

Abstract
Förster Resonance Energy Transfer (FRET) has become a powerful tool for monitoring protein folding, interaction and localization in single cells. Biosensors relying on the principle of FRET have enabled real-time visualization of subcellular signaling events in live cells with high temporal and spatial resolution. Here, we describe the application of a genetically encoded Bile Acid Sensor (BAS) that consists of two fluorophores fused to the farnesoid X receptor ligand binding domain (FXR-LBD), thereby forming a bile acid sensor that can be activated by a large number of bile acids species and other (synthetic) FXR ligands. This sensor can be targeted to different cellular compartments including the nucleus (NucleoBAS) and cytosol (CytoBAS) to measure bile acid concentrations locally. It allows rapid and simple quantitation of cellular bile acid influx, efflux and subcellular distribution of endogenous bile acids without the need for labeling with fluorescent tags or radionuclei. Furthermore, the BAS FRET sensors can be useful for monitoring FXR ligand binding. Finally, we show that this FRET biosensor can be combined with imaging of other spectrally distinct fluorophores. This allows for combined analysis of intracellular bile acid dynamics and i) localization and/or abundance of proteins of interest, or ii) intracellular signaling in a single cell.
Keywords: Bioengineering, Issue 107, Bile Acid, Bile Acid Sensor, Live Cell imaging, Förster Resonance Energy Transfer (FRET), Confocal Microscopy, FACS, FXR, Spatiotemporal, Homeostasis, Metabolism, Cellular Dynamics
Introduction
Förster Resonance Energy Transfer (FRET) is widely used to gain a better understanding of cellular functions in living cells with high temporal and spatial resolution1. In FRET, energy from an excited donor fluorophore is transferred to an acceptor fluorophore. FRET efficiency is strongly dependent on the distance between the donor and acceptor fluorophore and their orientation and is therefore a sensitive readout of conformational changes that affect the two fluorophores. This phenomenon is exploited to generate FRET-based biosensors for the imaging of small molecules. Changes in their concentration can be monitored as increases/decreases in the ratio of emission intensity of the acceptor versus the donor fluorophore2. For instance, FRET-based calcium biosensors allow for fast and stable detection of free calcium concentrations in living cells3. Other advantages of FRET-based biosensors are imaging in single living cells, their non-invasiveness, their ability to be targeted to different cell types and cellular compartments4.
Many aspects of intracellular bile acid dynamics are still poorly understood. For example, little is known about the mechanism underlying regulation of conjugated and unconjugated bile acid transport. Existing techniques to monitor this transport primarily make use of luciferase-based reporters, radiolabeled bile acids, or fluorescent bile acid analogs. The latter requires modification of bile acids, possibly affecting their properties. Luciferase-based reporters have poor time resolution. Besides, these techniques result in loss of the sample and are not applicable for imaging in single cells. Therefore, it would be beneficial to use methods that allow live single cell imaging of transport activity using FRET biosensors, especially since it includes the advantage of ratiometric detection5,6. While variants of CFP/YFP form most frequently used FRET pairs, new strategies using mOrange and mCherry carrying self-association-inducing mutations have led to an expansion of the FRET toolbox with novel sensors, including a red-shifted bile acid sensor7.
We previously created a genetically-encoded FRET bile acid sensor (BAS), that consists of a donor fluorophore (cerulean) and an acceptor fluorophore (citrine) that are fused with the farnesoid X receptor (FXR) ligand binding domain (FXR-LBD) and a peptide containing an LXXLL motif8. This peptide associates with the FXR-LBD in a bile acid-dependent manner. Upon FXR activation, the distance between citrine and cerulean will alter due to a conformational change. In mammalian cell lines, FXR activation results in a clearly detectable increase in the citrine/cerulean ratio, while the purified sensor works in the opposite direction and leads to a decreased FRET ratio upon FXR activation. This sensor (CytoBAS) allows monitoring of cytosolic bile acid dynamics. By carboxyl-terminal addition of subcellular targeting motifs, the BAS construct can be targeted to the nucleus (NucleoBAS) and peroxisomes (PeroxiBAS), allowing measurements of bile acid concentrations in different cellular compartments. Although the addition of the peroxisomal targeting motif does not impair its responsiveness to bile acids, cell permeable FXR-ligands did not induce any FRET changes of PeroxiBAS inside peroxisomes8. As the nature of this discrepancy is unknown, the protocol below is focused on CytoBAS and NucleoBAS.
The use of this genetically encoded FRET sensor was recently demonstrated in cells containing the hepatic bile acid transporters Na+/taurocholate co-transporting polypeptide (NTCP) and organic solute transporter alpha / beta (OSTαβ)8. NTCP is the principal hepatic bile acid importer and OSTαβ is a basolateral intestinal bile acid transporter that can function both as an importer and exporter dependent on the electrochemical bile acid concentration gradient9,10. Recent data showed that upon bile acid transport by NTCP and/or OSTαβ, robust and fast responses in FRET ratio as a result of ligand-FXR-LBD interaction can be observed.
Here, we describe detailed protocols for methods to measure FRET such as confocal microscopic analysis and fluorescence activated cell sorting (FACS), highlight critical steps, address potential problems and discuss alternative methods. Using this genetically encoded FRET sensor, bile acid interaction with FXR-LBD can be quantified and monitored directly in living cells and provides a rapid and simple method of visualizing bile acid transport and dynamics in real-time. Mammalian expression plasmids encoding CytoBAS and NucleoBAS are available commercially. Therefore, this biosensor can further contribute to the understanding of bile acid transporters or compounds that activate FXR and provide a deeper insight into bile acid biology and signaling.
Protocol
1. Transient Transfection
Note: CytoBAS and NucleoBAS (Please see Materials Table) are successfully used in several cell types, (U2OS, Huh7, HepG2, H69, MDCK and HEK293T cells). The main requirement to use the sensor is that it needs to be expressed, requiring the encoding DNA to enter the cell.
Harvest cells from an 80% confluent 25 cm2 flask. Dilute cells in complete culture medium suitable for the specific cell line (10% FBS, 1% L-glutamine, 1% pen/strep for U2OS and Huh7 cells).
Plate cells at the desired density into a sterile 8 well chambered cover glass (0.8 cm2). Aim for a sub-confluent (60-80% confluency) layer of cells on the day of the experiment, although this is not essential.
Add complete culture medium to a final volume of 400 µl per well. Allow cells to attach for 24 hr.
- Prepare the transfection mix in a sterile tube by adding 0.5 µg of NucleoBAS or CytoBAS DNA to 100 µl of culture medium (serum/antibiotic free). Vortex well. Add a transfection reagent and mix by vortexing. Transfection reagents that give optimal efficiency are cell type dependent and might require prior testing.
- For instance, use 2.5 µl of 1 mg/ml polyethylenimine (PEI) and incubate the DNA/PEI mix for 15 min at RT.
Add the transfection mixture in droplets to the cells and mix by gently shaking the well plate by hand. Add 100 µl of the mixture for a well of ~9.6 cm2. Use 10 µl of the transfection mixture for individual imaging chambers for transiently transfected cells. Note: After 24-48 hr, cells will express the bile acid sensor and can be used for experiments.
2. Stable Transfection
Harvest cells from an 80% confluent 25 cm2 flask. Dilute pellet in complete culture medium suitable for the specific cell line (10% Fetal Bovine Serum, 1% L-glutamine, 1% pen/strep for U2OS and Huh7 cells) to a concentration of 150,000 cells per ml and add around 300,000 cells per well in a 6-well culture plate (2 ml end volume).
Allow cells to attach for 24 hr.
- Prepare the transfection mix in a sterile tube by adding 0.5 µg of CytoBAS or NucleoBAS DNA (Please see Materials Table) to 100 µl of culture medium (serum/antibiotic free). Vortex well. Add a transfection reagent and mix by vortexing.
- For instance, use 2.5 µl of 1 mg/ml polyethylenimine (PEI) and incubate the DNA/PEI mix for 15 min at RT.
Add 100 µl of the transfection mixture in droplets to the cells and mix by gently shaking the well plate by hand. Always include an untransfected control when creating a stable cell line.
Grow cells O/N at 37 °C, 5% CO2.
Trypsinize cells according to manufacturer's protocol (Incubate cells at 37 °C with 1.5 ml prewarmed trypsin per 25 cm2 cell culture) and spin them down at 250 x g at RT for 5 min. Remove supernatant and resuspend cells in 13 ml of complete culture medium. Divide cells over various 10 cm diameter culture dishes: 0.5 ml (low density), 1.5 ml (medium density), and 4.5 ml (high density). Do the same for remaining 6.5 ml.
Add culture medium to a final volume of 10 ml. For U2OS cells, use 800 µg/ml G418 for plasmids with the neomycin resistance cassette such as the CytoBAS and NucleoBAS constructs to select for positive cells. This concentrations is cell type dependent and can be determined by selecting the lowest antibiotic (G418) concentration where untransfected cells died within 2 to 10 days (800 µg/ml G418 for U2OS cells).
Refresh the culture medium with the appropriate antibiotic (G418) every 72 hr until the untransfected cells are dead.
Examine the culture plate under the microscope with a 20X objective for positive colonies (fluorescence can be monitored using most filter sets used for green, cyan or yellow fluorescence).
Mark the positive colonies that lie isolated from other colonies with a pen on the exterior bottom of the culture dish.
Wash the plate twice with Phosphate Buffered Saline (PBS) and aspirate it. Take around 15 sterile cloning cylinders and dip them into sterile silicone. Apply them around the selected colonies by pressing lightly against the petri-dish and make sure that not more than one colony is included in the cloning ring.
Pipet 20 µl of pre-warmed trypsin (1x, 0.25%) in the cloning cylinder and incubate at 37 °C until most of the cells have rounded up (observe with a microscope).
Add 150 µl of culture medium with serum and the appropriate antibiotic (10% FBS and 800 µg/ml G418 for U2OS and Huh7 cells) in the cloning cylinder and pipet up and down several times to resuspend the cells and transfer to a 96-well plate. Refresh medium after 4-24 hr and allow cells to grow confluent in the next few days (37 °C, 5% CO2for U2OS and Huh7 cells).
Refresh medium with antibiotic every 3 or 4 days. When the cells have grown confluent, transfer them to a larger well plate. Repeat this step until the culture is large enough for experiments. Cryopreserve some vials for backup. Cryopreservation medium consists of 20% FBS and 20% DMSO in media (DMEM) without supplements. Note: Now, the cell line is monoclonal and considered stable for expressing the Bile Acid Sensor. Half the concentration of the antibiotic G418 (400 µg/ml) is now sufficient to maintain expression in the stable cell line.
3. Lentiviral Transduction
Note: Some cell lines are considered difficult to transfect by more traditional methods such as the polyethylenimine (PEI) method. Viral transduction of cells is an efficient alternative tool for gene-delivery and stable transgene expression.
Harvest cells from an 80% confluent 25 cm2 flask. Dilute cells in complete culture medium suitable for the specific cell line (10% FBS, 1% L-glutamine, 1% pen/strep for U2OS and Huh7 cells) and add around 200,000 cells per well in a 6-well culture plate.
Add culture medium to a final volume of 2 ml per well. Allow cells to attach for 24 hr.
Perform viral transductions by incubating cells for 4-6 hr with 500 µl CytoBAS lentivirus containing medium (available upon request) filled up to a total amount of 1 ml with complete culture medium containing 10 µg/ml diethylaminoethyl (DEAE)-dextran or another lentiviral transduction enhancer. Do not forget to include a well with untransduced control cells.
Remove medium and add 2 ml of complete culture medium containing the desired antibiotic (500 µg/ml hygromycin). Use a lentiviral construct for CytoBAS transduction that provides hygromycin resistance to cells. Grow cells under desired conditions.
- Refresh medium with (500 µg/ml hygromycin) antibiotic every 3 days and split the cells when 80% confluent, until all negative control cells are dead (takes about 1-2 weeks for most cell lines). The cell line is now considered stable for expressing the Bile Acid Sensor.
- Alternatively, use cells for experiments 1-3 days after viral transduction. As live virus might be present, this requires FRET-readout equipment in rooms with the appropriate safety level.
- Alternatively, for rapid isolation of stable cell lines, perform fluorescence activated cell sorting (FACS).
- Harvest cells from a confluent 160 cm2 flask.
- Dilute cells in Leibovitz culture medium (L-15 medium) with <5% serum to a concentration of maximum 5 x 106 cells per ml
- For collection of sorted cells, fill FACS tubes with 1 ml of collection medium (L-15 medium + 1.5% pen/strep, 1% L-glut and 20% serum).
- Sort cells for citrine (520-580 nm) or cerulean (450-520 nm), excited with 405 laser.
- After sorting approximately 500,000 cells, spin down cells (5 min, 250 x g) and resuspend them in 5 ml normal complete culture medium (10% FBS, 250 µg/ml Hygromycin for U2OS and Huh7 CytoBAS cells) and plate them in a T25 culture flask. Grow cells under desired conditions (37 °C, 5% CO2 for U2OS and Huh7 cells).
4. Live Cell Imaging of the Bile Acid Sensor
Note: Cells containing bile acid transporters can be cultured in medium with 1-10% charcoal-filtered serum that removes lipophilic compounds. Normal serum often contains bile acids that could lead to intracellular bile acid accumulation and saturation of the Bile Acid Sensor.
- FRET measurements using the confocal microscope
- Plate the cells stably or transiently expressing the sensor at the desired density into a sterile 8 well coverslip bottomed chamber slide (0.8 cm2). Grow cells in complete culture medium (using charcoal-filtered serum) so that on the day of the experiment, confluency is around 60-70%.
- Wash the adherent cells in chamber slide once with 200 µl 1x PBS or Leibovitz's L-15 culture medium.
- Aspirate and replace with 300 µl Leibovitz's L-15 culture medium so that no CO2control is necessary. Note: DMEM without phenol red can be used as well but requires CO2buffered conditions. The bile acid sensor shows (some) pH-sensitivity so aim to keep the pH constant during the data-collection.
- Dilute the compounds to be used in the confocal imaging experiment also in L-15 culture medium. Take into account that during imaging, relatively large amounts of liquid in the chambers have to be added to secure rapid mixing of the sample (50-100 µl), so make 3-5x solutions.
- Start the imaging software of a confocal microscope with a 37 °C incubation chamber and turn on the violet 405 nm laser. Put a drop of immersion oil onto the objective and place the 8-chamber on top of it. For single cell imaging of FRET, use the 63X oil objective. The sensor has been successfully used at RT, but cell behavior might be altered.
- Set the settings of the confocal microscope properly.
- Set Acquisition mode: xyt (time-lapse imaging of single focal plane).
- Acquire images at 20 sec intervals to allow compounds to be added between acquisitions without pausing the experiment.
- Set spectral range for emission detection: Cerulean: 450-520 nm; Citrine: 520-580 nm.
- Focus on the single cell layer using transillumination light. Start imaging and adjust the z-position more precisely. Adjust gain and offset for each channel to distinguish signal from background while remaining well below saturation for all pixels in cells of interest.
- Draw circles to define the region of interest (ROI). Select cells that show similar fluorescence intensity. Avoid cells that obviously differ from the average cell in size and shape. Expose the cells to light for a time as short as possible to minimize photobleaching of the sensor. It is advisable to draw ROIs not very close to the cell perimeter. This area is most sensitive to changes in fluorescence intensity due to focal drift (cells moving in z-direction) making it more difficult to monitor changes in fluorescence ratio during the experiment.
- Start measurements with the confocal microscope. Wait until the cerulean and citrine fluorescence is stable.
- Add 50-100 µl bile acids or other compounds at chosen time points during measuring. The relatively high amounts of liquid (one-fifth to one-third of the final volume) are necessary to mix the fluids well (within 10 sec) without shaking/pipetting up and down. Make sure not to touch the edge of the 8-well chamber with the pipette tip and add the liquid slowly so that the cells do not get out of focus. Do not add new compounds before the plateau phase is reached. This takes about 200 sec.
- End each experiment with the addition of 100 µl GW4064, end concentration 5-10 µM (sensor saturating dose) and wait until sensor fluorescence is stable.
- Save the experiment and export the data as an .AVI file.
- Open in ImageJ both AVI files (channel 00, cerulean; channel 01, citrine) by selecting Plugins > Stacks > Stack interleaver. Alternatively, import confocal file (e.g., .lsm or .lif files) directly in Image J using appropriate plugins (available at http://www.openmicroscopy.org) and move to step 4.1.14.
- For Stack 1: use Channel 01 (citrine).
- For Stack 2: use Channel 00 (cerulean).
- Click on Edit > Selection > Add to manager, to open the ROI manager window. Check the checkbox 'Show All'.
- Draw a few regions of interest (ROIs) covering specific cells with the oval selection tool. Also draw one circle in an area outside cells or inside a cell that does not express the sensor to determine the background signal. It is advisable to draw ROIs not very close to the cell perimeter in experiments when changes in fluorescence intensity due to focal drift (cells moving in z-direction) or cell migration were obvious.
- Select one cell. Click on Plugins > Ratio Profiler. This will result in 3 screens: RAW, ratio and Ratio_Profile. The RAW window shows the increase in intensity of citrine (blue line) and a decrease in cerulean (red line) if there is FRET. The Ratio window gives information about the ratio citrine/cerulean, which will increase with an increase in FRET. The Ratio_Profile window gives the actual numbers of fluorescence intensity measured in both channels. If microscope setup-specific files (e.g., .lsm or .lif instead of .avi) files are used the channel order might be reversed.
- Copy the data from the Ratio_Profile window in the spreadsheet attached as supplementary data. Do the same for all the other cells (and background ROI). Note: In the online spreadsheet, all data is normalized to the condition at which maximum BAS activation is expected. Given that GW4064 is the most potent activator of FXR, the fluorescence ratio after incubation with a surplus of GW4064 is set to 1. It is therefore important to end all of the experiments with addition of GW4064. The advantage of this is that the data is no longer dependent on laser intensity or detector gain and experiments on different days can be compared more easily. Furthermore, in the bottom graph of the spreadsheet, a running average can be used to smooth the curves for experimental noise. However, do not use this graph when analyzing kinetic data, since the running average will also smooth fast kinetic responses.
- FRET measurements using Fluorescence Activated Cell Sorting (FACS)
- Dilute all compounds for the FACS experiment in sterile FACS uptake buffer (0.3 mM EDTA, 0.5% BSA, 0.01% NaN3and 10 mM D-glucose).
- Harvest cells from an 80% confluent T-160 cm2 cell culture flask using 5 mM EDTA in PBS. Centrifuge cells at 250 x g for 5 min. Wash cell pellet 2x in 5 ml FACS uptake buffer at RT.
- Count cells using the coulter counter or a counting chamber.
- Dilute pellet in FACS uptake buffer to a concentration of 1 x 106 cells/ml. Pipette up and down to create a homogeneous suspension of single cells. If cells are difficult to disaggregate, put the samples through a cell strainer before sorting to minimize nozzle clogs.
- Pipet 200 µl cells per FACS tube and protect them from light.
- Add the desired concentration of the compound (e.g., bile acids, synthetic FXR ligands, transporter inhibitors). Vortex. Incubate for 20-30 min at RT while shaking (in the dark).
- Meanwhile, start the FACS (the lasers need time to warm up).
- Set the flow cytometry gating parameters for the experiment (see Figure 3):
- Load around 100,000-200,000 NucleoBAS or CytoBAS transfected cells to determine the gates.
- First adjust the FSC and SSC voltages to plot the cells in the center of the plot.
- Using the violet laser, adjust the cerulean (450/40 nm) voltage value and citrine (525/20 nm) voltage and ensure that all NucleoBAS or CytoBAS positive cells are plotted within the scatter plot.
- Set the correct gates (Gate P1 up to Gate P4), see Figure 4.
- Use gate P1 to exclude dead cells, usually displayed in the lower left corner by selecting the main population in the middle of the SSC-A/FSC-A plot.
- Use gate P2 in the FSC-H/FSC-A window to remove duplets from analysis. Single cells are presented in a more diagonal line than doublets.
- Gate P3 in the citrine (525/20 nm)/cerulean (450/40 nm) window can be used to select for NucleoBAS/CytoBAS positive cells, by gating citrine and cerulean high cells, a population displayed diagonally in the upper right corner of the plot.
- Draw gate P4 at the top left of the population, as close as possible and make sure that no more than 5% of the cells fall within this gate.
- Load some untransfected cells (no expression of CytoBAS or NucleoBAS) to determine auto-fluorescence. The untransfected population may not be located in gate P3. Adjust gate P3 when necessary to exclude auto-fluorescent cells.
- Vortex the samples before placing the tubes in the FACS. For each sample, measure at least 10,000 cells in gate P3. Note: The Population Hierarchy Window gives the number of events being displayed for each gate and % of the parent gate. In gate 4, the % of the parent states cells with an increased citrine/cerulean ratio as a percentage of all NucleoBAS positive cells.
Representative Results
The FRET-BAS sensor presented is based on the ligand binding domain of FXR (LBD-FXR) attached to two fluorophores citrine and cerulean) and an LXXLL motif. This sensor allows investigations into bile acid transport in living cells with high spatial and temporal resolution (Figure 1A). Mutations in cerulean and citrine were applied to promote the formation of the intramolecular complex (Figure 1B). Bile acids and other FXR ligands bind to FXR and thereby alter the distance between citrine and cerulean by a conformational change. In living mammalian cells, this results in an increased FRET efficiency (citrine increase, cerulean decrease) (Figure 1C, 1D). During a live cell quantitative intensity-based FRET experiment with the confocal microscope, it was observed that taurochenodeoxycholic acid (TCDCA)-treated (30 µM) U2OS cells expressing NucleoBAS lacking bile acid transporters do not show any changes in FRET, while the average citrine/cerulean ratio increased remarkably after treatment with GW4064 (5 µM) (Figure 1C). In contrast, U2OS cells co-expressing NucleoBAS, NTCP and OSTαβ did show a clear increase in citrine/cerulean ratio upon addition of TCDCA, and an even larger increase in ratio after addition of GW4064 (Figure 1D). This demonstrates that TCDCA is unable to pass through the cell membrane and requires specific transporters to enter the cell.
Cellular localization of the sensor is determined by the presence of specific localization signals. NucleoBAS is targeted to the nucleus by the nuclear localization signal (Figure 2B), while CytoBAS does not contain any localization signal and therefore remains in the cytosol (Figure 2A). The biosensor can also be combined with imaging of other fluorescent proteins, enabling measurements of protein expression/localization and FXR activation in the same cell. For instance, Figure 2C shows U2OS cells co-transfected with NucleoBAS and NTCP-mKate2. Additionally, the sensor can also be combined with other sensors for subcellular imaging of more than one parameter simultaneously. For instance, NucleoBAS was expressed along with TGR5 (EST clone IMAGE #5221127) and a cAMP cytosolic sensor (TEpacVV)11 to measure TGR5 and FXR activation in the same cell at the same time. Upon TGR5 binding, cAMP levels increased in the cytosol which was detected by the cAMP sensor, while FXR activation in the nucleus was visualized simultaneously. Figures 2D-F show a time series composed of 6 confocal images of NucleoBAS-TGR5-TEpacvv cells after treatment with GW4064 (5 µM) (Figure 2D), with TCDCA (10 µM) (Figure 2E) or with chenodeoxycholic acid (CDCA) (10 µM) (Figure 2F) at t = 0. The green color represents regions where 525/450 nm emission ratio is decreased (representing cAMP elevation) and the pink color represents an increase in emission ratio (FXR activation), compared to the ratios at the start of the experiment.
In addition, flow cytometry can be used to screen a pool of cells on single cell level or as high-throughput screening on the whole population. U2OS cells expressing NucleoBAS were excited with a violet 405 nm laser and the fluorescence was collected in the 450/40 range for cerulean and 525/20 range for citrine. In order to improve the efficiency of FACS-based FRET experiments, proper gates must be set (Figure 3). The P1 gate excludes cell debris and the P2 gate removes duplets from analysis. Next, the analysis is restricted to citrine (excited with 488 laser) and cerulean (excited with 405 laser) positive cells by gate 3. Finally, gate P4 represents the percentage of cells with a high citrine/cerulean emission ratio (using excitation of cerulean with 405 nm laser). For each sample, at least 10,000 cells that fell within gate 3 were measured.
Subsequently, FACS-based FRET flow cytometry was used to calculate an increase in FRET in living cells after 30 min incubation with TCDCA and GW4064. Non-treated U2OS cells expressing NucleoBAS showed <5% cells in gate P4 (citrine/cerulean-high cells). In absence of any bile acid transporters, a similar percentage was observed in 30 µM TCDCA treated cells (Figure 4A). In contrast, cell co-expressing NucleoBAS, NTCP and OSTαβ did show an increased amount of citrine/cerulean-high cells of around 38%. After addition of 10 µM GW4064, almost 90% of the cells were citrine/cerulean-high irrespective of expression of OSTαβ or NTCP (Figure 4B). Data can be presented in bar graphs (% cells in gate 4, population 4) (Figure 4C, D) or as population histograms of citrine-cerulean ratio (Figure 4E, F).
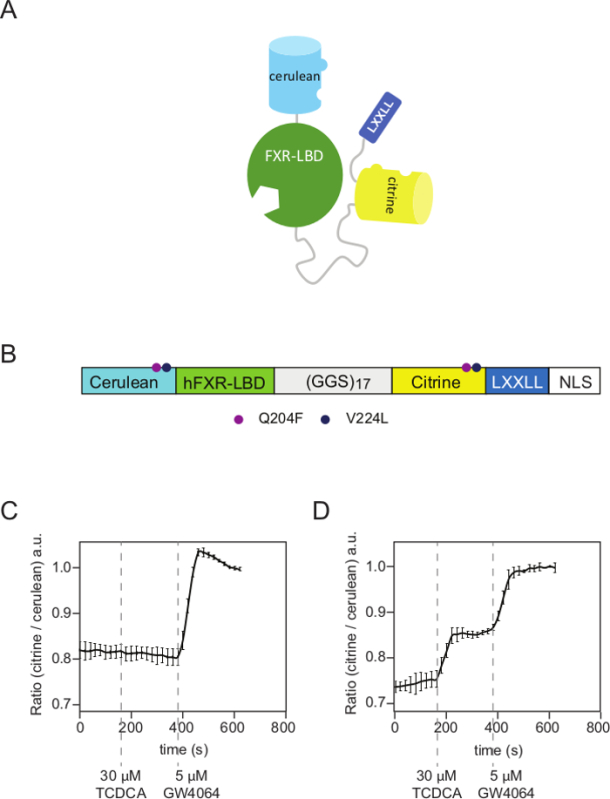
Figure 1. Design of the Bile Acid Sensor (BAS). (A) Structural representation of the mode of action of a genetically encoded Bile Acid Sensor. It consists of a donor fluorophore (cerulean) and an acceptor fluorophore (citrine) linked via the FXR-LBD and fused to a FXR-cofactor peptide (LXXLL motif). (B) Schematic domain architecture of the BAS. The Q204F/V224L fluorophore mutations promote the formation of intramolecular interactions between cerulean and citrine. The NucleoBAS construct contains a c-terminal nuclear localization signal (NLS) and therefore accumulates in the nucleus, whereas the CytoBAS construct lacks any targeting sequence and therefore shows cytosolic localization. (C) Representative confocal-based FRET experiment with BAS as a tool to monitor conjugated bile acid transport. No change in citrine/cerulean ratio is observed after 30 µM TCDCA addition in cells only expressing NucleoBAS. When 5 µM GW4064 was added, a strongly increased citrine/cerulean ratio was observed. (D) Import of TCDCA (30 µM) results in a considerable activation of the sensor in NTCP and OSTαβ co-transfected cells. Subsequent GW4064 (5 µM) treatment leads to a further increase in citrine/cerulean ratio. Results are expressed as mean ± SD, n = 6 individual cells. Please click here to view a larger version of this figure.
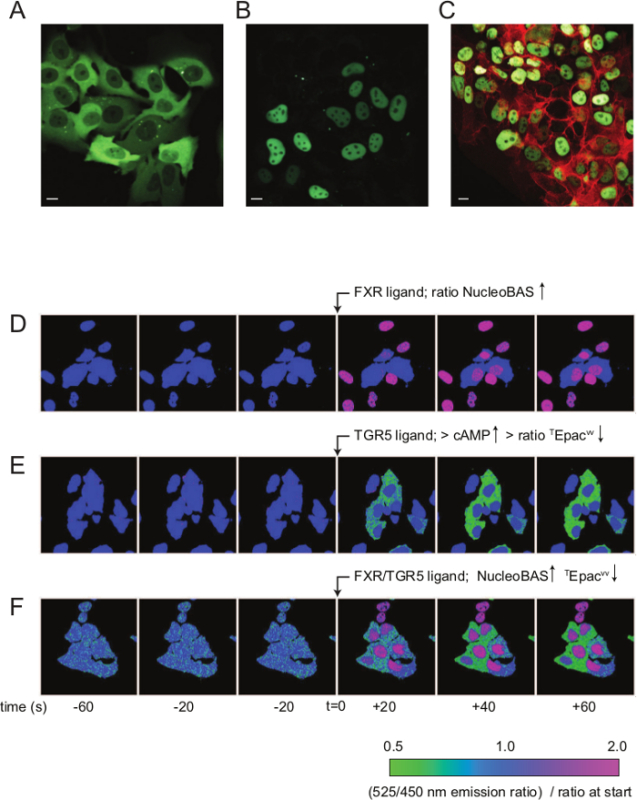
Figure 2. Localization FRET-BAS construct in living cells. Confocal microscope images of U2OS cells transfected with CytoBAS (A) or NucleoBAS (B). (C) U2OS cells co-transfected with NucleoBAS and NTCP-mKate2. The scale bar represents 10 µm. (D-F) Time series of NucleoBAS-TGR5-TEPACVV transfected cells. (D) GW4064 activates FXR (increased 525/450 nm ratio), but not TGR5. (E) TCDCA activates TGR5 on the plasma membrane (decreased 525/450 nm ratio), but is not able to enter the cell to activate FXR in the nucleus. (F) CDCA activates TGR5 on the plasma membrane as well as FXR in the nucleus (increased 525/450 nm ratio in the nucleus, decreased in the cytoplasm). Please click here to view a larger version of this figure.
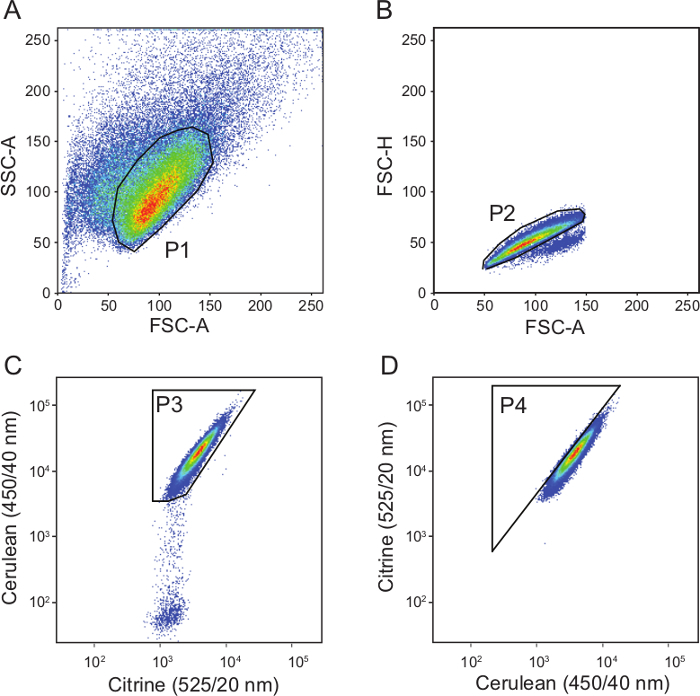
Figure 3. Flow cytometry gating parameters. (A) The P1 gate in the SSC-A/FSC-A window is used to exclude the dead cells. (B) In the FSC-H/FSC-A window, the P2 gate is drawn to eliminate doublet events from analysis. (C) In the citrine (525/20 nm)/cerulean (450/40 nm) window, NucleoBAS (and CytoBAS) positive cells are selected (Gate P3). (D) Finally, in the citrine (525/20 nm)/cerulean (450/40 nm) window, the P4 gate contains citrine/cerulean-high cells of the FACS experiment. Please click here to view a larger version of this figure.
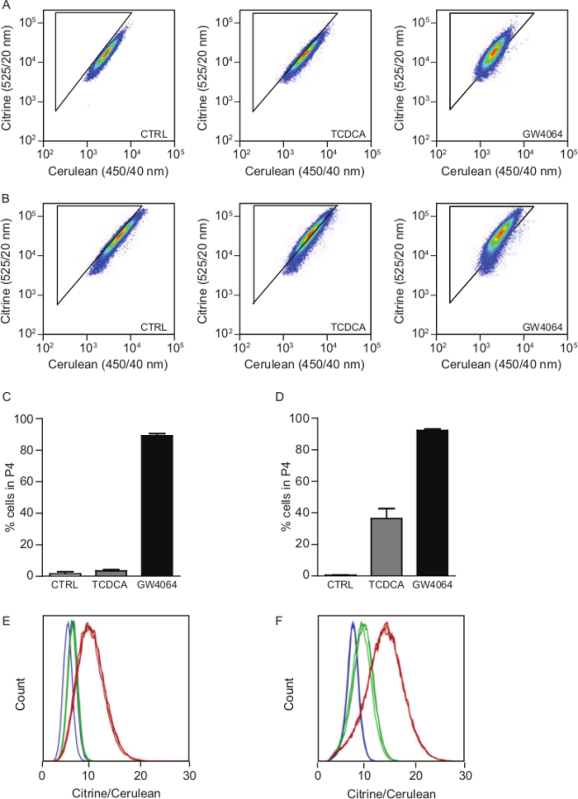
Figure 4. Representative FACS experiment measuring bile acid transport using NucleoBAS. (A, B) FACS-plots showing the amount of citrine/cerulean-high living NucleoBAS-expressing U2OS cells (A) and U2OS cells co-expressing NucleoBAS, OSTαβ and NTCP (B) after treatment with control buffer (left), 30 µM TCDCA (middle) or 10 µM GW4064 (right). (C, D) Bar graph showing percentage of citrine/cerulean-high cells after 30 min incubation with TCDCA or GW4064 in U2OS cells expressing NucleoBAS and co-transfected with (D) or without (C) OSTαβ and NTCP. (E, F) Histogram showing the distribution of citrine/cerulean ratio in a population of U2OS cells expressing NucleoBAS with or without OSTαβ and NTCP after 30 min incubation with TCDCA (yellow), GW4064 (red) or control buffer (blue). All conditions were measured three times. Bars represent mean values + standard deviation (SD) (n = 3). Please click here to view a larger version of this figure.
Supplemental File 1: Template BAS general. Please click here to download this file.
Supplemental File 2: Template BAS Zeiss Leica imported data. Please click here to download this file.
Discussion
Here we present a detailed protocol for the use of a novel genetically encoded bile acid sensor capable of monitoring the spatiotemporal dynamics of bile acid transport in living cells. This biosensor consists of cerulean and citrine fluorescent proteins that are fused to FXR-LBD, thereby forming a FRET-based bile acid sensor (BAS).
The Bile Acid Sensor is relatively simple and convenient in use when having basic experience with cell culture and FACS or (confocal) microscopy. However, some aspects might require some trouble shooting. When using the bile acid sensor in combination with bile acid transporters, it is recommended to culture cells in medium with charcoal-filtered serum. Charcoal further reduces bile acids levels by adsorption from the serum. Especially in the presence of importers such as NTCP, this is crucial to prevent saturation of the sensor during culturing, since this is the most common cause of an insensitive sensor during experiments. Therefore, another sensor was created (NucleoBAS N354K/I372V) containing mutations altering the sensitivity for bile acids, creating a larger dynamic range8. To define whether the sensor is already saturated, the citrine/cerulean ratio can be compared to the ratio in control cells not expressing any bile acid transporters, since those cells are assumed to have low intracellular bile acid levels. Alternatively, fluorescence-lifetime imaging microscopy (FLIM) measurements can be performed to investigate FRET in an intensity-independent manner12. This technique is beyond the scope of this paper, and requires more specialized equipment. Other reasons for an insensitive sensor include the use of cells that are auto-fluorescent and/or have lost NucleoBAS expression. Of note, fluctuations in intensity of cerulean and citrine (for instance due to focal shifts) during the experiment can obscure direct visualization of FRET changes during imaging, while the ratiometric nature of the sensor still allows for monitoring of bile acid dynamics. Often absolute intensity changes are modest for the individual fluorophores upon addition of FXR ligands, while the increase in ratio is evident.
For FRET analysis in cells transfected with the BAS sensor, appropriate laser light and filters should be selected. The increase of citrine intensity during FRET has to be measured with the violet diode (405 nm) laser with emissions monitored over 450-520 (cerulean) and 520-580 (citrine). An important aspect to take into account is the sensitivity of the sensor to photobleaching. When the intensity of one or both fluorophores slowly declines in time without addition of ligands, it often indicates photobleaching. This phenomenon mainly occurs when the laser power is too high. To avoid this unwanted effect, the lasers power should not exceed 5% of the full power and the cells have to be kept in dark as much as possible. Limit the time to search for the right cells. Fluctuations in fluorescence intensity can also be caused by focal drift in the z-axis. To counteract this, the pinhole size can be increased and it is advisable to draw ROIs not very close to the cell perimeter.
The FRET sensor for bile acids has been tested in multiple cell types (U2OS, Huh7, HepG2, H69, MDCK and HEK293T cells) that can be transfected and have a stable phenotype. However, primary and differentiated cells such as hepatocytes are difficult to transfect and to maintain stable in terms of characteristics. Viral transduction can help with difficult-to-transfect cells (construct available on request). We are currently generating a mouse line to allow use of the sensor in freshly isolated primary cells that rapidly dedifferentiate upon isolation.
To perform the confocal experiment described, it is imperative that the selected cell line is adherent to a culture dish and grows in a single cell layer. However, cells that grow in suspension, or adherent cells that can be trypsinized rather easily, can also be measured using the FACS. Finally, it is advised to select a cell line without endogenous bile acid transport or synthesis when analyzing a specific transport pathway. i.e., when measuring the activity of certain transfected (mutant) transporter proteins.
The presented genetically encoded fluorescent biosensor BAS is a valuable tool for monitoring single live cell bile acid transport. The BAS contains a FXR-LBD which is activated by a large variety of bile acids, allowing imaging of subcellular dynamics of bile acids8. This gives a great advantage in relation to the use of other techniques. For instance, in promoter-driven luciferase reporter assays, the luciferase signal is dependent on the stability of the reporter within cells, does not support subcellular information, and requires the destruction of the sample13. Another approach to measure bile acid transport is the use of fluorescent labeled or radiolabeled ligands. However, the fluorescent labeling of bile acids can alter bile acid transport kinetics and the availability of radiolabeled bile acids is limited. Additionally, combining the sensor with expression of bile acid transporters, cellular influx and efflux of specific bile acids can be addressed. For instance, increased intracellular levels of conjugated bile acids like TCDCA provide insight about transport activity. This also makes it possible to use BAS to examine the effect on bile acid transport after interference with specific pathways, for instance by using molecular or compound inhibitor screens, or examining the effect of other FXR ligands. Finally, another application of the sensor is combining it with other fluorescent proteins for quantitative analysis or localization of proteins, for example imaging of plasma membrane transporters expression and FXR activation in the same cell.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by ERC starting grants (ERC-2011-StG 280255 and ERC-2013-StG 337479) and by the Netherlands Organization for Health Research and Development (Vidi 91713319).
References
- Aoki K, Kamioka Y, Matsuda M. Fluorescence resonance energy transfer imaging of cell signaling from in vitro to in vivo: basis of biosensor construction, live imaging, and image processing. Dev. Growth Differ. 2013;55(4):515–522. doi: 10.1111/dgd.12039. [DOI] [PubMed] [Google Scholar]
- Jares-Erijman EA, Jovin TM. FRET imaging. Nature Biotechnol. 2003;21(11):1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- Mank M, et al. A FRET-based calcium biosensor with fast signal kinetics and high fluorescence change. Biophys. J. 2006;90(5):1790–1796. doi: 10.1529/biophysj.105.073536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li IT, Pham E, Truong K. Protein biosensors based on the principle of fluorescence resonance energy transfer for monitoring cellular dynamics. J. Biotechnol. Lett. 2006;28(24):1971–1982. doi: 10.1007/s10529-006-9193-5. [DOI] [PubMed] [Google Scholar]
- Stephens DJ, Allan VJ. Light microscopy techniques for live cell imaging. Science. 2003;300(5616):82–86. doi: 10.1126/science.1082160. [DOI] [PubMed] [Google Scholar]
- Merkx M, Golynskiy MV, Lindenburg LH, Vinkenborg JL. Rational design of FRET sensor proteins based on mutually exclusive domain interactions. Biochem. Soc. Trans. 2013;41(5):1201–1205. doi: 10.1042/BST20130128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenburg LH, et al. Quantifying Stickiness: Thermodynamic Characterization of Intramolecular Domain Interactions To Guide the Design of Förster Resonance Energy Transfer Sensors. Biochem. 2014;53(40):6370–6381. doi: 10.1021/bi500433j. [DOI] [PubMed] [Google Scholar]
- van der Velden LM, et al. Monitoring bile acid transport in single living cells using a genetically encoded Förster resonance energy transfer sensor. J. Hepatol. 2013;57(2):740–752. doi: 10.1002/hep.26012. [DOI] [PubMed] [Google Scholar]
- Dawson PA, et al. The heteromeric organic solute transporter α-β, Ostα-Ostβ, is an ileal basolateral bile acid transporter. J. Biol. Chem. 2005;280(8):6960–6968. doi: 10.1074/jbc.M412752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier PJ, Stieger B. Bile salt transporters. Annu. Rev. Physiol. 2002;64(1):635–661. doi: 10.1146/annurev.physiol.64.082201.100300. [DOI] [PubMed] [Google Scholar]
- Klarenbeek JB, Goedhart J, Hink MA, Gadella TW, Jalink K. A mTurquoise-based cAMP sensor for both FLIM and ratiometric read-out has improved dynamic range. PLoS One. 2011;6(4):e19170. doi: 10.1371/journal.pone.0019170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrabe H, Periasamy A. Imaging protein molecules using FRET and FLIM microscopy. Curr. Opin Biotechnol. 2005;16(1):19–27. doi: 10.1016/j.copbio.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Plass JR, et al. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. J. Hepatol. 2002;35(3):589–596. doi: 10.1053/jhep.2002.31724. [DOI] [PubMed] [Google Scholar]