

Abstract
Population aging simultaneously highlights the remarkable advances in science, medicine, and public policy, and the formidable challenges facing society. Indeed, aging is the primary risk factor for many of the most common chronic diseases and frailty, which have profound social and economic costs. Population aging also reveals an opportunity; that is, interventions to disrupt the fundamental biology of aging could significantly delay the onset of age-related conditions as a group, and as a result, extend healthy lifespan, or healthspan. There is now considerable evidence that cellular senescence is an underlying mechanism of aging and age-related conditions. Cellular senescence is a process in which cells lose the ability to divide and damage neighboring cells by the factors they secrete, collectively referred to as the senescence-associated secretory phenotype (SASP). Herein, we discuss the concept of cellular senescence, review the evidence that implicates cellular senescence and the SASP in age-related deterioration, hyperproliferation, and inflammation, and propose that this underlying mechanism of aging may play a fundamental role in the biology of frailty.
Introduction
Population aging results from the many remarkable achievements in science, medicine, and public policy. Indeed, in developed countries, innovations in antibiotics, vaccines, medications, sanitation, hygiene, and working conditions have increased average life expectancy at birth from roughly 45 years in the early 1900s to approximately 80 years today. By 2050, over 21% of the global population, or two billion persons, will be over the age of 60 [1].
This remarkable accomplishment also poses a formidable challenge to public health. Aging is the major risk factor for cardiovascular disease, cancer, Alzheimer’s disease, diabetes, arthritis, osteoporosis, blindness, and several other chronic conditions with profound consequences and enormous medical costs. For many, aging also leads to the incapacity to respond to stress and return to homeostasis (Figure 1A). The age-related loss of resiliency and increased vulnerability is now widely considered a geriatric health condition, termed frailty [2, 3]. The clinical definition and measurement of frailty are not universally agreed upon; however, it is clear that patients deemed frail by the existing tools suffer a host of adverse health outcomes, including perioperative complications, increased length of hospital stay, disability, loss of independence, institutionalization, and death [4–7].
Figure 1. Aging and Healthspan.
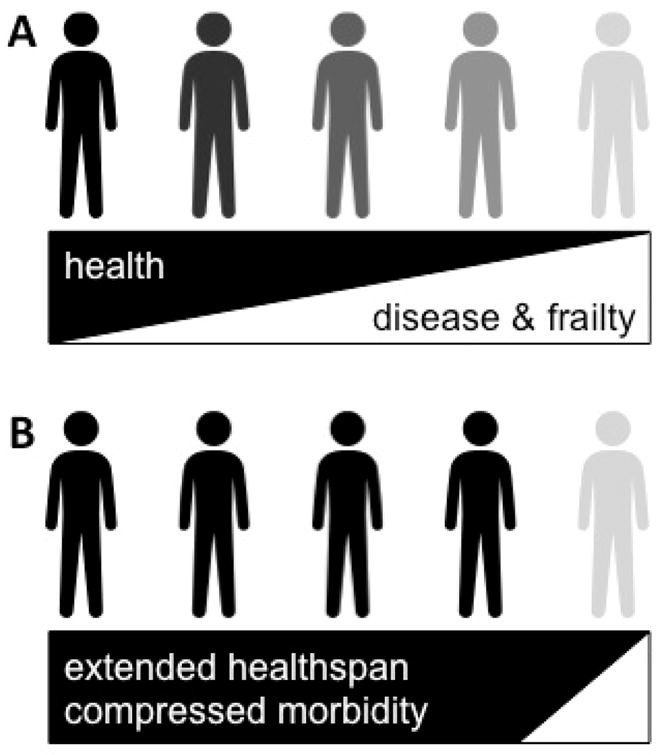
Aging is the primary risk factor of the majority of chronic diseases and frailty. As a result, the period of life in good health and relatively free from the burden of disease, disability, and frailty, termed healthspan, progressively declines (A). Interventions that target the underlying biology of aging, as opposed to individual diseases, have the potential to delay the onset of age-related conditions as a group. This would have a profound impact on extending healthspan and dramatically compress the period of morbidity at the end of life (B).
On the one hand, age-related chronic disease and frailty are enormous financial and social threats to society. On the other, with aging at their root, they represent an exciting opportunity. Specifically, interventions that disrupt the fundamental biology of aging have the potential to i) delay the onset of age-related chronic diseases and frailty as a group, ii) significantly extend healthy lifespan, and iii) compress end-of-life morbidity (Figure 1B). This concept has recently developed into the interdisciplinary field of geroscience. The objective of this manuscript is to outline the evidence that supports the biological process of cellular senescence as a unifying mechanism of aging, age-related disease, and frailty. The potential for therapies that target senescent cells to delay aging and the onset of age-related disease and frailty will also be discussed.
Cellular Senescence, the SASP, and Aging
Aging is a consequence of gradual life-long accumulation of molecular and cellular damage that is manifest in deterioration (e.g., as evident in the cardiovascular, metabolic, musculoskeletal, and nervous systems), hyperproliferation (i.e., as in the aberrant growth of malignant cells), and chronic, low grade, sterile inflammation (inflammaging). These phenotypes of aging have fueled a search for a unifying mechanism. In the 1960’s, Hayflick and Moorehead introduced the term cellular senescence to describe the state of permanent cellular growth arrest that occurred in normal human fibroblasts after extensive serial passaging in culture [8]. Although they postulated that replicative senescence might play a role in aging, it has been more widely appreciated as a fundamental anticancer defense. Subsequent studies have demonstrated that in addition to replication-induced senescence, cellular senescence is associated with diverse stimuli including telomere erosion, DNA lesions, reactive oxygen species (ROS), and other mitogenic and metabolic stressors [9]. The induction of “stable” growth arrest is associated with activation of the tumor suppressor pathways, p16INK4a/retinoblastoma protein (Rb) and/or p53/p21. Together these mechanisms of senescence limit excessive or aberrant growth of malignant cells. Intriguingly, several senescence-inducing stressors are also the foundation of several theories of aging (i.e., telomere erosion, DNA damage and mutation, protein aggregation, and ROS). However, instead of preventing the growth of cancers, the accumulation of senescent cells with advancing age negatively affects tissue structure and function, and ultimately leads to tissue pathology, as discussed herein. Thus, cellular senescence is an example of antagonistic pleiotropy; that is, a biological mechanism that was selected through evolution to enhance early life fitness that has late life costs.
How do senescent cells lead to age-associated tissue deterioration, hyperproliferation, and inflammation? The abundance of senescent cells increases in multiple tissues with chronological aging [10, 11]. Senescent cells were first distinguished from quiescent and terminally differentiated cells by their expression of a pH-dependent β-galactosidase (SA-β-Gal) [12]. Now, elements of the Rb and p53 signaling pathways, including p16INK4a and p21Cip1/Waf1, encoded by Cdkn2a and Cdkn1a, respectively, are mediators and also biomarkers of senescence [13, 14]. Though they are quite modest in number, senescent cells contribute to aging through three paths. First, the net accumulation of senescent cells, which have a markedly altered morphology and gene expression profile, may reach a point that compromises a tissue’s functionality. Specifically, senescent cells reorganize chromatin, which results in heterochromatin formation, increased cell size and protein content, and changes in organelle and cell shape [15, 16]. Second, senescence limits the regenerative potential of stem cell pools and undifferentiated progenitor cell pools (thoughtfully reviewed in [17]). As a result, the capacity to regenerate damaged and atrophic tissues declines. Moreover, senescent cells could be of great consequence to tissues that require “homeostatic cellular replication”, including the bone marrow, thymus, and endocrine pancreas [9]. Third, and potentially most interesting with regards to the etiology of age-related diseases and frailty, senescent cells are active factories that secrete a broad repertoire of cytokines and chemokines, matrix remodeling proteases, and growth factors, collectively referred to as the senescence-associated secretory phenotype (SASP) (reviewed in [18]). Importantly, SASP factors vary in distinct cell types and under different senescence-inducing stressors [19]. This plasticity within SASP composition predicts variability with respect to the biological processes impacted by different kinds of senescent cells. Recent data further suggest that senescence likely spreads to neighboring cells through components of the SASP, which, in turn, intensifies age-related tissue deterioration [20]. Together, these consequences of cellular senescence lead to age-related tissue deterioration (due to accumulation and spread of senescent cells, the loss of regenerative potential, and degradation of the extracellular matrix), hyperproliferation, or the growth and spread of cancers (paradoxically, through components of the SASP including growth factors and matrix remodeling proteases), and inflammaging (as cytokines and chemokines are prominent features of the SASP). Thus, cellular senescence has emerged as a potential unifying mechanism of aging, age-related diseases, and frailty.
Cellular Senescence and Age-Related Disease and Frailty
Inflammation is a hallmark of age-related chronic diseases and frailty. Factors such as interleukin 6 (IL-6), tumor necrosis factor α (TNFα), and acute phase proteins such as C-reactive protein (CRP) are now recognized as pathogenic factors in the development of cardiovascular disease, diabetes, cancer, depression, and dementias, as well as frailty (e.g., [21–27]). The cause and source of inflammation in these conditions has remained elusive. Although the immune system plays a major role in modulating the levels of pro- and anti-inflammatory factors, it is not the only source of these factors. It is plausible that cellular senescence and the SASP could serve as a significant source of inflammation and play a substantial role in the genesis and/or progression of age-related diseases and frailty.
In support of this concept, biomarkers of senescent cells, including p16INK4a and SA-β-gal, and components of the SASP, including TNFα, IL-6, matrix metalloproteinases (MMPs), monocyte chemoattractant protein-1 (MCP-1), and insulin-like growth factor binding proteins (IGFBPs), increase in multiple tissues with chronological aging [10, 14, 18, 28]. Even more noteworthy is that SA-β-gal, p16ink4a and/or p21 are elevated in the affected tissues of patients with age-related conditions, including osteoarthritis, pulmonary fibrosis, atherosclerosis, and Alzheimer’s disease (extensively reviewed in [29]). However, the extent to which senescent cells residing at the site of active pathology account for the onset and/or progression of these diseases is unknown, but is an active area of investigation by our laboratories and others. With regard to frailty, ongoing work will help determine what tissues (e.g., adipose, spleen, etc.) and systems (e.g., immune, endocrine, etc.) are affected by senescence, and whether or not senescent cells and the SASP negatively affect resiliency, functional reserves, and systemic inflammation. Answers to these questions and others will help to determine the therapeutic potential of therapies to selectively remove senescent cells and/or suppress the SASP.
Interventions Targeting Senescent Cells and the SASP
In light of the growing body of data implicating senescent cells in age-related disease and frailty, a number of strategies are being considered to mitigate there deleterious effects. First, the cell stresses and signaling pathways that lead to senescence-associated growth arrest could be targeted by therapeutic interventions. On the one hand, preventing cell damage could be an effective means to limit senescent cell accumulation. On the other, however, interfering with tumor suppressor pathways such as Rb, p16INK4A or p53, would compromise fundamental anticancer mechanisms and pose significant risk. Second, interventions could be devised to selectively eliminate senescent cells and, in turn, reduce tissue dysfunction and inflammation. As proof of concept, we designed a transgenic mouse model in which senescent cells could be inducibly eliminated. In this model, which we termed INK-ATTAC, a transgene that encodes caspase 8 fused to a mutated version of an FKBP domain is selectively expressed in p16Ink4a-positive senescent cells. Upon administration of the synthetic drug AP20187, FKBP-caspase 8 fusion proteins homodimerize and activate a chain of events that triggers apoptosis of p16Ink4a-positive cells. Recently, we demonstrated that in a BubR1 progeroid (rapid aging) mouse background, INK-ATTAC removes p16Ink4a-positive senescent cells upon drug treatment [30]. In tissues –such as adipose tissue, skeletal muscle, and eye– in which p16Ink4a contributes to the acquisition of age-related pathologies, lifelong removal of p16Ink4a-expressing cells delayed onset of these phenotypes and improved physical function. Furthermore, late-life clearance attenuated progression of already established age-related disorders. The findings demonstrate that senescent cells and the SASP contribute to age-related tissue dysfunction. Importantly, we observed no overt side effects of senescent cell clearance in our model, even though it has been postulated that senescent cells might enhance certain types of tissue repair. The translation of these findings to humans is contingent upon the ability to specifically target senescent cells using biological or small molecule “senolytic” therapies. We have very recently shown that deletion of p16ink4a-expressing cells through a pharmacological strategy can improve multiple aging-related phenotypes, including cardiac function, vascular reactivity, bone microarchitecture, and exercise capacity, in mouse models of aging [31]. The unique morphology and gene expression profile of senescent cells suggest they may either harbor unique epitopes that could be exploited by antibody-based therapies, or be more sensitive than non-senescent cells to small molecule senolytic agents. The third approach to prevent the deleterious effects of senescent cells is to prevent or ameliorate the effects of the SASP. This is an attractive approach given senescent cells are a major source of tissue inflammation, which, in turn, is a fundamental component of several age-related diseases and frailty. Moreover, the SASP could theoretically be disrupted without interfering with the anti-oncogenic pathways activated in senescent cells.
In parallel with the research and development of interventions to eliminate senescent cells and diminish the SASP, the clinical opportunities to test the effectiveness of senolytic agents are being discussed. Frailty offers a unique indication, and we are actively researching the association, if not the fundamental contribution, of cellular senescence to frailty syndrome. If, in fact, patients with high compared to low senescent cell burden experience worse short-term outcomes (e.g., more complications, higher incidence of delirium, longer length of stay) and longer-term outcomes (more readmissions, greater dependence in activities of daily living, delayed recover of function, higher institutionalization) following a medical intervention, then the efficacy of senolytics at improving these clinically important and patient-centered metrics could be evaluated. While tantalizing, several obstacles need to be overcome before this can be achieved. For example, biomarkers of senescent cell burden and SASP activity in humans need to be developed and validated. Moreover, frequency and duration of treatment regimens to reduce senescent cell number and SASP activity, without unwanted side effects, need to be defined. In fact, it is plausible that senescent cell removal could actually impair an older person’s response to infection, wound healing, and other stressors. Nonetheless, frailty offers a possible scenario in which eliminating senescent cells or SASP components would result in a rapidly detectable, clinically meaningful response.
Conclusions
Aging has long been appreciated to be the major risk factor for most chronic diseases and frailty, but deemed unalterable. However, recent and transformative breakthroughs in the biology of aging have challenged this assumption. In particular, significant advances in our understanding of cellular senescence and the SASP along with proof of concept studies in preclinical models, suggest that interventions to disrupt this fundamental mechanism of aging could have a profound effect on healthspan by delaying age-related conditions as a group. By compressing late-life morbidity, such interventions could have a transformative effect on public health. While the search is on for pharmacological strategies to delay aging, significant work is needed to further understand the biology of cellular senescence and its role in the pathogenesis of age-related diseases and frailty. Moreover, understanding how lifestyle choices, including diet and physical activity, can be optimized to counter the biology of aging may simultaneously provide a readily implementable and scalable means to extend healthspan.
Acknowledgments
The authors would like to acknowledge the support of the National Institutes of Health (AG41122), the Paul F. Glenn Foundation for Medical Research, Mayo Clinic, the Hoeft Family, the Noaber Foundation, and a generous gift from Robert and Arlene Kogod.
References
- 1.Cohen JE. Human population: the next half century. Science. 2003;302:1172–5. doi: 10.1126/science.1088665. [DOI] [PubMed] [Google Scholar]
- 2.Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci. 2001;56:M146–56. doi: 10.1093/gerona/56.3.m146. [DOI] [PubMed] [Google Scholar]
- 3.Lipsitz LA. Physiological complexity, aging, and the path to frailty. Sci Aging Knowledge Environ. 2004;2004:pe16. doi: 10.1126/sageke.2004.16.pe16. [DOI] [PubMed] [Google Scholar]
- 4.Makary MA, Segev DL, Pronovost PJ, et al. Frailty as a Predictor of Surgical Outcomes in Older Patients. Journal of the American College of Surgeons. 2010;210:901–908. doi: 10.1016/j.jamcollsurg.2010.01.028. [DOI] [PubMed] [Google Scholar]
- 5.Ensrud Ke, ESKTBC, et al. Comparison of 2 frailty indexes for prediction of falls, disability, fractures, and death in older women. Archives of Internal Medicine. 2008;168:382–389. doi: 10.1001/archinternmed.2007.113. [DOI] [PubMed] [Google Scholar]
- 6.Mitnitski A, Graham J, Mogilner A, et al. Frailty, fitness and late-life mortality in relation to chronological and biological age. BMC Geriatrics. 2002;2:1. doi: 10.1186/1471-2318-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dasgupta M, Rolfson DB, Stolee P, et al. Frailty is associated with postoperative complications in older adults with medical problems. Arch Gerontol Geriatr. 2009;48:78–83. doi: 10.1016/j.archger.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 9.Newgard CB, Sharpless NE. Coming of age: molecular drivers of aging and therapeutic opportunities. J Clin Invest. 2013;123:946–50. doi: 10.1172/JCI68833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waaijer ME, Parish WE, Strongitharm BH, et al. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell. 2012;11:722–5. doi: 10.1111/j.1474-9726.2012.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herbig U, Ferreira M, Condel L, et al. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 12.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumont P, Burton M, Chen QM, et al. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic Biol Med. 2000;28:361–73. doi: 10.1016/s0891-5849(99)00249-x. [DOI] [PubMed] [Google Scholar]
- 14.Krishnamurthy J, Torrice C, Ramsey MR, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–56. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Cecco M, Jeyapalan J, Zhao X, et al. Nuclear protein accumulation in cellular senescence and organismal aging revealed with a novel single-cell resolution fluorescence microscopy assay. Aging (Albany NY) 2011;3:955–67. doi: 10.18632/aging.100372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–33. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Tchkonia T, Zhu Y, van Deursen J, et al. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966–72. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Acosta JC, Banito A, Wuestefeld T, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–90. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franceschi C, Bonafe M, Valensin S, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 22.Tuomisto K, Jousilahti P, Sundvall J, et al. C-reactive protein, interleukin-6 and tumor necrosis factor alpha as predictors of incident coronary and cardiovascular events and total mortality. A population-based, prospective study. Thromb Haemost. 2006;95:511–8. doi: 10.1160/TH05-08-0571. [DOI] [PubMed] [Google Scholar]
- 23.Cesari M, Penninx BW, Newman AB, et al. Inflammatory markers and cardiovascular disease (The Health, Aging and Body Composition [Health ABC] Study) Am J Cardiol. 2003;92:522–8. doi: 10.1016/s0002-9149(03)00718-5. [DOI] [PubMed] [Google Scholar]
- 24.Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31:37–49. doi: 10.1093/carcin/bgp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spranger J, Kroke A, Mohlig M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–7. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- 26.Walston J, McBurnie MA, Newman A, et al. Frailty and activation of the inflammation and coagulation systems with and without clinical comorbidities: results from the Cardiovascular Health Study. Arch Intern Med. 2002;162:2333–41. doi: 10.1001/archinte.162.20.2333. [DOI] [PubMed] [Google Scholar]
- 27.Collerton J, Martin-Ruiz C, Davies K, et al. Frailty and the role of inflammation, immunosenescence and cellular ageing in the very old: cross-sectional findings from the Newcastle 85+ Study. Mech Ageing Dev. 2012;133:456–66. doi: 10.1016/j.mad.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Freund A, Orjalo AV, Desprez PY, et al. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16:238–46. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naylor RM, Baker DJ, van Deursen JM. Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin Pharmacol Ther. 2013;93:105–16. doi: 10.1038/clpt.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu Y, Tchkonia T, Pirtskhalava T, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging cell. 2015;14:644–58. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]