz, 1H), 3.90 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.49 (d, JC-F = 246.7 Hz), 157.73, 142.24, 136.79, 132.34, 132.14, 130.30 (d, JC-F = 8.0 Hz), 123.47, 120.85, 115.78 (d, JC-F = 21.4 Hz), 114.70, 105.42, 55.80.
2-(4-fluorophenyl)benzo[b]thiophen-6-ol (11)
2-(4-fluorophenyl)-6-methoxybenzo[b]thiophene (9)32 (4.8 g, 18.6 mmol) was dissolved in 100 mL of anhydrous dichloromethane and cooled to −78 °C under a dry ice acetone bath. BBr3 (1.0 M in CH2Cl2, 55 mL, 55 mmol) was added dropwise to this solution. The reaction mixture was stirred until starting material was consumed as monitored by TLC and then quenched by saturated NaHCO3/ice water. The solution was extracted with ethyl acetate and washed with brine. The organic extracts were combined, dried over anhydrous Na2SO4, concentrated in vacuum, and then purified by flash chromatography (5% - 30% ethyl acetate in hexane) to give 3.3 g white solid (yield, 73%). 1H NMR (400 MHz, CD3OD) δ 7.72 – 7.65 (m, 2H), 7.60 (d, J = 8.6 Hz, 1H), 7.47 (s, 1H), 7.20 (d, J = 2.0 Hz, 1H), 7.14 (t, J = 8.8 Hz, 2H), 6.86 (dd, J = 8.6, 2.2 Hz, 1H). 13C NMR (100 MHz, CD3OD) δ 163.81 (d, JC-F = 246.1 Hz), 156.52, 142.33, 140.46, 135.45, 132.45, 128.77 (d, JC-F = 8.1 Hz), 125.39, 120.41, 116.74 (d, JC-F = 22.1 Hz), 115.73, 108.05. ESI-HRMS (m/z): [M-H]− calcd. for C14H9FOS: 243.0280; observed, 243.0289.
3-(4-fluorophenyl)benzo[b]thiophen-6-ol (12)
This compound was prepared using similar method as 11 and gave 780 mg white solid (yield, 82%). 1H NMR (400 MHz, CD3OD) δ 7.59 (d, J = 8.8 Hz, 1H), 7.47 (dd, J = 8.6, 5.5 Hz, 2H), 7.28 (d, J = 2.2 Hz, 1H), 7.15 – 7.10 (m, 3H), 6.90 (dd, J = 8.8, 2.2 Hz, 1H). 13C NMR (100 MHz, CD3OD) δ 163.58 (d, JC-F = 245.1 Hz), 156.35, 143.61, 137.62, 133.77 (d, JC-F = 3.3 Hz) 132.32, 131.23 (d, JC-F = 8.0 Hz), 124.15, 121.15, 116.39 (d, JC-F = 21.6 Hz), 115.54, 108.64. ESI-HRMS (m/z): [M-H]− calcd. for C14H8FOS: 243.0280; observed, 243.0276.
(2-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-3-yl)(4-(trifluoromethyl)phenyl)methanone (13b)
To an oven-dried flask charged with 2-(4-fluorophenyl)-6-methoxybenzo[b]thiophene (258 mg, 1 mmol) and 4-(trifluoromethyl)benzoyl chloride (250 mg, 1.2 mmol) in 10 ml dichloromethane, AlCl3 (400 mg, 3 mmol) was added in three portions over 10 minutes. The mixture was stirred until all the starting material was consumed and then poured into ice water. The layers were separated, and the aqueous layer was extracted three times with dichloromethane. The organic layers were combined, washed by brine and dried over Na2SO4. The crude product was purified by silica gel chromatography (1-10 % ethyl acetate in hexane) to give 160 mg yellow solid ( yield, 37%). 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.1 Hz, 2H), 7.68 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 8.1 Hz, 2H), 7.35 – 7.26 (m, 3H), 7.04 (dd, J = 9.0, 2.4 Hz, 1H), 6.90 (t, J = 8.6 Hz, 2H), 3.91 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 192.87, 163.07 (d, JC-F = 250.5 Hz), 158.30, 144.42, 140.47, 134.40 (q, JC-F = 32.6 Hz), 133.43, 131.25 (d, JC-F = 8.4 Hz), 130.72, 130.18, 129.43 (d, JC-F = 3.5 Hz), 125.46 (q, JC-F = 3.7 Hz), 124.55, 123.60 (q, JC-F = 272.9 Hz), 116.02, 115.70 (d, JC-F = 20.7 Hz), 104.65, 55.82.
(2-(4-fluorophenyl)-6-hydroxybenzo[b]thiophen-3-yl)(4-(trifluoromethyl)phenyl)methanone (14b)
This compound was prepared by a procedure identical with the preparation of 11 except a C18 reverse phase column was used to get the pure compound (2.5 g, yield 61%). 1H NMR (400 MHz, CD3OD) δ 7.77 (d, J = 8.1 Hz, 2H), 7.65 (d, J = 8.8 Hz, 1H), 7.57 – 7.49 (m, 2H), 7.32 – 7.28 (m, 3H), 6.97 – 6.86 (m, 3H). 13C NMR (100 MHz, CD3OD) δ 194.20, 164.30 (d, JC-F = 248.8 Hz), 157.41, 145.51, 142.29, 141.84, 134.95 (q, JC-F = 32.5 Hz), 133.65, 132.56 (d, JC-F = 8.5 Hz), 131.85, 131.29, 130.97 (d, JC-F = 3.4 Hz), 126.36 (q, JC-F = 3.8 Hz), 125.45, 125.02(q, JC-F = 272Hz), 116.67, 116.54 (d, JC-F = 22.3 Hz), 107.87. ESI-HRMS (m/z): [M-H]− calcd. for C22H11F4O2S: 415.0416; observed, 415.0409.
(2-(4-chlorophenyl)-6-hydroxybenzo[b]thiophen-3-yl)(4-(trifluoromethyl)phenyl)methanone (14c)
This compound was isolated from the preparation of 14b as a minor product, (130 mg, yield 3%). 1H NMR (400 MHz, CD3OD) δ 7.79 (d, J = 8.2 Hz, 2H), 7.65 (d, J = 8.8 Hz, 1H), 7.56 (d, J = 8.2 Hz, 2H), 7.31 (d, J = 2.2 Hz, 1H), 7.27 (d, J = 8.5 Hz, 2H), 7.19 (d, J = 8.5 Hz, 2H), 6.95 (dd, J = 8.8, 2.2 Hz, 1H). 13C NMR (100 MHz, CD3OD) δ 194.18, 157.53, 145.00, 142.23, 141.94, 135.98, 135.06 (q, JC-F = 32.5 Hz).133.65, 133.35, 132.13, 131.91, 131.32, 129.79, 126.44 (q, JC-F = 3.8 Hz), 125.50, 125.10 (q, JC-F = 272 Hz), 116.76, 107.88.. ESI-HRMS (m/z): [M-H]− calcd. for C22H11ClF3O2S: 431.0120; observed, 431.0113.
2-(4-fluorophenyl)-6-methoxy-3-(4-methoxyphenoxy)benzo[b]thiophene 1-oxide (17)
NaH (380 mg, 9.5 mmol, 60% dispersion in mineral oil) was added in three portions to a flask charged with a solution of 4-methoxyphenol (0.85 g, 6.9 mmol) in 10 mL anhydrous DMF under argon atmosphere at room temperature. After the mixture was stirred for 30 min, 3-bromo-2-(4-fluorophenyl)-6-methoxybenzo[b]thiophene 1-oxide32 (2.2 g, 6.3 mmol) was added in small portions. After the mixture was stirred for 2 h, ethyl acetate and water were added, and the organic layer was washed several times with water and brine, then dried over Na2SO4. The residue was purified by flash chromatography (5% - 60% ethyl acetate in hexane) to yield 2.4 g (yield, 93%) title compound.1H NMR (400 MHz, CDCl3) δ 7.78 – 7.72 (m, 2H), 7.50 (d, J = 2.3 Hz, 1H), 7.09 – 6.97 (m, 5H), 6.91 (dd, J = 8.5, 2.3 Hz, 1H), 6.83 – 6.76 (m, 2H), 3.88 (s, 3H), 3.76 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.68 (d, JC-F = 249.6 Hz), 161.12, 156.29, 150.13, 149.01, 144.76, 130.26 (d, JC-F = 8.2 Hz), 130.19, 126.40, 125.94, 124.11, 118.24, 118.18, 116.04 (d, JC-F = 21.8 Hz), 115.03, 112.08, 56.10, 55.84.
2-(4-fluorophenyl)-6-methoxy-3-(4-methoxyphenoxy)benzo[b]thiophene (18)
LiAlH4 (372 mg, 9.8 mmol) was added in small portions to a solution of 17b (2.6 g, 6.6 mmol) in 30 mL of anhydrous THF under argon atmosphere at 0 °C. After the mixture was stirred for 2 h, 0.4 ml water was slowly added followed by 0.4 ml of 15% NaOH to quench the reaction. The precipitate was then filtered through celite and washed with ethyl acetate. The filtrate was washed with water and brine for three times. The organic layers were combined, dried over anhydrous Na2SO4, and then purified by flash chromatography (1% - 30% ethyl acetate in hexane) to yield 1.8 g (yield, 71%) product. 1H NMR (400 MHz, CDCl3) δ 7.76 – 7.68 (m, 2H), 7.28 – 7.24 (m, 2H), 7.04 (t, J = 8.7 Hz, 2H), 6.94 – 6.85 (m, 3H), 6.83 – 6.75 (m, 2H), 3.87 (s, 3H), 3.75 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.30 (d, JC-F = 248.0 Hz), 158.27, 155.06, 151.73, 140.81, 137.24, 129.34 (d, JC-F = 8.0 Hz), 128.38, 128.01, 125.52, 122.63, 116.52, 115.91 (d, JC-F = 21.6 Hz), 114.95, 114.68, 105.54, 55.80, 55.78.
2-(4-fluorophenyl)-3-(4-hydroxyphenoxy)benzo[b]thiophen-6-ol (19)
This compound was prepared by a procedure identical to the preparation of 11 (1.3 g, white solid, yield 78%). 1H NMR (400 MHz, CD3OD) δ 7.77 – 7.66 (m, 2H), 7.22 – 7.15 (m, 2H), 7.07 (t, J = 8.7 Hz, 2H), 6.83 – 6.72 (m, 3H), 6.71 – 6.64 (m, 2H). 13C NMR (100 MHz, CD3OD) δ 163.43 (d, JC-F = 246.6 Hz), 157.29, 153.68, 152.10, 142.36, 138.54, 130.31, 130.24 (d, JC-F = 8.0 Hz), 128.33, 125.19, 123.45, 117.35, 117.04, 116.52 (d, JC-F = 21.9 Hz), 115.67, 108.73. ESI-HRMS (m/z): [M+H]+ calcd. for C20H13FO3S: 353.0648; observed, 353.0632.
(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-2-yl)(4-(methylsulfonyl)phenyl)methanone (20a)
This compound was prepared by a procedure identical to the preparation of 13b (180 mg, yield, 21%). 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 8.3 Hz, 2H), 7.55 (d, J = 9.0 Hz, 1H), 7.37 (d, J = 2.1 Hz, 1H), 7.17 – 7.13 (m, 2H), 7.04 (dd, J = 9.0, 2.1 Hz, 1H), 6.88 (t, J = 8.5 Hz, 2H), 3.94 (s, 3H), 2.97 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 189.78, 162.61 (d, JC-F = 249.3 Hz), 160.49, 143.73, 142.96, 142.68, 142.37, 135.72, 133.52, 132.38 (d, JC-F = 8.2 Hz), 130.45 (d, JC-F = 3.5 Hz), 129.93, 126.95, 126.43, 116.70, 115.45 (d, JC-F = 21.6 Hz), 104.40, 55.92, 44.54.
(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-2-yl)(4-methoxyphenyl)methanone (20b)
This compound was prepared by a procedure identical to the preparation of 13b (488 mg, yield 52%). 1H NMR (400 MHz, CDCl3) δ 7.63 – 7.57 (m, 3H), 7.35 (d, J = 2.2 Hz, 1H), 7.29 – 7.21 (m, 2H), 7.02 (dd, J = 9.0, 2.3 Hz, 1H), 6.94 (t, J = 8.7 Hz, 2H), 6.68 (d, J = 8.8 Hz, 2H), 3.91 (s, 3H), 3.78 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 189.74, 163.19, 162.48 (d, JC-F = 248.6 Hz), 159.49, 142.55, 139.65, 135.81, 133.37, 132.17, 131.98 (d, JC-F = 8.2 Hz), 130.86 (d, JC-F = 3.4 Hz), 130.51, 125.65, 115.96, 115.42 (d, JC-F = 21.6 Hz), 113.31, 104.41, 55.81, 55.53.
(4-(dimethylamino)phenyl)(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-2-yl)methanone (20c)
This compound was prepared by a procedure identical to the preparation of 13b (393 mg, yield 48%). 1H NMR (400 MHz, CDCl3) δ 7.66 – 7.58 (m, 3H), 7.36 – 7.30 (m, 3H), 7.09 – 6.92 (m, 3H), 6.44 (d, J = 9.0 Hz, 2H), 3.92 (s, 3H), 3.00 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 188.84, 162.37 (d, JC-F = 247.1 Hz), 159.00, 153.43, 142.02, 137.96, 136.20, 133.30, 132.52, 131.85 (d, JC-F = 8.1 Hz), 131.16 (d, JC-F = 3.3 Hz), 125.18, 125.05, 115.58, 115.41 (d, JC-F = 21.5 Hz), 110.37, 104.49, 55.80, 40.09.
(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-2-yl)(p-tolyl)methanone (20d)
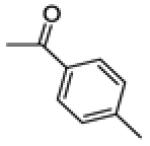
This compound was prepared by a procedure identical to the preparation of 13b (390 mg, yield 49%). 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 9.0 Hz, 1H), 7.50 (d, J = 8.1 Hz, 2H), 7.35 (d, J = 2.2 Hz, 1H), 7.25 – 7.19 (m, 2H), 7.06 – 6.96 (m, 3H), 6.93 (t, J = 8.7 Hz, 2H), 3.93 (s, 3H), 2.30 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 190.85, 162.51 (d, JC-F = 247.8 Hz), 159.62, 143.18, 142.74, 140.33, 135.82, 135.27, 133.46, 131.98 (d, JC-F = 8.2 Hz), 130.77 (d, JC-F = 3.4 Hz), 129.83, 128.64, 125.81, 116.02, 115.31 (d, JC-F = 21.6 Hz), 104.36, 55.79, 21.66.
(4-fluorophenyl)(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-2-yl)methanone (20e)
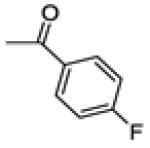
This compound was prepared by a procedure identical to the preparation of 13b (510 mg, yield 67%). 1H NMR (400 MHz, CDCl3) δ 7.64 – 7.54 (m, 3H), 7.36 (d, J = 2.2 Hz, 1H), 7.25 – 7.18 (m, 2H), 7.03 (dd, J = 9.0, 2.3 Hz, 1H), 6.94 (t, J = 8.6 Hz, 2H), 6.85 (t, J = 8.6 Hz, 2H), 3.92 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 189.72, 165.14 (d, JC-F = 254.4 Hz), 162.61 (d, JC-F = 248.7 Hz), 159.87, 142.96, 140.74, 135.54, 134.24 (d, JC-F = 3.0 Hz), 133.43, 132.16 (d, JC-F = 9.5 Hz), 132.07 (d, JC-F = 8.5 Hz), 130.61 (d, JC-F = 3.4 Hz), 125.96, 116.24, 115.49 (d, JC-F = 21.7 Hz), 115.11 (d, JC-F = 22.0 Hz), 104.42, 55.84.
Cyclopropyl(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-2-yl)methanone (20f)
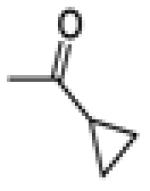
This compound was prepared by a procedure identical to the preparation of 13b (450 mg, yield 69%). 1H NMR (400 MHz, CDCl3) δ 7.48 – 7.41 (m, 2H), 7.37 (d, J = 9.0 Hz, 1H), 7.31 (d, J = 2.3 Hz, 1H), 7.20 (dd, J = 12.0, 5.4 Hz, 2H), 6.97 (dd, J = 9.0, 2.3 Hz, 1H), 3.91 (s, 3H), 1.84 – 1.73 (m, 1H), 1.22 – 1.10 (m, 2H), 0.73 (dq, J = 7.1, 3.5 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 195.69, 163.02 (d, JC-F = 248.0 Hz), 159.99, 142.64, 140.19, 138.65, 134.86, 131.96 (d, JC-F = 8.1 Hz), 131.35, 126.28, 116.02, 115.84 (d, JC-F = 21.6 Hz), 104.34, 55.83, 20.65, 12.58.
Cyclohexyl(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-2-yl)methanone (20g)
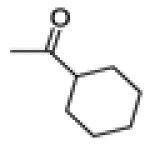
This compound was prepared by a procedure identical to the preparation of 13b (520 mg, yield 91%). 1H NMR (400 MHz, CDCl3) δ 7.37 – 7.31 (m, 2H), 7.28 (d, J = 2.2 Hz, 1H), 7.25 (d, J = 9.0 Hz, 1H), 7.21 – 7.17 (m, 2H), 6.93 (dd, J = 9.0, 2.3 Hz, 1H), 3.88 (s, 3H), 2.47 (ddd, J = 11.7, 7.3, 2.8 Hz, 1H), 1.77 – 1.60 (m, 4H), 1.54 (d, J = 13.2 Hz, 1H), 1.44 – 1.27 (m, 2H), 1.17 – 1.02 (m, 1H), 0.93 – 0.75 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 199.25, 162.96 (d, JC-F = 248.2 Hz), 159.99, 142.49, 139.95, 137.33, 135.15, 131.45, 131.43 (d, JC-F = 8.1 Hz), 126.33, 115.72 (d, JC-F = 21.6 Hz), 115.61, 104.25, 55.80, 48.44, 29.38, 25.81, 25.77.
(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-2-yl)(4-iodophenyl)methanone (20h)
This compound was prepared by a procedure identical to the preparation of 13b (982 mg, yield 26%). 1H NMR (400 MHz, CDCl3) δ 7.57 – 7.51 (m, 3H), 7.34 (d, J = 2.1 Hz, 1H), 7.26 (d, J = 8.3 Hz, 2H), 7.21 – 7.18 (m, 2H), 7.02 (dd, J = 8.7, 2.1 Hz, 1H), 6.94 (t, J = 8.5 Hz, 2H), 3.91 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 190.27, 162.69 (d, JC-F = 248.9 Hz), 159.94, 143.07, 141.17, 137.34, 137.18, 135.31, 133.43, 132.02 (d, JC-F = 8.2 Hz), 130.84, 130.46 (d, JC-F = 3.4 Hz), 126.06, 116.30, 115.50 (d, JC-F = 21.7 Hz), 104.34, 99.73, 55.83.
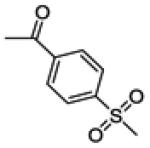
(3-(4-fluorophenyl)-6-hydroxybenzo[b]thiophen-2-yl)(4-(methylsulfonyl)phenyl)methanone (21a)
This compound was prepared by a procedure identical to the preparation of 11 (120 mg, light green solid, yield 85%). 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.4 Hz, 2H), 7.45 – 7.43 m, 2H), 7.23 (dd, J = 8.5, 5.6 Hz, 2H), 7.02 – 6.98 (m, 3H), 3.14 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 189.53, 161.80 (d, JC-F = 246.0 Hz), 158.49, 142.71, 142.69, 142.43, 142.27, 134.29, 132.50 (d, JC-F = 8.5 Hz), 132.15, 129.96, 129.35, 126.53, 126.26, 116.59, 114.94 (d, JC-F = 21.6 Hz), 107.22, 43.38. ESI-HRMS (m/z): [M-H]− calcd. for C22H14FO4S2: 425.0318; observed, 425.0326.
(3-(4-fluorophenyl)-6-hydroxybenzo[b]thiophen-2-yl)(4-hydroxyphenyl)methanone (21b)
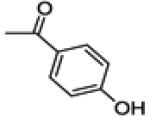
This compound was prepared by a procedure identical to the preparation of 11 (284 mg, white solid, yield 53%). 1H NMR (400 MHz, CD3OD) δ 7.51 – 7.44 (m, 3H), 7.29 (d, J = 2.0 Hz, 1H), 7.25 – 7.22 (m, 2H), 7.02 – 6.88 (m, 3H), 6.57 (d, J = 8.7 Hz, 2H). 13C NMR (100 MHz, CD3OD) δ 192.04, 163.79 (d, JC-F = 243.1 Hz), 163.31, 158.86, 143.82, 141.34, 135.99, 133.64, 133.48, 133.28 (d, JC-F = 8.3 Hz), 132.34 (d, JC-F = 3.4 Hz), 130.33, 126.72, 116.95, 116.13 (d, JC-F = 21.9 Hz), 115.75, 107.96. ESI-HRMS (m/z): [M+H]+ calcd. for C21H14FO3S, 365.0648; observed, 365.0638.
(4-(dimethylamino)phenyl)(3-(4-fluorophenyl)-6-hydroxybenzo[b]thiophen-2-yl)methanone (21c)
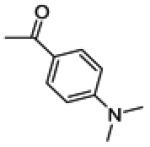
This compound was prepared by a procedure identical to the preparation of 11 (205 mg, white solid, yield 54%). 1H NMR (400 MHz, CD3OD) δ 7.54 (dd, J = 9.0, 2.9 Hz, 3H), 7.37 – 7.27 (m, 3H), 7.01 (t, J = 8.8 Hz, 2H), 6.96 (dd, J = 8.8, 2.2 Hz, 1H), 6.51 (d, J = 9.1 Hz, 2H), 3.00 (s, 6H). 13C NMR (100 MHz, CD3OD) δ 191.43, 163.72 (d, JC-F = 246.2 Hz), 158.47, 155.26, 143.39, 139.88, 136.14, 133.63, 133.52, 133.15 (d, JC-F = 8.3 Hz), 132.61, 126.30, 125.62, 116.76, 116.20 (d, JC-F = 21.9 Hz), 111.47, 107.99, 40.04. ESI-HRMS (m/z): [M+H]+ calcd. for C23H19FNO2S: 392.1121; observed, 392.1113.
(3-(4-fluorophenyl)-6-hydroxybenzo[b]thiophen-2-yl)(p-tolyl)methanone (21d)
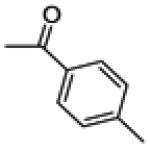
This compound was prepared by a procedure identical to the preparation of 11 (190 mg, white solid, yield 52%). 1H NMR (400 MHz, DMSO-d6) δ 10.18 (s, 1H), 7.52 – 7.38 (m, 4H), 7.32 – 7.27 (m, 2H), 7.11 – 7.02 (m, 4H), 6.99 (dd, J = 8.9, 2.1 Hz, 1H), 2.26 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 190.37, 162.18 (d, JC-F = 245.3 Hz), 158.18, 142.94, 142.32, 140.76, 135.37, 134.48, 132.57 (d, JC-F = 8.3 Hz), 132.27, 130.98 (d, JC-F = 3.1 Hz), 129.70, 128.90, 126.24, 116.74, 115.51 (d, JC-F = 21.6 Hz), 107.61, 21.50. ESI-HRMS (m/z): [M+H]+ calcd. for C22H16FO2S: 363.0855; observed, 363.0860.
(4-fluorophenyl)(3-(4-fluorophenyl)-6-hydroxybenzo[b]thiophen-2-yl)methanone (21e)
This compound was prepared by a procedure identical to the preparation of 11 (211 mg, green solid, yield 61%). 1H NMR (400 MHz, Acetone-d6) δ 7.68 – 7.59 (m, 2H), 7.56 (d, J = 8.9 Hz, 1H), 7.47 (d, J = 2.2 Hz, 1H), 7.39 – 7.29 (m, 2H), 7.09 – 7.02 (m, 3H), 7.01 – 6.94 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 189.93, 165.28 (d, JC-F = 251.6 Hz), 163.04 (d, JC-F = 247.5 Hz), 158.70, 143.53, 141.59, 135.85, 135.49 (d, JC-F = 3.0 Hz), 133.55, 133.30 (d, JC-F = 8.3 Hz) 132.85 (d, JC-F = 9.3 Hz), 131.69 (d, JC-F = 3.3 Hz), 126.95, 116.95, 115.92 (d, JC-F = 21.8 Hz), 115.60 (d, JC-F = 22.2 Hz), 108.02. ESI-HRMS (m/z): [M-H]− calcd. for C21H11F2O2S: 365.0448; observed, 365.0444.
Cyclopropyl(3-(4-fluorophenyl)-6-hydroxybenzo[b]thiophen-2-yl)methanone (21f)
NaOtBu (280 mg, 3.6 mmol) was added to an oven-dried round-bottom flask charged with 2-(dimethylamino)ethanethiol hydrochloride (305 mg, 1.8 mmol) in 3 ml DMF in an ice bath. After 15 min, the mixture was allowed to equilibrate to room temperature, and one portion of 20f (400 mg, 1.2 mmol) was added and the mixture was heated to reflux. The reaction was monitored by TLC and quenched by ice water. Ethyl acetate and water were added, and the organic layer was washed several times with water and brine, then dried over Na2SO4. The residue was purified by flash chromatography (5% - 40% ethyl acetate in hexane) to yield 94 mg (yield, 25%) of title compound. 1H NMR (400 MHz, Acetone-d6) δ 7.62 – 7.51 (m, 2H), 7.39 (d, J = 2.1 Hz, 1H), 7.38 – 7.31 (m, 3H), 7.00 (dd, J = 8.9, 2.2 Hz, 1H), 1.80 (tt, J = 7.8, 4.6 Hz, 1H), 1.05 – 0.97 (m, 2H), 0.75 – 0.69 (m, 2H). 13C NMR (100 MHz, Acetone-d6) δ 195.38, 163.89 (d, JC-F = 246.2 Hz), 158.91, 143.34, 141.12, 138.83, 135.01, 133.14 (d, JC-F = 8.3 Hz), 132.43 (d, JC-F = 3.4 Hz), 127.34, 116.86, 116.57 (d, JC-F = 21.7 Hz), 108.01, 20.97, 12.32. ESI-HRMS (m/z): [M+H]+ calcd. for C18H14FO2S: 313.0699; observed, 313.0696.
Cyclohexyl(3-(4-fluorophenyl)-6-hydroxybenzo[b]thiophen-2-yl)methanone (21g)
20g (6.56 g, 18 mmol) was dissolved in 50 mL of anhydrous dichloromethane and cooled to −78 °C in a dry ice acetone bath. BF3•SMe2 (23.4 g, 180 mmol) was added dropwise to this solution. The reaction mixture was stirred until starting material was consumed as monitored by TLC and then quenched by saturated NaHCO3/ice water. The reaction mixture was extracted with ethyl acetate and washed with brine. The organic extracts were combined, dried over anhydrous Na2SO4, concentrated in vacuo, and then purified by flash chromatography (5%-60% ethyl acetate in hexane) to give 4.8 g white powder (yield, 76%). 1H NMR (400 MHz, CD3OD) δ 7.46 – 7.38 (m, 2H), 7.31 (t, J = 8.8 Hz, 2H), 7.23 (d, J = 2.2 Hz, 1H), 7.19 (d, J = 8.9 Hz, 1H), 6.88 (dd, J = 8.9, 2.2 Hz, 1H), 2.51 (tt, J = 11.5, 2.9 Hz, 1H), 1.68 – 1.64 (m, 4H), 1.57 – 1.54 (m, 1H), 1.37 – 1.23 (m, 2H), 1.19 – 1.05 (m, 1H), 0.92 – 0.77 (m, 2H). 13C NMR (100 MHz, CD3OD) δ 201.15, 164.43 (d, JC-F = 246.9 Hz), 159.63, 144.07, 142.15, 137.60, 135.51, 133.00, 132.74 (d, JC-F = 8.2 Hz), 127.58, 117.02, 116.64 (d, JC-F = 21.9 Hz), 107.78, 49.50, 30.40, 26.79, 26.76. ESI-HRMS (m/z): [M+H]+ calcd. for C21H20FO2S: 355.1168; observed, 355.1150.
(4-ethynylphenyl)(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-2-yl)methanone (22)
To a mixture of 20h (244 mg, 0.5 mmol) and ethynyltrimethylsilane (108 mg, 0.55 mmol) in dry trimethylamine (5 ml) was added Pd(PPh3)2Cl2 (5% mol) and CuI (5% mol) under argon atmosphere. The mixture was heated at 70 °C. After cooling the mixture to room temperature, ethyl acetate was added and the suspension was filtered through a pad of silica gel. The filtrate was collected and the crude product was treated with 1M TBAF (1.5 equiv.) in THF. After 30 min, the reaction was quenched by pouring into ice water and extracted with ethyl acetate. The organic phase was washed with brine and dried over Na2SO4. The mixture was purified by flash chromatography (1% - 30% ethyl acetate in hexane) to give the title product (96 mg, yield 50%). 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 9.0 Hz, 1H), 7.51 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 2.2 Hz, 1H), 7.28 (d, J = 8.3 Hz, 2H), 7.24 – 7.16 (m, 2H), 7.03 (dd, J = 9.0, 2.3 Hz, 1H), 6.93 (t, J = 8.6 Hz, 2H), 3.92 (s, 3H), 3.18 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 190.35, 162.67 (d, JC-F = 248.8 Hz), 159.93, 143.10, 141.10, 137.90, 135.53, 133.46, 132.06 (d, JC-F = 8.2 Hz), 131.61, 130.51 (d, JC-F = 3.5 Hz), 129.41, 126.06, 125.94, 116.29, 115.49 (d, JC-F = 21.7 Hz), 104.37, 82.92, 80.08, 55.84.
(4-ethynylphenyl)(3-(4-fluorophenyl)-6-hydroxybenzo[b]thiophen-2-yl)methanone (23)
This compound was prepared by a procedure identical to the preparation of 21g (704 mg, yellow solid, yield 56%). 1H NMR (400 MHz, Acetone-d6) δ 7.57 – 7.52 (m, 3H), 7.47 (d, J = 2.2 Hz, 1H), 7.37 – 7.28 (m, 4H), 7.08 – 7.00 (m, 3H), 3.81 (s, 1H). 13C NMR (100 MHz, Acetone-d6) δ 190.56, 163.39 (d, JC-F = 246.4 Hz), 158.76, 143.71, 141.93, 139.09, 135.87, 133.64, 133.31 (d, JC-F = 8.3 Hz), 132.15, 131.67 (d, JC-F = 3.4 Hz), 130.12, 127.06, 126.43, 116.99, 115.93 (d, JC-F = 21.8 Hz), 108.03, 83.44, 81.75. ESI-HRMS (m/z): [M+H]+ calcd. for C23H14FO2S: 373.0699; observed, 373.0686.
2-bromo-3-(4-fluorophenyl)-6-methoxybenzo[b]thiophene (24)
N-Bromoacetamide (1.46 g, 10.5 mmol) was added in small portions to a solution of 10 (2.58 g, 10 mmol) in 300 mL of CH2Cl2 and 20 mL of ethanol at room temperature. After the mixture was stirred for 1 h, the solvent was removed in vacuo. The residue was titrated with pure ethanol and filtered to give 3.0 g (yield, 89%) white powder. 1H NMR (400 MHz, CDCl3) δ 7.47 – 7.41 (m, 2H), 7.39 (d, J = 8.9 Hz, 1H), 7.24 (d, J = 2.3 Hz, 1H), 7.23 – 7.17 (m, 2H), 6.94 (dd, J = 8.9, 2.4 Hz, 1H), 3.87 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.63 (d, JC-F = 247.5 Hz), 157.89, 157.88, 141.13, 135.81, 132.81, 131.79 (d, JC-F = 8.1 Hz), 130.10 (d, JC-F = 3.3 Hz), 123.51, 115.78 (d, JC-F = 21.6 Hz), 114.72, 104.66, 55.80.
(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophen-2-yl)(4-(trifluoromethyl)phenyl)methanone (25a)
To an oven dried flask charged with 24 (7.4 g, 21.8 mmol) in 40 ml THF in a dry ice acetone bath, n-BuLi (1.6 M, 15 ml) was added dropwise under argon atmosphere. After stirring for 30 min, a solution of 4-(trifluoromethyl)benzoyl chloride (5 g, 24 mmol) in 10 ml THF was added dropwise. The reaction was allowed to warm to room temperature and was monitored by TLC. Upon the consumption of the starting material, the reaction was poured into ice water. The mixture was extracted with ethyl acetate, washed with brine and then dried over anhydrous Na2SO4. The crude product was purified by flash chromatography (1%-30% ethyl acetate in hexane) to give the title product (5.3 g, yield 57%). 1H NMR (400 MHz, CDCl3) δ 7.59 – 7.55 (m, 3H), 7.42 – 7.36 (m, 3H), 7.18 – 7.13 (m, 2H), 7.03 (dd, J = 9.0, 2.1 Hz, 1H), 6.88 (t, J = 8.5 Hz, 2H), 3.93 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 190.14, 162.71 (d, JC-F = 249.5 Hz), 160.27, 143.46, 142.08, 141.28, 135.58, 133.59, 133.20 (q, JC-F = 32.7 Hz), 132.13 (d, JC-F = 8.2 Hz), 130.29 (d, JC-F = 3.4 Hz), 129.51, 126.35, 124.84 (q, JC-F = 3.7 Hz), 123.63 (q, JC-F = 272.9 Hz), 116.50, 115.46 (d, JC-F = 21.8 Hz), 104.37, 55.87.
4-(3-(4-fluorophenyl)-6-methoxybenzo[b]thiophene-2-carbonyl)benzonitrile (25b)
This compound was prepared by a procedure identical to the preparation of 25a (420 mg, yield 54%). 1H NMR (400 MHz, CDCl3) δ 7.56 (t, J = 8.1 Hz, 3H), 7.44 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 2.3 Hz, 1H), 7.20 – 7.13 (m, 2H), 7.04 (dd, J = 9.0, 2.3 Hz, 1H), 6.95 – 6.88 (m, 2H), 3.93 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 189.50, 162.61 (d, JC-F = 250.0 Hz), 160.22, 143.41, 141.99, 141.66, 135.04, 133.30, 132.03 (d, JC-F = 8.2 Hz), 131.52, 130.09 (d, JC-F = 3.5 Hz), 129.51, 126.21, 117.87, 116.48, 115.46 (d, JC-F = 21.7 Hz), 114.91, 104.23, 55.75.
(3-(4-fluorophenyl)-6-hydroxybenzo[b]thiophen-2-yl)(4-(trifluoromethyl)phenyl)methanone (26a)
This compound was prepared by a procedure identical to the preparation of 11 (3.8 g, light green powder, yield 82%). 1H NMR (400 MHz, Acetone-d6) δ 7.65 (d, J = 8.0 Hz, 2H), 7.53 (t, J = 9.1 Hz, 3H), 7.48 (d, J = 2.2 Hz, 1H), 7.33 – 7.22 (m, 2H), 7.06 (dd, J = 8.9, 2.2 Hz, 1H), 6.97 (t, J = 8.8 Hz, 2H). 13C NMR (100 MHz, Acetone-d6)) δ 190.61, 163.52 (d, JC-F = 246.9 Hz), 159.21, 144.17, 143.13, 142.97, 136.13, 133.92, 133.55 (d, JC-F = 8.4 Hz), 132.93 (q, JC-F = 32.2 Hz) 131.52 (d, JC-F = 3.4 Hz), 130.47, 127.51, 125.65 (q, JC-F = 3.9 Hz), 124.93 (q, JC-F = 271.1 Hz), 117.22, 115.99 (d, JC-F = 21.9 Hz), 108.13. ESI-HRMS (m/z): [M+H]+ calcd. for C22H13F4O2S: 417.0572; observed, 417.0563.
4-(3-(4-fluorophenyl)-6-hydroxybenzo[b]thiophene-2-carbonyl)benzonitrile (26b)
This compound was prepared by a procedure identical to the preparation of 11 (95 mg, yield 23%). 1H NMR (400 MHz, CDCl3) δ 7.60 – 7.54 (m, 3H), 7.45 (d, J = 8.5 Hz, 2H), 7.34 (d, J = 2.2 Hz, 1H), 7.21 – 7.13 (m, 2H), 6.98 (dd, J = 8.9, 2.3 Hz, 1H), 6.96 – 6.86 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 189.77, 162.81 (d, JC-F = 250.1 Hz), 156.43, 143.41, 142.17, 141.78, 135.24, 133.72, 132.17 (d, JC-F = 8.2 Hz), 131.72, 130.21, 129.68, 126.82, 117.99, 116.22, 115.65 (d, JC-F = 21.7 Hz), 115.14, 107.73. ESI-HRMS (m/z): [M-H]− calcd. for C22H11FNO2S: 372.0495; observed, 372.0487.
2-bromo-3-(4-fluorophenyl)-6-methoxybenzo[b]thiophene 1-oxide (27)
Trifluoroacetic acid (13 mL) was added dropwise to a solution of 24 (2.4 g, 7 mmol) in 13 mL anhydrous CH2Cl2 in an ice bath. The mixture was stirred for 5 min, H2O2 (1.0 mL, 7 mmol, 30% aqueous solution) was added dropwise, and the resulting mixture was allowed to warm to room temperature and stirred for 2 h. Upon the consumption of starting material, sodium bisulfite (0.3 g) was added to the solution followed by 5 mL of water. The mixture was stirred vigorously for 30 min and then concentrated in vacuo. The residue was partitioned between CH2Cl2 and saturated aqueous NaHCO3 solution. The layers were separated, and the organic layer was washed with water, saturated NaHCO3, and water, and then dried over anhydrous Na2SO4. The residue was titrated with diethyl ether and filtered to give 2.1 g (yield, 84%) pale yellow powder. 1H NMR (400 MHz, CDCl3) δ 7.52 – 7.45 (m, 3H), 7.25 – 7.20 (m, 2H), 7.16 (d, J = 8.5 Hz, 1H), 6.97 (dd, J = 8.5, 2.4 Hz, 1H), 3.89 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.45 (d, JC-F = 250.6 Hz), 160.84, 146.91, 144.07, 130.87 (d, JC-F = 8.7 Hz), 130.86, 127.36 (d, JC-F = 3.5 Hz), 125.09, 124.75, 118.07, 116.35 (d, JC-F = 21.9 Hz), 112.84, 56.13.
3-(4-fluorophenyl)-6-methoxy-2-(4-methoxyphenoxy)benzo[b]thiophene 1-oxide (28a)
This compound was prepared by a procedure identical to the preparation of 17 (2.47g, yellow solid, yield 89%). 1H NMR (400 MHz, CDCl3) δ 7.51 – 7.48 (m, 2H), 7.41 (d, J = 2.3 Hz, 1H), 7.26 (d, J = 8.5, 1H), 7.17 – 7.05 (m, 4H), 6.99 (dd, J = 8.5, 2.3 Hz, 1H), 6.81 (d, J = 9.0 Hz, 2H), 3.88 (s, 3H), 3.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.14 (d, JC-F = 249.7 Hz), 160.47, 157.89, 156.42, 150.83, 142.13, 130.80 (d, JC-F = 8.3 Hz), 129.34, 128.00, 126.13 (d, JC-F = 3.4 Hz), 124.35, 118.76, 118.01, 116.14 (d, JC-F = 21.7 Hz), 114.92, 112.92, 56.06, 55.80.
2-(4-chlorophenoxy)-3-(4-fluorophenyl)-6-methoxybenzo[b]thiophene 1-oxide (28b)
To an oven-dried flask charged with 4-chlorophenol in 10 ml DMF, CsCO3 (1.3 g, 4 mmol) was added in a portion. After 15 min, 27 (702 mg, 2 mmol) was added slowly. The reaction was heated to 80 °C until the consumption of starting material. The mixture was quenched by ice water and extracted with ethyl acetate and washed with water and brine. The crude material was then purified by flash chromatography (5%-40% ethyl acetate in hexane) to give 403 mg (yield, 50%) title compound. 1H NMR (400 MHz, CDCl3) δ 7.50 – 7.44 (m, 3H), 7.31 – 7.24 (m, 3H), 7.20 – 7.08 (m, 4H), 7.03 (dd, J = 8.5, 2.5 Hz, 1H), 3.91 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.29 (d, JC-F = 250.32 Hz), 160.82, 156.39, 155.68, 142.42, 130.72 (d, JC-F = 8.36 Hz), 130.10, 129.90, 129.35, 128.75, 125.76 (d, JC-F = 4Hz) , 124.73, 118.48, 118.18, 116.28 (d, JC-F = 21.8 Hz), 112.89, 56.10.
2-(4-fluorophenoxy)-3-(4-fluorophenyl)-6-methoxybenzo[b]thiophene 1-oxide (28c)
This compound was prepared by a procedure identical to the preparation of 28b (240 mg, yield 63%). 1H NMR (400 MHz, CDCl3) δ 7.49 – 7.44 (m, 2H), 7.43 (d, J = 2.4 Hz, 1H), 7.27 (d, J = 8.6 Hz, 1H), 7.18 – 7.07 (m, 4H), 7.04 – 6.91 (m, 3H), 3.88 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.20 (d, JC-F = 250.1 Hz), 160.67, 159.28 (d, JC-F = 242.7 Hz), 157.04, 152.95 (d, JC-F = 2.5 Hz), 142.22, 130.73 (d, JC-F = 8.3 Hz), 129.24, 128.92, 125.84 (d, JC-F = 3.5 Hz), 124.59, 118.72 (d, JC-F = 8.4 Hz), 118.10, 116.44 (d, JC-F = 24.2 Hz), 116.21 (d, JC-F = 22.3 Hz), 112.88, 56.07.
3-(4-fluorophenyl)-6-methoxy-2-(4-methoxyphenoxy)benzo[b]thiophene (29a)
This compound was prepared by a procedure identical to the preparation of 18 (1.2 g, white solid, yield 67%). 1H NMR (400 MHz, CDCl3) δ 7.56 – 7.48 (m, 3H), 7.20 (d, J = 2.4 Hz, 1H), 7.15 – 7.10 (m, 2H), 7.05 – 7.01 (m, 2H), 6.97 (dd, J = 8.8, 2.4 Hz, 1H), 6.85 – 6.77 (m, 2H), 3.86 (s, 3H), 3.78 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.19 (d, JC-F = 246.5 Hz), 157.42, 156.22, 153.10, 152.53, 134.57, 131.21 (d, JC-F = 8.0 Hz), 131.08, 129.05, 123.11, 120.80, 118.61, 115.71 (d, JC-F = 21.4 Hz), 114.80, 114.22, 106.03, 55.86, 55.82.
2-(4-chlorophenoxy)-3-(4-fluorophenyl)-6-methoxybenzo[b]thiophene (29b)
This compound was prepared by a procedure identical to the preparation of 18 (206 mg, yield 53%). 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 8.8 Hz, 1H), 7.49 – 7.43 (m, 2H), 7.26 – 7.21 (m, 3H), 7.11 (t, J = 8.8 Hz, 2H), 7.02 – 6.96 (m, 3H), 3.88 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.32 (d, JC-F = 247.2 Hz), 157.80, 157.28, 150.75, 135.01, 131.12 (d, JC-F = 8.1 Hz), 130.78, 129.75, 128.77, 128.62 (d, JC-F = 3.4 Hz), 123.50, 122.84, 118.08, 115.83 (d, JC-F = 21.5 Hz), 114.52, 105.98, 55.87.
2-(4-fluorophenoxy)-3-(4-fluorophenyl)-6-methoxybenzo[b]thiophene (29c)
This compound was prepared by a procedure identical to the preparation of 18 (340 mg, yield 83%). 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 8.8 Hz, 1H), 7.51 – 7.42 (m, 2H), 7.23 (d, J = 2.4 Hz, 1H), 7.12 (t, J = 8.7 Hz, 2H), 7.06 – 6.92 (m, 5H), 3.87 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.10 (d, JC-F = 247.0 Hz), 158.87 (d, JC-F = 242.2 Hz), 157.51, 154.46 (d, JC-F = 2.5 Hz), 151.58, 134.63, 130.99 (d, JC-F = 8.0 Hz), 130.71, 128.58 (d, JC-F = 3.3 Hz), 123.19, 121.90, 118.19 (d, JC-F = 8.3 Hz), 116.12 (d, JC-F = 23.6 Hz), 115.60 (d, JC-F = 21.5 Hz), 114.25, 105.83, 55.68.
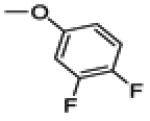
2-(3,4-difluorophenoxy)-3-(4-fluorophenyl)-6-methoxybenzo[b]thiophene (29d)
This compound was prepared by a procedure identical to the preparation of 18 (330 mg, yield 73%). 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 8.8 Hz, 1H), 7.47 – 7.44 (m, 2H), 7.24 (d, J = 2.1 Hz, 1H), 7.15 – 7.03 (m, 3H), 7.00 (dd, J = 8.9, 2.1 Hz, 1H), 6.92 – 6.83 (m, 1H), 6.77 (d, J = 9.1 Hz, 1H), 3.88 (s, 3H).
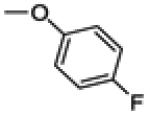
3-(4-fluorophenyl)-2-(4-hydroxyphenoxy)benzo[b]thiophen-6-ol (30a)
This compound was prepared by a procedure identical to the preparation of 11 (1.1 g, white powder, yield 75%).1H NMR (400 MHz, Acetone-d6) δ 7.62 – 7.57 (m, 2H), 7.46 (d, J = 8.7 Hz, 1H), 7.30 – 7.19 (m, 3H), 7.05 – 6.97 (m, 2H), 6.95 (dd, J = 8.7, 2.3 Hz, 1H), 6.86 – 6.78 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 162.57 (d, JC-F = 245.0 Hz). 155.67, 154.69, 153.54, 152.12, 135.01, 131.97 (d, JC-F = 8.1 Hz), 130.64, 130.04 (d, JC-F = 3.2 Hz), 123.50, 120.60, 119.42, 116.76, 116.03 (d, JC-F = 21.5 Hz), 115.35, 108.84. ESI-HRMS (m/z): [M-H]− calcd. for C20H12FO3S: 351.0491; observed, 351.0490.
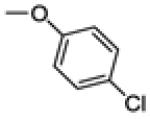
2-(4-chlorophenoxy)-3-(4-fluorophenyl)benzo[b]thiophen-6-ol (30b)
This compound was prepared by a procedure identical to the preparation of 11 (140 mg, white powder, yield 73%). 1H NMR (400 MHz, CDCl3) 7.51 (d, J = 8.7 Hz, 1H), 7.49 – 7.42 (m, 2H), 7.26 – 7.21 (m, 2H), 7.20 (d, J = 2.3 Hz, 1H), 7.11 (t, J = 8.7 Hz, 2H), 7.03 – 6.97 (m, 2H), 6.91 (dd, J = 8.7, 2.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 162.34 (d, JC-F = 247.3 Hz), 157.20, 153.43, 150.89, 135.05, 131.13 (d, JC-F = 8.1 Hz) , 129.76, 128.85, 128.55 (d, JC-F = 3Hz), 123.68, 122.71, 118.14, 115.84 (d, JC-F = 21.5 Hz), 114.61, 108.53. ESI-HRMS (m/z): [M+H]+ calcd. for C20H11ClFO2S: 369.0152; observed, 369.0144.
2-(4-fluorophenoxy)-3-(4-fluorophenyl)benzo[b]thiophen-6-ol (30c)
This compound was prepared by a procedure identical to the preparation of 11 (248 mg, white solid, yield 79%). 1H NMR (400 MHz, CDCl3) δ 7.56 – 7.41 (m, 3H), 7.18 (d, J = 2.3 Hz, 1H), 7.12 (t, J = 8.7 Hz, 2H), 7.06 – 6.92 (m, 4H), 6.90 (dd, J = 8.7, 2.4 Hz, 1H), 4.83 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 162.12 (d, JC-F = 247.1 Hz), 158.91 (d, JC-F = 242.4 Hz), 154.39, 153.15, 151.73, 134.68, 131.00, 130.99 (d, JC-F = 8.1 Hz), 128.53 (d, JC-F = 3.4 Hz), 123.36, 121.76, 118.27 (d, JC-F = 8.3 Hz), 116.14 (d, JC-F = 23.6 Hz), 115.61 (d, JC-F = 21.5 Hz), 114.37, 108.33. ESI-HRMS (m/z): [M-H]− calcd. for C20H11F2O2S: 353.0448; observed, 353.0451.
2-(3,4-difluorophenoxy)-3-(4-fluorophenyl)benzo[b]thiophen-6-ol (30d)
This compound was prepared by a procedure identical to the preparation of 11 (83 mg, yield 61%). 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.7 Hz, 1H), 7.45 (dd, J = 8.2, 5.9 Hz, 2H), 7.21 (d, J = 2.1 Hz, 1H), 7.17 – 7.00 (m, 3H), 6.97 – 6.83 (m, 2H), 6.79 – 6.75(m, 1H), 4.85 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 162.4 (d, JC-F = 247.4 Hz), 154.46 (dd, JC-F = 8.0, 2.4 Hz), 153.59, 150.60 (dd, JC-F = 249.9, 14.2 Hz), 150.59 (d, JC-F = 249.7 Hz), 146.91 (dd, JC-F = 244.0, 13.0 Hz), 135.07, 131.13 (d, JC-F = 8.1 Hz), 130.97, 128.41 (d, JC-F = 3.4 Hz), 123.81, 123.05, 117.61 (dd, JC-F = 18.9, 1.1 Hz), 115.9 (d, JC-F = 21.5 Hz), 114.73, 112.32 (dd, JC-F = 6.0, 3.8 Hz), 108.54, 106.7 (d, JC-F = 20.9 Hz). ESI-HRMS (m/z): [M-H]− calcd. for C20H10F3O2S: 371.0354; observed, 371.0346.
Supplementary Material
Acknowledgements
For financial support: NIH R01 CA188017 and R01 CA10259, the University of Illinois Cancer Center, UIC Center for Clinical and Translational Science Grant UL1RR029879. We would also like to thank Dr. Bernard D. Santarsiero from the Center for Pharmaceutical Biotechnology and the Department of Medicinal Chemistry and Pharmacognosy at UIC for his help with X-ray crystallography experiments.
Abbreviations
- ShERPAs
selective human estrogen receptor partial agonists
- ERα
estrogen receptor α
- ERβ
estrogen receptor β
- E2
estradiol
- SERM
selective estrogen receptor modulator
- DES
diethylstilbesterol
- LBD
ligand-binding domain
- BPAF
bisphenol AF
- PPA
polyphosphoric acid
- FP
fluorescence polarization
- ERE
estrogen response element
- RBAral
relative binding affinity to raloxifene
- RBA
relative binding affinity
- SRC3
steroid receptor coregulator-3
- AIB1
Amplified in Breast Cancer-1
- TR-FRET
Time-resolved fluorescence (or Förster) resonance energy transfer
- SEMs
selective estrogen mimics
- ChIP
Chromatin immunoprecipitation
- SA-Tb
streptavidin-terbium
- CMC
carboxymethyl cellulose
- TLC
thin layer chromatography
Footnotes
Supporting information
X-ray crystallography data for 29a, Effect of GPER antagonist, G15, on ShERPA luciferase activity.
Author contributions:
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116:561–570. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sommer S, Fuqua SA. Estrogen receptor and breast cancer. Semin Cancer Biol. 2001;11:339–352. doi: 10.1006/scbi.2001.0389. [DOI] [PubMed] [Google Scholar]
- 3.Jordan VC. Fourteenth Gaddum Memorial Lecture. A current view of tamoxifen for the treatment and prevention of breast cancer. Br J Pharmacol. 1993;110:507–517. doi: 10.1111/j.1476-5381.1993.tb13840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan VC. Tamoxifen: toxicities and drug resistance during the treatment and prevention of breast cancer. Annu Rev Pharmacol Toxicol. 1995;35:195–211. doi: 10.1146/annurev.pa.35.040195.001211. [DOI] [PubMed] [Google Scholar]
- 5.Ragaz J, Coldman A. Survival impact of adjuvant tamoxifen on competing causes of mortality in breast cancer survivors, with analysis of mortality from contralateral breast cancer, cardiovascular events, endometrial cancer, and thromboembolic episodes. J Clin Oncol. 1998;16:2018–2024. doi: 10.1200/JCO.1998.16.6.2018. [DOI] [PubMed] [Google Scholar]
- 6.Ingle J, Ahmann D, Green S, Edmonson J, Bisel H, Kvols L, Nichols W, Creagan E, Hahn R, Rubin J, Frytak S. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. New Engl J Med. 1981;304:16–21. doi: 10.1056/NEJM198101013040104. [DOI] [PubMed] [Google Scholar]
- 7.Ingle JN. Estrogen as therapy for breast cancer. Breast Cancer Res. 2002;4:133–136. doi: 10.1186/bcr436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schafer JM, Lee ES, Dardes RC, Bentrem D, O'Regan RM, De Los Reyes A, Jordan VC. Analysis of cross-resistance of the selective estrogen receptor modulators arzoxifene (LY353381) and LY117018 in tamoxifen-stimulated breast cancer xenografts. Clin Cancer Res. 2001;7:2505–12. [PubMed] [Google Scholar]
- 9.Roodi N, Bailey LR, Kao WY, Verrier CS, Yee CJ, Dupont WD, Parl FF. Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. J Natl Cancer Inst. 1995;87:446–51. doi: 10.1093/jnci/87.6.446. [DOI] [PubMed] [Google Scholar]
- 10.Jordan NJ, Gee JM, Barrow D, Wakeling AE, Nicholson RI. Increased constitutive activity of PKB/Akt in tamoxifen resistant breast cancer MCF-7 cells. Breast Cancer Res Treat. 2004;87:167–80. doi: 10.1023/B:BREA.0000041623.21338.47. [DOI] [PubMed] [Google Scholar]
- 11.Schiff R, Massarweh SA, Shou J, Bharwani L, Arpino G, Rimawi M, Osborne CK. Advanced concepts in estrogen receptor biology and breast cancer endocrine resistance: implicated role of growth factor signaling and estrogen receptor coregulators. Cancer Chemother Pharmacol. 2005;56(Suppl 1):10–20. doi: 10.1007/s00280-005-0108-2. [DOI] [PubMed] [Google Scholar]
- 12.Jordan VC, Lewis JS, Osipo C, Cheng D. The apoptotic action of estrogen following exhaustive antihormonal therapy: A new clinical treatment strategy. The Breast. 2005;14:624–630. doi: 10.1016/j.breast.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 13.Craig Jordan V, Lewis-Wambi J, Kim H, Cunliffe H, Ariazi E, Sharma CG, Shupp HA, Swaby R. Exploiting the apoptotic actions of oestrogen to reverse antihormonal drug resistance in oestrogen receptor positive breast cancer patients. Breast. 2007;16:S105–S113. doi: 10.1016/j.breast.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chalasani P, Stopeck A, Clarke K, Livingston R. A Pilot Study of Estradiol Followed by Exemestane for Reversing Endocrine Resistance in Postmenopausal Women With Hormone Receptor-Positive Metastatic Breast Cancer. Oncologist. 2014;19:1127–1128. doi: 10.1634/theoncologist.2014-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwase H, Yamamoto Y, Yamamoto-Ibusuki M, Murakami KI, Okumura Y, Tomita S, Inao T, Honda Y, Omoto Y, Iyama KI. Ethinylestradiol is beneficial for postmenopausal patients with heavily pre-treated metastatic breast cancer after prior aromatase inhibitor treatment: a prospective study. Br J Cancer. 2013;109:1537–1542. doi: 10.1038/bjc.2013.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, Dickler MN, Silverman P, Fleming GF, Kommareddy A, Jamalabadi-Majidi S, Crowder R, Siegel BA. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA. 2009;302:774–780. doi: 10.1001/jama.2009.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 18.Merrell KW, Crofts JD, Smith RL, Sin JH, Kmetzsch KE, Merrell A, Miguel RO, Candelaria NR, Lin CY. Differential recruitment of nuclear receptor coregulators in ligand-dependent transcriptional repression by estrogen receptor-alpha. Oncogene. 2011;30:1608–14. doi: 10.1038/onc.2010.528. [DOI] [PubMed] [Google Scholar]
- 19.Madak-Erdogan Z, Charn TH, Jiang Y, Liu ET, Katzenellenbogen JA, Katzenellenbogen BS. Integrative genomics of gene and metabolic regulation by estrogen receptors alpha and beta, and their coregulators. Mol Syst Biol. 2013;9:676. doi: 10.1038/msb.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maximov PY, Lee TM, Jordan VC. The Discovery and Development of Selective Estrogen Receptor Modulators (SERMs) for Clinical Practice. Curr Clin Pharmacol. 2013;8:135–55. doi: 10.2174/1574884711308020006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sporn MB. Arzoxifene: a promising new selective estrogen receptor modulator for clinical chemoprevention of breast cancer. Clin. Cancer. Res. 2004;10:5313–5315. doi: 10.1158/1078-0432.CCR-04-1377. [DOI] [PubMed] [Google Scholar]
- 22.Baselga J, Llombart-Cussac A, Bellet M, Guillem-Porta V, Enas N, Krejcy K, Carrasco E, Kayitalire L, Kuta M, Lluch A, Vodvarka P, Kerbrat P, Namer M, Petruzelka L. Randomized, double-blind, multicenter trial comparing two doses of arzoxifene (LY353381) in hormone-sensitive advanced or metastatic breast cancer patients. Ann. Oncol. 2003;14:1383–1390. doi: 10.1093/annonc/mdg368. [DOI] [PubMed] [Google Scholar]
- 23.Chan S. Arzoxifene in breast cancer. Eur. J. Cancer. 2002;38(Suppl 6):S55–S56. doi: 10.1016/s0959-8049(02)00286-1. [DOI] [PubMed] [Google Scholar]
- 24.Dardes RC, Bentrem D, O'Regan RM, Schafer JM, Jordan VC. Effects of the new selective estrogen receptor modulator LY353381.HCl (Arzoxifene) on human endometrial cancer growth in athymic mice. Clin. Cancer Res. 2001;7:4149–4155. [PubMed] [Google Scholar]
- 25.McMeekin DS, Gordon A, Fowler J, Melemed A, Buller R, Burke T, Bloss J, Sabbatini P. A phase II trial of arzoxifene, a selective estrogen response modulator, in patients with recurrent or advanced endometrial cancer. Gynecol. Oncol. 2003;90:64–69. doi: 10.1016/s0090-8258(03)00203-8. [DOI] [PubMed] [Google Scholar]
- 26.Suh N, Glasebrook AL, Palkowitz AD, Bryant HU, Burris LL, Starling JJ, Pearce HL, Williams C, Peer C, Wang Y, Sporn MB. Arzoxifene, a new selective estrogen receptor modulator for chemoprevention of experimental breast cancer. Cancer Res. 2001;61:8412–8415. [PubMed] [Google Scholar]
- 27.Taylor HS. Approaching the ideal selective estrogen receptor modulator for prevention and treatment of postmenopausal osteoporosis. Formulary. 2010;45:52–61. [Google Scholar]
- 28.Pickar JH. The endometrium - from estrogens alone to TSEC's. Climacteric. 2009;12:463–477. doi: 10.3109/13697130903042790. [DOI] [PubMed] [Google Scholar]
- 29.Lobo RA. Evaluation of bazedoxifene/conjugated estrogens for the treatment of menopausal symptoms and effects on metabolic parameters and overall safety profile. Fertil and Steril. 2009;92:1025–1038. doi: 10.1016/j.fertnstert.2009.03.113. [DOI] [PubMed] [Google Scholar]
- 30.Toader V, Xu X, Nicolescu A, Yu L, Bolton JL, Thatcher GRJ. Nitrosation, nitration, and autoxidation of the selective estrogen receptor modulator raloxifene by nitric oxide, peroxynitrite, and reactive nitrogen/oxygen species. Chem Res Toxicol. 2003;16:1264–76. doi: 10.1021/tx025641h. [DOI] [PubMed] [Google Scholar]
- 31.Liu J, Li Q, Yang X, van Breemen RB, Bolton JL, Thatcher GRJ. Analysis of protein covalent modification by xenobiotics using a covert oxidatively activated tag: raloxifene proof-of-principle study. Chem Res Toxicol. 2005;18:1485–96. doi: 10.1021/tx0501738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu H, Liu J, van Breemen RB, Thatcher GRJ, Bolton JL. Bioactivation of the selective estrogen receptor modulator desmethylated arzoxifene to quinoids: 4'-fluoro substitution prevents quinoid formation. Chem Res Toxicol. 2005;18:162–73. doi: 10.1021/tx049776u. [DOI] [PubMed] [Google Scholar]
- 33.Bolton JL, Yu L, Thatcher GRJ. Quinoids formed from estrogens and antiestrogens. Methods Enzymol. 2004;378:110–123. doi: 10.1016/S0076-6879(04)78006-4. [DOI] [PubMed] [Google Scholar]
- 34.Liu H, Bolton JL, Thatcher GRJ. Chemical modification modulates estrogenic activity, oxidative reactivity, and metabolic stability in 4'F-DMA, a new benzothiophene selective estrogen receptor modulator. Chem Res Toxicol. 2006;19:779–87. doi: 10.1021/tx050326r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu B, Dietz BM, Dunlap T, Kastrati I, Lantvit DD, Overk CR, Yao P, Qin Z, Bolton JL, Thatcher GRJ. Structural modulation of reactivity/activity in design of improved benzothiophene selective estrogen receptor modulators: induction of chemopreventive mechanisms. Mol Cancer Ther. 2007;6:2418–28. doi: 10.1158/1535-7163.MCT-07-0268. [DOI] [PubMed] [Google Scholar]
- 36.Overk CR, Peng KW, Asghodom RT, Kastrati I, Lantvit DD, Qin Z, Frasor J, Bolton JL, Thatcher GRJ. Structure-activity relationships for a family of benzothiophene selective estrogen receptor modulators including raloxifene and arzoxifene. ChemMedChem. 2007;2:1520–6. doi: 10.1002/cmdc.200700104. [DOI] [PubMed] [Google Scholar]
- 37.Liu H, Qin Z, Thatcher GRJ, Bolton JL. Uterine peroxidase-catalyzed formation of diquinone methides from the selective estrogen receptor modulators raloxifene and desmethylated arzoxifene. Chem Res Toxicol. 2007;20:1676–84. doi: 10.1021/tx7001367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin Z, Kastrati I, Ashgodom RT, Lantvit DD, Overk CR, Choi Y, van Breemen RB, Bolton JL, Thatcher GRJ. Structural modulation of oxidative metabolism in design of improved benzothiophene selective estrogen receptor modulators. Drug Metab Dispos. 2009;37:161–9. doi: 10.1124/dmd.108.023408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kastrati I, Edirisinghe PD, Hemachandra LP, Chandrasena ER, Choi J, Wang YT, Bolton JL, Thatcher GR. Raloxifene and desmethylarzoxifene block estrogen-induced malignant transformation of human breast epithelial cells. PLoS One. 2011;6:e27876. doi: 10.1371/journal.pone.0027876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel HK, Siklos MI, Abdelkarim H, Mendonca EL, Vaidya A, Petukhov PA, Thatcher GR. A chimeric SERM-Histone deacetylase inhibitor approach to breast cancer therapy. ChemMedChem. 2014;9:602–13. doi: 10.1002/cmdc.201300270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hemachandra LP, Patel H, Chandrasena RE, Choi J, Piyankarage SC, Wang S, Wang Y, Thayer EN, Scism RA, Michalsen BT, Xiong R, Siklos MI, Bolton JL, Thatcher GR. SERMs attenuate estrogen-induced malignant transformation of human mammary epithelial cells by upregulating detoxification of oxidative metabolites. Cancer Prev Res (Phila) 2014;7:505–15. doi: 10.1158/1940-6207.CAPR-13-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molloy ME, White BE, Gherezghiher T, Michalsen BT, Xiong R, Patel H, Zhao H, Maximov PY, Jordan VC, Thatcher GR, Tonetti DA. Novel selective estrogen mimics for the treatment of tamoxifen-resistant breast cancer. Mol Cancer Ther. 2014;13:2515–26. doi: 10.1158/1535-7163.MCT-14-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katzenellenbogen J. A. The 2010 Philip S. Portoghese Medicinal Chemistry Lectureship: addressing the "core issue" in the design of estrogen receptor ligands. J Med Chem. 2011;54:5271–82. doi: 10.1021/jm200801h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids. 1997;62:268–303. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 45.Nettles KW, Bruning JB, Gil G, O'Neill EE, Nowak J, Guo Y, Kim Y, DeSombre ER, Dilis R, Hanson RN, Joachimiak A, Greene GL. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep. 2007;8:563–568. doi: 10.1038/sj.embor.7400963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–8. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 47.Dang ZC, Audinot V, Papapoulos SE, Boutin JA, Lowik CW. Peroxisome proliferator-activated receptor gamma (PPARgamma ) as a molecular target for the soy phytoestrogen genistein. J Biol Chem. 2003;278:962–967. doi: 10.1074/jbc.M209483200. [DOI] [PubMed] [Google Scholar]
- 48.Hsieh CY, Santell RC, Haslam SZ, Helferich WG. Estrogenic effects of genistein on the growth of estrogen receptor-positive human breast cancer (MCF-7) cells in vitro and in vivo. Cancer Res. 1998;58:3833–8. [PubMed] [Google Scholar]
- 49.Rao A, Coan A, Welsh JE, Barclay WW, Koumenis C, Cramer SD. Vitamin D receptor and p21/WAF1 are targets of genistein and 1,25-dihydroxyvitamin D3 in human prostate cancer cells. Cancer Res. 2004;64:2143–7. doi: 10.1158/0008-5472.can-03-3480. [DOI] [PubMed] [Google Scholar]
- 50.Min J, Wang P, Srinivasan S, Nwachukwu JC, Guo P, Huang M, Carlson KE, Katzenellenbogen JA, Nettles KW, Zhou HB. Thiophene-core estrogen receptor ligands having superagonist activity. J Med Chem. 2013;56:3346–66. doi: 10.1021/jm400157e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang P, Min J, Nwachukwu JC, Cavett V, Carlson KE, Guo P, Zhu M, Zheng Y, Dong C, Katzenellenbogen JA, Nettles KW, Zhou HB. Identification and structure-activity relationships of a novel series of estrogen receptor ligands based on 7-thiabicyclo[2.2.1]hept-2-ene-7-oxide. J Med Chem. 2012;55:2324–41. doi: 10.1021/jm201556r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Minutolo F, Bellini R, Bertini S, Carboni I, Lapucci A, Pistolesi L, Prota G, Rapposelli S, Solati F, Tuccinardi T, Martinelli A, Stossi F, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA, Macchia M. Monoaryl-substituted salicylaldoximes as ligands for estrogen receptor beta. J Med Chem. 2008;51:1344–51. doi: 10.1021/jm701396g. [DOI] [PubMed] [Google Scholar]
- 53.Hanson RN, Hua E, Hendricks JA, Labaree D, Hochberg RB. Synthesis and evaluation of 11beta-(4-substituted phenyl) estradiol analogs: transition from estrogen receptor agonists to antagonists. Bioorg Med Chem. 2012;20:3768–3780. doi: 10.1016/j.bmc.2012.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Delfosse V, Grimaldi M, Pons JL, Boulahtouf A, le Maire A, Cavailles V, Labesse G, Bourguet W, Balaguer P. Structural and mechanistic insights into bisphenols action provide guidelines for risk assessment and discovery of bisphenol A substitutes. Proc Natl Acad Sci U S A. 2012;109:14930–14935. doi: 10.1073/pnas.1203574109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steffan RJ, Matelan E, Ashwell MA, Moore WJ, Solvibile WR, Trybulski E, Chadwick CC, Chippari S, Kenney T, Eckert A, Borges-Marcucci L, Keith JC, Xu Z, Mosyak L, Harnish DC. Synthesis and activity of substituted 4-(indazol-3-yl)phenols as pathway-selective estrogen receptor ligands useful in the treatment of rheumatoid arthritis. J Med Chem. 2004;47:6435–8. doi: 10.1021/jm049194+. [DOI] [PubMed] [Google Scholar]
- 56.Bruning JB, Parent AA, Gil G, Zhao M, Nowak J, Pace MC, Smith CL, Afonine PV, Adams PD, Katzenellenbogen JA, Nettles KW. Coupling of receptor conformation and ligand orientation determine graded activity. Nat Chem Biol. 2010;6:837–43. doi: 10.1038/nchembio.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones CD, Jevnikar MG, Pike AJ, Peters MK, Black LJ, Thompson AR, Falcone JF, Clemens JA. Antiestrogens .2. Structure Activity Studies in a Series of 3-Aroyl-2-Arylbenzo[B]Thiophene Derivatives Leading to [6-Hydroxy-2-(4-Hydroxyphenyl)Benzo[B]Thien-3-Yl][4-[2-(1-Piperidinyl)Ethoxy]-Phenyl]Methanone Hydrochloride (Ly156758), a Remarkably Effective Estrogen Antagonist with Only Minimal Intrinsic Estrogenicity. J Med Chem. 1984;27:1057–1066. doi: 10.1021/jm00374a021. [DOI] [PubMed] [Google Scholar]
- 58.Hillver SE, Bjork L, Li YL, Svensson B, Ross S, Anden NE, Hacksell U. (S)-5-fluoro-8-hydroxy-2-(dipropylamino)tetralin: a putative 5-HT1A-receptor antagonist. J Med Chem. 1990;33:1541–1544. doi: 10.1021/jm00168a002. [DOI] [PubMed] [Google Scholar]
- 59.Krapcho AP, Menta E, Oliva A, Di Domenico R, Fiocchi L, Maresch ME, Gallagher CE, Hacker MP, Beggiolin G, Giuliani FC, Pezzoni G, Spinelli S. Synthesis and antitumor evaluation of 2,5-disubstituted-indazolo[4, 3-gh]isoquinolin-6(2H)-ones (9-aza-anthrapyrazoles) J Med Chem. 1998;41:5429–5444. doi: 10.1021/jm9804432. [DOI] [PubMed] [Google Scholar]
- 60.Liu XF, Bagchi MK. Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J Biol Chem. 2004;279:15050–8. doi: 10.1074/jbc.M311932200. [DOI] [PubMed] [Google Scholar]
- 61.Hayakawa T, Nakayama J. Physiological roles of class I HDAC complex and histone demethylase. J Biomed Biotechnol. 2011;2011:129383–129392. doi: 10.1155/2011/129383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 63.Onate SA, Tsai SY, Tsai MJ, Omalley BW. Sequence and Characterization of a Coactivator for the Steroid-Hormone Receptor Superfamily. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 64.Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 65.Gunther JR, Du YH, Rhoden E, Lewis I, Revennaugh B, Moore TW, Kim SH, Dingledine R, Fu HA, Katzenellenbogen JA. A Set of Time-Resolved Fluorescence Resonance Energy Transfer Assays for the Discovery of Inhibitors of Estrogen Receptor-Coactivator Binding. J Biomol Screen. 2009;14:181–193. doi: 10.1177/1087057108329349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jeyakumar M, Carlson KE, Gunther JR, Katzenellenbogen JA. Exploration of dimensions of estrogen potency: parsing ligand binding and coactivator binding affinities. J Biol Chem. 2011;286:12971–82. doi: 10.1074/jbc.M110.205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ariazi EA, Cunliffe HE, Lewis-Wambi JS, Slifker MJ, Willis AL, Ramos P, Tapia C, Kim HR, Yerrum S, Sharma CG, Nicolas E, Balagurunathan Y, Ross EA, Jordan VC. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc Natl Acad Sci U S A. 2011;108:18879–86. doi: 10.1073/pnas.1115188108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tonetti DA, Chisamore MJ, Grdina W, Schurz H, Jordan VC. Stable transfection of protein kinase C alpha cDNA in hormone-dependent breast cancer cell lines. Br J Cancer. 2000;83:782–91. doi: 10.1054/bjoc.2000.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jordan VC, Lewis-Wambi JS, Patel RR, Kim H, Ariazi EA. New hypotheses and opportunities in endocrine therapy: amplification of oestrogen-induced apoptosis. Breast. 2009;18:S10–S17. doi: 10.1016/S0960-9776(09)70266-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewis-Wambi JS, Jordan VC. Estrogen regulation of apoptosis: how can one hormone stimulate and inhibit? Breast Cancer Res. 2009;11:206. doi: 10.1186/bcr2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grese TA, Cho S, Finley DR, Godfrey AG, Jones CD, Lugar CW, 3rd, Martin MJ, Matsumoto K, Pennington LD, Winter MA, Adrian MD, Cole HW, Magee DE, Phillips DL, Rowley ER, Short LL, Glasebrook AL, Bryant HU. Structure-activity relationships of selective estrogen receptor modulators: modifications to the 2-arylbenzothiophene core of raloxifene. J Med Chem. 1997;40:146–67. doi: 10.1021/jm9606352. [DOI] [PubMed] [Google Scholar]
- 72.Dutertre M, Smith CL. Molecular mechanisms of selective estrogen receptor modulator (SERM) action. J Pharmacol Exp Ther. 2000;295:431–437. [PubMed] [Google Scholar]
- 73.Johnson AB, O'Malley BW. Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol Cell Endocrinol. 2012;348:430–439. doi: 10.1016/j.mce.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dai SY, Burris TP, Dodge JA, Montrose-Rafizadeh C, Wang Y, Pascal BD, Chalmers MJ, Griffin PR. Unique ligand binding patterns between estrogen receptor alpha and beta revealed by hydrogen-deuterium exchange. Biochemistry. 2009;48:9668–76. doi: 10.1021/bi901149t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Katzenellenbogen JA, Johnson HJ, Jr., Myers HN. Photoaffinity labels for estrogen binding proteins of rat uterus. Biochemistry. 1973;12:4085–4092. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]
- 76.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry. 1997;36:14897–905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 77.Chisamore MJ, Ahmed Y, Bentrem DJ, Jordan VC, Tonetti DA. Novel antitumor effect of estradiol in athymic mice injected with a T47D breast cancer cell line overexpressing protein kinase Calpha. Clin Cancer Res. 2001;7:3156–3165. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.