

Abstract
The α-fluorination of α- and β-C-ethanals of galactose using Jørgensen catalysts and NFSI was investigated. The crude reaction products were transformed to their primary alcohol or methylenated derivatives, which are versatile precursors to biologically interesting fluorinated glycomimetics. The α-C-glycoside substrate gave moderate to high yields of fluorinated α-C-glycosides with minor amounts of β-C-glycoside analogues. The reactions on the β-C-glycoside were lower yielding but gave exclusively fluorinated β-C-glycosides. For both α– and β-C-glycoside substrates (R) and (S) catalyst showed complementary stereoselectivity. The preparation of difluorinated materials required the use of racemic catalyst as enantiomerically pure catalyst gave intractable mixtures of products. These results are in line with the results for simple achiral aldehydes, and suggest that stereochemistry in the reactions of these chiral, highly substituted, carbohydrate-derived aldehydes are controlled primarily by the chirality in the catalyst.
Graphical Abstract

The fluorination of α- and β-C-linked-galactosyl ethanals using NFSI and Jørgensen catalyst, followed by reduction or methylenation of the derived aldehydes provided versatile precursors to flluorinated glycomimetics. The (R) and (S) catalysts gave stereochemically complementary results.
Introduction
Fluorination of bioactive materials is an active area of research in medicinal chemistry because the electronic and conformational effects of fluorine and the stability of the C-F bond may enhance binding in receptor sites and improve drug absorption, distribution, metabolism and excretion (ADME).1,2,3 Accordingly a variety of strategies for introduction of fluorine into biomolecules have been developed.4,5,6 However, while these methods have met much success in simple substrates, their application to more highly functionalized substrates like carbohydrates is often problematic.7,8,9 In this context, fluorinated C-glycosides like 2 and versatile derivatives thereof that can be transformed to complex analogues like 3 and 4, are of interest (Fig. 1).10,11,12,13,14,15,16,17,18,19,20,21,22,23,24 The organocatalytic α-fluorination of simple C-glycoside aldehydes 1 is an appealing route to such precursors because of the mild reaction conditions, operational simplicity and easy access to reaction substrates.25,26,27 Herein, we describe the application of this strategy to C-glycosides of galactose. While the proline catalyzed amination of related C-glycoside aldehydes has been studied, to the best of our knowledge the mechanistically similar α-fluorination reaction has not been reported.28
Figure 1.
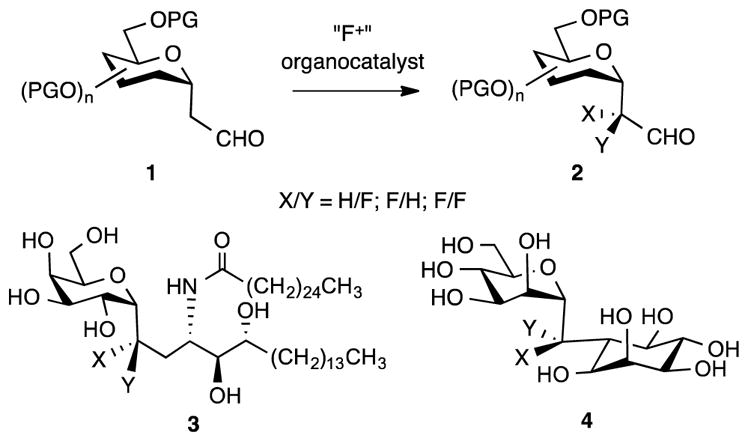
Fluorinated precursors for complex C-glycosides
Results and Discussion
The substrates for this study were the α- and β-C-linked ethanals 5a and 5b, which were obtained from their readily available C-allyl derivatives.20,21,29 Initial experiments were performed on 5a (Scheme 1). This material was treated with two molar equivalents of NFSI in THF in the presence of 2.5 mol % of (S) or (R) Jørgensen catalyst.25 Because of the instability of the fluorinated aldehyde products, the crude aldehyde was treated with NaBH4 or methylenetriphenylphosphorane to give the primary alcohol or terminal alkene derivative.30 NaBH4 reduction of the product from the fluorination with the (S) catalyst afforded α-C-monofluorinated glycoside 6, and a β-C-glycoside 7 of undetermined configuration at the fluorinated carbon, in 75 and 10% yield respectively from 5a. The fluorination/reduction sequence with the (R) catalyst produced the α-C-fluorohydrin 8 as the major product, together with 6, the β-C-fluorohydrin 9 that was epimeric to 7 at the fluorinated carbon, and the α-C-difluorinated product 10 in 68, 6, 3 and 20% yield respectively.
Scheme 1.

Fluorination-reduction on α-C-ethanal 5a
Wittig methylenation on the crude fluorination product with the (S) catalyst gave the α-C-alkene 11 and the β-C-product 12 in 73 and 9% yields, respectively (Scheme 2). The fluorination/methylenation sequence with the (R) catalyst afforded 13 (53%), the α-C-glycoside that was epimeric to 11 at the fluorinated carbon, and an intractable mixture of other fluorinated alkenes (31%).
Scheme 2.

Fluorination-methylenation on α-C-ethanal 5a
Towards more stereoselective syntheses of the β-C-fluorohydrins 7 and 9, the fluorination-reduction protocol on the β-C-ethanal 5b, was next investigated (Scheme 3). For optimal results, these reactions were performed at higher catalyst loading and shorter reaction times, compared to the reactions on 5a. With 30 mol% catalyst 7 and 9 were obtained as the only observed mono-fluorinated products with the (S) and (R) catalysts, in 44 and 50 % yields, respectively.
Scheme 3.
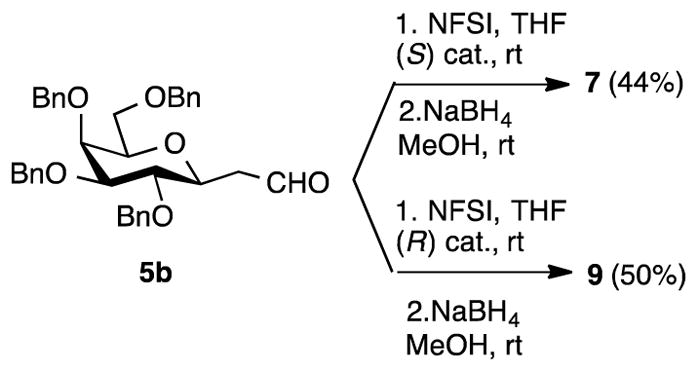
Fluorination-reduction on β-C-ethanal 5b
The synthesis of difluorinated derivatives was next examined more carefully (Scheme 4). Reactions on 5a using excess NFSI, extended reaction times and increased loading of the (R) or (S) catalyst, gave complex mixtures with low yields of fluorinated materials and other unidentified products. Experiments with pyrrolidine instead of the chiral catalyst gave similar results. However when the reaction was performed with a 20 mol% of a racemic mixture of the catalyst, and the crude product treated with NaBH4, or subjected to methylenation, the difluorinated products 10 and 14 were obtained in 83 and 40% yield, respectively. The alkene 14 was also prepared by oxidation of a purified sample of 10 with Dess-Martin periodinane followed by Wittig olefination on the crude aldehyde. The overall yield of 14 from 10 following this procedure was 66%. As for the mono-fluorination reactions, the difluorination-reduction protocol on 5b was lower yielding compared to case of 5a, giving a 16% yield of the difluorinated β-C-glycoside 15 with 30 mol% of the racemic catalyst.
Scheme 4.

Synthesis of difluorinated derivatives
Stereochemical assignment of the fluorinated compounds rests on X-ray analysis of alkene 11, JH,H values and chemical correlation between alkene and alcohol derivatives (Fig. 2, Supporting Information, Scheme 2). The X-ray structure of 11 indicated the α-C1 orientation and the R configuration at the fluorinated carbon. Interestingly, the molecule adopted the 1C4 chair conformation. A similar result has been reported for a related mono-fluorinated α-C-galactoside. A J1,2 of 2.4 Hz also supported the α-C1 orientation, but is ambiguous in relation to assignment of ring conformation. Similarly J1,2 values between 0 – 4.5 and close to 10 Hz were consistent with the α-C1 in 6, 8, 10, 13, 14, and a β-C1 in 7, 9, 12 and 15 respectively. The configuration at the fluorinated carbon in the α-C-glycosides 6, 8, 11 and 13 rests on correlation to 11. Thus, alkene 13 was deduced as S on the basis of its epimeric relationship to 11. The α-C- fluorohydrins 6 and 8 were assigned by correlating them to 11 and 13 respectively, through an ozonolysis/aldehyde reduction sequence. The configurations at the fluorinated carbons in the β-C-glycosides 7, 9 and 12 were tentatively assigned using the stereochemical model proposed for the Jørgensen catalyst (vide infra). That the (S) v.s. (R) catalyzed reactions selectively gave epimeric fluorides, 6 vs. 8, 7 vs. 9, 11 vs. 13 as the major products provides a measure of self-consistency to this argument.
Figure 2.

X-ray for 11
The stereoselectivity in the fluorination of α-C-ethanal 5a is in agreement with the model proposed for the Jørgensen catalysts (Scheme 5). Thus, the (S) and (R) catalyst lead to E enamines 16 and 20, which should favour fluorination on the Re and Si face respectively, which is in line with the experimental result, stereoselective formation of 6 and 8. Similarly high stereoselectivities have been observed in the proline catalysed amination on related carbohydrate aldehydes. The apparently better performance of the (S) catalyst compared to the (R), on the α-C-glycosoide substrate may represent a “matched/mistmatched” scenario. Thus 16 and 20 are expected to adopt the “exo-anomeric” conformation with respect to the pseudoglyconic torsion (i.e. the aminated carbon of the enamine is gauche to O5 and H1 of the sugar ring).31 The facial selectivity imposed by the (R) catalyst (i.e. on 20), is opposed by steric hindrance from O5 of the sugar. For 16, the enamine from the (S) catalyst, the facial selectivity of the catalyst is matched to that imposed by the sugar residue. Therefore, fluorination on 20 may be less stereoselective and side reactions more likely compared to 16.
Scheme 5.

Mechanism for fluorination
The results for the difluorination reactions also appear to be consistent with the Jørgensen model. Thus, for a given enantiomer of the catalyst, α-deprotonation on the mono-fluorination iminium ion (i.e. 17, 21), is sterically hindered by the bis-arylcarbinol side chain in the catalyst residue.25 A racemic mixture of catalyst allows for amine exchange on 17 and 21 to give diastereomeric iminum ions in which α-deprotonation is not sterically blocked, thereby facilitating formation of the di-fluorinated product. As in the initial fluorination step, conformational effects may also impact on difluorination and explain why the (S) catalyst apparently shows a lesser propensity for difluorination compared to the (R) catalyst. Thus, “exo-anomeric” conformations for 17 and 21, suggests that deprotonation in 17 (the favored mono-fluorinated iminium ion with the (S) catalyst), is hindered by both the sugar ring and the catalyst moiety, whereas the hindrance to deprotonation on 21, the corresponding species with the (R) catalyst, comes from the catalyst moiety only.
Without definitive assignment of the configuration at the fluorinated carbons in the β-C-glycosides, it is impossible to tell whether the Jørgensen model extends to the fluorination of 5b. However, the finding that enantiomeric catalysts exhibit opposite stereoselectivity in the fluorination of 5b, supports this notion. This observation is also relevant to the mechanism by which the fluorinated β-glycosides 7 and 12 are formed from the α-C-glycoside 5a. A pathway in which 5a or 16 first epimerizes to 5b or the β-C-glycoside of 16 respectively (via an retro-Michael-Michael mechanism), followed by fluorination, seems more likely than epimerization of a first formed α-C-fluorinated species.32 In the latter scenario, the individual reactions of 5a with the enantiomeric catalysts might have been expected to give mixtures of the β-C-glycosides 7 and 12.
Conclusions
The α-fluorination of C-linked galactosyl ethanals using the Jørgensen catalyst and NFSI is a promising route to versatile precursors for complex fluorinated glycomimetics. Enantiomeric catalysts influenced complementary facial selectivity in line with the results for simple achiral aldehydes. The high diastereoselectivity observed for these stereochemically complex substrates is similar to that observed for the organocatalyzed amination on related substrates, and point to the powerful stereodirecting effects of these proline-based catalysts in enamine reactions. These findings provide insight on the effects of conformation and remote substituents in such complex substrates, and provide the basis for the wider application of this chemistry in carbohydrate synthesis.
Experimental section
Solvents were purified by standard procedures or used from commercial sources as appropriate. Petroleum ether refers to the fraction of petroleum ether boiling between 40 and 60 °C. Ether refers to diethyl ether. Unless otherwise stated thin layer chromatography (TLC) was done on 0.25 mm thick precoated silica gel 60 (HF-254, Whatman) aluminium sheets and flash column chromatography (FCC) was performed using Kieselgel 60 (32–63 mesh, Scientific Adsorbents). Elution for FCC usually employed a stepwise solvent polarity gradient, correlated with TLC mobility. Chromatograms were observed under UV (short and long wavelength) light, and/or were visualized by heating plates that were dipped in a solution of ammonium (VI) molybdate tetrahydrate (12.5 g) and cerium (IV) sulfate tetrahydrate (5.0 g) in 10% aqueous sulphuric acid (500 mL), or a solution of 20% sulfuric acid in ethanol. 1H and 13C and 19F NMR spectra were recorded using Bruker Ultra Shield 400 MHz, Bruker Ultra Shield 500 MHz and Bruker Ultra Shield Plus 600 MHz instruments, in CDCl3 or C6D6 solutions with residual CHCl3 or C6H6 as internal standard (δH 7.27, 7.16 and δC 77.2, 128.4 ppm). Chemical shifts are quoted in ppm relative to tetramethysilane (δH 0.00) and coupling constants (J) are given in Hertz. First order approximations are employed throughout. High resolution mass spectrometry was performed on an Ultima Micromass Q-TOF or Waters Micromass LCT Premier mass spectrometers.
General procedure for fluorination-reduction sequence on aldehydes: 2-(2′, 3′, 4′, 6′-Tetra-O-benzyl-α-D-galactopyranosyl)-2-R-fluoroethanol (6) and 2-(2′, 3′, 4′, 6′-Tetra-O-benzyl-β-D-galactopyranosyl)-2-R-fluoroethanol (7)
To a solution of aldehyde 5a (250 mg, 0.44 mmol) in THF (2.2 mL) was added (S)-Jørgensen catalyst (7.0 mg, 0.012 mmol). The mixture was stirred at rt for 15 min at which time NFSI (275 mg, 0.87 mmol) was added, and stirring continued at rt for 6 h, or after NMR indicated disappearance of the starting material. The reaction mixture was then concentrated at rt under reduced pressure and the residue filtered over a short column of silica gel. The filtrate was evaporated in vacuo at rt, the residue taken up in methanol (10 mL) and NaBH4 (250 mg, 6.8 mmol) slowly added at 0 °C to the mixture. The reaction was stirred for 30 min, then pH adjusted to 6 with 1M HCl, and the volatiles removed under reduced pressure at rt. The residue was extracted with EtOAc and the organic phase washed with saturated aqueous NaHCO3, and brine, dried (Na2SO4), and evaporated in vacuo. FCC of the crude product gave 6 (195 mg, 75%) and 7 (27 mg, 10%).
For 6: white amorphous solid, m.p = 79–83 °C; Rf 0.50 (60% hexanes: 40% EtOAc); [ ]D25 + 40 (c 1.6, CH2Cl2); 1HNMR (500 MHz, CDCl3) δ 7.38–7.16 (m, 20H), 4.65 (ddt, J = 2.8, 8.9, 48 Hz, 1H), 4.58–4.42 (m, 8H), 4.43 (m, partially buried, 1H), 4.33 (m, 1H), 4.20 (t, J = 8.1 Hz, 1H), 3.98 (dd, J = 3.2, 6.3 Hz, 1H), 3.95–3.78 (m, 2H), 3.75 (t, J = 3.5 Hz, 1H), 3.69 (m, 1H), 3.60 (dd, J = 3.1, 11.6 Hz, 1H), 2.91 (br s, 1H); 13CNMR (125 MHz, CDCl3) δ 138.3, 138.2, 138.0, 137.8, 128.7, 128.6, 128.4, 128.3, 128.1, 128.0, 127.9, 127.8, 90.4 (d, JC-F = 169 Hz), 74.2, 73.7, 73.5, 73.4 (two peaks), 73.3, 73.2, 71.7, 65.5, 65.2 (d, JC-F = 32 Hz), 63.1 (d, JC-F = 21 Hz); 19FNMR (377 MHz, CDCl3) δ −198.5 (s); HRMS (ESI, MNa+) m/z calc for C36H39FNaO6 609.2628, found 609.2618.
For 7: colorless oil; Rf = 0.28 (60% hexanes: 40% EtOAc); [ ]D25 +24 (c 0.7, CH2Cl2); 1HNMR (600 MHz, CDCl3) δ 7.32–7.22 (m, 20H), 5.00 (m, 2H), 4.91 (dt, J = 3.8, 51.7 Hz, 1H), 4.78 (m, 1H), 4.82–4.65 (m, 4H), 4.45 (ABq, J = 11.6 Hz, Δδ = 0.06 ppm, 2H), 4.19 (t, J = 9.6 Hz, 1H), 4.05 (ddd, J = 6.2, 12.6, 19.6 Hz, 1H), 3.97 (bs, 1H), 3.85 (m, 1H), 3.66 (dd, J = 2.2, 9.5 Hz, 1H), 3.61 (dd, J = 6.3, 8.7 Hz, 1H), 3.58 (t, J = 6.1 Hz, 1H), 3.48 (dd, J = 5.8, 8.8 Hz, 1H), 3.43 (dd, J = 9.2, 28.4 Hz, 1H), 2.64 (m, 1H); 13CNMR (150 Mz, CDCl3) δ 138.5, 138.2, 138.1, 137.7, 128.7, 128.4, 128.3, 128.2, 128.1, 127.9, 127.8, 127.7, 90.0 (d, JC-F = 178 Hz), 84.6, 79.2 (d, JC-F = 19 Hz), 77.8, 75.8, 74.4, 74.3 (d, JC-F = 4.4 Hz), 73.8, 72.9, 72.4, 69.3, 62.9 (d, JC-F = 23 Hz); 19FNMR (377 MHz, CDCl3) δ −206.9 (s); HRMS (ESI, MNa+) m/z calc for C36H39FNaO6 609.2628, found 609.2625
2-(2′, 3′, 4′, 6′-Tetra-O-benzyl-α-D-galactopyranosyl)-2-S-fluoroethanol (8), 2-(2′, 3′, 4′, 6′-Tetra-O-benzyl-β-D-galactopyranosyl)-2-R-fluoroethanol (9) and 2-(2′, 3′, 4′, 6′-Tetra-O-benzyl-α-D-galactopyranosyl)-2,2-difluoroethanol (10)
Aldehyde 5a (150 mg, 0.265 mmol) was subjected to the standard fluorination-reduction procedure with (R)-Jørgensen catalyst, as described for the synthesis of 6 and 7. FCC of the product after the reduction step afforded 6 (5 mg, 3%), 8 (105 mg, 68%), 9 (9 mg, 6%) and 10 (31 mg, 20%).
For 8: colorless oil; Rf = 0.38 (40% EtOAc/60% hexanes); [ ]D25 +39 (c 1.3, CH2Cl2); 1HNMR (500 MHz, CDCl3) δ 7.27–7.18 (m, 20H), 4.87 (dq, J = 4.3, 46.7 Hz, 1H), 4.76 (app d, J = 12.0 Hz, 1H), 4.71 (app d, J = 10.5 Hz, 1H), 4.67 (s, 2H), 4.53 (app d, J = 11.5 Hz, 1H), 4.48 (app d, J = 11.5 Hz, 1H), 4.40 (ABq, J = 12.0 Hz, Δδ = 0.08 ppm, 2H), 4.14 (m, 1H), 4.05 (br t, J = 7.1 Hz, 1H), 3.97 (ddd, J = 3.0, 5.9, 34.1 Hz, 1H), 3.96 (dd, J = 2.0, 9.5 Hz 1H), 3.84 (t, J = 2.6 Hz, 1H), 3.81–3.76 (m, 2H), 3.59 (t, J = 9.5 Hz, 1H), 3.31 (dd, J = 4.4, 10.2 Hz, 1H), 2.43 (br s, 1H); 13CNMR (125 MHz, CDCl3) δ 138.7, 138.6, 138.3, 138.2, 128.7, 128.6, 128.5, 128.3, 128.1, 128.0, 127.8, 93.5 (d, JC-F = 178 Hz), 79.1, 75.7, 75.0 (d, JC-F = 4 Hz), 74.6, 74.2, 73.5 (d, JC-F = 35 Hz), 69.2, 63.1 (d, JC-F = 25 Hz); 19FNMR (377 MHz, CDCl3) δ −198.5 (s); HRMS (ESI, MNa+) m/z calc for C36H39FNaO6 609.2628, found 609.2618.
For 9: colorless oil; Rf = 0.48 (40% EtOAc/60% hexanes); [ ]D25 +32 (c 0.1, CH2Cl2); 1HNMR (500 MHz, C6D6) δ 7.03–7.38 (m, 20H), 4.94 (m, 2H), 4.88 (dt, J = 4.5, 45 Hz, 1H partially buried under m at 4.94 ppm), 4.55 (m, 2H), 4.30 (ABq, J = 11.8 Hz, Δδ = 0.06 ppm, 2H), 4.24 (ABq, J = 11.8 Hz, Δδ = 0.05, 2H), 4.08 (t, J = 9.6 Hz, 1H), 3.85 (m, 3H), 3.70 (ddd, J = 1.8, 10.0, 11.8 Hz, 1H), 3.63 (t, J = 8.1 Hz, 1H), 3.52 (dd, J = 5.4, 8.9 Hz, 1H), 3.36 (m, 1H), 3.33 (dd, J = 2.7, 8.2 Hz, 1H); 13CNMR (125 MHz, C6D6) δ 139.7, 138.2, 139.2, 139.1, 129.0 (two peaks), 128.9, 128.5 128.3 (two peaks), 128.1, 128.0, 93.4 (d, JC-F = 174 Hz), 85.7, 80.5 (d, JC-F = 21 Hz), 77.5, 75.5 (d, JC-F = 8 Hz), 75.4, 75.3, 74.1, 73.9, 72.4, 69.1, 62.3 (d, JC-F = 25 Hz); 19FNMR (377 MHz, CDCl3) δ −196.5 (s); HRMS (ESI, MNa+) m/z calc for C36H39FNaO6 609.2628, found 609.2626.
For 10: colorless oil; Rf = 0.63 (40% EtOAc/60% hexanes); [ ]D26 +44 (c 0.6, CH2Cl2); 1HNMR (500 MHz, C6D6) δ 7.22–7.07 (m, 20H), 4.62 (ddd, J = 2.1, 8.9, 18.0 Hz, 1H), 4.52 (apparent d, J = 11.5 Hz, 1H), 4.47 (m, 1H), 4.38 (br m, 5H), 4.18 (ABq, J = 11.8 Hz, Δδ = .05 ppm, 1H), 4.10 (br m, 1H), 4.05 (dd, J = 2.2, 5.0 Hz, 1H), 3.99 (dd, J = 3.1, 5.3 Hz, 1H), 3.93 (bm, 1H), 3.80 (dd, J = 3.1, 5.0 Hz, 1H), 3.71 (dd, J = 2.9, 11.2 Hz, 1H), 3.27 (t, J = 7.5 Hz, 1H); 13CNMR (125 MHz, C6D6) δ 139.0, 138.8, 138.7, 132.4, 132.2, 131.9, 131.7, 128.8, 128.1, 127.9, 127.8, 127.7, 127.6, 127.5, 127.4, 127.2, 125.6, 125.2, 123.6 (t, JC-F = 243 Hz), 75.6, 75.0, 74.9, 74.3, 73.9, 73.8, 73.7, 72.6, 68.3 (dd, JC-F = 25, 34 Hz), 66.6, 64.1 (dd, JC-F = 28, 34 Hz); 19FNMR (377 MHz, CDCl3) δ −114.6 (ABq, J = 256 Hz, Δδ = 9.8 ppm, 2F); HRMS (ESI, MNa+) m/z calc for C36H38F2NaO6 627.2534, found 627.2523.
Synthesis of 7 from 5b
Aldehyde 5b (46 mg, 0.081 mmol) was subjected to the standard fluorination procedure with NFSI (51 mg, 0.16 mmol) and (S)-Jørgensen catalyst (15 mg, 0.024 mmol) for 1.5 h. Reduction of the crude product with NaBH4 following the standard procedure and FCC of the product afforded 7 (21 mg, 44%).
Synthesis of 9 from 5b
Aldehyde 5b (46 mg, 0.081 mmol) was subjected to the standard fluorination procedure with NFSI (51 mg, 0.16 mmol) and (R)-Jørgensen catalyst (15 mg, 0.024 mmol) for 1.5 h. Reduction of the crude product with NaBH4 following the standard procedure and FCC of the product afforded 9 (24 mg, 50%).
Synthesis of 10 using racemic catalyst
Aldehyde 5a (330 mg, 0.58 mmol) was subjected to the fluorination procedure described for 6, with NFSI (730 mg, 2.30 mmol), (R)-Jørgensen catalyst (35 mg, 0.058 mmol) and and (S)-Jørgensen catalyst (35 mg, 0.058 mmol) for 18 h. Reduction of the crude product with NaBH4 following the standard procedure and FCC of the product afforded 10 (290 mg, 83%).
General procedure for fluorination-Wittig methylenation sequence on aldehydes: 3-(2′, 3′, 4′, 6′-Tetra-O-benzyl-α-D-galactopyranosyl)-3-R-fluoropropene (11) and 3-(2′, 3′, 4′, 6′-Tetra-O-benzyl-β-D-galactopyranosyl)-3-R-fluoropropene (12)
To a solution of aldehyde 5a (490 mg, 0.87 mmol) in THF (5.0 mL) was added (S)-Jørgensen catalyst (25 mg, 0.022 mmol). The mixture was stirred at rt for 15 min at which time NFSI (540 mg, 1.7 mmol) was added, and stirring continued at rt for 6 h. The reaction was monitored and processed as described for the synthesis of 6. The crude fluoro-aldehyde was used directly in the next step. To a solution of Ph3PCH3Br (932 mg, 2.6 mmol) in anhydrous THF (10 mL) was added 2M NaHMDS in THF (1.3 mL, 2.6 mmol) at −78 °C. The resulting bright orange solution was stirred for 1 h at this temperature and then a solution of the product from the previous step in THF (10 mL) was added. The reaction mixture was warmed to rt over 2 h, then poured into saturated aqueous NH4Cl and extracted with EtOAc. The organic phase was washed with brine, dried (Na2SO4), filtered, and evaporated under reduced pressure. FCC of the residue afforded 11 (423 mg, 73%) and 12 (51 mg, 9%).
For 11: White needles (ether:methanol); m.p = 97–98 °C; Rf = 0.35 (20 % ethyl ether: 80% hexanes); [ ]D25 +53 (c 0.5, CH2Cl2); 1HNMR (500 MHz, CDCl3) δ 7.27–7.10 (m, 20H), 6.05 (m, 1H), 5.43 (br d, J = 17.5 Hz, 1H), 5.28 (d, J = 11.0 Hz, 1H), 5.02 (ddd, J = 5.5, 7.0, 46.8 Hz, 1H), 4.40–4.59 (m, 8H), 4.33 (t, J = 7.2 Hz, 1H), 4.07 (dd, J = 8.5, 11.5 Hz, 1H), 3.99 (dd, J = 2.4, 7.0 Hz, 1H), 3.72 (m, 3H); 13CNMR (125 MHz, CDCl3) δ 138.8, 138.5, 138.3, 138.0, 134.8 (d, JC-F = 17 Hz), 128.6, 128.5, 128.4, 128.2, 127.9, 127.8, 127.7, 118.0 (d, JC-F = 13 Hz), 90.2 (d, JC-F = 168 Hz), 75.3, 74.7, 74.0, 73.6, 73.5, 73.3, 73.2, 71.8, 69.6 (d, JC-F = 29 Hz), 65.8; 19FNMR (377 MHz, CDCl3) δ −190.5 (s); HRMS (ESI, MNa+) m/z calc for C37H39FNaO6 605.2679, found 605.2668.
For 12: colorless oil; Rf = 0.27 (20 % ethyl ether: 80% hexanes); [ ]D26 + 30 (c 1.1, CH2Cl2); (500 MHz, CDCl3) δ 7.30–7.15 (m, 20H), 6.01 (m, 1H), 5.32 (ddd, J = 1.0, 2.5, 17.0 Hz, 1H), 5.21 (d, J = 10.5 Hz, 1H), 5.09 (dd, J = 8.0, 47.0 Hz, 1H), 4.91 (app d, J = 10.5, 1H), 4.87 (app d, J = 11.5, 1H), 4.70–4.55 (m, 4H), 4.37 (ABq, J = 11.5, Δδ = 0.05 ppm, 2H), 4.08 (t, J = 9.5 Hz, 1H), 3.90 (d, J = 2.0 Hz, 1H), 3.53 (m, 3H), 3.46 (t, J = 6.3 Hz, 1H), 3.20 (ddd, J = 1.7, 9.7, 27.7 Hz, 1H); 13CNMR (125 MHz, CDCl3) δ 138.9, 138.5 (two peaks), 138.2, 133.6 (d, JC-F = 20 Hz), 128.7, 128.6, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 118.9 (d, JC-F = 12 Hz), 90.7 (d, JC-F = 174 Hz), 85.0, 80.9 (d, JC-F = 20 Hz), 78.0, 75.7, 74.9 (d, JC-F = 4 Hz), 74.6, 73.8, 73.5, 72.5, 69.2; 19FNMR (377 MHz, CDCl3) δ −193.7 (s); HRMS (ESI, MNa+) m/z calc for C37H39FNaO6 605.2679, found 605.2679.
3-(2′, 3′, 4′, 6′-Tetra-O-benzyl-α-D-galactopyranosyl)-3-(S)-fluoropropene (13)
Aldehyde 5a (540 mg, 0.95 mmol) was subjected to the standard fluorination-Wittig methylenation procedure with (R)-Jørgensen catalyst, as described for the synthesis of 11 and 12. FCC of the product after the methylenation step afforded 13 (309 mg, 53 %) and an unseparated mixture of fluorinated products (180 mg, ca 31%).
For 13: colorless oil; Rf = 0.06 (CHCl3); [ ]D25 +19 (c 5.5, CH2Cl2); 1HNMR (500 MHz, CDCl3) δ 7.25–7.17 (m, 20H), 5.82 (m, 1H), 5.30 (dt, J = 1.6, 18.6 Hz, 1H), 5.20 (d, J = 10.7 Hz, 1H), 5.19 (dt, J = 5.0, 47.8 Hz, 1H), 4.69–4.40 (m, 8H), 4.19 (m, 1H), 3.92 (m, 3H), 3.82 (dt, J = 4.5, 29.0 Hz, 1H), 3.70 (dd, J = 7.2, 10.3 Hz, 1H), 3.52 (dd, J = 5.1, 10.3 Hz, 1H); 13CNMR (125 MHz, CDCl3) δ 138.8, 138.7, 138.6, 138.4, 133.2 (d, JC-F = 19 Hz), 128.7, 128.6, 128.5, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 119.0 (d, JC-F = 11 Hz), 94.1 (d, JC-F = 174 Hz), 77.9, 76.2, 74.9, 74.5, 73.8, 73.6, 73.5, 73.4, 73.3 (d, JC-F = 14 Hz), 73.2, 67.9; 19FNMR (377 MHz, CDCl3) δ −184.7 (s); HRMS (ESI, MNa+) m/z calc for C37H39FNaO6 605.2679, found 605.2671.
3-(2′, 3′, 4′, 6′-Tetra-O-benzyl-α-D-galactopyranosyl)-3,3-difluoropropene (14)
To a solution of aldehyde 5a (5.30 g, 9.35 mmol) in THF (50 mL) was added (S)-Jørgensen (250 mg, 0.47 mmol) and (R)-Jørgensen catalyst (250 mg, 0.47 mmol). The mixture was stirred at rt for 15 min at which time NFSI (7.00 g, 22.4 mmol) was added, and stirring continued at rt for 12 h. The reaction mixture was concentrated at rt under reduced pressure and the residue filtered over a short column of silica gel. The filtrate was evaporated in vacuo at rt, and the crude aldehyde was used in the next step without further purification. To a solution of Ph3PCH3Br (15.0 g, 37.4 mmol) in anhydrous THF (90 mL) was added 2M NaHMDS in THF (23 mL, 46.0 mmol). The resulting bright orange solution was stirred for 1 h at this temperature at which time a solution of the crude aldehyde in THF (30 mL) was added. The reaction mixture was warmed to rt over 2 h, then processed following the general procedure for the Wittig reaction, as described for the synthesis of 11 and 12. FCC of the crude product afforded 14 (2.23 g, 40%): colorless oil; Rf = 0.17 (CHCl3); [ ]D25 +37 (c 0.4, CH2Cl2); 1HNMR (600 MHz, C6D6) δ 7.04–7.38 (m, 20H), 6.36 (m, 1H), 5.71 (bd, J = 17.5 Hz, 1H), 5.10 (d, J = 11.2 Hz, 1H), 4.57 (m, 1H), 4.35–4.50 (m, 6H), 4.31 (ABq, J = 11.0 Hz, Δδ = 0.03 ppm, 2H), 4.22 (app d, J = 11.5 Hz, 1H), 4.20 (dd, J = 7.9, 11.3 Hz, 1H), 4.06 (dd, J = 2.4, 5.3 Hz, 1H), 4.00 (m, 1H), 3.97 (dd, J = 3.5, 11.3 Hz, 1H), 3.82 (dd, J = 2.8, 4.4 Hz, 1H); 13CNMR (150 MHz, C6D6) δ 139.7, 139.2 (two peaks), 138.8, 132.9 (t, JC-F = 24 Hz), 129.3, 129.2, 129.1, 129.0, 128.9, 128.8, 128.7, 128.5, 128.1, 128.0, 127.9, 127.8, 120.9 (t, JC-F = 242 Hz), 119.3 (t, JC-F = 10 Hz), 76.1 (two peaks), 76.0, 75.9, 74.5, 73.7 (two peaks), 72.9, 72.3 (t, JC-F = 31 Hz), 67.0; 19FNMR (377 MHz, CDCl3) δ −103.2 (ABq, J = 264 Hz, Δδ = 7.3 ppm, 2F); HRMS (ESI, MNa+) m/z calc for C37H38F2NaO6 623.2585, found 623.2571.
Synthesis of difluoroalkene 14 from alcohol 10
To a solution of alcohol 10 (40 mg, 0.066 mmol) in dry DCM (1 mL) was added Dess-Martin periodinane solution (0.1 mL of a 15% wt/vol in DCM, 0.035 mmol). The reaction mixture was stirred at rt for 20 min, then concentrated at rt under reduced pressure. Next, the residue was filtered over a short column of silica gel, and the filtrate evaporated in vacuo at rt. The crude aldehyde product was used in the next step without further purification. To a solution of Ph3PCH3Br (400 mg, 1.1 mmol) in anhydrous THF (1 mL) was added 2M NaHMDS in THF (0.7 mL, 1.4 mmol). The resulting bright orange solution was stirred for 1 h at this temperature at which time a solution of the crude aldehyde in THF (1.0 mL) was added. The reaction mixture was warmed to rt over 2 h, then processed following the standard procedure for Wittig reactions. FCC of the crude product afforded 14 (32 mg, 80%).
2-(2′, 3′, 4′, 6′-Tetra-O-benzyl-β-D-galactopyranosyl)-2,2-difluoroethanol (15)
Aldehyde 5b (65 mg, 0.115 mmol) was subjected to the fluorination procedure described for 6, with NFSI (181 mg, 0.570 mmol), (R)-Jorgensen catalyst (10 mg, 0.017 mmol) and and (S)-Jorgensen catalyst (10 mg, 0.017 mmol) for 18 h. Reduction of the crude product with NaBH4 following the standard procedure and FCC of the product afforded 15 (11 mg, 16%): colorless oil; Rf = 0.60 (5% acetone: 95% CH2Cl2); [ ]D20 +20 (c 0.1, CHCl3); 1HNMR (600 MHz, C6D6) δ 7.40–7.05 (m, 20H), 4.84 (ABq, J = 10.7 Hz, Δδ = 0.14 ppm, 2H), 4.74 (ABq, J = 11.6 Hz, Δδ = 0.40 ppm, 2H), 4.45 (t, J = 9.4 Hz, 1H), 4.41 (ABq, J = 11.8 Hz, Δδ = 0.02 ppm, 2H), 4.38 (br m, 5H), 4.22 (ABq, J = 11.8 Hz, Δδ = 0.10 ppm, 2H), 3.91 (br m, 1H), 3.78 (br m, 2H), 3.70 (d, J = 1.90 Hz, 1H), 3.52 (dd, J = 6.3, 9.2 1H), 3.47 (dd, J = 6.6, 9.2 Hz, 1H), 3.34 (dd, J = 2.8, 9.3 Hz, 1H), 3.30 (t, J = 6.5 Hz, 1H), 2.15 (t, J = 6.5 Hz, 1H); 13CNMR (150 MHz, C6D6) δ 139.6, 139.5, 139.2, 138.9, 130.0–127.0 (several lines buried under C6D6 triplet), 121.9 (t, JC-F = 245 Hz), 85.2, 78.2 (t, JC-F = 28 Hz), 77.9, 75.5, 75.3, 75.1 (d, JC-F = 3 Hz), 74.3, 74.0, 73.0, 69.5, 64.2 (t, JC-F = 32 Hz); 19FNMR (377 MHz, CDCl3) δ −114.3 (ABq, J = 264 Hz, Δδ = 6.6 ppm, 2F); HRMS (ESI, MNa+) m/z calc for C36H38F2NaO6 627.2534, found 627.2521.
General procedure for correlation of allylic fluorides and fluorohydrin derivatives: Transformation of 11 to 6
A stream of O3/O2 was bubbled through a solution of 11 (50 mg, 0.09 mmol) in DCM 20 mL at −78 °C, for 5 min. PPh3 (50 mg, 0.19 mol) was then added to the mixture and stirring continued at rt for an additional 2 h. The volatiles were evaporated in vacuo, and NaBH4 (30 mg, 0.8 mmol) was then slowly added at 0 °C to a solution of the residue in MeOH (2 mL). The reaction mixture was processed following the standard procedure for NaBH4 reduction. FCC of the crude product provided 6 (31 mg, 62%).
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (#1301330). A Research Centers in Minority Institutions Program grant from the National Institute of Health Disparities (MD007599) of the National Institutes of Health (NIH), which supports the infrastructure at Hunter College, is also acknowledged. We thank Dr. Matthew Devany for help with NMR experiments.
Footnotes
Electronic Supplementary Information (ESI) available: Copies of 1H, 13C, 2D NMR spectra and tabulated peak assignments for 0 and 00 – 00, X-ray data for 00,. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Hagmann WK. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 2.O’Hagan D. Chem Soc Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 3.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 4.Champagne PA, Desroches J, Hamel J-D, Vandamme M, Paquin J-F. Chem Rev. 2015;115 doi: 10.1021/cr500706a. ASAP. doi:1021/cr500706a. [DOI] [PubMed] [Google Scholar]
- 5.Yang X, Wu T, Phipps RJ, Toste DF. Chem Rev. 2015;115:826–870. doi: 10.1021/cr500277b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valero G, Companyó X, Rios R. Chem Eur J. 2011:17, 2018–2037. doi: 10.1002/chem.201001546. [DOI] [PubMed] [Google Scholar]
- 7.Schedler D, Baker DC. Carbohydr Res. 2004;339:1585–1595. doi: 10.1016/j.carres.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 8.Cheng Y, Guo A-L, Guo D-S. Cur Org Chem. 2010;14:977–999. [Google Scholar]
- 9.Leclerc E, Pannecoucke X, Ethève-Queiquejeu M, Sollogoub M. Chem Soc Rev. 2013;42:4270–4283. doi: 10.1039/c2cs35403a. [DOI] [PubMed] [Google Scholar]
- 10.Plantier-Royon R, Portella C. Carbohydr Res. 2000;327:119–146. doi: 10.1016/s0008-6215(00)00034-3. [DOI] [PubMed] [Google Scholar]
- 11.Jiménez-Barbero J, Demange R, Schenk K, Vogel P. J Org Chem. 2001;66:5132–5138. doi: 10.1021/jo0102462. [DOI] [PubMed] [Google Scholar]
- 12.Marcotte S, D’Hooge F, Ramadas S, Feasson C, Pannecoucke X, Quirion J-C. Tetrahedron Lett. 2001;42:5879–5882. [Google Scholar]
- 13.Picard J, Lubin-Germain JN, Uziei J, Augé J. Synthesis. 2006:979–982. [Google Scholar]
- 14.Tony KA, Denton RW, Dilhas A, Jiménez-Barbero J, Mootoo DR. Org Lett. 2007;9:1441–1444. doi: 10.1021/ol070169i. [DOI] [PubMed] [Google Scholar]
- 15.Pérez-Castells J, Hernández-Gay JJ, Denton RW, Tony KA, Mootoo DR, DR, Jiménez-Barbero J. Org Biomol Chem. 2007;5:1087–1092. doi: 10.1039/b615752a. [DOI] [PubMed] [Google Scholar]
- 16.Poulain F, Serre A-L, Lalot J, Leclerc E, Quirion J-C. J Org Chem. 2008;73:2435–2438. doi: 10.1021/jo702466h. [DOI] [PubMed] [Google Scholar]
- 17.Colombel S, Van Hijfte N, Poisson T, Leclerc E, Pannecoucke X. Chem Eur J. 2013;19:12778–12787. doi: 10.1002/chem.201302070. [DOI] [PubMed] [Google Scholar]
- 18.Colombel S, Van Hijfte N, Poisson T, Pannecoucke X, Monneaux F, Leclerc E. J Fluorine Chem. 2015;173:84–91. [Google Scholar]
- 19.For examples of general strategies for transforming C- glycoside alkenes and aldehydes to more complex C-glycosides, Gustafsson T, Saxin M, Kihlberg J. J Org Chem. 2003;68:2506–2509. doi: 10.1021/jo026758d., and ref. 19–23.
- 20.Wipf P, Pierce JG. Org Lett. 2006;8:3375–3378. doi: 10.1021/ol0613057. [DOI] [PubMed] [Google Scholar]
- 21.Pu J, Franck RW. Tetrahedron. 2008;64:8618–8629. doi: 10.1016/j.tet.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thota VN, Gervay-Hague J, Kulkarni SS. Org Biomol Chem. 2012;10:8132–8139. doi: 10.1039/c2ob26078f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thota VN, Brahmaiah M, Kulkarni SS. J Org Chem. 2013;78:12082–12089. doi: 10.1021/jo402115w. [DOI] [PubMed] [Google Scholar]
- 24.Altiti AS, Mootoo DR. Org Lett. 2014;16:1466–1469. doi: 10.1021/ol5002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marigo M, Fielenbach D, Braunton A, Kjærsgaard A, Jørgensen KA. Angew Chem Int Ed. 2005;44:3703–3706. doi: 10.1002/anie.200500395. [DOI] [PubMed] [Google Scholar]
- 26.Steiner DD, Mase N, Barbas CF., III Angew Chem Int Ed. 2005;44:3706–3710. doi: 10.1002/anie.200500571. [DOI] [PubMed] [Google Scholar]
- 27.Beeson TD, MacMillan DWC. J Am Chem Soc. 2005;127:8826–8828. doi: 10.1021/ja051805f. [DOI] [PubMed] [Google Scholar]
- 28.Nuzzi A, Massi A, Dondoni A. Org Lett. 2008;10:4485–4488. doi: 10.1021/ol801685x. [DOI] [PubMed] [Google Scholar]
- 29.Nolen EG, Watts MM, Fowler DJ. Org Lett. 2002;4:3963–3965. doi: 10.1021/ol026839w. [DOI] [PubMed] [Google Scholar]
- 30.For related one-pot reactions on simple aldehydes using the Jørgensen catalyst, Jiang H, Falcicchio A, Jensen KL, Paixão MW, Bertelsen S, Jørgensen KA. J Am Chem Soc. 2009;131:7153–7157. doi: 10.1021/ja901459z.
- 31.Jimenez-Barbero J, Espinosa JF, Asensio JL, Cañada FJ, Poveda A. Adv Carbohydr Chem Biochem. 2001;56:235–284. doi: 10.1016/s0065-2318(01)56006-0. [DOI] [PubMed] [Google Scholar]
- 32.Massi A, Nuzzi A, Dondoni A. J Org Chem. 2007;72:10279–10282. doi: 10.1021/jo701959b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.