

Abstract
Background/Aims
Statins act as antineoplastic agents through the inhibition of cell proliferation. This study sought to demonstrate the effects of statins on extrahepatic bile duct cancer cell apoptosis and to document the changes in protein expression involved in tumor growth and suppression.
Methods
Human extrahepatic bile duct cancer cells were cultured. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were performed to determine the effect of statins on cell proliferation. Apoptosis was measured by a cell death detection enzyme-linked immunosorbent assay and caspase-3 activity assay, and flow cytometry was used to determine the percentage of cells in each phase of the cell cycle. The protein expression of Bax, Bcl-2, insulin-like growth factor 1 (IGF-1) receptor, extracellular signal-regulated kinase 1/2 (ERK1/2), and Akt was measured by Western blot analysis.
Results
Simvastatin suppressed cell proliferation by inducing G1 phase cell cycle arrest in bile duct cancer cells. Furthermore, it induced apoptosis via caspase-3 activation, downregulated the expression of the Bcl-2 protein, and enhanced the expression of the Bax protein. Moreover, simvastatin suppressed the expression of the IGF-1 receptor and IGF-1-induced ERK/Akt activation.
Conclusions
Simvastatin induces apoptosis in bile duct cancer cells, which suggests that it could be an antineoplastic agent for bile duct cancer.
Keywords: Simvastatin, Bile duct neoplasms, Apoptosis, Receptor, IGF type 1, Bax
INTRODUCTION
Cholangiocarcinoma is the most common malignant tumor in the extrahepatic bile duct, and its worldwide incidence and mortality rate are increasing.1 Early diagnosis through screening is not easy, because most bile duct cancers arise in the absence of underlying risk factors, and subjective symptoms usually appear in late stages. Accordingly, the 5-year survival rate is approximately 20% to 40%, even in patients with tumor-free margins after surgical resection.2 Palliative therapies, including biliary drainage, provide patients with less than 18 months survival, and untreated patients usually die within 6 months to a year after diagnosis.3
Conventional chemotherapy and radiation therapy for bile duct cancer were not remarkably effective in improving survival.1 However, recent studies showed that adjuvant gemcitabine-based chemotherapy can improve survival after surgical resection,4 and that combination chemotherapy with gemcitabine and cisplatin has a modest effect in patients with metastatic or unresectable bile duct cancer.5 New agents, such as cyclooxygenase 2 (COX-2) inhibitors, COX-2 gene expression inhibitors, epidermal growth factor receptor inhibitors, cetuximab, Raf kinase inhibitors, peroxisome proliferator-activated receptor γ (PPARγ) agonists, and tumor necrosis factor (TNF) family cytokines, are on trial to develop novel chemopreventive and adjuvant therapeutic agents, based on selective molecular targets that may affect the growth and behavior of cholangiocarcinoma.1,6,7 However, more additional trials are still needed before clinical application.
Recently, statins have received attention, because they are known to be anti-inflammatory and anticancer agents through the inhibition of cell proliferation, induction of apoptosis, or inhibition of angiogenesis, beyond its lipid-lowering effects. These effects are due to the suppression of the mevalonate pathway,8–11 and 3-hydroxy-3-methylglutaryl-coenzyme A reductase expression has been demonstrated in cholangiocarcinoma cell lines and the normal human cholangiocyte cell line.12 Furthermore, in an epidemiological study, statins were associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes.13 Another study indicated that the use of statins is also associated with a small to considerable reduction in the risk of colon cancer.14,15
Our study using biliary epithelial cells revealed that statins activate PPARγ and inhibit TNF-α production,16 which are known biomarkers and therapeutic targets for cholangiocarcinoma. This study showed that statins could be used as antineoplastic agents for bile duct cancer. However, the effect of statins on the growth and progression of bile duct cancer remains unclear. Moreover, the effects of statins on genes and proteins associated with tumor growth and suppression is unknown.
The insulin-like growth factor 1 (IGF-1)/IGF-1 receptor (IGF-1R) system plays a crucial role in the carcinogenesis and progression of gastrointestinal cancers.17 Ligand binding to the external subunit of the IGF-1R activates multiple signaling pathways, including the mitogen-activated protein kinase (MAPK, also known as extracellular signal-regulated kinase [ERK]) and phosphatidylinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway. These pathways have antiapoptotic effects, and can induce transcription, metabolism, proliferation, and growth.18,19 It is also known that human intrahepatic cholangiocarcinomas express IGF-1R, which modulates cell growth and apoptosis. Accordingly, modulation of IGF-1R signaling could represent a strategy for the management of cholangiocarcinoma.20 However, the effect of statins on IGF-1 signaling in the modulation of growth and progression of bile duct cancer is unknown.
This study evaluated the effects of statins on bile duct cancer cell growth and apoptosis, and documented the changes in protein and gene expression involved in tumor growth and suppression.
MATERIALS AND METHODS
1. Materials
RPMI 1640 medium, fetal bovine serum (FBS), trypsin/ethylenediaminetetraacetic acid (EDTA), and penicillin/streptomycin were from Gibco (Grand Island, NY, USA). Pravastatin and simvastatin were from Calbiochem (Gibbstown, NJ, USA). Dimethyl sulfoxide (DMSO) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were from Sigma Aldrich (St. Louis, MO, USA). The phospho-p44/42 MAPK, p44/42 MAPK, phospho-Akt, Akt, and β-actin antibodies were from Cell Signaling Technology (Danvers, MA, USA). The IGF-1Rβ antibody and goat antirabbit IgG-horseradish peroxidase (HRP) were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The Amersham ECLTM Advance Western Blotting Detection Kit was from Amersham Biosciences (Buckinghamshire, UK).
2. Cell culture
Bile duct cancer cells (SNU-245) harvested from well-differentiated common bile duct cancer without major gene mutations were purchased from the Korean Cell Line Bank.21 Cells were cultured in RPMI 1640 medium with 2 mM glutamine, 25 mM sodium bicarbonate, and 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) supplemented with 10% FBS, 100 IU/mL penicillin, and 100 μg/mL streptomycin. The medium was changed twice per week, and the cells were maintained in 37°C incubator with 5% CO2. The cells were sub-cultured when confluent (every 5 to 7 days) using trypsin (2.5 g/L) and EDTA (1 g/L).
3. MTT assay
Cell proliferation was measured by the MTT assay. Cells were seeded at a density of 5×109 cells/mL in a 96-well plate. After incubation for 24 hours, cells were treated with various concentration of pravastatin or simvastatin in serum-free medium for 24 or 48 hours. MTT (0.5 mg/mL) was added to each well and incubated for an additional 4 hours at 37°C. After the medium was removed, 100 μL DMSO was added to each well. The plate was gently shaken for 10 minutes, and the color intensity (proportional to the number of live cells) was evaluated with a DTX 880 Multimode Detector (Beckman Coulter, Brea, CA, USA) at 570 nm wavelength.
4. Cell cycle analysis
Cells were seeded at a concentration of 1×104 cells/well in a 6-well plate and incubated for 48 or 72 hours. Cells were treated with various concentrations of simvastatin for 24 or 48 hours. Cells were harvested with 0.25% trypsin-EDTA and washed with phosphate buffered saline (PBS). Cells were then centrifuged twice at 1,400 g for 5 minutes and fixed with 70% ethanol (1 mL) at −20°C for 1 hour. After centrifugation at 1,400 g for 5 minutes, the supernatant was removed, and cells were incubated with 1 mL PBS containing 10 μL RNAse (10 μg/mL; Sigma Aldrich) and 20 μL propidium iodine (1 mg/mL; Sigma Aldrich) at 37°C for 1 hour. The cell cycle was analyzed using a FACS-Calibur flow cytometer and CellQuest software (BD Biosciences, San Jose, CA, USA). Flow cytometry was used to determine the percentage of cells in each phase of the cell cycle.
5. Cell apoptosis assay
Cell apoptosis was assessed by the detection of mono-oligo-nucleosomes (histone-associated DNA fragments) using the cell death detection ELISA plus kit (Roche Applied Science, Mannheim, Germany), according to the manufacturer’s instructions. Cells were seeded in a 96-well plate at a density of 1×104 cells/well and incubated for 24 hours. Cells were treated with various concentrations of simvastatin for 24 or 48 hours. After the medium was removed, cells were treated with 100 μL lysis buffer included in the kit for 30 minutes and centrifuged at 200 g at 4°C for 10 minutes. The supernatant (cell lysate solutions) was placed in the wells of a streptavidin-coated plate supplied by the manufacturer. A mixture of anti-histone-biotin and anti-DNA-peroxidase antibodies was added to the cell lysate and incubated for 2 hours. After washing, 100 μL 2,2′-azinobis-3-ethyl-benzothiazoline-6-sulfonic acid (ABST) substrate was added to each well for 20 minutes. Absorbance at 405 nm was measured with a DTX 880 Multimode Detector (Beckman Coulter).
6. Caspase-3 activity assay
A caspase-3 activity assay kit (BioVision, Mountain View, CA, USA) was used to measure caspase-3 activity according to the manufacturer’s instructions. Cells were plated on 60 mm dishes at a density of 2×106 cells/mL and treated with various concentrations of simvastatin for 24 or 48 hours. Cells were washed with PBS and harvested with lysis buffer included in the kit. Cells were maintained on ice for 10 minutes. Cell lysates were centrifuged at 4°C and 12,000 g, and the supernatant was transferred to a new tube and stored on ice. Protein content was analyzed using the Bradford assay (Sigma Aldrich). Assays were performed in 96-well plates containing 90 μg of protein in 50 μL lysis buffer, followed by the addition of 4 mM DEVD-pNA (5 μL). The samples were incubated at 37°C for 2 hours. Absorbance was measured at 405 nm using a DTX 880 Multimode Detector (Beckman Coulter).
7. Western blotting
Cells were washed with PBS and harvested with lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 1 μM phenylmethylsulfonyl fluoride, 5 μg/mL aprotinin, 5 μg/mL leupeptin). Protein content was analyzed by the Bradford assay. SDS-polyacrylamide gel electrophoresis was performed with a 4% stacking gel and a 10% resolving gel, followed by transfer to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked for 1 hour at room temperature in blocking solution (5% skim milk in Tris-buffer with Tween-20 [TBS-T]: 200 mM Tris, 500 mM NaCl, pH 7.5, 0.05% v/v Tween-20) and incubated overnight at 4°C in 5% bovine serum albumin in TBS-T with antibodies targeting Bax, Bcl-2, IGF-1Rβ, phospho-p44/42 MAPK, p44/42 MAPK, phospho-Akt, Akt, or β-actin. The membranes were washed with TBS-T and incubated with goat antirabbit IgG-HRP for 1 hour at room temperature. The membranes were then washed again, incubated with Amersham ECLTM Advance Western Blotting Detection Kit (Amersham Biosciences) for 2 minutes, and autoradiography was performed. The signal intensities for specific bands were quantified using Image J density analysis software version 1.43 (National Institutes of Health, Bethesda, MD, USA).
8. Statistical analyses
The results of each experiment were expressed as the mean± standard deviation (SD) of duplicate cultures, and all results are representative of at least three separate experiments. The one-way analysis of variance (ANOVA) for three or more unpaired groups or the Student t-test for two unpaired groups was used, and p-values less than 0.05 were considered statistically significant.
RESULTS
1. Simvastatin suppressed cell proliferation by inducing G1 phase cell cycle arrest in bile duct cancer cells
Statin-induced inhibition of bile duct cancer cell proliferation was measured by the MTT assay after 24 or 48 hour incubation. Pravastatin, a hydrophilic statin, dose-dependently suppressed bile duct cancer cell proliferation (Fig. 1A). In contrast, simvastatin, a lipophilic statin, was more effective than pravastatin, and time- and dose-dependently suppressed bile duct cancer cell proliferation (p<0.05 vs untreated control and the same concentration of pravastatin) (Fig. 1B). Based on these results, we decided to use simvastatin for the subsequent experiments.
Fig. 1.

Simvastatin and pravastatin dose-dependently suppress cell proliferation. MTT assays following 24 hours (upper graph) or 48 hours (lower graph) of treatment with pravastatin (A) or simvastatin (B) were performed to evaluate the inhibitory effect of statins on bile duct cancer cell proliferation. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. *p<0.05 vs untreated control; †p<0.05 vs untreated control and the same concentration of pravastatin.
Cell cycle analysis by flow cytometry was performed on bile duct cancer cells after 48 or 72 hours 50 μM simvastatin treatment. After 48 hours treatment, the cell population in the G1 phase was significantly increased, as compared to control cells (p<0.05). Furthermore, after 72 hours treatment, the induction of G1 and G0 phases was significantly increased, as compared to control cells (p<0.05) (Fig. 2). Taken together, these results demonstrate that simvastatin induces the arrest of bile duct cancer cells mainly in G1 phase in a time-dependent manner.
Fig. 2.

(A) Dot plot (upper) and histogram (lower) of flow cytometry. (B) Graphic representaion of the flow cytometry data. Simvastatin suppresses cell proliferation via the induction of G1 phase cell cycle arrest in bile duct cancer cells. Cell cycle analysis by flow cytometry was performed using bile duct cancer cells after 48 or 72 hours of treatment with 50 μM simvastatin. These results represent three separate experiments.
SV, simvastatin 50 μM. *p<0.05 vs untreated control; †p<0.05 vs untreated control and 48 hours of treatment.
2. Simvastatin induces apoptosis via caspase-3 activation, downregulation of Bcl-2 expression, and enhancement of Bax expression
The effect of simvastatin on apoptosis was evaluated by cell death detection ELISA and caspase-3 activity assays. Simvastatin significantly and dose-dependently induced bile duct cancer cell apoptosis at doses greater than 2.5 μM (Fig. 3A). Caspase-3 activity, which plays a central role in the execution-phase of cell apoptosis, was also significantly and dose-dependently increased by simvastatin (Fig. 3B). We also analyzed expression of the proapoptotic protein, Bax and antiapoptotic protein, Bcl-2 by Western blot. Simvastatin significantly enhanced the expression of Bax and downregulated the expression of Bcl-2 (Fig. 4), as compared to control cells. These results indicate that simvastatin induces apoptosis and modulates proteins directly involved in bile duct cancer cell apoptosis.
Fig. 3.

Simvastatin dose-dependently induces apoptosis and caspase-3 activation. The effect of simvastatin on apoptosis was directly evaluated by a cell death detection enzyme-linked immunosorbent assay (A) and a caspase-3 activity assay (B). *p<0.05 compared to the untreated control; †p<0.01 compared to the untreated control.
Fig. 4.
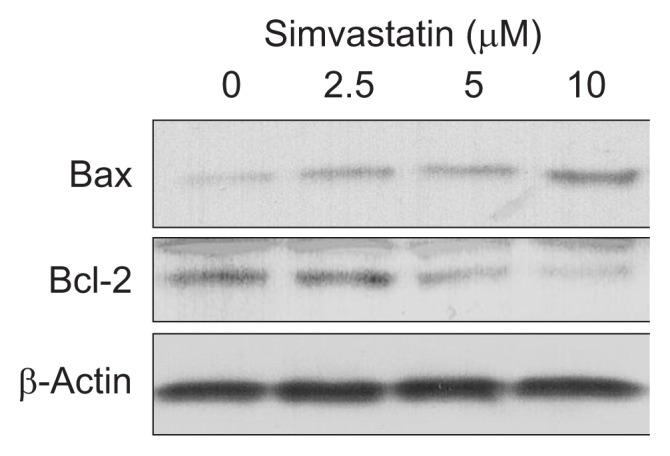
Simvastatin dose-dependently downregulates Bcl-2 expression and enhances Bax expression. Western blot analyses of Bax and Bcl-2 in bile duct cancer cells were performed following treatment with various concentrations of simvastatin. These results are representative of three separate experiments.
3. Simvastatin suppresses IGF-1R expression and IGF-1-induced ERK/Akt activation
We evaluated the effects of simvastatin on IGF-1R and the IGF-1R signaling pathway in bile duct cancer cells. First, we confirmed that simvastatin significantly downregulated the expression of IGF-1Rβ, as compared to untreated control cells (Fig. 5). We next examined the effect of simvastatin on the IGF-1R signaling pathway. Bile duct cancer cells were pretreated with simvastatin (10 μM) for 24 hours, and then stimulated with IGF-1 (100 nM) for 15 minutes. IGF-1 treatment phosphorylated ERK, which was significantly abolished by simvastatin treatment (Fig. 6A). We further evaluated Akt activation following IGF-1 and simvastatin treatment. IGF-1 induced Akt phosphorylation, which was also abolished by simvastatin treatment (Fig. 6B). These results indicate that simvastatin suppresses the expression of IGF-1R and IGF-1-induced ERK and Akt activation in bile duct cancer cells.
Fig. 5.
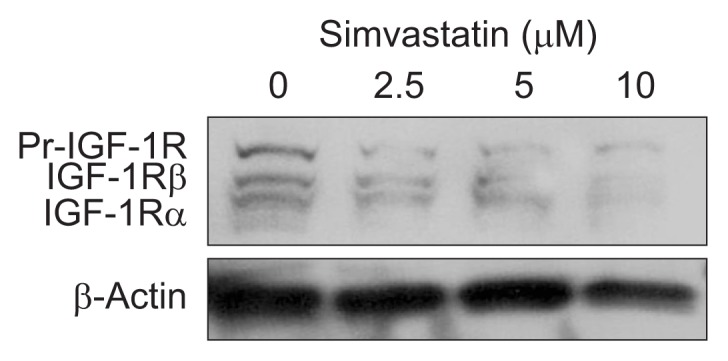
Simvastatin dose-dependently suppresses insulin-like growth factor 1 receptor (IGF-1R) expression. The effect of simvastatin on IGF-1R in bile duct cancer cells was evaluated by Western blot analysis. According to the manufacturer’s datasheet, three bands (IGF-1R precursor, IGF-1Rβ, and IGF-1Rα) are reported to appear after IGF-1Rβ antibody treatment. These results are representative of three separate experiments.
Fig. 6.

Simvastatin suppresses insulin-like growth factor 1 (IGF-1)-induced extracellular signal-regulated kinase (ERK)/Akt activation. A Western blot analysis of phosphorylated ERK and Akt in bile duct cancer cells was performed following treatment with or without IGF-1 (100 nM) or simvastatin (10 μM), as indicated. These results are representative of three separate experiments.
DISCUSSION
Stains are involved in immunomodulation and antioxidant activity, and have anti-inflammatory and antineoplastic effects through their ability to induce apoptosis, suppress cell proliferation, and prevent metastasis and angiogenesis. These effects are mediated by inhibition of the mevalonate pathway. Geranyl-geranyl pyrophosphate and farnesyl pyrophosphate, derivatives of the mevalonate pathway, are important for the activation of several cellular proteins, including K-ras and the Rho family, which are linked with activation of ERK, Akt, and/or p38 MAPK, thereby regulating cancer cell proliferation.8–11 In this study, simvastatin inhibited the proliferation of bile duct cancer cells, increased apoptosis, and suppressed IGF-1-induced ERK/Akt activation. Thus, statins have antineoplastic effects in bile duct cancer cells.
The lipophilicity of statins is associated with cytotoxicity in a number of cancer cell lines. A study demonstrated that lipophilic, but not hydrophilic, statins induce cell death in melanoma cells via inhibition of hypoxia-inducible factor 1α expression, nonenzymatic antioxidant levels, and superoxide dismutase activity.22 A different study revealed that lipophilic statins induce apoptosis via activation of caspase-8 and caspase-9, BH3 interacting-domain death agonist cleavage, cytochrome C release, and poly (ADP-ribose) polymerase cleavage.23 Our study also showed that simvastatin, a lipophilic statin, more effectively and dose- and time-dependently suppressed bile duct cancer cell proliferation, whereas pravastatin, a hydrophilic statin, only suppressed proliferation at very high doses. Taken together, lipophilic statins, such as simvastatin and atorvastatin, may be more suitable antineoplastic agents.
A study evaluating the effects of statins on the expression of Bax, Bad, Bcl-2, and Bcl-XL in esophageal adenocarcinoma cells revealed that statin treatment increased mRNA and protein expression of the proapoptotic proteins Bax and Bad, while protein levels of the antiapoptotic proteins Bcl-2 and Bcl-XL were unchanged.24 However, other studies using other malignant cells indicated that statins suppressed Bcl-2 expression. Additionally, other studies reported that statins had no effect on Bax expression.25 Our results showed that simvastatin activated Bax and suppressed Bcl-2 in bile duct cancer cells, suggesting that statins are a powerful agent to control cell death in bile duct cancer cells.
IGF-1R is composed of two α and β subunits, which are synthesized from a single mRNA precursor. The precursor is then glycosylated and cross-linked by cysteine bonds to form a functional transmembrane αβ chain. In response to ligand binding, the α subunits induce tyrosine autophosphorylation of the β subunits.17–19 These events activate multiple signaling pathways, including the MAPK and PI3K/Akt-1 pathway. Overexpression of IGF-IR was demonstrated in many solid cancers, and a number of studies showed strong evidence that activation of IGF-IR is associated with cancer progression and metastasis.26,27 Human intrahepatic cholangiocarcinomas are also known to express IGF-1R, which is associated with aggressive forms of cancer. Therefore, modulation of IGF-1R may be an important strategy for the management of cholangiocarcinoma.20 In a study with prostate cancer cells, statins were potent inhibitors of the IGF-1/IGF-1R system.28 Our study also demonstrated that simvastatin suppressed IGF-1R expression and IGF-1-induced ERK/Akt activation in bile duct cancer cells. These results raise the possibility that statins effectively control the IGF-1R signaling pathway, which is major target for bile duct cancer treatment.
In a xenograft model study, the effect of statins on inoculated colon cancer growth was evaluated. Tumors treated with simvastatin had smaller volumes, larger necrotic areas, lower expression of vascular endothelial growth factor, and higher apoptotic scores. The authors suggested that simvastatin could be a potential chemopreventive and therapeutic agent for colon cancer.29 Besides, combinatorial treatment with statin and enzastaurin inoculated with hepatocellular carcinoma cells in mice was demonstrated to enhance antitumor efficacy.30 In some epidemiological studies, statins are associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes,13 and a small to considerable reduction in the risk of colon cancer.14,15 However, there are few clinical studies on the synergistic effect of statins in patients with cancers. In a phase 3 clinical study in patients with untreated advanced gastric cancer, addition of 40 mg simvastatin to capecitabine-cisplatin failed to show an increase in the median progression-free survival.31 On the contrary, some in vitro cell studies have demonstrated that statins induce apoptosis and additive suppression of proliferation in combination with pre-existing anticancer drugs, including gemcitabine, cisplatin, 5-fluorouracil, and S-1, in cholangiocarcinoma cells.32,33 We hope that various clinical studies will evaluate the synergistic effect of statins with proven chemotherapeutics in bile duct cancer.
This study has some limitations. First, additional studies using tumor-bearing animal models are needed to confirm the actual effects of statins on the suppression of bile duct cancer growth, and to evaluate the synergistic antineoplastic effects of statins and pre-existing chemotherapeutics. Second, although apoptotic effect was confirmed by the detection of histone-associated DNA fragments using ELISA, caspase-3 activity assay, and flow cytometry, transcriptional mechanism involved in simvastatin-induced apoptosis in bile duct cancer cell was not fully documented. Accordingly, we expect our future study will define this mechanism.
On the other hand, 1.0 μM simvastatin loading in the media corresponds to the dose of 0.4586 mg/kg of bodyweight. Therapeutic dose of simvastatin to lower cholesterol level is up to 1.0–2.0 mg/kg, which means that simvastatin doses in our experiments were approximately 1 to 3 times higher than general cholesterol lowering therapeutic dose. Considering that we had to demonstrate definite change in experiments for short-term period (24 or 48 hours) and had to prove antineoplastic effects (not cholesterol lowering effect), the concentrations we loaded can be acceptable. Doses for future animal study need to be adjusted to lower concentration because animal experiments will need longer time than in vitro study.
In conclusion, the current study demonstrated that simvastatin inhibits cell proliferation by the induction of G1 phase cell cycle arrest, activates the proapoptotic protein Bax, and suppressed the antiapoptotic protein Bcl-2 in bile duct cancer cells. Additionally, simvastatin suppressed IGF-1R expression and IGF-1 induced ERK/Akt activation in bile duct cancer cells. This study indicated that statins could be used as an antineoplastic agent, a chemopreventive agent in patients with a high risk of cancer, and/or a therapeutic option which enhances other chemotherapeutics.
ACKNOWLEDGEMENTS
This study was supported by Hallym University Academic-Industrial Cooperation Program (HL-11-31, 2011).
Footnotes
CONFLICTS OF INTEREST
No conflict of interest relevant to this article was reported.
REFERENCES
- 1.Sirica AE. Cholangiocarcinoma: molecular targeting strategies for chemoprevention and therapy. Hepatology. 2005;41:5–15. doi: 10.1002/hep.20537. [DOI] [PubMed] [Google Scholar]
- 2.Jarnagin WR, Fong Y, DeMatteo RP, et al. Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann Surg. 2001;234:507–517. doi: 10.1097/00000658-200110000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ustundag Y, Bayraktar Y. Cholangiocarcinoma: a compact review of the literature. World J Gastroenterol. 2008;14:6458–6466. doi: 10.3748/wjg.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murakami Y, Uemura K, Sudo T, et al. Gemcitabine-based adjuvant chemotherapy improves survival after aggressive surgery for hilar cholangiocarcinoma. J Gastrointest Surg. 2009;13:1470–1479. doi: 10.1007/s11605-009-0900-0. [DOI] [PubMed] [Google Scholar]
- 5.Lee GW, Kang JH, Kim HG, Lee JS, Lee JS, Jang JS. Combination chemotherapy with gemcitabine and cisplatin as first-line treatment for immunohistochemically proven cholangiocarcinoma. Am J Clin Oncol. 2006;29:127–131. doi: 10.1097/01.coc.0000203742.22828.bb. [DOI] [PubMed] [Google Scholar]
- 6.Blechacz B, Gores GJ. Cholangiocarcinoma: advances in pathogenesis, diagnosis, and treatment. Hepatology. 2008;48:308–321. doi: 10.1002/hep.22310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosen ED, Spiegelman BM. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276:37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 8.Gauthaman K, Fong CY, Bongso A. Statins, stem cells, and cancer. J Cell Biochem. 2009;106:975–983. doi: 10.1002/jcb.22092. [DOI] [PubMed] [Google Scholar]
- 9.Blanco-Colio LM, Villa A, Ortego M, et al. 3-Hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitors, atorvastatin and simvastatin, induce apoptosis of vascular smooth muscle cells by downregulation of Bcl-2 expression and Rho A prenylation. Atherosclerosis. 2002;161:17–26. doi: 10.1016/S0021-9150(01)00613-X. [DOI] [PubMed] [Google Scholar]
- 10.Danesh FR, Sadeghi MM, Amro N, et al. 3-Hydroxy-3-methyl-glutaryl CoA reductase inhibitors prevent high glucose-induced proliferation of mesangial cells via modulation of Rho GTPase/p21 signaling pathway: implications for diabetic nephropathy. Proc Natl Acad Sci U S A. 2002;99:8301–8305. doi: 10.1073/pnas.122228799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-meth-ylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–1719. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 12.Miller T, Yang F, Wise CE, et al. Simvastatin stimulates apoptosis in cholangiocarcinoma by inhibition of Rac1 activity. Dig Liver Dis. 2011;43:395–403. doi: 10.1016/j.dld.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Serag HB, Johnson ML, Hachem C, Morgana RO. Statins are associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes. Gastroenterology. 2009;136:1601–1608. doi: 10.1053/j.gastro.2009.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hachem C, Morgan R, Johnson M, Kuebeler M, El-Serag H. Statins and the risk of colorectal carcinoma: a nested case-control study in veterans with diabetes. Am J Gastroenterol. 2009;104:1241–1248. doi: 10.1038/ajg.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poynter JN, Gruber SB, Higgins PD, et al. Statins and the risk of colorectal cancer. N Engl J Med. 2005;352:2184–2192. doi: 10.1056/NEJMoa043792. [DOI] [PubMed] [Google Scholar]
- 16.Lee J, Hong EM, Koh DH, et al. HMG-CoA reductase inhibitors (statins) activate expression of PPARalpha/PPARgamma and ABCA1 in cultured gallbladder epithelial cells. Dig Dis Sci. 2010;55:292–299. doi: 10.1007/s10620-009-0734-3. [DOI] [PubMed] [Google Scholar]
- 17.Adachi Y, Yamamoto H, Ohashi H, et al. A candidate targeting molecule of insulin-like growth factor-I receptor for gastrointestinal cancers. World J Gastroenterol. 2010;16:5779–5789. doi: 10.3748/wjg.v16.i46.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baserga R. The insulin-like growth factor I receptor: a key to tumor growth? Cancer Res. 1995;55:249–252. [PubMed] [Google Scholar]
- 19.Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst. 2000;92:1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 20.Alvaro D, Barbaro B, Franchitto A, et al. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am J Pathol. 2006;169:877–888. doi: 10.2353/ajpath.2006.050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ku JL, Yoon KA, Kim IJ, et al. Establishment and characterisation of six human biliary tract cancer cell lines. Br J Cancer. 2002;87:187–193. doi: 10.1038/sj.bjc.6600440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alupei MC, Licarete E, Cristian FB, Banciu M. Cytotoxicity of lipophilic statins depends on their combined actions on HIF-1α expression and redox status in B16.F10 melanoma cells. Anticancer Drugs. 2014;25:393–405. doi: 10.1097/CAD.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 23.Kato S, Smalley S, Sadarangani A, et al. Lipophilic but not hydrophilic statins selectively induce cell death in gynaecological cancers expressing high levels of HMGCoA reductase. J Cell Mol Med. 2010;14:1180–1193. doi: 10.1111/j.1582-4934.2009.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogunwobi OO, Beales IL. Statins inhibit proliferation and induce apoptosis in Barrett’s esophageal adenocarcinoma cells. Am J Gastroenterol. 2008;103:825–837. doi: 10.1111/j.1572-0241.2007.01773.x. [DOI] [PubMed] [Google Scholar]
- 25.Wood WG, Igbavboa U, Muller WE, Eckert GP. Statins, Bcl-2, and apoptosis: cell death or cell protection? Mol Neurobiol. 2013;48:308–314. doi: 10.1007/s12035-013-8496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 27.Gallagher EJ, LeRoith D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol Metab. 2010;21:610–618. doi: 10.1016/j.tem.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sekine Y, Furuya Y, Nishii M, Koike H, Matsui H, Suzuki K. Simvastatin inhibits the proliferation of human prostate cancer PC-3 cells via down-regulation of the insulin-like growth factor 1 receptor. Biochem Biophys Res Commun. 2008;372:356–361. doi: 10.1016/j.bbrc.2008.05.043. [DOI] [PubMed] [Google Scholar]
- 29.Cho SJ, Kim JS, Kim JM, Lee JY, Jung HC, Song IS. Simvastatin induces apoptosis in human colon cancer cells and in tumor xenografts, and attenuates colitis-associated colon cancer in mice. Int J Cancer. 2008;123:951–957. doi: 10.1002/ijc.23593. [DOI] [PubMed] [Google Scholar]
- 30.Kim W, Yoon JH, Kim JR, et al. Synergistic anti-tumor efficacy of lovastatin and protein kinase C-beta inhibitor in hepatocellular carcinoma. Cancer Chemother Pharmacol. 2009;64:497–507. doi: 10.1007/s00280-008-0897-1. [DOI] [PubMed] [Google Scholar]
- 31.Kim ST, Kang JH, Lee J, et al. Simvastatin plus capecitabine-cisplatin versus placebo plus capecitabine-cisplatin in patients with previously untreated advanced gastric cancer: a double-blind randomised phase 3 study. Eur J Cancer. 2014;50:2822–2830. doi: 10.1016/j.ejca.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Kamigaki M, Sasaki T, Serikawa M, et al. Statins induce apoptosis and inhibit proliferation in cholangiocarcinoma cells. Int J Oncol. 2011;39:561–568. doi: 10.3892/ijo.2011.1087. [DOI] [PubMed] [Google Scholar]
- 33.Cai JP, Chen W, Hou X, Liang LJ, Hao XY, Yin XY. Simvastatin enhances the chemotherapeutic efficacy of S-1 against bile duct cancer: E2F-1/TS downregulation might be the mechanism. Anti-cancer Drugs. 2013;24:1020–1029. doi: 10.1097/CAD.0b013e328364f935. [DOI] [PubMed] [Google Scholar]