

Abstract
The pattern of ovarian cancer metastasis is markedly different from that of most other epithelial tumors, because it rarely spreads hematogenously. Instead, ovarian cancer cells exfoliated from the primary tumor are carried by peritoneal fluid to metastatic sites within the peritoneal cavity. These sites, most notably the abdominal peritoneum and omentum, are organs covered by a mesothelium-lined surface. To investigate the processes of ovarian cancer dissemination, we assembled a complex three-dimensional culture system that reconstructs the lining of the peritoneal cavity in vitro. Primary human fibroblasts and mesothelial cells were isolated from human omentum. The fibroblasts were then mixed with extracellular matrix and covered with a layer of the primary human mesothelial cells to mimic the peritoneal and omental surfaces encountered by metastasizing ovarian cancer cells. The resulting organotypic model is, as shown, used to examine the early steps of ovarian cancer dissemination, including cancer cell adhesion, invasion, and proliferation. This model has been used in a number of studies to investigate the role of the microenvironment (cellular and acellular) in early ovarian cancer dissemination. It has also been successfully adapted to high throughput screening and used to identify and test inhibitors of ovarian cancer metastasis.
Keywords: Medicine, Issue 106, microenvironment, ovarian cancer, adhesion, invasion, proliferation, 3D culture, modeling, model
Introduction
Ovarian cancer is the deadliest gynecologic malignancy1. The majority of patients are diagnosed after the cancer has disseminated throughout the peritoneal cavity. Once the cancer has spread throughout the peritoneal cavity, cytoreductive surgery and chemotherapy are often not sufficient treatment to prevent cancer recurrence and chemoresistance, resulting in a less than 30% 5-year survival rate. Ovarian cancer metastasis is predominantly limited to the peritoneal cavity, and several other cancer types, including gastric, pancreatic, and colon cancers, metastasize to the same anatomic sites in the peritoneal cavity. In general, ovarian cancer cells detach from the in situ carcinoma in the fallopian tube or the primary ovarian tumor, travel in peritoneal fluid as single cells or spheroids, and attach to mesothelium-lined surfaces of the omentum, bowel, and abdominal wall2.
The tumor microenvironment plays an important role in disease progression and chemoresistance in many cancers3-6. The peritoneal cavity is a unique microenvironment, with a mesothelial cell monolayer covering the majority of surfaces (Figure 1A)7. The mesothelial lining acts as a barrier that creates a low-friction surface, which tends to be protective against cancer cell adhesion8. Immediately underneath this mesothelial-lined surface is a layer made predominantly of fibroblasts and extracellular matrix (ECM), which promote cancer cell adhesion and invasion8. Ovarian cancer cells secrete factors that induce changes in the mesothelial cell lining that enhance ovarian cancer cell adhesion, invasion, and metastasis9,10. Ovarian cancer cells adhere to the mesothelial surface via integrin and CD44-mediated mechanisms (Figure 1B)11-16.
Historically, several 3D models have been developed to investigate ovarian cancer interactions with the microenvironment. Some of the first models studied ovarian cancer-ECM interface17-21, ovarian cancer-mesothelial cell communication13,14,21-24, or both25 (reviewed by us 26). Niedbala et al. discovered that ovarian cancer cells display a quicker and firmer adhesion to ECM than to mesothelial cells or to plastic alone25. However, these models did not histologically resemble the peritoneal microenvironment. Therefore, we established a 3D organotypic model to more thoroughly replicate the ovarian cancer microenvironment. In order to better understand the role of the microenvironment and the interaction between cancer and peritoneal cells in the peritoneal dissemination of ovarian cancer, we have developed a 3D organotypic in vitro culture model of the peritoneal cavity lining (schematic in Figure 1C). The proposed model is composed of primary human fibroblasts and ECM, covered with a layer of primary human mesothelial cells-each cell type is isolated from human omentum. Histologically, this model resembles the normal peritoneal or omental lining, and provides a surface on which we can study the tumor microenvironment, the interaction between cancer cells and normal tissue, and the processes of cancer cell adhesion, invasion, and proliferation8.
Protocol
All research protocols described have been reviewed by the University of Chicago Institutional Review Board (IRB). Informed consent was obtained from each patient before surgery and the study was approved by the University of Chicago IRB. A biological safety cabinet type 2 and gloves should be used when handling human tissue for protection and to reduce risk of contaminating cells.
1. Isolation and Culture of Primary Untransformed Stromal Cells
- Human tissue collection and preparation.
- Obtain specimens of human omentum, 2 cm3, removed during an abdominal surgery, and immediately immerse tissue in RT phosphate-buffered saline (PBS) (Figure 2A). A piece of omentum 3 cm x 2 cm typically yields 1 million primary human mesothelial cells (HPMC) and 200,000 primary human fibroblasts or normal omental fibroblasts (NOF).
- Spin down the PBS the tissue was immersed in at 0.5 x g for 3 min as soon as possible after collection (< 2 hr in PBS) and transfer the tissue to fresh 20 ml PBS in a 50 ml conical tube. Cut up tissue into 5mm3 pieces by mincing with scalpels in a 15 cm diameter, sterile culture dish (Figure 2B).
- Primary human mesothelial cell isolation8
- To isolate HPMC, transfer the minced tissue into 50 ml conical tubes and wait 1 min for the solid pieces to float to the top (Figure 2C & D). HPMC and red blood cells (RBC) will be present in the liquid on the bottom. Use a pipet to remove the liquid from the bottom of the tube and into a new conical tube. Spin the liquid down at 0.5 x g for 3 min and aspirate the supernatant from the HPMC/RBC pellet.
- Repeat the process three times: add 20 ml PBS to the minced tissue, allow tissue to rise, remove liquid from the bottom with a pipette and into the HPMC/RBC tube, spin down, and remove the supernatant. After all spins are completed and the supernatant is aspirated, the pellet will contain HPMC and RBC.
- Plate all cells from the pellet into a 75 cm2 flask in 15 ml of full growth media (DMEM with 10% fetal bovine serum [FBS],1% MEM vitamins [93 mg/L], 1% MEM nonessential amino acids [81.4 mg/L], 1% penicillin streptomycin [Pen-Strep, 100 U/ml penicillin and 100 µg/ml streptomycin]).
- Perform a secondary PBS wash to isolate additional HPMCs.
- For the secondary PBS wash, shake the remaining solid tissue at 200 rpm, 37 °C for 10 min in 20 ml of PBS, then wait 1 min for the tissue to float to the top. Remove the liquid from the bottom of the tube into a new tube, centrifuge at 0.5 x g for 3 min, and aspirate the supernatant.
- Plate all the HPMC/RBC from the secondary PBS wash in a separate 75 cm2 flask in 15 ml of full growth media.
- To isolate any remaining HPMC, shake the minced tissue at 200 rpm, 37 °C degrees for 10 min in 20 ml of a 1:1 0.25% trypsin 25 mM EDTA:PBS solution by volume. Allow the tissue to rise, collect the liquid at the bottom of the tube into a new 50 ml tube, and spin down at 0.5 g x 3 min.
- Aspirate the supernatant and plate all the HPMC/RBC in a separate 75 cm2 flask in 15 ml of full growth media.
- Culture the plated cells at 37°C and 5% CO2 in a humidified environment. Feed plated cells by adding 15 ml of full growth media on days 3 and 5 without removing the spent media. HPMC can be cultured for 5-7 days before splitting.
- Use low-passage HPMC (up to passage 2) in all experiments to minimize dedifferentiation and modification of original phenotype. Use a wide-field microscope preferably with a 20x objective with plastic differential interference contrast capabilities or 4-20x objective with phase contrast capabilities (phase contrast filters on microscope) for taking all images of cells, and a 10x objective ()in bright field to analyze immunohistochemical 3,3'-Diaminobenzidine (DAB) staining.
- Confirm that the HPMC are cuboidal and express cytokeratin 8 and vimentin by performing immunohistochemistry8 (Figure 3A, C, E).
- NOF isolation8
- Prepare 10 ml of a stock of collagenase type III solution (10x) by combining a 1:1:1 mixture of 100x Pen-Strep: 1,500 units activity/ml of collagenase type III: 714 units activity/ml of hyaluronidase in PBS.
- Shake the minced omentum tissue that remains after HPMC isolation at 200 rpm, 37 °C for 6 hr in 10 ml of the 10x collagenase type III solution diluted in serum-free media (DMEM with 1% MEM vitamins [93 mg/L], 1% MEM nonessential amino acids [81.4 mg/L], 1% penicillin streptomycin [Pen-Strep, 100 U/ml penicillin and 100 µg/ml streptomycin]). Digested tissue will look opaque and may have some fibrous debris (Figure 2E).
- Centrifuge the solution containing NOF cellsin suspension at 0.5 x g for 3 min at RT, aspirate the supernatant and plate the pellet in a 75 cm2 flask in 13 ml of full growth media.
- Remove old media and replace with 15 ml of fresh full growth media after 24 hr. NOF can be cultured for 1-3 days before splitting. Note proliferation of the NOF will cease when the cells reach confluency.
- Confirm that the primary human fibroblasts are flat, elongated cells and express vimentin but not cytokeratin 8 by performing immunohistochemistry8 (Figure 3B, D, F). Use low-passage NOF for experiments (ideally before passage 3) to minimize dedifferentiationand modification of original phenotype. Use a wide-field microscope with a 20x objective with plastic DIC capabilities for taking all phase images of cells, and a 10x objective in bright field to analyze immunohistochemical DAB staining.
2. Plating the Organotypic Culture8
Release NOF from the 75 cm culture flask by rinsing with 10 ml of PBS followed by 3 ml of 0.25% trypsin/25 mM EDTA for no more than 5 min. Neutralize trypsin with at least 3 times the volume of full growth media.
Transfer trypsinized cells into a 50 ml conical tube and spin down at 0.5 g x 3 min. Remove supernatant and bring cells back up in 5 ml of full growth media. Use a cell counter to count the cells recovered from the culture flask.
Dilute cells in the appropriate volume of full growth media to plate 2,000-4,000 NOF per 100 µl onto a black, clear-bottomed, 96-well plate. Add 0.5 µg/100 µl of rat-tail collagen I to the cell mixture before plating. Let cells sit at 37 °C in 5% CO2 in a humidified environment for at least 4 hr, or until the cells to adhere to the plate surface.
Release HPMC from the culture flask using the same methods described in steps 2.1 and 2.2. Dilute cells in the appropriate amount of full growth media to plate 10,000-20,000 HPMC per 50 µl on top of the NOF already plated with collagen I in 96-well plates. Let the cells sit at 37 °C in 5% CO2 in a humidified environment for at least 18 hr before beginning experiments.
Culture green fluorescent protein (GFP)-expressing HeyA8 ovarian cancer cells in full growth media. HeyA8 ovarian cancer cells are fluorescently labeled by expression of a lentiviral vector expressing copepod cGFP (pCDH-CMV-MCS-EF1). HeyA8 ovarian cancer cells should be 80-90% confluent on the day of use (logarithmic growth phase). Release cells from the culture plate and count using the same methods described in steps 2.1-2.2 for primary cells.
Allow ovarian cancer cells to recover in full growth media for 15-20 min at 37°C in a conical tube with the lid loosely sealed.
3. Adhesion Assay8
After recovery, pellet the ovarian cancer cells by spinning for 3 min at 0.5 x g and then remove the supernatant and dilute the cells in the appropriate volume of serum-free media to achieve a concentration of 50,000 cells/100 µl.
Invert the 96-well plates with the HPMC/NOF organotypic cultures to remove spent media, and add 100 µl of the cancer cell suspension in serum-free media to each well. Incubate the plate at 37 °C in 5% CO2 in a humidified environment for 30 min to 4 hr.
Invert the 96-well plate to remove media and non-adherent cells. Use a multichannel pipet at low power to carefully and gently pipet 100 µl of PBS into each well, and invert plate again. Repeat the PBS wash once more.
Invert to remove PBS and add 100 µl 4% paraformaldehyde to each well. Allow the plate to sit for 20 min to fix the cells. After 20 min, invert the plate and add 100 µl PBS.
- Note that the total fluorescence of wells can be reported or the number of cells can be quantified. For quantification, first plate a standard curve in duplicate using HeyA8 cells at a concentration of 1 million cells/ml.
- Make the standard curve by spinning down 1 million cells, removing media, and allowing them to sit in 1 ml 4% paraformaldehyde for 20 min.
- Spin cells again and bring up in 1 ml PBS (recount cells to confirm 1 million/ml concentration). Plate 50,000, 40,000, 30,000, 20,000, 10,000, 5000, 1,000, and 0 cells into each well in duplicate, and add PBS for a final total volume of 50 µl in each well.
Bottom read the culture plate on a fluorescent plate reader (excitation = 488 nm, emission = 528 nm), scaling high wells (50,000 on standard curve). Use the standard curve to calculate how many ovarian cancer cells adhered to the organotypic culture in each well (Figure 4A-C).
4. Proliferation Assay10
After the ovarian cancer cells recover (see steps 2.5 and 2.6 above), pellet the cells by spinning for 3 min at 0.5 x g and then dilute the cells in the appropriate volume of 1% FBS media (DMEM with 1% FBS, 1% MEM vitamins [93 mg/L], 1% MEM nonessential amino acids [81.4 mg/L], 1% penicillin streptomycin [Pen-Strep, 100 U/ml penicillin and 100 µg/ml streptomycin]) to achieve a concentration of 4,000 cells/150 µl.
Invert the 96-well plate with the HPMC/NOF organotypic cultures to remove spent media, and add 150 µl of the cancer cell suspension in 1% FBS media to each well. Incubate the plate at 37°C in 5% CO2 in a humidified environment for 72 hr.
Bottom read the culture plate on a fluorescent plate reader once a day (excitation = 488 nm, emission = 528 nm) to evaluate the relative proliferation of cells. Continue the assay for 96 hr, changing the media at 48 hr (Figure 5A-C). Be careful not to disturb the culture when changing media (inverting the plate is preferable).
5. Invasion Assay8
Before plating the 3D culture, add 7.5 µg rat-tail collagen I in 200 µl PBS to a 24-well culture plate insert (8 µm pore size). Let the plate incubate O/N, then carefully remove the PBS with a pipet immediately before plating the HPMC/NOF organotypic culture onto the collagen-coated inserts (step 2, above).
Prepare the ovarian cancer cells as in step 3.1. To plate the ovarian cancer cells onto the organotypic cultures on the inserts, carefully use a pipet to remove excess media from the top of the inserts. Add 100 µl of the ovarian cancer cell suspension in serum-free media to each well.
Add 750 µl of full growth media into the well below the insert to induce cancer cell invasion into the organotypic cultures. Incubate the plate at 37 °C in 5% CO2 in a humidified environment for 24 hr. Note: Ovarian cancer cells that invade the organotypic culture will cross the insert and remain on the bottom side of the insert.
After 24 hr, grasp the insert with tongs and briefly rinse it in a beaker of PBS, then move the insert into an empty well. Scrub the top of each insert with both sides of a cotton swab for a total of 1 min. Quickly place the insert into a plate containing 750 µl 4% paraformaldehyde in each well. Allow each insert to sit in paraformaldehyde for 10 min before transferring the inserts into wells filled with 750 µl PBS.
View the invaded cells (adhered and fixed to the bottom of inserts) with a microscope at 2.5x magnification. When the entire well is within the field of view, adjust the magnification to 10x and take 5 pictures, one near the center and 1 in each quadrant of the well, avoiding taking images that are too close to the edges. Follow the same pattern for each well.
Use the manufacturer's program to count the number of cells in each image to get an average number of cells invaded per field in each well (Figure 6A-C) according to their protocol.
Representative Results
The organotypic culture was assembled by first mixing primary human fibroblasts with collagen type I and then overlaying this culture with 5 times the number of mesothelial cells. The culture was incubated for at least 18 hr before ovarian cancer cells were added to study adhesion, invasion or proliferation. Each assay was repeated with multiple (n=3-5) 3D cultures obtained from different patients and numerous wells were tested in each condition for the adhesion (n=5), proliferation (n=5) and invasion assays (n=3). Ovarian cancer cells adhered to the 3D organotypic culture after 4 hr (Figure 4), proliferated on the culture after 72 hr (6 times the HeyA8 cell doubling time) (Figure 5), and invaded after 24 hr (Figure 6). The adhesion, proliferation and invasion of HeyA8 ovarian cancer cells was integrin-dependent (Figure 4-6). Specifically, the RGD peptide and blocking antibodies to α5-integrin, β1-integrin and αvβ3-integrin inhibited the ovarian cancer cell adhesion to the organotypic culture, while the RAD peptide, control antibody and β4-integrin antibody did not (Figure 4)10. In the proliferation assay, the RGD peptide and blocking antibodies to α5- and β1-integrin inhibited ovarian cancer cell proliferation on the organotypic culture, while the RAD peptide, control antibody, αvβ3-integrin antibody and β4-integrin antibody did not (Figure 5). In the invasion assay, the RGD peptide and blocking antibodies to α5- and β1-integrin inhibited, while the αvβ3-integrin antibody enhanced ovarian cancer cell invasion through the 3D organotypic culture. The RAD peptide, control antibody, and β4-integrin antibody did not affect invasion (Figure 6). Rarely, these assays will be un-interpretable (i.e., α5-integrin blocking antibody will not inhibit adhesion, proliferation or invasion when compared to the control antibody). Previously, we have found that certain organotypic cultures will wash away upon changing of the media or PBS washing during the assays. In addition, if the ovarian cancer cells are dead or have not recovered long enough from the trypsin digest to re-express integrins, the assays will not work. Therefore, it is always important to have a positive and negative control in each assay so the quality of the organotypic culture and ovarian cancer cell assay can be assessed. As shown here, the RGD peptide, α5-integrin and β1-integrin blocking antibodies all inhibited ovarian cancer cell adhesion, invasion and proliferation to the organotypic culture and could be used as positive controls in future assays. Likewise, β4-integrin blocking antibody did not affect ovarian cancer cell adhesion, invasion and proliferation to the organotypic culture and could be used as a negative control in future assays.
 Figure 1. Histology of human omental metastasis. Hematoxylin and eosin stain of (A) normal human omentum (MC, mesothelial cells, Fib, fibroblasts, Adi, adipocytes) and (B) ovarian cancer omental metastasis (Met, ovarian cancer metastasis). Bar in panels = 100 μm in length. (C) Schematic of the 3D organotypic model of ovarian cancer metastasis mimicking early ovarian cancer cell metastasis to the surface of the human omentum. Please click here to view a larger version of this figure.
Figure 1. Histology of human omental metastasis. Hematoxylin and eosin stain of (A) normal human omentum (MC, mesothelial cells, Fib, fibroblasts, Adi, adipocytes) and (B) ovarian cancer omental metastasis (Met, ovarian cancer metastasis). Bar in panels = 100 μm in length. (C) Schematic of the 3D organotypic model of ovarian cancer metastasis mimicking early ovarian cancer cell metastasis to the surface of the human omentum. Please click here to view a larger version of this figure.
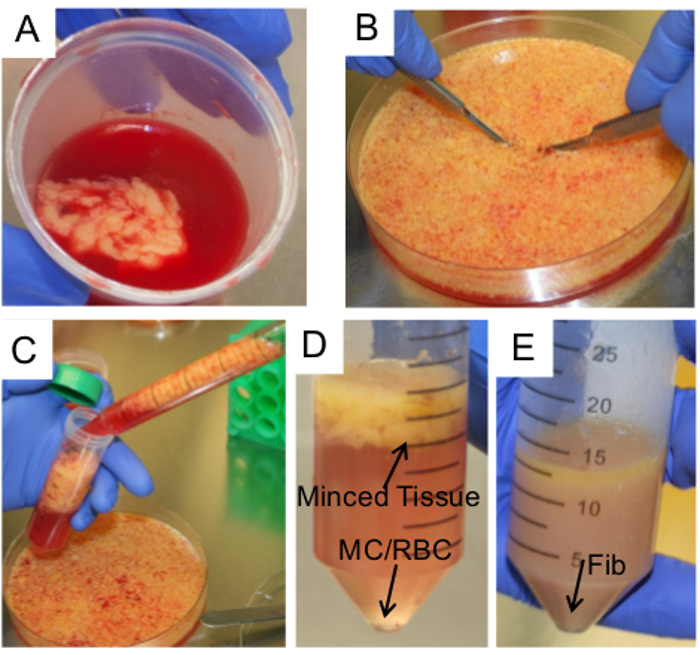 Figure 2. Isolation of primary human omental cells. (A) A piece of omentum is collected at surgery in PBS and transported to the laboratory. The tissue is pelleted at 0.5 x g x 3 min and transferred to fresh PBS. (B) The piece of omentum is transferred to a 10 cm petri dish, washed with PBS, and scalpels are used to cut the omentum into small pieces (0.5 cm3). (C-D)The tissue pieces are collected using a 25 ml serological pipette and transferred into a 50 ml conical tube for isolation of HPMC. PBS is removed from the tube and HPMC are pelleted from the PBS a 0.5 x g at 37°C. (MC, mesothelial cells, RBC, red blood cells, Fib, fibroblasts) (E) NOF are isolated from the remaining omental tissue after incubation with hyaluronidase and collagenase type III. The image shows the tissue after a 6- to 12-hr digestion. NOF are pelleted by spinning at 0.5 x g at RT. Please click here to view a larger version of this figure.
Figure 2. Isolation of primary human omental cells. (A) A piece of omentum is collected at surgery in PBS and transported to the laboratory. The tissue is pelleted at 0.5 x g x 3 min and transferred to fresh PBS. (B) The piece of omentum is transferred to a 10 cm petri dish, washed with PBS, and scalpels are used to cut the omentum into small pieces (0.5 cm3). (C-D)The tissue pieces are collected using a 25 ml serological pipette and transferred into a 50 ml conical tube for isolation of HPMC. PBS is removed from the tube and HPMC are pelleted from the PBS a 0.5 x g at 37°C. (MC, mesothelial cells, RBC, red blood cells, Fib, fibroblasts) (E) NOF are isolated from the remaining omental tissue after incubation with hyaluronidase and collagenase type III. The image shows the tissue after a 6- to 12-hr digestion. NOF are pelleted by spinning at 0.5 x g at RT. Please click here to view a larger version of this figure.
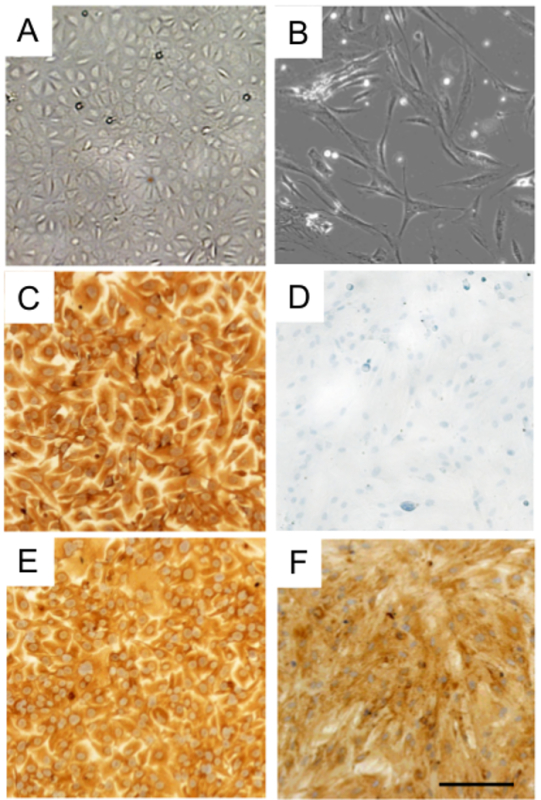 Figure 3. Characterization of HPMC and NOF isolated from human omentum tissue. Phase-contrast images of (A) mesothelial cells (HPMC) and (B) fibroblasts (NOF) after isolation from omentum tissue. Human HPMC stained for (C) cytokeratin 8 and (E) vimentin. Human NOF stained for (F) vimentin, but did not stain for (D) cytokeratin 8. Bar = 100 µm, applies to all panels. Please click here to view a larger version of this figure.
Figure 3. Characterization of HPMC and NOF isolated from human omentum tissue. Phase-contrast images of (A) mesothelial cells (HPMC) and (B) fibroblasts (NOF) after isolation from omentum tissue. Human HPMC stained for (C) cytokeratin 8 and (E) vimentin. Human NOF stained for (F) vimentin, but did not stain for (D) cytokeratin 8. Bar = 100 µm, applies to all panels. Please click here to view a larger version of this figure.
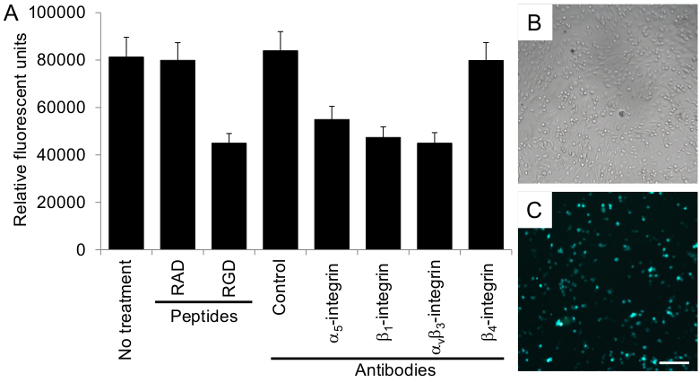 Figure 4. Ovarian cancer cell adhesion to the organotypic culture is integrin-dependent. (A) The adhesion assay was performed with 50,000 fluorescently-labeled ovarian cancer cells, HeyA8 cells, as described in the Protocol section. The total fluorescence in each well is reported. The control antibody is mouse IgG. Each bar represents the mean +/- standard error mean. Representative (B) phase-contrast and (C) fluorescent images of ovarian cancer cell adhesion to the organotypic culture. Bar = 100 µm, applies to both panels. Please click here to view a larger version of this figure.
Figure 4. Ovarian cancer cell adhesion to the organotypic culture is integrin-dependent. (A) The adhesion assay was performed with 50,000 fluorescently-labeled ovarian cancer cells, HeyA8 cells, as described in the Protocol section. The total fluorescence in each well is reported. The control antibody is mouse IgG. Each bar represents the mean +/- standard error mean. Representative (B) phase-contrast and (C) fluorescent images of ovarian cancer cell adhesion to the organotypic culture. Bar = 100 µm, applies to both panels. Please click here to view a larger version of this figure.
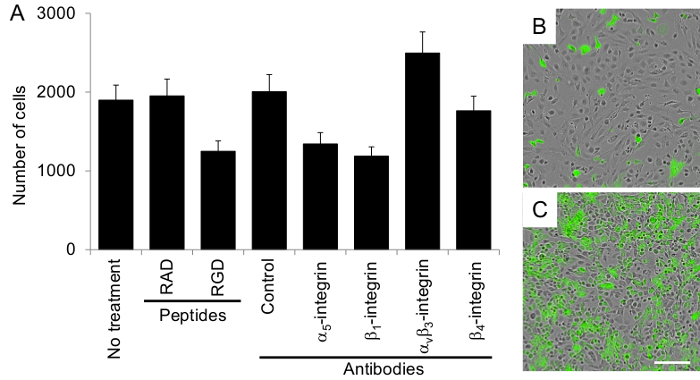 Figure 5. Ovarian cancer cell proliferation on the organotypic culture is integrin-dependent. (A) The proliferation assay was performed with 4,000 fluorescently-labeled ovarian cancer cells, HeyA8 cells, as described in the Protocol section. The total number of cells is reported. The control antibody is mouse IgG. Each bar represents the mean +/- standard error mean. Merged phase-contrast and fluorescent images of ovarian cancer cell proliferation on the organotypic culture at (B) 24 and (C) 96 hr. Bar = 100 µm, applies to both panels. Please click here to view a larger version of this figure.
Figure 5. Ovarian cancer cell proliferation on the organotypic culture is integrin-dependent. (A) The proliferation assay was performed with 4,000 fluorescently-labeled ovarian cancer cells, HeyA8 cells, as described in the Protocol section. The total number of cells is reported. The control antibody is mouse IgG. Each bar represents the mean +/- standard error mean. Merged phase-contrast and fluorescent images of ovarian cancer cell proliferation on the organotypic culture at (B) 24 and (C) 96 hr. Bar = 100 µm, applies to both panels. Please click here to view a larger version of this figure.
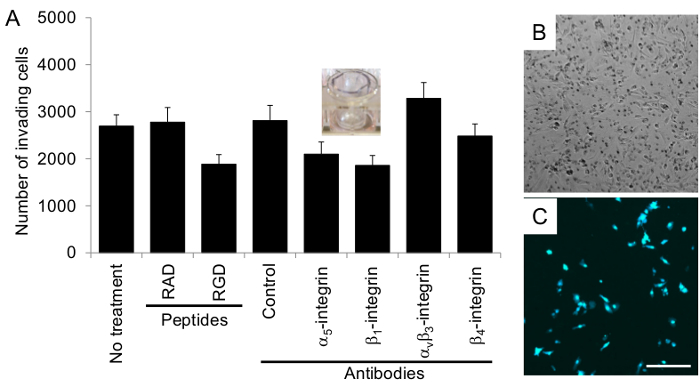 Figure 6. Ovarian cancer invasion through the organotypic culture is integrin-dependent. (A) The invasion assay was performed with 40,000 fluorescently-labeled ovarian cancer cells, HeyA8 cells, as described in the Protocol section. The number of invading cells (moved through the 3D culture to the bottom of the insert) is reported. The control antibody is mouse IgG. Each bar represents the mean +/- standard error mean. (B) Phase-contrast and (C) fluorescent images of ovarian cancer cells that invaded through the organotypic culture. Bar = 100 µm, applies to all panels. Please click here to view a larger version of this figure.
Figure 6. Ovarian cancer invasion through the organotypic culture is integrin-dependent. (A) The invasion assay was performed with 40,000 fluorescently-labeled ovarian cancer cells, HeyA8 cells, as described in the Protocol section. The number of invading cells (moved through the 3D culture to the bottom of the insert) is reported. The control antibody is mouse IgG. Each bar represents the mean +/- standard error mean. (B) Phase-contrast and (C) fluorescent images of ovarian cancer cells that invaded through the organotypic culture. Bar = 100 µm, applies to all panels. Please click here to view a larger version of this figure.
Discussion
An organotypic model of the peritoneal microenvironment was established to assess the individual and collective function(s) of both the cellular and innate components of the microenvironment in ovarian cancer dissemination. The specific protocols for plating and customizing the 3D organotypic culture to investigate ovarian cancer cell adhesion, proliferation, and invasion are provided. Primary human omental mesothelial cells and fibroblasts were isolated from patients and used at an early passage to preserve normal morphology and patient variation. In this model, normal omental fibroblasts and ECM (typically collagen type I) were layered with a primary human mesothelial cell monolayer. This model histologically mimics the peritoneal lining, and allows us to recreate and examine key events in ovarian cancer metastasis including ovarian cancer cell adhesion, proliferation, and invasion8,27.
Previously, limited 3D models have been established to investigate ovarian cancer interactions with the microenvironment13,14,21-25, while this 3D organotypic model is the first to recreate the construction of the mesothelial-lining of the peritoneal cavity. The human omentum is collected from patients varying in age and health. The health of the tissue directly affects the mesothelial cell and fibroblast isolation techniques. The healthier tissue appears lighter in color with very small lobules (1-3 mm) compared to tissue that is more orange in color with large lobules present (>5 mm). The majority of HPMC slough off in the initial PBS as the tissue is carried from surgery to the laboratory. The healthier the tissue, however, the more resilient the HPMC are to removal, and additional PBS washes and the 1:1 PBS:trypsin digest may result in a mixed culture of HPMC and NOF cells. In the healthier tissues, some HPMC can survive the 6-hr collagenase digest, and a digest O/N in 1x collagenase solution in full growth media is recommended to obtain a pure NOF culture. Note the omentum must be placed immediately in PBS upon removal or no HPMCs will be recovered from tissue. Furthermore, if the tissue is stored in PBS for an extended period of time (>2 hr) at RT or in a refrigerator, the number of viable HPMCs isolated is significantly reduced.
A 1:5 ratio of fibroblasts to mesothelial cells is plated to mimic the ratio of fibroblasts to mesothelial cells in vivo8. It is important to note that the size of isolated mesothelial cells varies from patient to patient and this may require adjustment to the plating ratio. The mesothelial cells of some patients are much smaller; in these cases, we plate double the number of mesothelial cells (20,000). In addition, the primary cells are always used at an early (1-2) passage to preserve morphology. Mesothelial cells can be cuboidal or spindly, and cuboidal mesothelial cells become more spindly as they are passaged, or with greater exposure to conditioned media from ovarian cancer cells or treatment with TGFβ10.
Ovarian cancer cells may adhere to and invade through the mesenchymal mesothelial cells more efficiently than the epithelial mesothelial cells from the same or different patients. Typically, we set up the organotypic culture for 18 hr before starting the functional assays with ovarian cancer cells. The longer the mesothelial cells and fibroblasts are cultured together, the more time they have to secrete different proteins including ECM proteins, which can affect the behavior of the ovarian cancer cells. Another important factor to take into account when performing these assays is the differential proliferation rate and expression level of proteins associated with increased adhesion, invasion, and/or proliferation, including integrins, proteases, and growth factors, in each cancer cell line or primary human cancer cell types. The time and number of cancer cells used in each of the functional assays with the organotypic culture needs to be optimized for each cancer cell line or type of primary cancer cell. For example, in the described adhesion assay, ovarian cancer cells could be invading the culture during this time.
Clearly, this organotypic model does not contain all components of the normal peritoneal microenvironment such as immune, endothelial, and adipocyte cells. Additionally, the model depends on the accessibility and availability of primary human tissue, as the primary cells are only used within the first three passages. However, the model has many advantages in its use of primary human cells and incorporation of multiple cell types that have been demonstrated to play important roles in metastasis. This model has been used to investigate gene and protein regulation in the individual cell types (i.e., cancer cells, normal mesothelial or fibroblast cells) after co-culture. In particular, the roles of c-Met28, MMP-29,29, E-cadherin30, β1-integrin10, β3-integrin16, α5-integrin10,31, and fibronectin10 in early metastasis have been explored. Additionally, the model has the potential to expand to include more cell types, such as the incorporation of adipocytes6. The model has been modified and miniaturized for use in high-throughput screening to identify small molecule inhibitors of ovarian cancer cell adhesion/invasion to the peritoneal microenvironment27. Future applications of this model will include the incorporation of immune cells into the assay, including macrophages for mechanistic studies and high-throughput screening. In summary, the organotypic cell culture provides a useful model to evaluate the interactions between the human peritoneal microenvironment and ovarian cancer cells, with the goal of elucidating important molecular mechanisms of dissemination and disease progression, and identifying potential therapeutic targets.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank all residents and attending physicians, notably Dr. A.F. Haney (the University of Chicago, Department of Obstetrics and Gynecology) for collecting omental biopsies. Also, we thank Stacey Tobin and Gail Isenberg for carefully editing this manuscript. This work was supported by Bears Care, the charitable beneficiary of the Chicago Bears Football Club, the National Institute of Neurological Disorders and Stroke (NINDS) R21 NS075702, and the National Cancer Institute grant R01 CA111882 to E.L.
References
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics. CA Cancer J. Clin. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- Lengyel E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010;177:1053–1064. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia AL, Bissell MJ. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat. 2012;15:39–49. doi: 10.1016/j.drup.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Coussens LM. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Nieman KM, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daya D, McCaughy WT. Pathology of the peritoneum: A review of selected topics. Semin. Diagn. Pathol. 1991;8:277–289. [PubMed] [Google Scholar]
- Kenny HA, Krausz T, Yamada SD, Lengyel E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells. Int. J. Cancer. 2007;121:1463–1472. doi: 10.1002/ijc.22874. [DOI] [PubMed] [Google Scholar]
- Kenny HA, Kaur S, Coussens LM, Lengyel E. The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J. Clin. Invest. 2008;118:1367–1379. doi: 10.1172/JCI33775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny HA, et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J. Clin. Invest. 2014;124:4614–4628. doi: 10.1172/JCI74778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobel T, Cannistra SA. β1-integrins partly mediate binding of ovarian cancer cells to peritoneal mesothelium in vitro. Gynecol. Oncol. 1999;73:362–367. doi: 10.1006/gyno.1999.5388. [DOI] [PubMed] [Google Scholar]
- Strobel T, Swanson L, Cannistra SA. In vivo inhibition of CD44 limits intra-abdominal spread of a human ovarian cancer xenograft in nude mice: A novel role for CD 44 in the process of peritoneal implantation. Cancer Res. 1997;57:1228–1232. [PubMed] [Google Scholar]
- Iwanicki M, et al. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011;1:144–157. doi: 10.1158/2159-8274.CD-11-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessan K, Aguiar D, Oegema TR, Siebenson L, Skubitz AP. CD44 and β1 integrin mediate ovarian carcinoma cell adhesion to peritoneal mesothelial cells. Am. J. Pathol. 1999;154:1525–1537. doi: 10.1016/s0002-9440(10)65406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N, Riley C, Rice G, Quinn M. Role of integrin receptors for fibronectin, collagen and laminin in the regulation of ovarian carcinoma functions in response to a matrix microenvironment. Clin. Exp. Metastasis. 2005;22:391–402. doi: 10.1007/s10585-005-1262-y. [DOI] [PubMed] [Google Scholar]
- Kaur S, et al. β3-integrin expression on tumor cells inhibits tumor progression, reduces metastasis, and is associated with a favorable prognosis in patients with ovarian cancer. Am. J. Pathol. 2009;175:2184–2196. doi: 10.2353/ajpath.2009.090028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedbala MJ, Crickard K, Bernacki R. In vitro degradation of extracellular matrix by human ovarian carcinoma cells. Clin. Exp. Metastasis. 1987;5:181–197. doi: 10.1007/BF00058063. [DOI] [PubMed] [Google Scholar]
- Kanemoto T, Martin GR, Hamilton TC, Fridman R. Effects of synthetic peptides and protease inhibitors on the interaction of a human ovarian cancer cell line (NIH:OVCAR-3) with a reconstituted basement membrane (matrigel) Invasion Metastasis. 1991;11:84–92. [PubMed] [Google Scholar]
- Rieppi M, et al. Mesothelial cells induce the motility of human ovarian carcinoma cells. Int. J. Cancer. 1999;80:303–307. doi: 10.1002/(sici)1097-0215(19990118)80:2<303::aid-ijc21>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Barbolina MV, Adley BP, Ariztia EV, Liu Y, Stack MS. Microenvironmental regulation of membrane type 1 matrix metalloproteinase activity in ovarian carcinoma cells via collagen-induced EGR1 expression. J. Biol. Chem. 2007;282:4924–4931. doi: 10.1074/jbc.M608428200. [DOI] [PubMed] [Google Scholar]
- Burleson KM, et al. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol. Oncol. 2004;93:170–181. doi: 10.1016/j.ygyno.2003.12.034. [DOI] [PubMed] [Google Scholar]
- Suzuki N, et al. HMOCC-1, a human monoclonal antibody that inhibits adhesion of ovarian cancer cells to human mesothelial cells. Gynecol. Oncol. 2004;95:290–298. doi: 10.1016/j.ygyno.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Kishikawa T, et al. Two distinct pattern of peritoneal involvement shown by in vitro and in vivo ovarian cancer dissemination models. Invas. Metast. 1995;15:11–21. [PubMed] [Google Scholar]
- Casey RC, et al. Establishment of an in vitro assay to measure the invasion of ovarian carcinoma cells through mesothelial cell monolayers. Clin. Exp. Metastasis. 2003;20:343–356. doi: 10.1023/a:1024009131191. [DOI] [PubMed] [Google Scholar]
- Niedbala MJ, Crickard K, Bernacki R. Interactions of human ovarian tumor cells with human mesothelial cells grown on extracellular matrix. Exp. Cell Res. 1985;160:499–513. doi: 10.1016/0014-4827(85)90197-1. [DOI] [PubMed] [Google Scholar]
- White EA, Kenny HA, Lengyel E. Three-Dimensional modeling of ovarian cancer. Adv. Drug Deliv. Rev. 2014;79-80:184–192. doi: 10.1016/j.addr.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny HA, et al. Quantitative high throughput screening using a primary human three-dimensional organotypic culture predicts in vivo efficacy. Nature Comm. 2015. [DOI] [PMC free article] [PubMed]
- Sawada K, et al. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 2007;67:1670–1680. doi: 10.1158/0008-5472.CAN-06-1147. [DOI] [PubMed] [Google Scholar]
- Kenny HA, Lengyel E. MMP-2 functions as an early response protein in ovarian cancer metastasis. Cell cycle. 2009;8:683–688. doi: 10.4161/cc.8.5.7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada K, et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008;68:2329–2339. doi: 10.1158/0008-5472.CAN-07-5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra AK, et al. Ligand-independent activation of c-Met by fibronectin and α(5)β(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene. 2011;30:1566–1576. doi: 10.1038/onc.2010.532. [DOI] [PMC free article] [PubMed] [Google Scholar]