

Abstract
The adequate procurement and preservation of high-quality tissue specimens from patients with melanoma is a critical clinical issue as patients’ tumor samples are now used not only for pathological diagnosis but are also necessary to determine the molecular signature of the tumor to stratify patients who may benefit from targeted melanoma therapy. Tissue resources available for physicians and investigators include formalin-fixed paraffin-embedded (FFPE) tissue and frozen tissue, either preserved in optimal cutting temperature (OCT) media or snap frozen. Properly preserved tissue may be used to evaluate melanoma biomarkers by immunohistochemistry (IHC) with tissue microarray (TMA) technology, to perform genetic and genomic analyses, and for other types of translational research in melanoma.
Keywords: Frozen section, Prognostic markers, Therapy targets, Eligibility criteria, Tissue preservation, Tissue microarray analysis
1 Introduction
Recent insights into the genetic aberrations and other molecular underpinnings of melanoma, in addition to an improved understanding of the role of the immune system, have ushered in a new and exciting era of rapidly evolving targeted [1, 2] and immune-based treatments for this disease. The use of tissue in contemporary translational melanoma investigation is essential to identify biomarkers that improve treatments and personalize the management of patients [3]. Examples of overarching themes in this arena include tissue-based research initiatives that: (1) enhance prognostic assessment, (2) predict response to a specific therapy, (3) predict resistance to a specific therapy, or (4) elucidate mechanisms of resistance—all of which are critical to improve treatments for patients with melanoma. In this chapter, we focus our attention on the tissue resources that may be employed for such efforts.
1.1 Use of Tissue in the Era of Targeted Therapy in Melanoma
Focused profiling has demonstrated that oncogenic driver mutations in BRAF and NRAS are present in ~70 % of patients with cutaneous melanoma, while other genetic events have been identified in other melanoma subtypes (i.e., KIT, GNαQ, and GNα11). Vemurafenib, a small molecule inhibitor that potently and selectively inhibits the most common mutation that occurs in melanoma (BRAF V600E), improved clinical responses rates, progression-free survival, and overall survival compared to chemotherapy in a randomized phase III clinical trial of metastatic melanoma patients whose tumors harbored this mutation [4]. On the other hand, vemurafenib, and other mutant-selective BRAF inhibitors (i.e., dabrafenib), appear to cause increased growth of melanomas that do not have activating BRAF mutations [5–7]. Thus, the currently approved use of vemurafenib is limited to patients with mutations that result in amino acid substitutions at the V600 residue of the BRAF protein. As a result of these findings, as well as the demonstration of activity of other targeted therapies in molecularly defined subpopulations (e.g., imatinib in metastatic melanoma patients with KIT mutations), molecular testing is becoming part of the standard evaluation for any patient with metastatic disease, as well as in patients with unresectable, clinically localized disease [8]. As molecular testing rapidly evolves from testing of single genes to panel-based testing using massively parallel sequencing, the need for high-quality tissue specimens has become a critical clinical issue. Molecular testing platforms vary significantly in their sensitivity for mutation detection; thus, characterization of specimens being analyzed (i.e., percent tumor cells, presence of necrosis, amount of melanin pigment) is critical to the interpretation of testing results. The type of specimens collected, and the amount of DNA and/or RNA isolated from them, is crucial to determine what types of molecular testing can be done, based on the requirements of the different testing platforms.
In addition to clinical testing implications, the development of the BRAF inhibitors has demonstrated in multiple ways the essential role played by tissue-based research. Although there are multiple examples demonstrating the critical nature of pretreatment molecular testing for patient selection, generally less attention is paid to the pharmacodynamic effects of agents. However, in the initial study of imatinib in patients with metastatic melanoma, biopsies were taken both pretreatment and again 2 weeks later. This study demonstrated a direct effect of imatinib on the pattern of expression of five known imatinib targets: ABL, BCR-ABL, KIT, and PDGF receptors alpha and beta [9]. The viable tumor in the post-treatment biopsies showed a marked decrease in the percentage and intensity of expression of the targets, thus suggesting that the “sensitive” melanoma cells were killed by the therapy, whereas the “resistant” cells were able to survive, similar to the effects of antibiotics on bacteria. In a more recent study, a phase I trial of vemurafenib, a cohort of 15 patients also agreed to allow tumor biopsies to be performed both pretreatment and after approximately 1–2 weeks of therapy. The specimens were analyzed for the expression of phosphorylated (active) ERK (P-ERK), a downstream effector of the BRAF-MEK-ERK pathway that vemurafenib is designed to inhibit. This analysis demonstrated a nearly linear relationship in these patients between the degree of P-ERK inhibition that was achieved with vemurafenib treatment, and the degree of tumor shrinkage that was achieved clinically [10]. Thus, this analysis demonstrated not only the importance of selecting the correct patients, but also that the dose of medication that the patients received and that reached the tumor was critical to the antitumor activity, which has significant implications for the management and adjustment of dosing in patients.
Although vemurafenib and other structurally unrelated V600 mutant-selective BRAF inhibitors achieve clinical responses in ~50 % of patients, the duration of these responses can be quite transient, lasting on average 5–6 months [4, 11]. A growing number of studies have been reported describing the molecular changes that are detected in samples of tumor tissue at the time of disease progression. Although there is no universal agreement, these studies have reported that many of the progressing lesions demonstrate reactivation of the BRAF-MEK-ERK pathway, as exemplified by recovery of P-ERK expression [12, 13]. Tissue-based analyses have demonstrated multiple mechanisms by which this reactivation can occur, including alternative splicing of BRAF, copy-number gain of BRAF, and acquired mutations in NRAS or MEK1 [14–17]. Laboratory experiments performed on cell lines established from these progressing lesions demonstrated that the resistance to the selective BRAF inhibitors was maintained in vitro, allowing for functional testing of various strategies to overcome this resistance. One strategy that emerged from these studies was to combine a selective BRAF inhibitor with an MEK inhibitor. A randomized phase II study has now demonstrated that combined treatment with trametinib (an MEK inhibitor) and dabrafenib (a BRAF inhibitor) compared to treatment with dabrafenib alone in BRAF-mutant melanoma patients who have not previously been treated with a BRAF inhibitor results in significant improvements in clinical response rate (76 vs. 54 %), progression-free survival (median 9.4 vs. 5.8 months), and the duration of clinical responses (median 10.5 vs. 5.6 months) [18]. This combination also achieved clinical responses in ~20 % of patients who had previously developed disease progression on selective BRAF inhibitors [19]. Analyses of patient-derived specimens are ongoing to better understand the heterogeneity of the responses to this combination regimen and to characterize resistance mechanisms in order to develop other rational therapeutic approaches [2, 20].
In summary, the development of the BRAF inhibitors demonstrates the marked and rapid clinical impact that tissue-based research can have on patient care and outcomes. As the pattern of metastases in melanoma often allows for repeated biopsies to be performed safely, there is a tremendous opportunity to use tissue-based research approaches to rapidly develop rational, more effective treatments for patients. However, the types and quality of the research that can be performed critically depend upon the amount and quality of the tissue samples that are obtained for research. The amount of DNA needed for genetic and genomic analyses can vary widely depending on the type. Additional DNA always is needed beyond the amount for the actual technique for quality control purposes, so it is crucial to account for that extra amount in any planned investigation. It also is important to recognize that although technologies, such as Ion Torrent (Life Technologies™), can be used to perform massively parallel sequencing on as little as 10 ng of DNA, they can be particularly error-prone when using low quantities of DNA. The data obtained from massively parallel sequencing are more reliable with large amounts of DNA, usually 250–500 ng. For techniques such as array-based comparative genomic hybridization or whole exome sequencing, 1 μg (1,000 ng) of DNA is generally needed. Massively parallel sequencing has the advantage that it is more sensitive than other techniques, generally requiring approximately 20 % tumor to detect mutations, so that macrodissection may not need to be done, depending on specific tumor architecture. Such endeavors therefore require careful planning before tissue specimens are collected to ensure that the methods used will yield analytes that are both sufficient and appropriate for the planned analyses. Considerations include the sensitivity of the analytical platform to determine the level of tumor purity that will be required, as well as the amount of material needed for different assays. Thus, translational research using tissue specimens critically depends upon close communication and collaboration between scientists and physicians to maximize opportunities to advance our understanding of melanoma tumor biology and care of patients [2, 20].
1.2 Fresh Frozen Tissue for Clinical Use
Tissue samples, both tumor and normal, may be obtained in various ways: fine needle aspiration (FNA), image-guided core needle biopsy, punch biopsy, skin ellipse, and other surgical excision specimens. It should be stressed that core biopsy—whether collected as freshly frozen or FFPE—is superior to fine needle aspiration (FNA), since it usually provides more tissue for analysis (see Notes 1 and 2) [21]. FNAs can be used for targeted mutation analysis, specifically BRAF V600E mutations, but are currently not particularly useful for larger research studies. Preferred sizes for punch biopsies of cutaneous and subcutaneous lesions are 4–6 mm when feasible. A single punch biopsy, skin ellipse, or other surgical excision specimen-containing high-quality tumor is generally sufficient for most targeted research purposes at a single point in time.
Of great importance is the establishment of standard operating procedures (SOPs) to optimally archive biospecimens for research use, a principal goal of which is to maximize tissue preservation for contemporary or possible future molecular-based translational studies that include extraction of DNA, RNA, and protein (the latter of which is particularly important for assessment of phosphoprotein expression and cell-signal pathways). Strategies to collect and archive FFPE tissue are well documented elsewhere [22, 23]. FFPE tissue, although generally the most widely collected and most commonly employed for clinical DNA-based mutational testing, will not be further discussed since it is such a common fixture in all CLIA (Clinical Laboratory Improvement Amendments) certified clinical pathology laboratories.
Optimal Cutting Temperature (OCT) compound is especially useful for embedding frozen tissue, and since the tissue is not fixed in formalin when this procedure is used, it improves RNA and DNA recovery and quality. A key advantage of the OCT approach is that it facilitates histological evaluation of all samples prior to molecular analyte preparation. A frozen section H&E obtained from an OCT-embedded specimen, albeit of lesser quality than that from permanent (i.e., FFPE) H&E sections, enables histological assessment pertaining to the presence, quality and quantity of tumor. This consideration is particularly important since fibrosis, necrosis, hemorrhage, melanin pigmentation, and significant lymphocytic infiltrate can all “contaminate” melanoma tumor specimens and may adversely impact the yield and quality of analytes (e.g., secondary to fibrosis, necrosis, hemorrhage) or the extent to which the expression profiles are related to the tumor (e.g., from heavy lymphocytic infiltrates or melanin pigmentation). OCT also prevents tissue from desiccation and crumbling, acts as an insulator from thermal change and limits ice crystal formation (see Note 3). If tissue samples are obtained by core (punch or needle) biopsies, each core should be embedded in OCT as separate blocks.
Controlled snap freezing (in isopentane (2-methylbutane) prechilled in liquid nitrogen) is a viable alternative when OCT processing is not feasible, and is a preferred approach by some investigators. During surgery, fresh tissue may be acquired by the transfer of the surgical specimen from the operating room to the pathology department in a tumor container without formalin fixative. Once it arrives in the pathology department, the tissue specimen may be sectioned (tissue cores may also be obtained with a punch biopsy instrument) and snap frozen; this processing should be performed as a coordinated effort with the clinical/research pathology team so that diagnostic material is not compromised. Alternatively, this process can also sometimes be coordinated so that it is performed in the operating room once the specimen has been removed, which may achieve further reduction in ischemic time. If necessary, to obtain H&E sections of the specimen, it is possible to later transfer the frozen tissue to OCT without thawing the tissue and then obtain frozen sections.
Fresh-tissue samples should be frozen as soon as possible. If they cannot be frozen immediately, the tissue should be refrigerated and frozen as soon as possible. Time is critical as the longer a specimen is at room temperature the greater the opportunity for RNAses and proteases to degrade their respective macromolecules, especially important if the downstream application will depend on RNA or proteins. RNA integrity is reduced in direct proportion to delayed tissue processing in freshly harvested tissues [24]. Ischemia time affects the expression of proteins, which is more enhanced at room temperature and mainly leads to a decrease of signal intensity of a resected specimen [25]. Snap-frozen tissue samples should be transferred to either liquid nitrogen (preferred) or to a −80 °C freezer. Tissue samples processed in OCT should be transferred as soon as possible to a −80 °C freezer. Frozen tissue samples should be transported on dry ice in a closed but non-sealed Styrofoam or other ultralow temperature container (see Note 4). Frozen tissue samples should be securely stored long-term either in liquid nitrogen or in at −80 °C or colder. If future uses of the tissue are unknown, storing the tissue in the vapor phase of liquid nitrogen will help to ensure long-term viability. Storage equipment may include: small (2″ × 3″) plastic, zip-top bags; mega-cassettes (for example, with each tissue or cryomold wrapped in aluminum foil); cryoboxes; and plastic or metal storage freezer racks (e.g., for cryovial and cryomold storage). The freezer should be on an electrical emergency power line and be alarmed and centrally monitored. We use both a digital (usually wired to unit) and an analog (a thermometer probe in the interior that is independently connected to the alarm system) alarm approach for operational redundancy. Time intervals between collection and freezing should be documented, and is particularly helpful for troubleshooting since preservation of protein and other molecular analyte expression may vary over time.
1.3 Labeling and Quality Assurance for Fresh Frozen Samples
Accurate labeling is essential. Ideally, labels should include a specimen ID that is guaranteed to be unique across all samples and linked to a complete sample description by electronic means—specific procedures for labeling specimens which may include use of unique sample IDs and/or barcodes should be standardized and clearly be defined in the protocol to ensure accuracy and uniformity. The label must not include patient-identifying information and must be compliant with the Health Insurance Portability and Accountability Act (HIPAA) and any institutional and other State and Federal regulations. The type of label media, and ink used must be selected with care for appropriate resistance to chemicals, temperature extremes, storage media, and transport conditions.
To assess the quality of collected frozen tissue, an H&E-stained, cryostat section should be prepared from the frozen OCT block to confirm the presence of tumor and identify potential contaminants and/or other potential confounding material. Histological features that should ideally be recorded include assessment of percent tumor cells, preservation of tumor morphology, presence of any confounding material, such as necrotic, fibrotic, and/or hemorrhagic regions, amount of melanin pigment, and/or inflammatory infiltrate. The H&E slide may be used as a guide to isolate or “macrodissect” desirable portions of the sample for molecular analyses (i.e., viable tumor and adjacent normal tissue) and remove areas that may confound subsequent analyses and/or analyte preparation (e.g., areas of fibrosis, necrosis, hemorrhage, and excessive melanin pigment) (see Fig. 1).
Fig. 1.
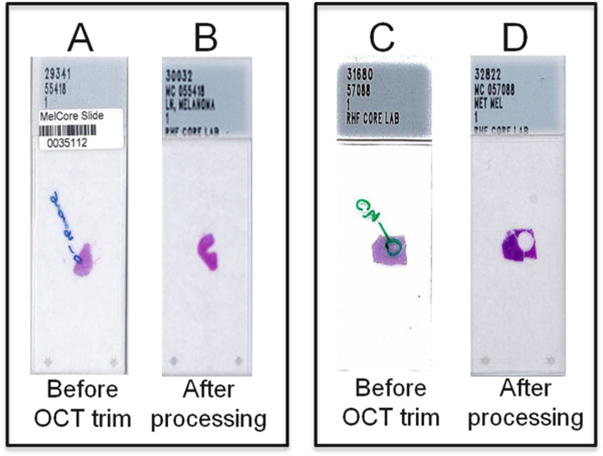
H&E-based “trimming” of OCT specimens. Examples of H&E-stained OCT tumor sections before (A and C) and following (B and D) trimming of OCT blocks to maximize tumor cell nuclei and to remove areas of necrosis. Note that circled areas (A and C) were identified for trimming
1.4 Tissue Microarray Technology
Tissue microarrays (TMAs) offer an efficient, effective laboratory tool to analyze dozens of formalin-fixed, paraffin-embedded (FFPE) tissue samples on a single microscope slide and provide a very valuable research tool for immunohistochemical (IHC) screening of known or putative cancer biomarkers. TMAs are assembled by obtaining tissue cores containing 0.6–2.0 mm diameter samples of patient tumors from donor FFPE tissue blocks and placing these cores in predrilled holes of recipient (master) paraffin blocks. These master blocks are arrayed to a grid that is linked to pathological and clinical data (see Fig. 2) [26]. Manual or automated tissue arrayers provide precise, reproducible quality transfer of tissue cores or histospots from donor blocks to the master TMA blocks. One TMA block may provide up to 500 or more (depending on the diameter of tissue cores and how frequently the TMA block is refaced) 5-μm thick, FFPE sections available for high-throughput profiling, enabling on each slide the equivalent of over 100 assays (depending on the core size) [27].
Fig. 2.
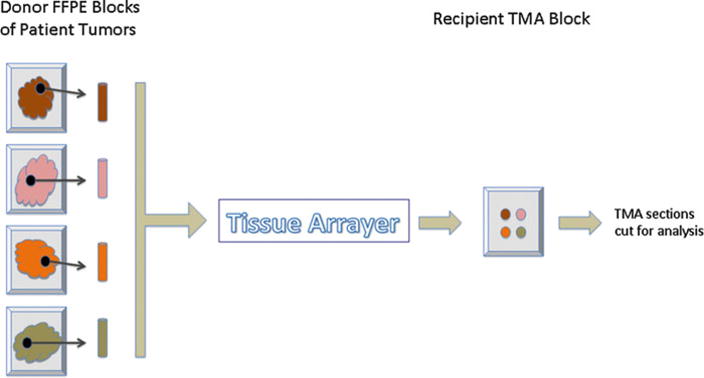
Diagram of assembled TMA. Tissue cores of patient’s tumors obtained from formalin-fixed paraffin-embedded (FFPE) donor blocks are placed in recipient TMA block by a tissue arrayer. This process can place dozens of donor tissue cores in one TMA block that can be cut and evaluated on a single microscopic slide
Traditional IHC evaluation of hundreds of tumor samples by whole slide sections requires that they be stained in batches and requires days to complete. Each batch is de facto exposed to inherent variables during the course of the processing and staining procedures. In contrast, TMAs allow evaluation of tissue samples in a single, consistent IHC procedure, and can usually be completed in one day. TMAs also conserve tissue samples and preserve much of the tissue within each block since only small cores of tumor tissue are used for analysis. In summary, when compared with IHC evaluation of a whole slide section of tissue, TMAs save valuable time, reagents, and cost in analyzing biomarkers, and in addition control for many inherent immunohistochemical variables of tissue-based research efforts.
The accuracy of TMAs in elucidating underlying biology has been raised as a concern since protein expression in cancer may be heterogeneous and TMAs by their design examine only a fraction of any given tumor. However, several groups have demonstrated significant correlation in IHC results obtained from TMA histospots or whole-tissue sections [27–29]. Increasing the number of tissue cores from donor FFPE tumor block on the array has been shown to reduce error rate of negative IHC results that may result from tumor heterogeneity. In general, two 0.6 mm histospots (i.e., two 0.6-mm tissue cores from a single donor FFPE tumor block) adequately represent IHC staining of entire histological section in most tumors [26–28]. However, fibroblastic tumors may require three 0.6 mm tissue cores from a donor FFPE tumor block to achieve a concordance of 91–96 % when compared to whole-tissue sections; four 0.6 mm tissue cores may be necessary for some melanocytic tumors to reach a concordance of 96.6 % [30, 31]. In some cases, use of multiple cores may significantly reduce the number of available sections. It is also important to note that the layout of the cores on the block should be arranged so as to make the block asynchronous in both (i.e., x and y) axes to minimize confusion in orienting the block.
In summary, TMAs provide a way of efficiently analyzing multiple tumor samples while also preserving tissue. Although some tumors show heterogeneous immunogenicity, use of more than one core per sample markedly improves the correlation of immunohistochemical results between TMAs and whole-tissue sections. Although details of individual studies employing TMAs in the melanoma biomarker arena are beyond the scope of this chapter, the interested reader is directed to recent studies that have successfully employed TMAs, many of which were constructed as institutional or inter-institutional initiatives through the NIH-funded SPORE (Specialized Program of Research Excellence) programs that have focused research efforts in melanoma. Recent relevant studies that have utilized TMAs to evaluate the prognostic significance of biomarkers in melanoma include the following: (1) phosphorylated STAT3 (p-STAT3) expression in patients with stage IV melanoma did not convey risk for CNS metastasis [32]; (2) NEDD9 protein expression was shown to be significantly upregulated in metastatic melanoma compared to melanocytic nevi [33]; (3) detection of mitosis and melanoma cells in G2 phase of the cell cycle with histone H3K79me3T80ph defined a subset of primary melanomas with risk for metastasis [34]; (4) confirmation of expression of imatinib targets in melanoma patients, particularly those with acrallentiginous melanoma [9, 26]; (5) expression of galectin 3 in melanoma lesions and its possible relationship to sun exposure[35]; and (6) decreased expression of retinoid receptors in primary and metastatic melanoma, and its possible correlation with impaired prognosis [36].
2 Materials
2.1 General Materials
Safety glasses or face shield, disposable latex gloves, laboratory coat, protective shoes.
2.2 Labeling and Storage
Laser Cryo-Labels (we use 1.42″ × 0.55″ removable Laser Labels), Histoprep Marker, permanent ink pen (e.g., Sharpie©), #2 pencil (see Note 5).
Cryovials, racks for cryovials.
Tissue-Tek ® OCT™ compound, cryomold, Parafilm ®, heavy-duty aluminum foil.
Cryo/freezer boxes (5″ × 5″ × 2″, 9 × 9 grid usually works best) (Fisher Scientific). For cryomold storage, we convert a 9 × 9 (81 cell) grid insert by removing dividers to form a 3 × 9 grid: remove the 1st two dividers, leave a divider, then remove another two dividers, leave a divider, then remove the last two dividers) (see Fig. 3i).
Liquid nitrogen freezer or permanent Dewar storage space, and/or −80 °C freezer.
Fig. 3.

Frozen tissue preparation. (a) Example of liquid nitrogen cryomold freezing stand (LN2 Stand). (b) LN 2 stand in ice bucket with liquid nitrogen. (c) Examples of: OCT compound; labeled cryomold; and prepped tissue ready for freezing (not on ice; for demonstration purposes only). (d) Process of rapid freezing of tissue in OCT (partially hardened) using LN2 stand. (e) Process of rapid freezing of tissue in OCT (fully hardened) using slurry method (see Note 15). (f) Example of labeled aluminum foil OCT archival sample wrapper. (g) Example of frozen OCT sample just prior to wrapping. (h) Examples of wrapped sample (1) backside and (2) front side. (i) Example of wrapped labeled sample placed in archival cryobox
2.3 Preparing Biospecimen for Cryo-Storage
Liquid nitrogen (LN2) in approved LN2 transport carrier (see Notes 6–7). Alternatively, 100 % isopropanol/dry ice slurry (slurry method) in a non-leaking ice bucket or other ultralow temperature container may also be employed in clinical pathology laboratories (see Note 8).
Freezer/cryo-gloves.
Liquid nitrogen cryomold freezing stand (LN2 Stand) in ice bucket—outside the clinical pathology laboratory, we use a custom-built double metal cylinder/can welded together with an outside top-edge handle. The interior solid can is 2″ shorter and about half the diameter of the outer open-top can and provides a horizontal metal-disc surface with a raised lip to hold the cryomold (see Fig. 3a) Liquid nitrogen is placed into the outer ring between the two cylinders and between the outer cylinder and ice bucket (see Fig. 3b).
Disposable, preferably sterile, scalpels or scalpel blades (note—autoclaved single-edge razor blades can also be used).
Forceps, disposable petri dish, glass petri dish (to autoclave unwrapped single-edge razor blades or when slurry method is used), Kimwipes©, Bunsen burner.
PBS, isopropyl alcohol, Tissue-Tek ® OCT compound.
3 Methods
3.1 Initial Specimen Preparation
Employ universal precautions consistent with regulatory requirements at all times.
Preprepare labels when possible; we find that printed cryo-labels work best. When hand-preparing labels, markers that support cryo-temperatures are recommended (i.e., alcohol-based permanent markers may smudge or fade and will not stay if using slurry method for freezing).
Remove any excess blood from the tissue using paper towel, gauze, or equivalent. If necessary, rinse in sterile PBS and blot dry (see Notes 9–13).
Tissue should be cut on a dissection board or similar set-up using a clean/sterile razor blade or scissors (see Note 9). For the tissue to “fit” in either cryovials or cryomolds, at least one of the tissue dimensions should be no greater than the maximum thickness of 0.5 cm. When sterile technique is required/desired, we often place the tissue in a sterile Petri dish and trim with sterile blades (if possible, perform in a laminar air flow hood).
Use separate forceps for each specimen to minimize the possibility of cross-contamination.
Discard any unused tissue according to procedures for disposal of human biological waste material.
Discard the blade(s) according to procedures for disposal of biohazard sharps.
If possible, weigh sectioned specimen prior to freezing (see also below).
- When possible, the following should also be recorded:
- Time (in minutes) before tissue has been frozen.
- Freezing temperature and/or conditions employed.
3.2 OCT Embedding
- Pre-procedure preparation:
- Prepare foil wrappers—cut heavy-duty aluminum foil into rectangles (approximately 5″ × 3″).
- Label cryomolds with printed cryolabel and/or permanent marker, and prepare (preferably print) sticker labels for foil wrappers. Labels should include: study ID, sample ID, tissue type/description, and date. Place cryomolds on a flat surface in a manner that will allow for easy identification of planned contents.
When sample is available, fill the bottom of each cryomold with a thin layer (less than 1 mm) of OCT by slowly and carefully filling the mold. It is important to avoid formation of bubbles and creation of uneven surfaces. If necessary, carefully tapping the bottom of the cryomold on a flat hard surface facilitates distribution of OCT.
If possible, weigh and record each section of the specimen prior to placing it in a cryomold.
If possible, prechill the prepared cryomold (step 2). This can be done by: (a) setting on ice for 2–5 min or (b) partially freezing the cryomold just prior to transferring specimen (step 5) by holding cryomold over the liquid nitrogen with forceps until OCT starts to become cloudy. Do not let the OCT fully harden at this time.
Using forceps, transfer the specimen to the prechilled OCT-filled cryomold.
Cover the exposed tissue with OCT; ensure the top surface of the OCT compound completely covers the tissue and is level. If the tissue section is taller than the cassette, the OCT can be layered (see Note 14).
The OCT can be hardened by placing the filled cryomold flatly on the interior metal-disc surface of the Liq N2 Stand (see Fig. 3a, d) that has been acclimated in liquid nitrogen (see Fig. 3b). Avoid allowing the cryomold to come in direct contact with the liquid nitrogen (see Notes 7–15). Specimen(s) are sufficiently frozen when the OCT has become completely white and firm. One may use the Slurry Method as an alternative approach to OCT processing (see Fig. 3e) (see Note 8).
While the OCT is hardening, adhere the appropriate sample label to a precut foil wrap in the center of the foil (see Fig. 3f).
After the OCT has hardened (i.e., has become firm and solid white in appearance), place the cryomold face down onto the non-labeled side of the appropriately pre-labeled foil wrap (Fig 3g). Fold the long side of aluminum wrap over the long edges to the back of the cryomold. Tuck short ends over short edges to the back of the cryomold (i.e., similar to wrapping a gift). After completing these folding maneuvers (see Fig. 3 h1), the label should be positioned in the middle of the front of the wrapped OCT tissue (see Fig. 3 h2).
Place samples on dry ice in a Styrofoam container or other suitable device with lid until transferred to −80 °C freezer for storage.
The wrapped OCT specimen is then placed into the 3 × 9 divided (27 cell) 5″ × 5″ × 2″ cryo-storage box (see Fig. 3i) and stored in a −80 °C freezer rack. We also suggest that the rack, box and grid position of the OCT specimen be documented in the study database.
3.3 Snap Freezing
Pre-procedure preparation: label 1.8 ml or similar cryovials using a permanent cryomarker (i.e., ethanol and freezer-resistant).
To determine specimen section weight: record weight of the empty vial, add tissue, and then reweigh. Subtract weights to determine the weight of the specimen and record.
Place individual specimen section in a 1.8 ml cryovials using forceps. Use separate forceps for each specimen to minimize the possibility of cross-contamination.
Tightly secure the cap and submerge in liquid nitrogen for “snap freezing” (see Notes 5–8).
Place samples on dry ice in Styrofoam container or other suitable device with lid until transferred to −80 ° freezer for storage.
The snap-frozen specimen-containing cryovial is then placed in a 9 × 9 (i.e., 81 cell) 5″ × 5″ × 2″ cryo-storage box and stored at −80 °C or colder in freezer racks. We also suggest that the rack, box, and grid position of the snap-frozen specimen be notated in the study database.
If cryostat sections will be performed on snap-frozen tissue samples (and not embedded in OCT), mount tissue samples to gum tragacanth to ensure desired tissue specimen orientation (e.g., skin punch tissue specimens) and snap-freeze. Wrap labeled snap-frozen tissue samples in Parafilm ® then cover in aluminum foil and store at −80 °C [37].
Acknowledgments
This work was supported by NIH grant P50 CA093459 University of Texas MD Anderson Cancer Center SPORE in Melanoma and an NIH/SAIC Cancer Genome Atlas Project for melanoma contract, also awarded to MD Anderson.
Footnotes
While both core biopsies and FNA biopsies are commonly employed for diagnostic purposes, core biopsies usually provide more tissue and are preferred for research purposes. The minimum size for fresh/frozen tissue collection is generally 0.25 cubic cm, which is generally achieved with approximately four passes of a 14-gauge needle. In the diagnostic setting, the collection of 2–4 fresh/frozen cores for research is recommended, in addition to two cores for diagnosis. The fraction of cores that will contain viable tumor will decrease if/as the tumor shrinks in response to therapy. Therefore, fewer cores may be obtainable as lesions become smaller. If a previous biopsy site is noted in the specimen to be sampled, if at all possible, biopsies should not be taken from near that site. Core biopsies may alter the biology of adjacent tissue—e.g., they introduce inflammatory material from wound reaction and biomolecules involved in wound healing—which can be problematic if a subsequent core taken from that tissue is used. Notably, many genes involved in wound healing are also noted to be involved in cancer progression [38].
If possible, representative adjacent normal tissue should be collected in addition to the tumor tissue. Normal tissue should be maximally distant from the tumor, although clear definitions of what constitutes normal control tissue in the context of primary melanoma research (e.g., is normal skin a good normal control?) are not well established. Nonetheless, if normal skin is to be collected, it should be collected as far from the primary site as practical. Collection of germline DNA (i.e., extracted from peripheral blood mononuclear cells (PBMC)) should be considered in all protocols that include the collection of tumor tissue for DNA isolation, as defining truly “normal” tissue for melanoma research is indeed challenging.
Do not slow-freeze. Samples in cryovial or OCT/cryomolds should be snap frozen. Slow freezing promotes the formation of ice crystals, which can damage nucleic acids (e.g., RNA) in the specimen. The slower a sample freezes, the larger the ice crystals that may form. Older models of cryostats that require significant specimen freeze time should be avoided whenever possible.
Containers used to transport samples with dry ice needs ventilation so there is no buildup of the CO2 within the container.
If a pen is used to label cryovials or other receptacles that will be stored in freezing conditions, ensure that the pen is waterproof/solvent-proof and can withstand long-term freezing conditions. For cryovials, if no cryomarker is available, pencil may work on the writing surface and withstand long-term LN2 conditions; it is advisable to confirm this functionality in advance of specimen processing.
Use freezer gloves and a face shield while working with liquid nitrogen. Take precautions to avoid accidental spillage or spattering of liquid nitrogen.
Do not place the specimen directly in liquid nitrogen. If the tissue is frozen too quickly, it may shatter, potentially causing difficulty with H&Es and IHC.
If liquid nitrogen is not available, the slurry method can be used by making an isopropanol/dry ice slurry. It is best to use crushed dry ice mixed with 100 % isopropanol until the ice is moistened throughout, but not so much as to have a layer of pure isopropanol over the ice. The slurry should be ready to use once the isopropanol stops forming new bubbles. It is not recommended to simply use dry ice for freezing.
Instruments should be changed or thoroughly cleaned between dissection of normal and tumor tissue to prevent cross-contamination—even a tiny amount of cross-contamination can interfere with analyte preparation and potentially adversely confound results.
If RNA analyte preparation is planned, use a sterile RNAse-free container. Rinse tissue in DEPC-treated water or RNAse-free PBS. We do not recommend RNAlater ® particularly with cutaneous/subcutaneous tissue as the RNAlater ® makes the connective tissue more rigid, and causes the homogenization process to be less efficient, giving lower yields with greater degradation of RNA.
Do not add serum to the specimen.
Do not directly touch the biopsy/specimen without sterile gloves.
Use sterile or disposable equipment for both dissection and snap freezing.
When tissue is taller than cryomold, the OCT can be layered in steps until the tissue is completely covered. Add OCT only up to the top of the tissue insert section of the cryomold. Partially freeze/harden the tissue in OCT—the OCT will become cloudy/white on the edges. Before the OCT that is next to the tissue becomes cloudy, remove from liquid nitrogen (or dry ice/alcohol slurry) and add another layer of OCT onto the partially hardened previous layer of OCT and reexpose to cold (until it gets cloudy) and repeat if necessary. Once the tissue is completed covered by OCT, let it freeze completely.
Alternative approach: OCT preparation without Liq N2 Stand— use of a petri dish to freeze OCT/tissue in cryomold: Carefully position a Petri dish at the surface of the liquid nitrogen using forceps or the isopropanol/dry ice slurry (see Fig. 2e); allow the petri dish to stabilize with the liquid nitrogen or slurry; place the filled cryomold in the Petri dish and freeze until the OCT media is completely hard.
References
- 1.Davies MA, Gershenwald JE. Targeted therapy for melanoma: a primer. Surg Oncol Clin N Am. 2010;20:165–180. doi: 10.1016/j.soc.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson M, Zhao F, Letrero R, et al. Somatic mutation status of melanomas and effect on clinical outcome in patients on ECOG 2603. Society for Melanoma Research 2012 Congress; Hollywood, CA. 2012. [Google Scholar]
- 3.Becker D, Mihm MC, Hewitt SM, et al. Markers and tissue resources for melanoma: meeting report. Cancer Res. 2006;66:10652–10657. doi: 10.1158/0008-5472.CAN-06-0921. [DOI] [PubMed] [Google Scholar]
- 4.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halaban R, Zhang W, Bacchiocchi A, et al. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woodman SE, Lazar AJ, Aldape KD, et al. New strategies in melanoma: molecular testing in advanced disease. Clin Cancer Res. 2012;18:1195–1200. doi: 10.1158/1078-0432.CCR-11-2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivan D, Niveiro M, Diwan AH, et al. Analysis of protein tyrosine kinases expression in the melanoma metastases of patients treated with Imatinib Mesylate (STI571, Gleevec) J Cutan Pathol. 2006;33:280–285. doi: 10.1111/j.0303-6987.2006.00432.x. [DOI] [PubMed] [Google Scholar]
- 10.Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 12.McArthur GA, Ribas A, Chapman PB, et al. Molecular analyses from a phase I trial of vemurafenib to study mechanism of action (MOA) and resistance in repeated biopsies from BRAF mutation-positive metastatic melanoma patients. J Clin Oncol. 2011:29. abstract 8502. [Google Scholar]
- 13.Sosman JA, Pavlick AC, Schuchter LM, et al. Analysis of molecular mechanisms of response and resistance to vemurafenib (vem) in BRAF V600E melanoma. J Clin Oncol. 2012;30:8503. [Google Scholar]
- 14.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can Be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF (V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flaherty KT, Infante JR, Falchook GS, et al. Phase I/II expansion cohort of BRAF inhibitor GSK2118436 + MEK inhibitor GSK1120212 in patients with BRAF mutant metastatic melanoma who progressed on a prior BRAF inhibitor. Pigment Cell Mel Res. 2011;24:1022. [Google Scholar]
- 20.Daud A, Sosman J, Weber J, et al. Mutation and copy number analysis in melanoma biopsies from a Phase I/II study evaluating the combination of dabrafenib and trametinib. Society for Melanoma Research 2012 Congress; Hollywood, CA. 2012. [Google Scholar]
- 21.Singh B. Preoperative therapy in invasive breast cancer: reviewing the state of the science and exploring new research directions. Bethesda; Maryland: 2007. Initial pathology assessment prior to preoperative therapy. [Google Scholar]
- 22.Hewitt SM, Lewis FA, Cao Y, et al. Tissue handling and specimen preparation in surgical pathology: issues concerning the recovery of nucleic acids from formalin-fixed, paraffin-embedded tissue. Arch Pathol Lab Med. 2008;132:1929–1935. doi: 10.5858/132.12.1929. [DOI] [PubMed] [Google Scholar]
- 23.Leyland-Jones BR, Ambrosone CB, Bartlett J, et al. Recommendations for collection and handling of specimens from group breast cancer clinical trials. J Clin Oncol. 2008;26:5638–5644. doi: 10.1200/JCO.2007.15.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ibberson D, Benes V, Muckenthaler MU, et al. RNA degradation compromises the reliability of microRNA expression profiling. BMC Biotechnol. 2009;9:102. doi: 10.1186/1472-6750-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gundisch S, Hauck S, Sarioglu H, et al. Variability of protein and phosphoprotein levels in clinical tissue specimens during the preanalytical phase. J Proteome Res. 2012;11:5748–5762. doi: 10.1021/pr300560y. [DOI] [PubMed] [Google Scholar]
- 26.Shen SS, Zhang PS, Eton O, et al. Analysis of protein tyrosine kinase expression in melanocytic lesions by tissue array. J Cutan Pathol. 2003;30:539–547. doi: 10.1034/j.1600-0560.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 27.Camp RL, Neumeister V, Rimm DL. A decade of tissue microarrays: progress in the discovery and validation of cancer biomarkers. J Clin Oncol. 2008;26:5630–5637. doi: 10.1200/JCO.2008.17.3567. [DOI] [PubMed] [Google Scholar]
- 28.Camp RL, Charette LA, Rimm DL. Validation of tissue microarray technology in breast carcinoma. Lab Invest. 2000;80:1943–1949. doi: 10.1038/labinvest.3780204. [DOI] [PubMed] [Google Scholar]
- 29.Torhorst J, Bucher C, Kononen J, et al. Tissue microarrays for rapid linking of molecular changes to clinical endpoints. Am J Pathol. 2001;159:2249–2256. doi: 10.1016/S0002-9440(10)63075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoos A, Urist MJ, Stojadinovic A, et al. Validation of tissue microarrays for immunohistochemical profiling of cancer specimens using the example of human fibroblastic tumors. Am J Pathol. 2001;158:1245–1251. doi: 10.1016/S0002-9440(10)64075-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pacifico MD, Grover R, Richman P, et al. Validation of tissue microarray for the immunohistochemical profiling of melanoma. Melanoma Res. 2004;14:39–42. doi: 10.1097/00008390-200402000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Lee I, Fox PS, Ferguson SD, et al. The expression of p-STAT3 in stage IV melanoma: risk of CNS metastasis and survival. Oncotarget. 2012;3:336–344. doi: 10.18632/oncotarget.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim M, Gans JD, Nogueira C, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125:1269–1281. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 34.Martinez DR, Richards HW, Lin Q, et al. H3K79me3T80ph is a novel histone dual modification and a mitotic indicator in melanoma. J Skin Cancer. 2012;2012:823534. doi: 10.1155/2012/823534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prieto VG, Mourad-Zeidan AA, Melnikova V, et al. Galectin-3 expression is associated with tumor progression and pattern of sun exposure in melanoma. Clin Cancer Res. 2006;12:6709–6715. doi: 10.1158/1078-0432.CCR-06-0758. [DOI] [PubMed] [Google Scholar]
- 36.Chakravarti N, Lotan R, Diwan AH, et al. Decreased expression of retinoid receptors in melanoma: entailment in tumorigenesis and prognosis. Clin Cancer Res. 2007;13:4817–4824. doi: 10.1158/1078-0432.CCR-06-3026. [DOI] [PubMed] [Google Scholar]
- 37.Curry JL, Qin JZ, Bonish B, et al. Innate immune-related receptors in normal and psoriatic skin. Arch Pathol Lab Med. 2003;127:178–186. doi: 10.5858/2003-127-178-IIRRIN. [DOI] [PubMed] [Google Scholar]
- 38.Riss J, Khanna C, Koo S, et al. Cancers as wounds that do not heal: differences and similarities between renal regeneration/repair and renal cell carcinoma. Cancer Res. 2006;66:7216–7224. doi: 10.1158/0008-5472.CAN-06-0040. [DOI] [PubMed] [Google Scholar]