

Abstract
The differentiation of mouse spermatids is one critical process for the production of a functional male gamete with an intact genome to be transmitted to the next generation. So far, molecular studies of this morphological transition have been hampered by the lack of a method allowing adequate separation of these important steps of spermatid differentiation for subsequent analyses. Earlier attempts at proper gating of these cells using flow cytometry may have been difficult because of a peculiar increase in DNA fluorescence in spermatids undergoing chromatin remodeling. Based on this observation, we provide details of a simple flow cytometry scheme, allowing reproducible purification of four populations of mouse spermatids fixed with ethanol, each representing a different state in the nuclear remodeling process. Population enrichment is confirmed using step-specific markers and morphological criterions. The purified spermatids can be used for genomic and proteomic analyses.
Keywords: Developmental Biology, Issue 106, Spermatogenesis, spermiogenesis, spermatid, flow cytometry cell sorting, DNA, chromatin remodeling
Introduction
Haploid round spermatids differentiate into spermatozoa by a process called spermiogenesis. This involves many different steps including the acquisition of a flagellum, chromatin and cytoskeleton remodeling, condensation of the nucleus as well as the loss of most of the cytoplasm. These unique cellular events must be finely regulated in order to produce a mature functional gamete with an intact genome suitable for fertilization. Spermiogenesis can hardly be studied in vitro since no reliable cell culture system has so far been able to support progression through the different steps of the process. Moreover, actual in vitro techniques lead to a poor yield1,2. In vivo, proper transitions through the different steps of spermiogenesis are crucial for the natural functional integrity of the male gamete. Successful purification of spermatids according to their differentiation steps has never been accomplished with a level of enrichment sufficient to allow molecular characterization of spermiogenesis. For instance, purification of key steps of the spermatidal differentiation would be especially useful to study the developing acrosome, formation of the midpiece3, cell junction dynamics4, RNA dynamics5, chromatin remodeling process6,7 or genomic stability8. Purification of spermatids has been hampered by their progressive morphological transformation, the lack of known stage-specific external biomarkers, and their peculiar shape and size.
Although most male germ cells display a direct relationship between DNA staining and ploidy (DNA content), we noticed that such positive correlation is no longer applicable to spermatids. This stems from our early observation that seminiferous tubule sections show variable intensity of DNA staining throughout the different spermiogenesis steps. Although DNA staining is consistent with their haploid set of chromosomes from spermiogenesis steps 1 to 7 (round spermatids), we observed a sharp increase in fluorescence intensity with DAPI or SYTO 16 around the onset of nuclear reorganization and chromatin remodeling (spermiogenesis step 8) reaching a peak at the onset of nuclear condensation (spermiogenesis steps 11-12). Following condensation of the nucleus, DNA staining intensity decreases until spermiation (spermiogenesis step 16). We surmised that this was likely associated with the formation of their peculiar chromatin structure transition where histones are replaced by protamines. We therefore developed a reliable flow cytometry method that allows the separation of spermatids using the variation of DNA intensity of spermatids as a main selection parameter.
A simple flow cytometry approach is described to separate mouse spermatids with high purity (95-100%) based on their apparent DNA content (SYTO16 staining), size and granulosity. Spermatids are separated into four populations; spermiogenesis steps 1-9, 10-12, 13-14 and 15-16. Purified spermatids are suitable for genetic/genomic analysis, as well as proteomic applications as described in a recent publication from our group9.
Protocol
Animal care was in accordance with the Université de Sherbrooke animal care and use committee.
1. Tube Preparation
The day before cell sorting, add 1-2 ml of heat-inactivated fetal bovine serum (FBS) to 5 ml polypropylene round bottom tubes, and to 15 ml and 50 ml polypropylene conical tubes. Critical step: Ensure that every tube used in the protocol is coated. Note: FBS coating prevents germ cells from sticking to tube walls.
Slowly mix O/N by inversion at 4 °C to coat the tubes uniformly using a rotator.
- The next day, remove FBS from coated tubes by decanting.
- *Critical step: Ensure that the 5 ml polypropylene round bottom tubes and the 15 and 50 ml tubes used for cell preparation (protocol steps 2.1 to 2.11) are completely emptied of FBS to prevent fanning (i.e., imprecise deviation of the side streams). Use a P200 pipet to remove the FBS. Note: Residual FBS in tubes prior to sorting may lead to a loss of cells or contamination of other tubes used during cell sorting,
- Leave a small residual volume of FBS (about 100-200 µl) in the 5 ml polypropylene round bottom tubes used for collecting purified cells.
2. Cell Preparation
Sacrifice one male mouse under anesthesia using O2/isoflurane (induction at 5% then maintained at 2%) followed by CO2 asphyxiation.
Excise and decapsulate both testes and mince with small scissors in 500 µl of sorting buffer into a 1.5 ml tube.
Flush germ cells from seminiferous tubules by gentle up and down pipetting in the 1.5 ml tube, first with a truncated 100-1,000 µl tip, and then with an intact 100-1,000 µl tip.
Remove debris and clumps by one filtration step using a 40 µm cell strainer and collect filtrate into the 50 ml conical tube previously coated with FBS in step 1.
Wash the filter once with sorting buffer up to a total volume of 3 ml and transfer filtrate into the 15 ml conical tube previously coated in step 1.
Add 16 µl of 50 mM EDTA pH 8 per milliliter of cell suspension (48 µl for 3 ml) so as to obtain a final concentration of 0.8 mM EDTA.
To fix germ cells, slowly add 3 volumes of ice-cold 100% ethanol using a 10 ml pipet with gentle agitation (vortex at low speed), which will produce a milky suspension. Critical step: Slowly adding ethanol is crucial and important to preserve the integrity of the cell preparation. We noted that rapid addition of ethanol causes cell lysis resulting in a major decrease of sorting efficiency. For a 3 ml cell suspension, 9 ml of ethanol should be added in approximately 1 min.
Incubate cells on ice for 15 min with occasional mixing by inversion.
Centrifuge cell suspension at 800 x g for 5 min and remove the clear supernatant.
Gently resuspend the fixed germ cells in 2 ml of sorting buffer.
Add 4 µl of SYTO16 DNA dye (1 mM solution in DMSO) and incubate for at least 30 min (protected from light). Note: Best sorting results are obtained using SYTO16 since it is a permeable DNA dye that is easily incorporated into the highly compacted nuclei of spermatids.
3. Cell Sorter Set Up
Note: Here, a 4-Laser (405 nm - violet, 488 nm - blue, 561 nm - yellow-green, 633 nm - red) 20-parameter BDFACSAria III cell sorter is used. The BD FACSDiva 6.1.3 software is used to visualize and analyze the data. The settings may vary depending on the type of sorter used.
Sterilize the FACS sorter by running 70% ethanol as sheath fluid during the fluidics shutdown procedure ("Cytometer" menu in the software) prior to sorting. Select "Cytometer" in the toolbar of the software and choose "Fluidic shutdown". Follow the steps mentioned by the processes.
Prepare and filter 1x PBS with a 0.22 µm filter so as to eliminate any crystals and contaminants. Fill the sheath tank to the upper weld line on the inside of the tank.
Start the software and log in. Start the FACS sorter to warm up the lasers for at least 30 min prior to sorting. Run two fluidic startup processes to flush out the ethanol from the fluidics. Select "Cytometer" in the toolbar of the software and choose "Fluidic startup". Follow the steps mentioned by the processes. Note: This restores fluidics with normal sheath fluid (1x PBS).
Clean the sorting block and the deflection plates with distilled water and dry them thoroughly. Sonicate a 100 µm nozzle placed in a tube of distilled water for 2 min in a sonication bath. Wipe the nozzle thoroughly.
Insert the 100 µm nozzle into the flow cell and turn the nozzle-locking lever clockwise to the 12 hr position. Set the sheath pressure to 20 psi.
Turn on the stream and adjust the amplitude to obtain target values for frequency (around 30), Drop 1 (around 150) and Gap (around 12) and to establish a stable droplet breakoff pattern (few satellite drops). Turn off attenuation.
Make sure that the stream is straight and hits the center of the waste aspirator by changing the angle of the sort block. Note: A 130 µm nozzle at 10 psi was also tested and no obvious difference was found.
Set laser delay to 0 for the blue, -77.39 for the red, 37.33 for the violet and -39.85 for the yellow-green lasers.
Set areas scaling to 1.14 for the blue, 1.0 for the red, 0.75 for the violet and 0.96 for the yellow-green lasers.
Using a Test Sort (in the "Side Stream" window of the software), make sure that side streams are not fanning. In the "Side Stream" window, adjust the voltage sliders of each side stream so that they hit the middle of test collection tube. If the center stream is not tight, adjust the 2nd, 3rd, and 4th drop settings to constrain the center stream.
Start the Cytometer Setup and Tracking (CS&T) application in the software ("Cytometer" menu).
Use the cytometer setup and tracking beads at 1 drop per 350 µl to automate the characterization and tracking of the cytometer performance using manufacturer's instructions.
Quit the CS&T application. Apply CS&T setting, make sure the stream parameters still allow a stable droplet breakoff pattern, and turn on the Sweet Spot button as described in manufacturer's instructions (in the "Breakoff window").
- Optimize the Drop Delay value using fluorescent beads (see Table of Materials).
- Install the collection tubes in the holder. Open the sort block door. In the "Side Stream" window, turn on voltage then select "Test sort" and click the Aspirator Drawer button to open the drawer and have access to the collection tubes.
- Optimize the side streams so they go into the center of the tube. Adjust values and sliders as needed.
- Close the sort block door and make sure to adjust the micrometer dial to obtain the brightest bead spot at the center. Unselect waste drawer, test sort and voltage. Remove the collection tubes.
- Optimize the drop delay according to manufacturers' instructions.
- Briefly, shake bead vial vigorously and dilute 1 drop of beads in 0.5 ml of PBS.
- Using manufacturers' instructions, set the drop delay using the Drop Delay experiment template available in the software. Alternatively, use the Auto Drop Delay feature to set the drop delay.
- Load the tube of fluorescent beads and adjust the flow rate until the threshold rate is ~3,000 to 4,000 events/second. Set the sort precision to "Initial" and finally to "Fine tune" in the "Sort layout" window. Select the optical filter and sort beads. Adjust the Drop Delay until ~100% are sorted to the left. Note: This step is critical to make sure that cells of interest sort into a side stream. The beads and the optical filter are used to achieve close to a 100% droplet deviation.
Place a FSC 1.5 ND (neutral density) filter at the left end of the FSC detector block. Set the window extension to 2.00 µs and the FSC Area scaling to 1.00 (in the "Laser" tab in the "Cytometer" window).
Before loading sample tube, place a sample filter line of 50 µm at the end of the sample line to avoid clumping. To do so, select "Change Sample Filter" in the "Cytometer" menu.
- Test the composition of the sample fluid to achieve good separation without fanning between side stream and to prevent the cells from sticking to the polypropylene tubes.
- Load the sample tube, turn on the deflection plates and select "Test sort" with the drawer closed. Look at side streams if separation occurs without too much fanning. Note: Fanning is likely due to a high protein concentration in the sample buffer.
Load the sample tube into the FACS instrument. Select a sample agitation of 300 rpm and a sample temperature of 4 °C. Select "Acquire Data" in the "Acquisition Dashboard".
If necessary, dilute the sample to achieve an appropriate event rate to a maximum of 5,000 events/sec (at a maximum flow rate of 3.0). Strictly respect these limits so as to maintain the purity of the sorted cells.
To analyze and sort the cells, optimize the FSC threshold value to eliminate debris without interfering with the population of interest. In logarithmic scale, adjust the voltage of the photo-multiplier tube (PMT) detector with the 530/30 filter in order to make sure that the fluorescence signal of the total population fits within the scale, and gate on the positive population (Figure 1, Gate 1).
Produce a Forward Scatter-area (FSC-A)/Side Scatter-area (SSC-A) plot of the SYTO16 positive population and adjust the FSC-A to around 110 V in linear scale, and the SSC-A to around 150 V in logarithmic scale to visualize all the events while excluding very large cells and cell aggregates.
In the "Sort Layout" window, set the "Sort Precision Mode" to "4-way purity" (Yield mask: 0, Purity Mask: 32, Phase Mask: 0). Select "Continuous" from the "Target Events" menu for continuous sorting.
Add populations to sort in the field corresponding to the tubes (in the "Sort Layout" window). Place populations with higher frequencies in the middle and those with lower frequencies at the ends. Set the voltage plates to 2,500 V. During sorting, keep the sample collection tubes on ice.
4. Cell Sorting
Immediately before sorting, filter cells using a 50 µm filter in a previously coated 5 ml polypropylene round bottom tube.
Wash the filter extensively with 1-2 ml of sorting buffer.
- Sort fixed germ cells using a 488 nm laser-equipped cell sorter following the gating scheme detailed in Figure 1. Collect purified fractions in 5 ml polypropylene round bottom tubes previously coated in section 1 on ice.
- Make sure that 100-200 µl of FBS is kept in the collection tubes to avoid sticking of the sorted cells to the tube wall.
- Make sure that the concentration of SYTO16 is saturating to avoid fluorescence fluctuations during the sorting period. SYTO16 is saturating when there is no further variation of cell staining intensity at the Alexa Fluor 488 parameter (DNA staining) in the sorting graphics. If necessary, add 1 µl of SYTO16 to the sorting tube to reach saturation.
- Display the total events on a graphic displaying the following parameters: number of events vs Alexa Fluor 488-A.
- Select the positive DNA staining cells with Gate 1, as shown in Figure 1.
- Display cells from gate 1 on a graphic using SSC-A vs FSC-A as parameters
- Select the cells with Gate 2, as showed in Figure 1.
- Display cells from Gate 2 on a graphic using FSC-A vs Alexa Fluor 488-A as parameters.
- Select cells with Gate 5 and Gate 6 on Gate 2 graphic, as showed in Figure 1.
- Display separately Gate 5 and Gate 6 on graphics using Alexa Fluor 488-W vs Alexa Fluor 488-A as parameters.
- Select spermiogenesis steps 1-9 spermatids on the Gate 5 graphic and spermiogenesis steps 10-12 spermatids on the Gate 6 graphics, as showed in Figure 1.
- Display cells from gate 1 on a graphic using FSC-A vs Alexa Fluor 488-A as parameters
- Select cells with Gate 3 and Gate 4, as showed in Figure 1.
- Display separately Gate 3 and Gate 4 on graphics using Alexa Fluor 488-W vs Alexa Fluor 488-A as parameters.
- Select spermiogenesis steps 13-14 spermatids on the Gate 3 graphic and spermiogenesis steps 15-16 spermatids on the Gate 4 graphic, as showed in Figure 1.
Representative Results
Gating strategy used with flow cytometry
Figure 1 represents the gating strategy used in flow cytometry to sort four highly pure spermatid populations. Briefly, cells with positive DNA staining (Alexa Fluor 488-A) are first selected with Gate 1. Spermatids from spermiogenesis steps 1-12 are selected (Gate 2) on a dot plot showing the granulosity (SSC-A) vs size (FSC-A) from Gate 1. Then, spermatids from spermiogenesis steps 1-9 and spermiogenesis steps 10-12 can be separated from each other according to the variation of DNA staining intensity (Gates 5 and 6). Spermatids from spermiogenesis steps 13-14 and 15-16 are directly selected from positively stained cells on a dot plot showing size (FSC-A) vs DNA staining (Alexa Fluor 488-A) (Gates 3 and 4). All sorted populations are redefined with Alexa Fluor 488-W vs Alexa Fluor 488-A dot blots to increase their purity.
Validation of the purification scheme by epifluorescence microscopy
Figure 2 represents DAPI staining of the sorted populations of spermatids by flow cytometry. The spermiogenesis steps 1-9 spermatids display the typical round shaped nucleus of the round spermatids and oval shaped nucleus of the spermiogenesis step 9 spermatids. The spermiogenesis steps 10-12 spermatids show a large hook-shaped elongating nucleus. Spermiogenesis steps 13-14 and 15-16 spermatids have a similar shaped nucleus, but differ slightly in DNA staining intensity, which allowed their sorting. The differentiation steps and purity of the different sorted populations were also ascertained using specific biomarkers as described in a previous study9. The purity of the sorted spermatid populations is depicted in Table 1.
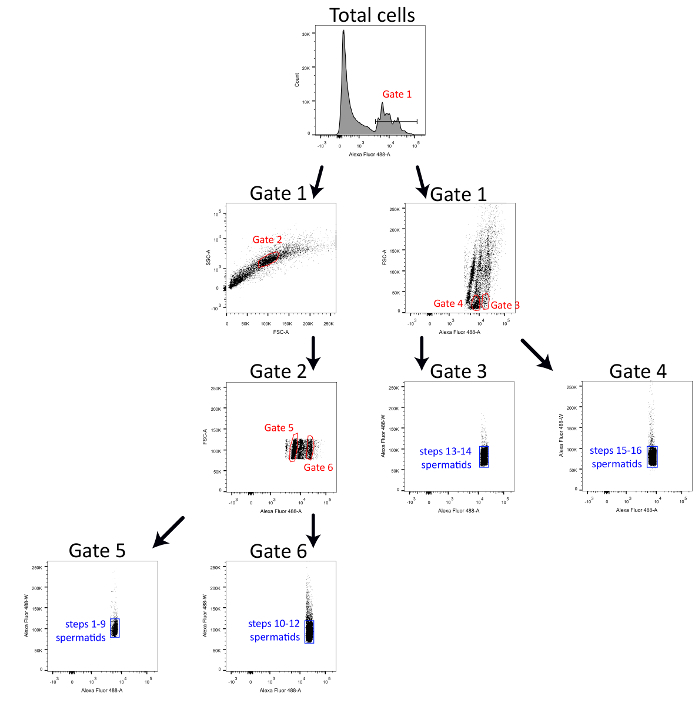 Figure 1. Detailed gating strategy to sort ultrapure spermatid populations. Schematic representation of the gating strategy used to sort spermiogenesis steps 1-9 spermatids, steps 10-12 spermatids, steps 13-14 spermatids and steps 14-16 spermatids simultaneously with a 488 nm laser-equipped cell sorter. Please click here to view a larger version of this figure.
Figure 1. Detailed gating strategy to sort ultrapure spermatid populations. Schematic representation of the gating strategy used to sort spermiogenesis steps 1-9 spermatids, steps 10-12 spermatids, steps 13-14 spermatids and steps 14-16 spermatids simultaneously with a 488 nm laser-equipped cell sorter. Please click here to view a larger version of this figure.
 Figure 2. Visualization of sorted spermatids by epifluorescence. Sorted spermatids are cytospined on microscope slides, stained with DAPI and visualized using an epifluorescence microscope with the appropriate filters for DAPI and equipped with a CCD camera. Cells are shown in inverted gray scale. Bar = 20 µm. Please click here to view a larger version of this figure.
Figure 2. Visualization of sorted spermatids by epifluorescence. Sorted spermatids are cytospined on microscope slides, stained with DAPI and visualized using an epifluorescence microscope with the appropriate filters for DAPI and equipped with a CCD camera. Cells are shown in inverted gray scale. Bar = 20 µm. Please click here to view a larger version of this figure.
| Cell type observed in majority | Purity | Approximate number of cells (8 hr sorting) | Contaminants | |
| Population 1 | Steps 1-9 spermatids | 99-100% | 4,000,000 | Step 10 spermatids |
| Population 2 | Steps 10-12 spermatids | 95-98% | 1,000,000 | Spermatocytes |
| Population 3 | Steps 13-14 spermatids | 99-100% | 800,000 | Spermatocytes, steps 1-9 spermatids |
| Population 4 | Steps 15-16 spermatids | 98-100% | 1,500,000 | Spermatocytes |
Table 1: Sorted spermatid populations and their contaminants.
Discussion
Spermatogenic cells have always been challenging to study given the complexity of the seminiferous epithelium, as well as the limited success of in vitro culture. Over the years, many approaches to purify germ cells from various species were developed. Sedimentation techniques using gravity purification with Percoll or bovine serum albumin gradients usually provide a good yield of intact germinal cells, but lack proper definition between some cell types such as meiotic tetraploid cells and spermatids10. Moreover, these techniques require special devices (often homemade) that may not be readily available to many laboratories, which renders them difficult to reproduce without cumbersome trial and error. These methods are also time-consuming, very sensitive to vibrations and inconvenient as the apparatus is usually kept at 4 °C to maintain cell viability. When successful, sedimentation methods however provide several million purified live cells per population. However, the low level of cell enrichment that can be achieved is the main drawback of these techniques as it rarely reaches more than 80-90% purity. In certain cases, a contamination of 10-20% from other cell types is present when measured by DNA, RNA or protein content, which represents a major impediment to molecular analyses.
When seeking to improve purity of cell populations, some investigators have combined gravity sedimentation to other methods. One of the most effective approaches is to use immature mice to limit the number of different cell populations representing later stages. One can take advantage of the first wave of spermatogenesis and obtain a cell preparation containing germinal cells up to a given differentiation stage based on their sequential appearance after birth. However, this combined approach requires more animals to compensate for the limited amount of starting material and yet does not resolve the lack of definition of the sedimentation methods. In addition, such an approach cannot be practically applied for later cells types such as spermatids, but is used mainly for spermatogonia and primary spermatocyte. Moreover, there is some evidence that the first wave of spermatogenesis may harbor cells with slightly different properties than that of the cycling epithelium of mature mice11,12.
Vitamin A synchronization of spermatogenesis was also used to improve germinal cell purification as this procedure narrows down the number of stages present in the testes of a treated animal. However, this procedure was mainly used to synchronize spermatogenesis in rats, with some success reported in mice13,14. From our own experience, vitamin A synchronization in mice is time-consuming, rarely gives reproducible results and produces harmful side effects raising concern about the cellular integrity of the seminiferous epithelium.
Other groups successfully used flow cytometry to purify germ cells from mouse15-18. However, none of them reported purification of spermatid populations past the round spermatids steps. Our gating strategy allows to purify four highly purified haploid cell populations representing important differentiation steps amenable to molecular studies. The main constraint of the method described in this paper may be the limited yield of sorted cells. The high level of purity is somewhat obtained at the expense of the number of sorted cells. However, to make sure that sorting conditions are optimal, we added few critical advices to the protocol. All these technical advices aim to increase either the number or the purity of sorted spermatids. For instance, the use of FBS coated tubes during sorting greatly diminishes the loss of cells that stick to the tubes. Also, using saturating concentration of SYTO16 helps reduce the variations of DNA staining intensity resulting in more stable populations as seen on FACS plots and a better yield and purity of sorted spermatids over a 8 hr sorting period. In addition, the provided technical advices for the FACS sorter set-up definitely help to increase the overall efficiency of sorting by avoiding cell clogs or cross-contamination in collecting tubes. Alternatively, gates can be expanded to increase the number of sorted cells, eventually resulting in a small decrease in population purity, which can be acceptable for some studies. Furthermore, ethanol-fixed germ cells stained with SYTO16 are very stable at 4 °C and can be sorted for more than 8 hr with limited gating adjustments and supervision. Ultimately, several days of sorting can be pooled to obtain a sufficient number of cells for any application.
Flow sorted spermatids obtained as described here can be used for a variety of experiments. DNA from these cells can easily be extracted using commercial genomic DNA purification kits and demonstrated to be of proper quality for PCR analyses9 and next generation sequencing (data not shown). Sorted spermatids can also be used for protein analyses9 where we performed several immunoblots against stage-specific markers to confirm the high purity of the sorted spermatids. We verified the cellular integrity of the sorted spermatids and found that the nucleus and cytoplasm of all populations remain intact. However, we found a proportion of steps 13-14 spermatids and a smaller proportion of steps 15-16 spermatids that are missing their flagellum (data not shown). Hence, highly pure germ cell populations sorted using this approach are suitable for proteomic and genomic analyses, as well as other applications.
Disclosures
The authors declare no competing financial interests.
Acknowledgments
The authors wish to thank Dr. Leonid Volkov and Éric Bouchard for their technical advice regarding epifluorescence microscopy.
Financial support
Funded by the Canadian Institutes of Health Research (grant #MOP-93781) to G.B.
References
- Sato T, et al. In vitro production of functional sperm in cultured neonatal mouse testes. Nature. 2011;471(7339):504–507. doi: 10.1038/nature09850. [DOI] [PubMed] [Google Scholar]
- Sato T, et al. In vitro production of fertile sperm from murine spermatogonial stem cell lines. Nat. Commun. 2011;2:472. doi: 10.1038/ncomms1478. [DOI] [PubMed] [Google Scholar]
- Sun X, Kovacs T, Hu Y-J, Yang W-X. The role of actin and myosin during spermatogenesis. Mol. Biol. Rep. 2011;38(6):3993–4001. doi: 10.1007/s11033-010-0517-0. [DOI] [PubMed] [Google Scholar]
- Cheng CY, Mruk DD. Cell junction dynamics in the testis: Sertoli-germ cell interactions and male contraceptive development. Physiol. Rev. 2002;82:825–874. doi: 10.1152/physrev.00009.2002. [DOI] [PubMed] [Google Scholar]
- Laiho A, Kotaja N, Gyenesei A, Sironen A. Transcriptome profiling of the murine testis during the first wave of spermatogenesis. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0061558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc F, Maquennehan V, Nkoma GB, Boissonneault G. DNA damage response during chromatin remodeling in elongating spermatids of mice. Biol. Reprod. 2008;78(2):324–332. doi: 10.1095/biolreprod.107.064162. [DOI] [PubMed] [Google Scholar]
- Meistrich ML, Mohapatra B, Shirley CR, Zhao M. Roles of transition nuclear proteins in spermiogenesis. Chromosoma. 2003;111(8):483–488. doi: 10.1007/s00412-002-0227-z. [DOI] [PubMed] [Google Scholar]
- Grégoire M-C, et al. Male-driven de novo mutations in haploid germ cells. Mol. Hum. Reprod. 2013;19(8):495–499. doi: 10.1093/molehr/gat022. [DOI] [PubMed] [Google Scholar]
- Simard O, et al. Instability of trinucleotidic repeats during chromatin remodeling in spermatids. Hum. Mutat. 2014. [DOI] [PubMed]
- Wykes SM, Krawetz S. Separation of spermatogenic cells from adult transgenic mouse testes using unit-gravity sedimentation. Mol. Biotechnol. 2003;25(2):131–138. doi: 10.1385/MB:25:2:131. [DOI] [PubMed] [Google Scholar]
- Yoshida S, et al. The first round of mouse spermatogenesis is a distinctive program that lacks the self-renewing spermatogonia stage. Development (Cambridge, England) 2006;133(8):1495–1505. doi: 10.1242/dev.02316. [DOI] [PubMed] [Google Scholar]
- Moreno RD, Lizama C, Urzúa N, Vergara SP, Reyes JG. Caspase activation throughout the first wave of spermatogenesis in the rat. Cell Tissue Res. 2006;325(3):533–540. doi: 10.1007/s00441-006-0186-4. [DOI] [PubMed] [Google Scholar]
- Meistrich ML, Trostle-Weige PK, Van Beek ME. Separation of specific stages of spermatids from vitamin A-synchronized rat testes for assessment of nucleoprotein changes during spermiogenesis. Biol. Reprod. 1994;51(2):334–344. doi: 10.1095/biolreprod51.2.334. [DOI] [PubMed] [Google Scholar]
- van Pelt AM, de Rooij DG. Synchronization of the seminiferous epithelium after vitamin A replacement in vitamin A-deficient mice. Biol. Reprod. 1990;43:363–367. doi: 10.1095/biolreprod43.3.363. [DOI] [PubMed] [Google Scholar]
- Getun IV, Torres B, Bois PRJ. Flow cytometry purification of mouse meiotic cells. J. Vis. Exp. 2011. [DOI] [PMC free article] [PubMed]
- Kovtun IV, McMurray CT. Trinucleotide expansion in haploid germ cells by gap repair. Nature Genet. 2001;27(4):407–411. doi: 10.1038/86906. [DOI] [PubMed] [Google Scholar]
- Bastos H, et al. Flow cytometric characterization of viable meiotic and postmeiotic cells by Hoechst 33342 in mouse spermatogenesis. Cytometry A. 2005;65(1):40–49. doi: 10.1002/cyto.a.20129. [DOI] [PubMed] [Google Scholar]
- Lassalle B, Ziyyat A, Testart J, Finaz C, Lefèvre A. Flow cytometric method to isolate round spermatids from mouse testis. Hum. Reprod. 1999;14(2):388–394. doi: 10.1093/humrep/14.2.388. [DOI] [PubMed] [Google Scholar]