

Abstract
Functional in vivo imaging has become a powerful approach to study the function and physiology of brain cells and structures of interest. Recently a new method of Ca2+-imaging using the bioluminescent reporter GFP-aequorin (GA) has been developed. This new technique relies on the fusion of the GFP and aequorin genes, producing a molecule capable of binding calcium and — with the addition of its cofactor coelenterazine — emitting bright light that can be monitored through a photon collector. Transgenic lines carrying the GFP-aequorin gene have been generated for both mice and Drosophila. In Drosophila, the GFP-aequorin gene has been placed under the control of the GAL4/UAS binary expression system allowing for targeted expression and imaging within the brain. This method has subsequently been shown to be capable of detecting both inward Ca2+-transients and Ca2+-released from inner stores. Most importantly it allows for a greater duration in continuous recording, imaging at greater depths within the brain, and recording at high temporal resolutions (up to 8.3 msec). Here we present the basic method for using bioluminescent imaging to record and analyze Ca2+-activity within the mushroom bodies, a structure central to learning and memory in the fly brain.
Keywords: Neuroscience, Issue 107, Ca2+-imaging, bioluminescent reporter, GFP-Aequorin, mushroom body, P2X2, Drosophila, spontaneous activity
Introduction
The fundamental patterning and function of activity within the brain and its discrete structures have long been an area of intense study within the field of neuroscience. Perhaps some of the earliest successful approaches used to address this issue were direct physiological measurement of the change in electrical activity — however alternate methods which allow live imaging of changes in voltage, pH or calcium concentrations (as proxy for activity) have brought additional capabilities to the study of neuronal activity 1. Imaging techniques carry a wide array of advantages such as reduced invasiveness and the ability to monitor activity within whole brain structures. Furthermore in animals with tractable genetics including the fruit fly Drosophila, development of genetically encoded calcium indicators, such as cameleon, GCaMPs and others1 have allowed researchers to monitor single neurons, neuronal populations and whole brain structures. While more traditional fluorescent calcium imaging methods using GCaMPs (GFP fused to calmodulin) or FRET (CFP and YFP fused to calmodulin) have proved to be useful techniques providing good spatial and temporal resolution, the underlying process of light excitation engenders some limitations on its experimental applications. Due to the nature of light excitation, autofluorescence, photo-bleaching, and phototoxicity are unavoidably produced, which consequently limits the depth of the structures that can be imaged, the duration of recordings as well as the real time continuity of recording. In other words, recordings must often be performed intermittently to avoid these undesired side effects.
Because of these challenges, additional alternative methods of calcium imaging have been developed. One of the most promising methods is bioluminescent imaging, which relies on light produced through an enzymatic reaction after calcium binding. Bioluminescent imaging does not require light excitation and consequently does not experience the same difficulties as conventional calcium imaging. The bioluminescent reporter Aequorin — isolated from Aequorea victoria, the same Jellyfish from which GFP was also isolated- binds calcium via its three EF hand structures and undergoes a conformational change, leading to the oxidation of its cofactor coelenterazine and the release of blue light (λ=469 nm)2,3. Aequorin has a very low affinity for calcium (Kd = 10 µM) which makes it ideal for monitoring calcium transients because it produces very little background noise and does not interfere with the intracellular calcium buffering system4. Although aequorin has been previously used to monitor the process of neural induction in the Xenopus egg5 the light produced is too dim to use for real time imaging of neuronal activity. In an effort to address this challenge, Philippe Brûlet and colleagues at the Pasteur Institute created a genetic construct fusing GFP and aequorin genes, mimicking the native state within the jellyfish4. This fusion gene product binds calcium again via the three EF hand structures of aequorin, which undergoes a conformational change leading to oxidation of the coelenterazine, which transfers energy non-radiatively to the GFP. This energy transfer results in the release of green light (λ=509 nm) from GFP, instead of blue light from aequorin. The fusion of GFP and aequorin also confers greater protein stability helping the fusion protein produce light 19 to 65 times brighter than aequorin alone4. Using a bioluminescent approach within the brain expands the current experimental application of calcium imaging both in terms of the duration of continuous recording and the number of structures within the brain that can be recorded. Broadly in the context of previous work it means that both global and a greater variety of regionalized “activity” of the brain can be monitored (through the proxy of calcium transients). More importantly activity can be monitored for longer, in a real time and continuous basis, which readily lends itself to the pursuit of a complete understanding of basal function in the brain.
In the last few years, our lab and others have developed transgenic flies expressing the GFP-aequorin under the control of the binary expression system (GAL4/UAS-GFP-aequorin)5,6. We have used GFP-aequorin bioluminescence to record activity produced by natural stimuli such as scent7,8, as well as studied the cellular components modulating Ca2+-activity in the mushroom bodies following acetylcholine receptor stimulation by direct nicotinic application9. In addition, we have also used GFP-aequorin to address a number of experimental questions that have not been able to be addressed previously, like long-term imaging of spontaneous calcium transients and visualization of deeper brain structures10. More recently, we have used the GAL4/UAS system to express the mammalian ATP receptor P2X211 a cation channel, in the projection neurons (PNs) and used the LexA system to express GFP-aequorin in the mushroom bodies (MB247-LexA,13XLexAop2-IVS-G5A-BP) (a courtesy of B. Pfeiffer, Janelia Farm, USA). This has allowed us to activate the PNs by application of ATP, which in turn activates the mushroom bodies (a downstream target of the PNs), mimicking more natural conditions, such as the response to an odor stimulus. Overall this technique combining P2X2 and GFP-aequorin expands the experimental possibilities. Broadly it offers the opportunity to better understand the patterns of activity in the brain, initiating new studies about how different stimuli and perturbations can alter the basal patterning of activity.
Protocol
1. Preparation of Samples
- Preparation of solutions and set up
- Maintain all Drosophila melanogaster lines at 24 °C on standard food medium. Rear and keep them at low density in the vial to generate standardized size and weight of flies.
- Add 10 virgin females with 10 males in a vial, let them mate and transfer them every 2 days to a fresh vial. Then 10 days later, when the flies start to eclose, harvest the flies every day. Keep a precise record of the age of the flies and keep them in good condition.
- At day 3, separate the males from the females and keep the females 20 flies per vial. Record the flies at 4 or 5 days-old.
- Prepare Ringer’s solution with the following concentrations: 130 mM NaCl, 5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 5 mM HEPES, and 36 mM sucrose and adjust the pH of the solution to precisely 7.3. Prepare perfusion solutions of 25 µM nicotine and 100 mM KCl in Ringer’s solution.
- Prepare 1,000 µl pipette tip: using a razor blade shape tip to a slant (approximately 35° from end of tip) and remove excess base beyond 1 ½ centimeters from tip.
- Prepare box for storage (23 cm x 17 cm x 8.5 cm) of sample during incubation. Place two sponges (12 cm x 8 cm x 3 cm) saturated with water in bottom [to prevent sample desiccation] below a small rack apparatus to place samples on as their preparation is completed.
- Prepare flies samples so that they contain at least 1 copy of the UAS-GFP-aequorin and one copy of a Gal4 line to drive expression of GFP-aequorin. OK107 Gal4 line (a line driving expression primarily in the mushroom bodies) is crossed with UAS-GFP-aequorin in this preparation. NOTE: The inside of box must be waterproof and the outside must block light from entering box to prevent degradation of coelenterazine during incubation.
- Preparation of samples
- Ice anesthetize Drosophila by transferring it to a glass vial and keep on ice for 2 min before being moved to the chilled Petri dish for positioning in the pipette tip. Prepare pipette tips on slightly dampened filter paper in 100 ml Petri dish on ice under the dissection microscope.
- Gently with forceps place fly inside of pipette tip.
- Using a brush gently push and align fly such that the head is completely past the edge of tip and the dorsal region is partially exposed by the section removed during tip shaping (Figure 2B).
- Combine a small (approximately 3-4 µl) portions of the two components of the dental glue. Apply the glue carefully around the front and back head and neck down to and around the pipet tip edge — avoiding the crown of the head. Let the glue dry for 2 min.
- Place pipette tip with glued fly through hole in recording chamber (Figure 2D) and gently press to secure in place.
- Combine the two components of silicon glue (approximately 3-4 µl). Apply glue on the flat side of chamber along the edge where the chamber and pipette tip meet to prevent leak of the Ringer’s solution. Let the glue dry for 2 min.
- Dissection
- Affix tape over the perfusion channel, short edge and extending to the back of the chamber.
- Place chamber with fly on dissection block under microscope turn on fluorescent lamp. Pipet 1 ml of Ringer’s solution into chamber.
- Use fine surgical knife to remove cuticle. Make parallel incisions from the back of the head to antennal region. Then cut along the edge of eye then make a perpendicular incision above antenna connecting the previous incisions. Make a final incision parallel to that at the back of the head. Use the fine sharp forceps to remove cuticle (Figure 2C).
- Use fine sharp forceps to gently grasp and clear away exposed respiratory tissue until the brain and [fluorescing] mushroom body is clearly visualized.
- Use ultra-sharp forceps to carefully grasp and pinch neuroepithelial tissue covering brain to allow for permeation of the coelenterazine. NOTE: Alternatively papain can be used to permeabilize the neuroepithelia9.
- Using a pipet, wash out twice with Ringer’s solution to remove any debris from dissection and place sample in the dark box.
- Pipet 1 ml of Ringer’s solution containing 5 µM benzyl-coelenterazine into the chamber. Close the box and allow incubation with coelenterazine at RT for a minimum of 2 hr.
2. Imaging
- Setup
- Start recording system: turn on microscope, computer, camera and drainage system and set room environmental controls to 25 °C.
- Open Measurement and Automation Explorer program on the computer. Click on “Devices and Interfaces”, then double click on “NI Motion Devices” and finally right click on “PCI-7334” and select “initialize device”.
- Open Photon Imager. Create new folder and name first recording file. Camera must reach -80 °C before system will function.
- Set up perfusion system — add KCl, nicotine, and Ringer’s solution to reservoirs and adjust so the each solution discharges at 2 ml/min. Wash with Ringer’s solution prior to starting experiment.
- Prepare sample
- Place imaging mount block dish on mount under microscope at 5X magnification and insert perfusion tube into channel through puncture.
- Set microscope to fluorescent mode then center and bring mushroom bodies (MBs) into focus. Adjust to 20X and re-center and focus.
- Position drainage apparatus over drainage pool, adjust height of the drainage shunt so that the tip is just above pool, and run Ringer’s solution for 30 sec to ensure adequate drainage.
- Pull down shield to seal off apparatus from light. Using the automated system in photon imager make fine adjustments to the focus and take a reference fluorescent image of the MBs.
- Recording
- Adjust photon speed to desired recording speed (from 50 msec up to 1 sec or more). Select photon mode and open shutter. Record genotype, sex, age, and sample number in log entries. Adjust position ROI boxes over calyx and cell bodies (CCB) and medial lobes by clicking on the ROI boxes and dragging while holding control.
- Record baseline for about 5 to 10 min (as desired).
- Apply nicotine for 1 min. Note start/stop in log. Wash with Ringer’s solution for 5 min.
- Wait 5 min for recovery and prepare KCl log entry and timer.
- Apply KCl for 1 min. Note start/stop in log. Wash with Ringer’s solution for 1 min.
- Switch to fluorescent mode, take final fluorescent image, and turn off Ringer’s solution.
- Press “Stop”. Dialog box will ask to shut off the system. Select “No” to continue recording.
- Open shield, move drainage system, switch lens objective to 5X, remove perfusion tube. Remove sample/recording dish from stage, clean off 20X lens by rinsing and wiping the lens twice with water.
- Press the white play button at the top of the program window to initiate the next recording.
3. Analysis and Video Creation
- Extract photon values
- Open the photon analysis program (e.g., Photon Viewer) and open first sample spreadsheet file.
- Select the display control tab. Select the ROI by clicking on the region while holding control. Adjust size, orientation and shape of regions of interest to encompass GFP illuminated CCB and medial lobes.
- Add an additional ROI by clicking “Define ROI” and clicking and dragging on screen to create the new ROI.
- Select the “Movie” tab to play photon video. Adjust “sec width” and “sec steps” as desired. Readjust ROI placement as necessary.
- Select representative screen shots of GFP fluorescence, nicotine response and KCl response. Crop screenshots and paste image into a presentation slide for later analysis and comparison.
- Adjust both the “sec width” and “sec step” to the recording speed in seconds. Select length of analysis by moving grey markers on top panel to bracket region before stimulus initiation and just after response end.
- Select “Suspend views while playing” press “<<REW” and press “PLAY>”. Select the tab “Count Rate Chart” and adjust units as desired and line colors to match ROI color.
- When analysis is finished, press “Export Count Rate Data”. Select screen shots of combined recordings CCB and medial lobe region of interest from each side. Crop screenshots and paste image into a slide with response images. This slide will be later used in final analysis.
- Example Analysis: one method for analysis of extracted photon data detailed
- Open the spreadsheet files in “Count Rate Exports” folder.
- Reformat the cells the first column labeled time to display the time in hour and minutes.
- Reference the corresponding slide for the sample. Remove all values except the columns for the time and the two representative ROIs.
- Determine nicotine stimulation start time — available in log entry file in main folder- remove additional data prior to start or highlight value.
- Find highest/ peak value for both CCB and medial lobe and mark/highlight.
- Determine response start and end time for both CCB and medial lobe by creating an estimate using time point from corresponding graphed response on slide and select actual value by change in values in proximity to these points. Highlight the region between these points in both the CCB and medial lobes. Alternatively, use a macro9.
- Combine all samples of one experimental group on a new spreadsheet.
- Align on peak by determining the row value is for each CCB peak value. Subtract the lower values from the highest row value to determine approximate row value where that sample data must be recopied to. Verify that all peak values are aligned.
- Copy the sheet twice and delete either the medial lobe columns or the CCB columns creating pages of only CCB or medial lobe values.
- Remove data after the all CCB responses have ended. Create four rows labeled Total Photons, Response Length (duration), Peak Amplitude, and Response Latency. Copy and paste this again below the originals.
- Use the SUM function to determine total photons during response. Use the COUNT function to determine length of response. Copy and link highest value cell to determine peak amplitude value. Use COUNT function — select from the beginning of the perfusion of nicotine to the start of the response — to determine Response Latency.
- Copy values by using the linking function and multiply (all except peak amplitude) by recording speed in seconds.
- Bar/box graphs may be created for calculated parameters such as Total Photons, Peak Amplitude, and Response Length, if so desired (ex.: Figure 5B, C, D).
- In the column after the raw data photon response, average the raw data points for each time point using average function. If desired follow with a column determining the standard error of the mean (SEM), for each of the averaged photon values at a time point.
- Use the average column to construct an average response profile [Graph] for the CCB sample group. To do this select the column specifying the time as the x-axis values and the column of the average photon values as the y-axis and finally select the scatterplot graph creation tool.
- Repeat this analysis procedure with the data from the medial lobes.
- Creating Images for the Video
- Open photon viewer.
- Select display control tab. Click on ROI while holding control to select, remove and/or minimize it.
- Modify “sec width” (accumulation time) as desired to determine the content of each frame.
- Modify “sec step” to define the overlap of each frame.
- Select “export views while playing” press “<<REW” then” PLAY>”. The images for the video will be exported to the “view exports” folder as a series of .png files.
- Using Virtual Dub to make video: one method for video creation detailed
- Create a new folder named “resultats” in the view exports folder containing the images.
- Bring script (renommer.VBS) into the folder of images.
- Double click on renommer.VBS to run.
- Images will automatically be renamed numerically and copied to “resultats” folder.
- Open VirtualDub.exe.
- Select open video file — select first image (subsequent images will automatically load).
- Select video — select frame rate adjust as desired.
- Select video — select compression select Cinepak Codec by radius.
- In file select Save as AVI the video will be created automatically.
Representative Results
The fusion of the GFP to aequorin allows us to visualize our region of interest prior to bioluminescent imaging through excitation of GFP in fluorescent mode on the microscope (Figure 3A, 4A, 6A, 6E). One of the simplest ways to stimulate the mushroom bodies is through activation of the ionotropic nicotinic acetylcholine receptors. Although acetylcholine is the endogenous ligand of this receptor we have found that nicotine produces more reproducible responses in the mushroom bodies. This is in part because they specifically express the nicotinic acetylcholine receptors, and thus by using nicotine we can avoid the activation of the muscarinic acetylcholine receptors expressed elsewhere in the brain. Moreover, nicotine is more stable than the acetylcholine, and thus allows better and more reliable stimulus control. A typical response commences with the fast activation of both the calyx, cell-bodies (CCB) and medial lobes (ML) (see Figure 4A for labeled image). Generally the CCB response lasts longer and exhibits a greater bioluminescent response than the lobes as shown in the sequential panels of the response (Figure 3C-H). Figure 4B shows a 10 sec accumulation of photons recorded at a temporal resolution of 100 msec (10 Hz) during the peak of a response to a 1 min perfusion of 25 µM nicotine. Following a washout and recovery period of 10 min a second stimulation of a 1 min perfusion of 100 mM KCl is initiated (Figures 4C, E) which allows us to confirm the viability of our sample. The quantitative responses can be analyzed by selecting ROI during analysis in photon viewer. Running the analysis in photon viewer produces both graphs displaying the number of photons emitted in selected ROI over time (Figure 4D and E) as well as exportable spreadsheets of those values. Figure 4D is an example of the graphs produced by photon viewer plotting the photons emitted in the ROI 2 [green] (right CCB) and ROI 4 [blue] (right medial lobes). Similar data for the KCl response represented in Figure 4E. The exported data can be used to reconstruct the curve of the response (Figure 5A) as well as to extract additional data about various parameters such as duration, peak amplitude, and total response (Figure 5B-D). Recently using the Gal4-UAS and LexA systems we have devised a method of eliciting a more natural response. Briefly, the ATP-gated P2X2 cation channel is expressed in the projection neurons (PNs) and GFP-aequorin (GA) is expressed in the mushroom bodies — a target of the PNs; this allows us to selectively excite the PNs though application of ATP, which can in turn, produce response in mushroom bodies (Figure 6).
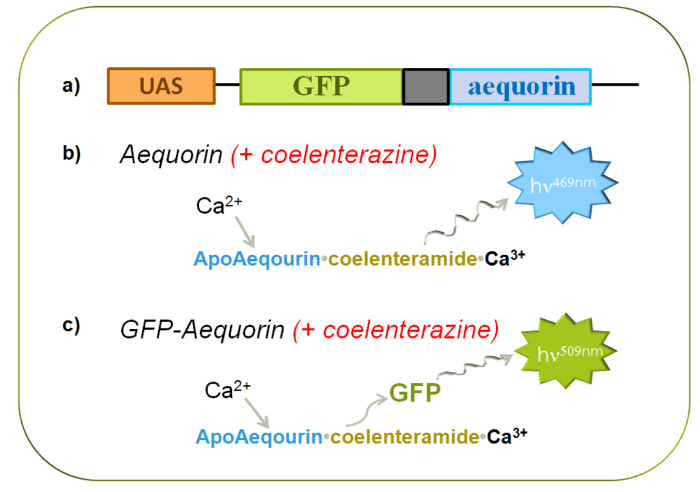 Figure 1. Characteristics of Bioluminescent Reporters (GFP-aequorin). (A) Schematic of GFP and aequorin fusion gene with upstream activation sequence. (B) Model of blue light emission by aequorin in response to high levels of Ca2+. (C) Model of green light emission by GFP-aequorin in response to high levels of Ca2+. Please click here to view a larger version of this figure.
Figure 1. Characteristics of Bioluminescent Reporters (GFP-aequorin). (A) Schematic of GFP and aequorin fusion gene with upstream activation sequence. (B) Model of blue light emission by aequorin in response to high levels of Ca2+. (C) Model of green light emission by GFP-aequorin in response to high levels of Ca2+. Please click here to view a larger version of this figure.
 Figure 2. Experimental set up. (A) Fly positioned in pipet tip. (B) An illustration of adequate gluing technique stabilizing the head and sealing the region between the fly body and pipet tip. (C) Schematic of dissection to open head capsule. (D) 1 ml chamber on holding block with perfusion tube inserted. Total chamber dimensions: length: 7.4 cm, width: 2.7 cm, height: 0.55 cm. Inner chamber dimensions: length: 1.7 cm, width: 1.4 cm, height: 0.35 cm, and the radius of the chamber hole: 0.1 cm. (E) View of fly head showing GFP-positive signal in the MBs: low dim light with fluorescence derived from OK107-driven GFP-aequorin after removal of cuticle. (F) Microscope with photon camera and perfusion system. Please click here to view a larger version of this figure.
Figure 2. Experimental set up. (A) Fly positioned in pipet tip. (B) An illustration of adequate gluing technique stabilizing the head and sealing the region between the fly body and pipet tip. (C) Schematic of dissection to open head capsule. (D) 1 ml chamber on holding block with perfusion tube inserted. Total chamber dimensions: length: 7.4 cm, width: 2.7 cm, height: 0.55 cm. Inner chamber dimensions: length: 1.7 cm, width: 1.4 cm, height: 0.35 cm, and the radius of the chamber hole: 0.1 cm. (E) View of fly head showing GFP-positive signal in the MBs: low dim light with fluorescence derived from OK107-driven GFP-aequorin after removal of cuticle. (F) Microscope with photon camera and perfusion system. Please click here to view a larger version of this figure.
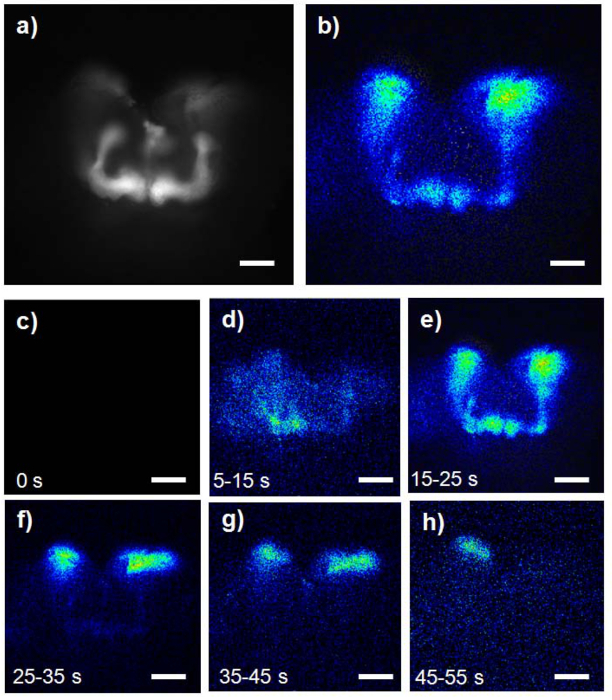 Figure 3. Representative response to 25 µM nicotine in mushroom bodies. (A) Fluorescent image of the GFP-aequorin expression in the mushroom bodies in GA2/+;OK107/+ control fly brain (scale bar = 50 μm). (B) Peak bioluminescent response in the mushroom bodies. (C) Prior to stimulation. (D) Start of response. (E) Peak response. (F) Continued CCB response. (G) Falling CCB response. (H) End of response (B-H: scale bar = 33 μm). Please click here to view a larger version of this figure.
Figure 3. Representative response to 25 µM nicotine in mushroom bodies. (A) Fluorescent image of the GFP-aequorin expression in the mushroom bodies in GA2/+;OK107/+ control fly brain (scale bar = 50 μm). (B) Peak bioluminescent response in the mushroom bodies. (C) Prior to stimulation. (D) Start of response. (E) Peak response. (F) Continued CCB response. (G) Falling CCB response. (H) End of response (B-H: scale bar = 33 μm). Please click here to view a larger version of this figure.
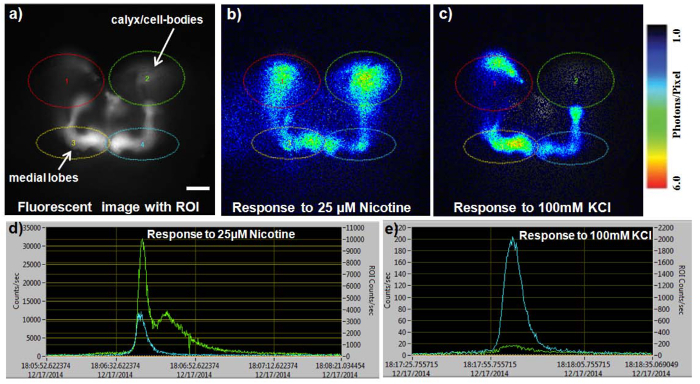 Figure 4. Representative analysis of sample recording. (A) Fluorescent image of the mushroom body of the GA2/+;OK107/+ control fly with four ROI’s selected over the CCB and medial lobes (scale bar = 50 μm). (B) 10 sec accumulation of the bioluminescent response to 25 µM nicotine — photon response recorded at 100 msec. (C) 10 sec accumulation of bioluminescent response to 100 mM KCl recorded at 100 msec. (D) Graph of recorded number of photons emitted during nicotine response within ROI’s 2 and 4 covering the right CCB and medial lobes respectively. (E) Graph of recorded number of photons emitted during KCl response within ROI’s 2 and 4 covering the right CCB and medial lobes, respectively. In d and e, the left Y-axis corresponds to the total number of photons within the whole field of view, while the right Y-axis corresponds to the photons within the ROI. Please click here to view a larger version of this figure.
Figure 4. Representative analysis of sample recording. (A) Fluorescent image of the mushroom body of the GA2/+;OK107/+ control fly with four ROI’s selected over the CCB and medial lobes (scale bar = 50 μm). (B) 10 sec accumulation of the bioluminescent response to 25 µM nicotine — photon response recorded at 100 msec. (C) 10 sec accumulation of bioluminescent response to 100 mM KCl recorded at 100 msec. (D) Graph of recorded number of photons emitted during nicotine response within ROI’s 2 and 4 covering the right CCB and medial lobes respectively. (E) Graph of recorded number of photons emitted during KCl response within ROI’s 2 and 4 covering the right CCB and medial lobes, respectively. In d and e, the left Y-axis corresponds to the total number of photons within the whole field of view, while the right Y-axis corresponds to the photons within the ROI. Please click here to view a larger version of this figure.
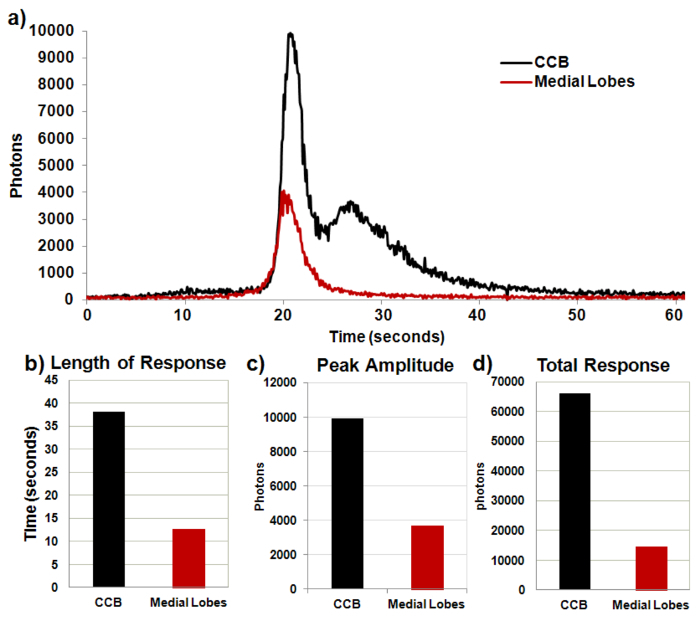 Figure 5. Representative parameters analysis of sample recording. Representative excel analysis of the bioluminescent response to nicotinic activation of the mushroom body in the GA2/+;OK107/+ control fly. (A) Photon response over time in CCB and medial lobes. (B) Duration of responses in the CCB and medial lobes. (C) Highest photon value (maximum amplitude) during response within the CCB and medial lobes. (D) Total photons within CCB and medial lobes of entire response. Please click here to view a larger version of this figure.
Figure 5. Representative parameters analysis of sample recording. Representative excel analysis of the bioluminescent response to nicotinic activation of the mushroom body in the GA2/+;OK107/+ control fly. (A) Photon response over time in CCB and medial lobes. (B) Duration of responses in the CCB and medial lobes. (C) Highest photon value (maximum amplitude) during response within the CCB and medial lobes. (D) Total photons within CCB and medial lobes of entire response. Please click here to view a larger version of this figure.
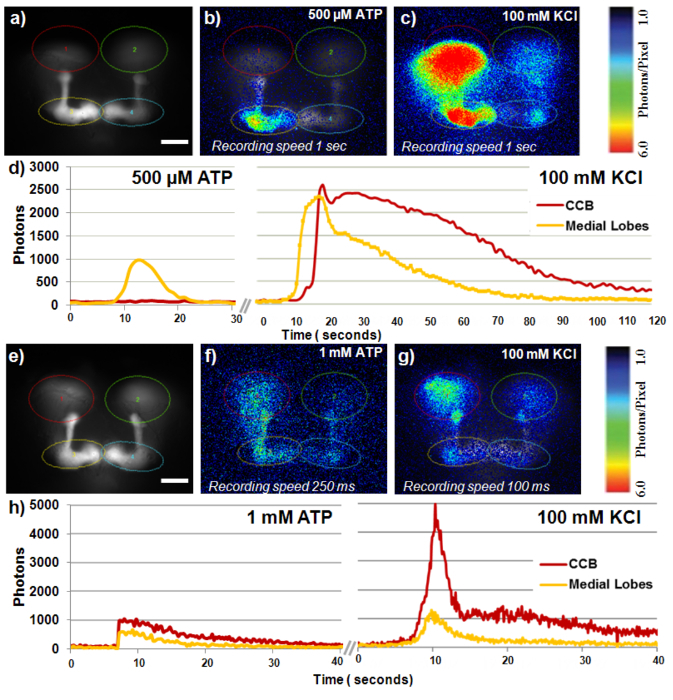 Figure 6. Two different response in mushroom bodies after stimulation of PNs through ATP and P2X2. Sample 1 fly expressing P2X2 in PNs and GFP-aequorin in mushroom bodies (GH146/+; P2X2/MB247-LexA,13XLexAop2-IVS-G5A-BP (a-d). (A) Fluorescent image of mushroom bodies with ROI’s (scale bar = 50 μm). (B) Bioluminescent image of the peak response in mushroom bodies following activation of the PNs by P2X2 stimulated with 500 μM ATP, observed primarily in medial lobes. (C) Bioluminescent image of peak KCl response. (D) Time course of photon emission within ROI regions, throughout the response. Sample 2 fly expressing P2X2 in PNs and GFP-aequorin in mushroom bodies (GH146/+; P2X2/MB247-LexA,13XLexAop2-IVS-G5A-BP (e-h). (E) Fluorescent image with ROI’s (scale bar = 50 μm). (F) Bioluminescent image of the induced response in the mushroom bodies following activation of the PNs by P2X2 stimulated with 1mM ATP, in both lobes and CCB. (G) Bioluminescent image of peak KCl response. (H) Time course of photon emission within ROI regions, throughout the response. Please click here to view a larger version of this figure.
Figure 6. Two different response in mushroom bodies after stimulation of PNs through ATP and P2X2. Sample 1 fly expressing P2X2 in PNs and GFP-aequorin in mushroom bodies (GH146/+; P2X2/MB247-LexA,13XLexAop2-IVS-G5A-BP (a-d). (A) Fluorescent image of mushroom bodies with ROI’s (scale bar = 50 μm). (B) Bioluminescent image of the peak response in mushroom bodies following activation of the PNs by P2X2 stimulated with 500 μM ATP, observed primarily in medial lobes. (C) Bioluminescent image of peak KCl response. (D) Time course of photon emission within ROI regions, throughout the response. Sample 2 fly expressing P2X2 in PNs and GFP-aequorin in mushroom bodies (GH146/+; P2X2/MB247-LexA,13XLexAop2-IVS-G5A-BP (e-h). (E) Fluorescent image with ROI’s (scale bar = 50 μm). (F) Bioluminescent image of the induced response in the mushroom bodies following activation of the PNs by P2X2 stimulated with 1mM ATP, in both lobes and CCB. (G) Bioluminescent image of peak KCl response. (H) Time course of photon emission within ROI regions, throughout the response. Please click here to view a larger version of this figure.
Discussion
The recently developed bioluminescent-based GFP-aequorin approach presented here allows in vivo functional recording of Ca2+-activity in different neurons, as well as if desired, in other kind of cells such as kidney stellate cells as reported in Cabrero et al., 20135.
Modifications, trouble shooting, additional components, and critical steps
Drosophila Husbandry
GFP-aequorin transgene (pG5A)4 was inserted into the pUAST vector to create three independent transformant lines6. All lines were tested for their bioluminescence, and all were able to respond to the in vivo Ca2+-activity6. However more recent experiments have pointed out that the GA2 line inserted on the third chromosome generates more robust signal than the GA1 line inserted on the second chromosome. Here, the representative results were obtained by using UAS-GA2 crossed with the MB-specific line OK107. Moreover, recently B. Pfeiffer has generated a GFP-aequorin construct placed under the control of 20XUAS regulatory element (20X-G5A-2) and this construct has been validated in our lab. Briefly, it gives a very strong signal, stronger than our standard GA2, which therefore should be more efficient and useful to further monitor the Ca2+-activity in deeper structures of the brain, such as the ellipsoid-body. Additionally, it may also be useful for imaging small quantities of neurons or in axon terminals (synaptic contacts).
Preparations for Imaging
Although the fly must be ice anesthetized during positioning and gluing, it is important to minimize the time on ice. Flies should be transferred to a glass vial and kept on ice for 2 min before being moved to the chilled Petri dish for positioning in the pipette tip. We have noticed that extended time period of anesthesia reduces the viability of the sample and may attenuate the response. Optimally, we keep the time of ice anesthesia below 5-10 min.
Taping and gluing are critical steps. It is crucial to secure the fly head in place for dissection and imaging as well as to prevent leakage of the Ringer’s solution into the pipet tip and/or out of the recording chamber. Dental glue (and to a lesser extent the silicon glue) will become sticky and subsequently begin to dry as soon as the two components are combined. Therefore it is critical to apply the glue expeditiously to avoid weak bonding and clumping of the glue. In some cases we have noticed that alternate varieties of tape and glue, when in contact with the Ringer’s solution, may release contaminants into the Ringer’s solution which can affect the fly and its response, particularly so for experiments dedicated to the study of olfactory system, using different odors. Consequently, use caution here if substituting materials (see materials for specific varieties we use).
Several different forms of coelenterazine have been developed in order to optimize its activity such as the speed of light emission, Ca2+-affinity (sensitivity), or the intensity of light emission12. For example, the native coelenterazine is more stable, thus more suitable of long term imaging as several hours, while the benzyl-coelenterazine (also named h-coelenterazine) results in a faster and higher light emission with shorter half-life. For additional information about different forms of coelenterazine, see the web address in the Materials List.
Imaging
Bioluminescence signals can be monitored with an electron multiplier CCD camera (EM-CCD, cooled to -80 ºC) fitted onto a microscope. This setup must be housed inside a tight dark box to avoid any undesired (ambient) light contamination. The setup described in this manuscript uses a 20X immersion-objective lens permitting a field of view of 400 x 400 µm (512 x 512 pixels). To improve signal to noise ratio, data were acquired with a 0.100 sec integration time (10 Hz), and 2 x 2 binning was used (1 pixel = 1.2 x 1.2 μm). To acquire and store data, each detected photon was assigned x,y-coordinates and a time point. Between 500 and 600 nm wavelength, the EM-CCD camera (see precise description in Materials List) has a quantum efficiency of 96%.
One of the most crucial parameters for the in vivo functional brain imaging is the temporal resolution. Up to now, the majority of fluorescence-based Ca2+-imaging technique such as GCaMP and cameleon provides a temporal resolution of about 4 Hz (250 msec/frame). Recently, with bioluminescence-based GFP-aequorin we have been able to raise the acquisition time up to 100 msec/frame with the EMCCD camera and even up to 120 frames/sec (8.3 msec/frame) with the electron multiplier ICDD camera10. At this temporal resolution approaches that of the electrophysiological techniques (in the range of about 1 msec). While high temporal resolution is both informative and crucial for the studies dedicated to synaptic transmission and stimulus induced activity, extended slower yet essential Ca2+-kinetics also occurs within neurons, for instance the Ca2+ release from the intracellular Ca2+-stores (for a review: Berridge et al., 200313). For this latter type, a slower temporal resolution is still acceptable, while inversely, they generally require a better Ca2+-affinity from the sensor to be detected. This requirement has been achieved with GFP-Aequorin to detect the release of the intracellular Ca2+-stores at the axon terminal of the olfactory receptors neurons7,8. Alternatively, depending on the desired experimental conditions, slower acquisition time could be used. For instance, in long O/N recordings, we have used a 2 sec integration times due to the large number of data points collected (Minocci et al., 2013). In brief, one practical feature with the bioluminescence is that the temporal resolution can be easily adjusted according to the desired experimental conditions.
Alternative chambers (fly’s setup) have been developed to facilitate the objectives of the experiment. For example, for the studies based on natural odor stimulus, the antenna must be kept free, therefore an approach like the one developed by A. Fiala14 is best suited (we used a similar approach for Murmu et al. 2010). Briefly, in this approach, the fly is tethered (glued) on a minutien pin at the neck, which is glued to a plastic cover slip containing a small hole. A seal is created around the top of the head of the fly. Thereafter, a drop of ringer is deposited on the head, and the head capsule is dissected. In such a way, the antennae are kept free and are available to sense odors. Similarly, for the long term recording, as O/N or even couple of days10 the fly needs to be kept as free of constraint as possible, and in some cases fed (for example: with a capillary paper soaked in a 5% glucose solution). In these circumstances Fiala’s approach is also more suitable than the approach in the blue pipet tip (snorkeling method).
All drug applications can be controlled externally using a 6 way multi-valves gravity perfusion system. The flow can be controlled using volumetric perfusion regulators calibrated prior to each recording session for a flow of 2 ml/min. Drainage apparatus extracts liquid at a rate of 2 ml/min from the recording chamber via peristaltic pump permitting a continuous flow.
In some conditions, due to expensive cost of the drug and/or the amount of drug to use, one would like to apply only small quantities of compounds. Thus, it is necessary to directly apply it in the bath rather than through perfusion system. In this case the shutter must be closed during this process to avoid the light contamination.
Analysis methods
Counts per sec or counts per sec per coordinate have both been used by our group for analysis. Counts per second is perhaps best suited for bright responses in whole structures whereas counts per sec and per coordinate (which represents a normalization of the responses to the size of the ROI) is best suited and accurate for small populations of cells. Both of them can be easily selected in the analysis software.
One of the main advantages of using the bioluminescence approach is the way that are acquired and stored the data (x,y,t parameters of each emitted photons). Indeed, while any other imaging techniques used standard mode as an acquired full image (generally in a tiff format), with this system, we acquires only the relevant and significant signal by storing only the pixel coordinates which have been activated by the light during a pre-fixed duration of time (which corresponds to the acquisition time or temporal resolution). Consequently, the storage of the data is really “light” compared to the storage a full image, and consequently, makes it easier to analyze the data. For data analysis, we use a related software (photon viewer), which allows setting of the accumulation time as desired. Then the display of an image of the signals can be adjusted as desired in a goal to convincingly present the data. In parallel the results are quantified at the same resolution time than their acquisition time.
Significance and biological applications of bioluminescent imaging
As mentioned above bioluminescent calcium imaging offers three principal advantages to fluorescent calcium imaging techniques: (1) a deeper regions of the brain may be imaged, (2) imaging can occur continuously for longer durations as several hours as O/N and even in some cases up to two days, and (3) more recently, with a higher temporal resolution, up to 120 frame/sec (8.3 msec). The main limitation of the bioluminescent approach is due to its incompatibility with confocal illumination, which consequently does not allow us to determine the precise depth (z-plan) of the activated structures or neurons.
The first advantage of bioluminescent imaging, imaging of deep brain regions, was previously demonstrated in 2007 by the imaging of a high potassium induced calcium response within the ellipsoid body (eb), a deep structure located in the middle of the brain, with no prior electrophysiological or functional reports6. Similarly, a more recent example is the recording of the spontaneous Ca2+-activity in the eb following application of picrotoxin (a blocker of GABAA-receptors) demonstrating that the GABAergic ring-neurons of the eb are indeed inhibitory neurons (described in: Diaper et al., article submitted).
The second benefit, longer and continuous time recording, has been exploited in several studies from our lab. Odor stimulation has been used to examine activity of brain neurons in previous studies11,15,16. Using longer term imaging, however, our lab has been able to show previously unreported changes in kinetics within the olfactory receptor neurons to odors. The first study by Murmu et al. (2010) showed that while short odor stimuli (1-3 sec) exhibited similar kinetics and a moderate difference in amplitude, a longer odor stimulus (5 sec) caused a much greater amplitude as well as a change in the kinetics/structure of the response which was attributed to intracellular calcium stores. A subsequent study showed that while 5 successive applications of 1 sec odor pulses at 5 min intervals did not change their calcium response, increasing the length of the stimulus to 5 sec led to progressive attenuation of the response, suggesting an adaptation process. Furthermore this adaptation phenomenon appeared to be initiated by GABAergic regulation of previously identified calcium stores8. Importantly this data also proves that, in this experimental condition, the GFP-aequorin’s Ca2+-sensor can detect and follow the calcium released from the intracellular Ca2+-stores. An additional unique study from our lab used bioluminescent imaging O/N to record basal (spontaneous) activity in the whole neuronal and glial populations in the brain as well as activity in the mushroom bodies10. These recordings revealed surprising spontaneous activity in antennal mechanosensory and motor center (AMMC) and antennal lobe. Furthermore, unexpected cell autonomous activity in the glia occurring throughout the night. Within the mushroom body the study identified both punctate activity and two distinctive peaks, one short and one long whose incidence changed with age10.
In the most recent study in our lab, we used bioluminescent imaging to examine the effects of the manipulation of secondary signaling molecules implicated in memory, and how they affect the calcium response within the mushroom bodies9. Indeed, although dunce and rutabaga were identified more than 30 years ago and are known to regulate the level of cAMP, their in vivo effects on cellular and physiological mechanisms and particularly on the Ca2+-response still remain largely unknown. With the GFP-aequorin approach, we have been able to simultaneously record both the calyx (the dendritic structures) and the cell-bodies, as well as the axonal projections at the level of the lobes, and thus allowing us to correlate the level and the kinetic of the response in the different compartments of this highly complex structure.
The most recent application we are developing mimics a natural response through artificially activating neuronal populations presynaptic to the structure of interest. To do this we express the P2X2 receptor, a ligand-gated cation channel which is responsive to ATP, in the presynaptic neuron and express GFP-aequorin in the post-synaptic neuronal population. The segregation of these two elements into different neuronal populations requires two separate expression systems. To this end a LexA construct containing GFP-aequorin has been created. In our preliminary experiments with this system we have expressed P2X2 in a subset of the PNs using the Gal4-GH146 with UAS-P2X2 and GFP-aequorin in the mushroom body using the LexA MB247 line combined with LexA-13X-GFP-aequorin. We have successfully recorded calcium responses in the mushroom body with stimulations of 1 mM ATP to the P2X2 receptor in the PNs (Figure 6).
In conclusion, the bioluminescence GFP-aequorin approach has proven to be useful in recording the stimulus induced Ca2+-activity, analogous to other Ca2+-indicators. But more importantly, it also permits the detection of spontaneous Ca2+-activity within the brain. This approach opens up future possibilities for long term experiments and whole brain imaging that can increase the depth of our understanding of the physiological phenomena and activity patterns within the brain.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We are indebted to E. Karplus, from Sciences Wares, USA, for his precious and useful help and advices. We thank E. Carbognin, A. Avet-Rochex, M. Murmu, P. Pavot, D. Minocci, G. Vinatier, and J. Stinnakre for their contribution to the development and improvement of this technique. We also thank B. Pfeiffer and G. Rubin, Janelia Farm, H.H.M.I., Ashburn, USA, for the pJFRC65-13XLexAop2-IVS-G5A-BP line. This work was supported by the Chateaubriand Fellowship, French Embassy, Washington, USA to A. Lark, by the National Science Foundation (IOS 1352882) to T. Kitamoto, and by the French ANRs (ANR-05-NEUR-009 Drosaequorin, 2005, and FlyBrainImaging, 2011), the Conseil Régional Ile-De France (NeRF and DIM), the IFR-144, the Physique-Chimie-Biologie Interface Program of the CNRS (2009), and by the CNRS, France to JR Martin.
References
- Martin J-R. In vivo brain imaging: fluorescence or bioluminescence, which to choose. J Neurogenet. 2008;22(3):285–307. doi: 10.1080/01677060802298517. [DOI] [PubMed] [Google Scholar]
- Shimomura O, Johnson FH. Peroxidized coelenterazine, the active group in the photoprotein aequorin. Proc Natl Acad Sci USA. 1978;75(6):2611–2615. doi: 10.1073/pnas.75.6.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura O, Johnson FH, Saiga Y. Further data on the bioluminescent protein, aequorin. J. Cell. Comp. Physiol. 1963;62(1):1–8. doi: 10.1002/jcp.1030620102. [DOI] [PubMed] [Google Scholar]
- Baubet V, et al. Chimeric green fluorescent protein-aequorin as bioluminescent Ca2+ reporters at the single-cell level. Proc Natl Acad Sci USA. 2000;97(13):7260–7265. doi: 10.1073/pnas.97.13.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrero P, Richmond L, Nitabach M, Davies SA, Dow JAT. A biogenic amine and a neuropeptide act identically: tyramine signals through calcium in Drosophila tubule stellate cells. Proc. Biol. Sci. 2013;280(1757):20122943. doi: 10.1098/rspb.2012.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc C, Webb SE, Daguzan C, Moreau M, Miller AL. Imaging patterns of calcium transients during neural induction in Xenopus laevis embryos. J. Cell Sci. 2000;113(19):3519–3529. doi: 10.1242/jcs.113.19.3519. [DOI] [PubMed] [Google Scholar]
- Martin J-R, Rogers KL, Chagneau C, Brûlet P. In vivo bioluminescence imaging of Ca2+ signalling in the brain of Drosophila. PLoS One. 2007;2(3):e275. doi: 10.1371/journal.pone.0000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murmu MS, Stinnakre J, Martin J-R. Presynaptic Ca2+ stores contribute to odor-induced responses in Drosophila olfactory receptor neurons. J. Exp. Biol. 2010;213(24):4163–4173. doi: 10.1242/jeb.046474. [DOI] [PubMed] [Google Scholar]
- Murmu MS, Stinnakre J, Réal E, Martin J-R. Calcium-stores mediate adaptation in axon terminals of Olfactory Receptor Neurons in Drosophila. BMC Neurosci. 2011;12:105. doi: 10.1186/1471-2202-12-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavot P, Carbognin E, Martin J-R. PKA and cAMP/CNG channels independently regulate the cholinergic Ca2+-response of Drosophila mushroom body neurons. eNeuro. 2015;2(2):0054–0068. doi: 10.1523/ENEURO.0054-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocci D, Carbognin E, Murmu MS, Martin J-R. In vivo functional calcium imaging of induced or spontaneous activity in the fly brain using a GFP-apoaequorin-based bioluminescent approach. BBA-Mol Cell Res. 2013;1833(7):1632–1640. doi: 10.1016/j.bbamcr.2012.12.017. [DOI] [PubMed] [Google Scholar]
- Lima SQ, Miesenböck G. Remote control of behavior through genetically targeted photostimulation of neurons. Cell. 2005;121(1):141–152. doi: 10.1016/j.cell.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Shimomura O, Musicki B, Kishi Y, Inouye S. Light-emitting properties of recombinant semisynthetic aequorins and recombinant fluorescein-conjugated aequorin for measuring cellular calcium. Cell Calcium. 1993;14(5):373–378. doi: 10.1016/0143-4160(93)90041-4. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Bio. 2003;4(7):517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Fiala A, Spall T. In vivo calcium imaging of brain activity in Drosophila by transgenic cameleon expression. Sci STKE. 2003;2003(174):PL6. doi: 10.1126/stke.2003.174.pl6. [DOI] [PubMed] [Google Scholar]
- Wang JW, Wong AM, Flores J, Vosshall LB, Axel R. Two-photon calcium imaging reveals an odor-evoked map of activity in the fly brain. Cell. 2003;112(2):271–282. doi: 10.1016/s0092-8674(03)00004-7. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Turner GC, Laurent G. Transformation of Olfactory Representations in the Drosophila Antennal Lobe. Science. 2004;303(5656):366–370. doi: 10.1126/science.1090782. [DOI] [PubMed] [Google Scholar]