

Abstract
Fibroblasts are the principle cell type responsible for secreting extracellular matrix and are a critical component of many organs and tissues. Fibroblast physiology and pathology underlie a spectrum of clinical entities, including fibroses in multiple organs, hypertrophic scarring following burns, loss of cardiac function following ischemia, and the formation of cancer stroma. However, fibroblasts remain a poorly characterized type of cell, largely due to their inherent heterogeneity. Existing methods for the isolation of fibroblasts require time in cell culture that profoundly influences cell phenotype and behavior. Consequently, many studies investigating fibroblast biology rely upon in vitro manipulation and do not accurately capture fibroblast behavior in vivo. To overcome this problem, we developed a FACS-based protocol for the isolation of fibroblasts from the dorsal skin of adult mice that does not require cell culture, thereby preserving the physiologic transcriptional and proteomic profile of each cell. Our strategy allows for exclusion of non-mesenchymal lineages via a lineage negative gate (Lin-) rather than a positive selection strategy to avoid pre-selection or enrichment of a subpopulation of fibroblasts expressing specific surface markers and be as inclusive as possible across this heterogeneous cell type.
Keywords: Developmental Biology, Issue 107, fibroblast, harvest, uncultured, cultured, cell isolation, cell culture, flow cytometry, lineage, FACS
Introduction
Fibroblasts are frequently defined morphologically as spindle-shaped cells that adhere to plastic substrates. Fibroblasts are the principle cell type responsible for synthesizing and remodeling the extracellular matrix in embryonic and adult organs1. Fibroblasts are thus critical to mammalian development and contribute substantially to the extracellular milieu that influences the behavior of neighboring cell types present in each tissue and organ.
Fibroblasts are also the principal cell type behind a diverse set of medical conditions that cause enormous clinical burden. Pathologic fibroblast activity impairs normal tissue function and includes tissue and organ fibrosis (such as the lung and liver), scarring following cutaneous wound healing, atherosclerosis, systemic sclerosis, and formation of atheromatous plaques after blood vessel injury2-5. Wound healing in particular, both acutely and chronically, involves deposition of scar tissue that neither resembles nor functions like the normal tissue surrounding it, and leads to significant morbidity across diverse pathologic states. Following injury, there is a transition of fibroblasts to myofibroblasts, which then secrete structural ECM components, exert paracrine effects on neighboring cell types, and restore mechanical stability by depositing scar tissue6.
In cutaneous tissues there exists significant variation in the quality of wound repair across developmental time and between anatomic sites. In the first two trimesters of life the fetus heals without scarring; however, from the third trimester on and throughout adulthood, humans heal with a scar. Site-specific, in addition to age-specific, differences in wound healing exist. Wounds in the oral cavity remodel with minimal scar formation7,8, while scar tissue deposition within cutaneous wounds is significant9. Controversy persists concerning the relative influence of the environment versus the intrinsic properties of local fibroblasts on the outcome of wound healing in regards to both age and location10,11. Given the significant differences in the healing of mouse oral vs. cutaneous dermis and earlier embryonic (E15) vs. later embryonic (E18) dermis, it is likely that intrinsic differences in the populations of fibroblasts at certain developmental ages and among various anatomic sites exist.
In 1986, Harold F. Dvorak posited tumors are wounds that do not heal12. Dvorak concluded that tumors behave like wounds in the body and induce their stroma by activating the wound healing response of the host. Numerous studies have since investigated the contribution of fibroblasts to the progression of carcinomas13-15, but as in the case of wound healing, the identity and embryonic origin of the fibroblasts that contribute to the stromal compartment of cutaneous carcinomas has not been adequately defined. The answer to this question bears medical relevance given recent studies exposing the tumor-associated fibroblast as a potentially effective target for anti-cancer therapy16.
Identifying and prospectively isolating the fibroblast lineages endowed with fibrogenic potential in vivo is an essential step towards effectively manipulating their response to injury across a wide range of acute and chronic disease states. In 1987, Cormack demonstrated two subpopulations of fibroblasts, one residing within the papillary and one within the reticular dermis17,18. A third subpopulation was found associated with hair follicles in the dermal papilla region of the follicle19,20. When cultured, these fibroblast subtypes exhibit differences in growth potential, morphology, and growth factor/cytokines profiles21-24.
To date, studies examining fibroblast heterogeneity have largely failed to adequately characterize developmental and functional diversity among fibroblasts in vivo. This is, in part, is a result of a reliance on cultured fibroblast populations and the homogenizing effect of cell culture or positive selection on the basis of a self surface receptor not expressed by all fibroblasts25. A recent study from our lab demonstrated a profound surface marker and transcriptional shift in cultured vs. uncultured fibroblasts isolated by the FACS-based isolation methodology presented in this manuscript26.
Subsequently, we identified a specific fibroblast lineage within the murine dorsal dermis and determined that this lineage, defined by embryonic expression of Engrailed-1, is primarily responsible for connective tissue deposition in the dorsal skin. The lineage functions during both acute and chronic forms of fibrosis including wound healing, cancer stroma formation, and radiation induced fibrosis27. The characterization of distinct fibroblast lineages has critical implications for therapies aimed at modulating fibrogenic behavior.
Rather than using existing protocols that rely upon in vitro manipulation to achieve cell isolation28,29, the harvest protocol (Figure 1) detailed here will help yield informative analyses of fibroblasts that more accurately capture phenotype and behavior in vivo.
Protocol
This protocol follows methods approved by the Stanford University Administrative Panel on Laboratory Animal Care.
1. Digestion of Murine Dermis
Euthanize mice by cervical dislocation after anesthesia with an intraperitoneal injection of ketamine 100 mg/kg + xylazine 20 mg/kg + acepromazine 3 mg/kg. Note: Various ages and backgrounds can be used.
Shave and depilate the dorsal skin. Approximately 100,000 cells can be isolated from a piece of dorsal skin 60 mm x 100 mm.
Submerge the mouse in 70% ethanol and place on a clean, sterile surface to dry.
Immediately harvest dorsal mouse skin using sterile dissecting scissors. In female mice, avoid including the mammary tissue.
Starting the base of the tail, use forceps to tent up the skin and make a transverse cut before dissecting along the supra-fascial plane.
Carefully avoid including any subcutaneous fat while harvesting the skin. Examine the harvested skin for any subcutaneous fat and carefully scrape it off using the blunt edge of a scalpel.
Rinse the harvested skin in betadine followed by 5x PBS washes on ice. Note: It is important to keep the skin as close to sterile as possible to avoid contamination.
Mince the skin using razor blades and dissecting scissors in a sterile dish until the sample is of a uniform consistency with 2-3 mm pieces.
Prepare 50 ml conical tubes containing 20 ml collagenase IV at a concentration of 1 mg/ml in DMEM. Divide the dermis into the tubes on the basis of five mice per tube.
Agitate samples vigorously while incubating at 37 °C for 1 hr in either a water bath or oven.
Remove samples from the incubator and pass through a 10 ml syringe without a needle 3-5x in a sterile hood.
Place the samples back into the incubator at 37 °C and shake vigorously for a further 30 min.
In a sterile hood, pipette the samples up and down 3-5x using a 10 ml pipette. Pipette the sample through a 100 µm filter into a new 50 ml conical tube.
Pass 20 ml of 10% FBS DMEM through the same filter to maximize cell yield and bring the total volume to 40 ml. Centrifuge at 300 g for 8 min at 4 °C.
Remove the supernatant using a sterile glass pipette, taking great care to first remove the upper fat layer prior to remaining supernatant. Note: This step is critical to reduce adipocyte contamination.
Resuspend the pellets in 20 ml 10% FBS DMEM.
Pass the cell/DMEM suspension through a 70 µm filter.
Rinse the filter with 10 ml 10% FBS DMEM and centrifuge the filtered suspension at 300 g for 8 min at 4 °C.
Remove the supernatant using a sterile glass pipette, again taking care to first remove any remaining fat layer.
If there is significant RBC contamination (the pellet is visibly red), re-suspend the pellets in 20 ml ACK lysis buffer and incubate for 5 min at RT. Otherwise skip to step 24.
Add an equal volume (20 ml) of FACS buffer (PBS, 10% FBS, 0.1% sodium azide), then mix, and keep aside a 5 ml aliquot as an unstained control. Centrifuge the remaining sample at 300 g for 8 min at 4 °C.
Remove supernatant and put pellet on ice. The cells may be frozen down at this point if FACS time is not available.
2. Isolation of Fibroblasts by FACS
Make 500 µl of lineage antibody incubation mix for each pellet. Do this by first adding 475 µl of FACS buffer containing DNase (10 µg/ml) to a tube, and then adding fluorophore-conjugated CD31 (1:100), CD45 (1:200), Tie2 (1:50), Ter-119 (1:200), and EpCAM (1:100) antibodies to achieve the respective dilution for each antibody.
Re-suspend each pellet in 500 µl of lineage antibody incubation mix and incubate this suspension on ice for 20 min.
Add 5 ml FACS buffer containing DNase (10 µg/ml) to the sample and gently mix. Centrifuge at 300 g for 8 min at 4 °C.
Remove the supernatant and wash the cell pellet with 5ml FACS buffer containing DNase (10 µg/ml) and centrifuge using the same conditions as in step 26.
Resuspend the pellet in 500 µl FACS buffer containing DNase (10 µg/ml) and put aside a 50 µl aliquot as a viability dye control.
Add viability dye of choice to the remaining sample in the concentration indicated for the chosen dye.
Perform FACS analysis31 and sorting for Viability Dye-/CD31-/CD45-/Tie2-/Ter-119-/EpCAM- cells (see Figure 2A). Sort directly into FACS buffer.
Representative Results
The validity of this approach (Figure 1) has been verified in a number of ways, which can be examined in detail in our recent publication27. These include immunocytochemistry of sorted cells and mass cell and single cell transcriptional analysis of freshly sorted cells. Sorting fibroblasts directly rather than relying on culture more accurately captures their in vivo phenotype. Using a lineage negative depletion approach (Figure 2A) rather than a positive selection approach avoids pre-selecting for particular subpopulations. The value of this approach was recently demonstrated by Rinkevich et al.27, identifying the presence of CD26 positive fibroblasts at a level of the dermis previously not described.
To confirm that the process of culturing fibroblasts leads to significant changes in gene expression, we used microarrays to compare cultured fibroblasts to uncultured fibroblasts. We cultured fibroblasts that were isolated by two techniques, the live harvest protocol detailed in this manuscript and a well-known tissue explant protocol28. Whole-transcriptome microarray analysis revealed that cultured fibroblasts isolated by the live harvest (C.LH) and by tissue explant (C.TE) methodologies have a high degree of similarity at a transcriptome-wide level with a Pearson product moment correlation coefficient (r) of 0.92 (Figure 2B). By comparison, cultured fibroblasts differed significantly from live harvested uncultured fibroblasts (U.LH). A comparison between C.LH versus U.LH yielded an r of 0.61, while a comparison of C.TE versus U.LH yielded an r of 0.64 (Figure 2B). These results establish the importance of analyzing live harvested fibroblasts rather than cultured fibroblasts. For a more complete analysis of transcriptional and proteomic differences between cultured and uncultured fibroblasts isolated using this protocol, refer to Walmsley et al.26
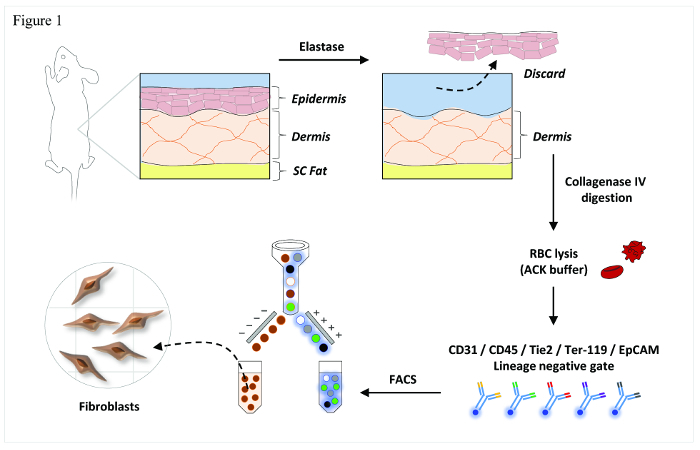 Figure 1. Overview of Fibroblast Isolation. Schematic representation of the primary steps involved in this FACS-based isolation protocol. Reused with permission from Walmsley, G. G. et al. Live Fibroblast Harvest Reveals Surface Marker Shift in vitro. Tissue engineering. Part C, Methods, doi:10.1089/ten.TEC.2014.0118 (2014). Please click here to view a larger version of this figure.
Figure 1. Overview of Fibroblast Isolation. Schematic representation of the primary steps involved in this FACS-based isolation protocol. Reused with permission from Walmsley, G. G. et al. Live Fibroblast Harvest Reveals Surface Marker Shift in vitro. Tissue engineering. Part C, Methods, doi:10.1089/ten.TEC.2014.0118 (2014). Please click here to view a larger version of this figure.
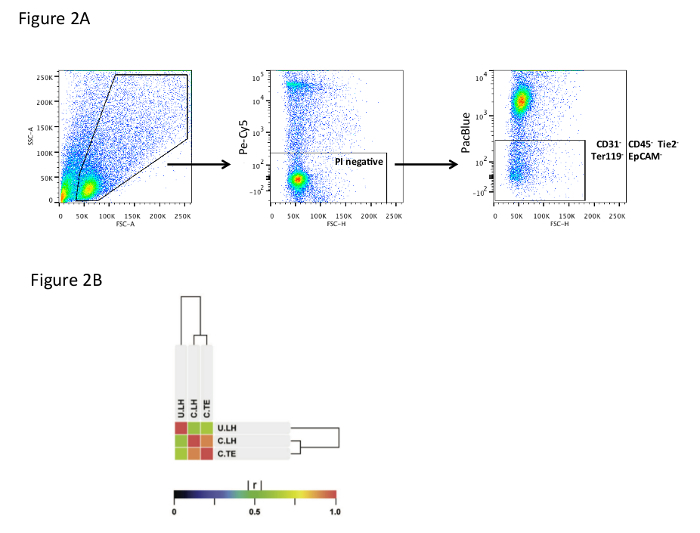 Figure 2. Flow Cytometry and Microarray Analysis. (A) FACS gating strategy showing selection of single cells (left plot), selection for viable cells based on propidium iodide staining (middle plot), and selection of fibroblasts (right plot) on the basis of lineage negativity for CD31, CD45, Tie2, Ter119, and EpCAM. (B) Microarray Analysis of Uncultured Live Harvested (U.LH) versus Cultured Live Harvested (C.LH) versus Cultured Tissue Explant (C.TE) Fibroblasts. Similarity of gene expression between U.LH (n = 3), C.LH (n = 3), and C.TE (n = 3) fibroblast populations as measured by the Pearson product-moment correlation coefficient (r). [C.LH vs. C.TE: r = 0.92]; [C.LH vs. U.LH: r = 0.61]; [C.TE vs. U.LH: r = 0.64]. Reused with permission from Walmsley, G. G. et al. Live Fibroblast Harvest Reveals Surface Marker Shift in vitro. Tissue engineering. Part C, Methods, doi:10.1089/ten.TEC.2014.0118 (2014). Please click here to view a larger version of this figure.
Figure 2. Flow Cytometry and Microarray Analysis. (A) FACS gating strategy showing selection of single cells (left plot), selection for viable cells based on propidium iodide staining (middle plot), and selection of fibroblasts (right plot) on the basis of lineage negativity for CD31, CD45, Tie2, Ter119, and EpCAM. (B) Microarray Analysis of Uncultured Live Harvested (U.LH) versus Cultured Live Harvested (C.LH) versus Cultured Tissue Explant (C.TE) Fibroblasts. Similarity of gene expression between U.LH (n = 3), C.LH (n = 3), and C.TE (n = 3) fibroblast populations as measured by the Pearson product-moment correlation coefficient (r). [C.LH vs. C.TE: r = 0.92]; [C.LH vs. U.LH: r = 0.61]; [C.TE vs. U.LH: r = 0.64]. Reused with permission from Walmsley, G. G. et al. Live Fibroblast Harvest Reveals Surface Marker Shift in vitro. Tissue engineering. Part C, Methods, doi:10.1089/ten.TEC.2014.0118 (2014). Please click here to view a larger version of this figure.
Discussion
The protocol described in this manuscript offers a means to isolate fibroblasts by FACS-based sorting, in comparison to existing methods, which either select for a subpopulation or require time in cell culture before subsequent analyses. The time required from harvesting of the skin to sorting of fibroblasts is approximately 6 hr; however, the number of mice used in the harvest will influence this estimate.
Several points in the protocol require particular care. The first is limiting adipocyte contamination by removal of fat from the skin before digestion and removal of the upper lipid layer of supernatant following digestion and centrifugation during the isolation process. It may also be helpful to change to fresh tubes after the cells are pelleted as some lipid components adhere to the plastic walls of the tubes and may contaminate subsequent pellet washings. A second point involves meticulous care to separate dermis from epidermis along the epidermal-dermal junction. Although contaminating epidermal cells will be removed by the FACS depletion strategy, an effort to limit contamination here should still be made.
The limitations of this approach include the potential presence of contaminating cells not captured by the current lineage panel. When choosing the fluorophore conjugated to the lineage antibodies (CD31, CD45, Tie2, Ter119, EpCAM), researchers must take care to consider other surface marker analyses they may wish to perform. Additional stains must be in distinct channels from the chosen lineage antibody fluorophore. In general, we found PacBlue to be an ideal conjugate that preserves a wide range of available wavelengths for further analysis. Matching the viability dye to the lineage antibody fluorophore preserves an additional range of wavelengths. For example, the viability dye DAPI excites and fluoresces at similar wavelengths to PacBlue. In this manner, all DAPI positive and lineage antibody positive cells can be effectively eliminated using a single gate, leaving only viable fibroblasts as the target population. It should also be noted that cells are exposed to FBS during the isolation procedure for limited amounts of time and this likely influences gene expression, to an unknown degree.
The ability to inclusively sort for all fibroblast populations represents an opportunity to truly interrogate the heterogeneity of this poorly understood cell type. This has application in the context of normal physiology as well as a variety of diseases that involve excessive fibrosis and aberrant fibroblast behavior.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported in part by a grant from NIH grant R01 GM087609 (to H.P.L.), a Gift from Ingrid Lai and Bill Shu in honor of Anthony Shu (to H.P.L.), NIH grant U01 HL099776 (to M.T.L.), the Hagey Laboratory for Pediatric Regenerative Medicine and The Oak Foundation (to M.T.L., G.C.G. and H.P.L.). G.G.W. was supported by the Stanford School of Medicine, the Stanford Medical Scientist Training Program, and NIGMS training grant GM07365. Z.N.M. was supported by the Plastic Surgery Foundation Research Fellowship Grant and the Hagey Family Fund. M.S.H. was supported by the California Institute for Regenerative Medicine (CIRM) Clinical Fellow training grant TG2-01159, the American Society of Maxillofacial Surgeons (ASMS)/Maxillofacial Surgeons Foundation (MSF) Research Grant Award, and the Transplant and Tissue Engineering Fellowship Award.
References
- Sorrell JM, Caplan AI. Fibroblasts-a diverse population at the center of it all. International review of cell and molecular biology. 2009;276:161–214. doi: 10.1016/S1937-6448(09)76004-6. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. The Journal of pathology. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DW, et al. Myofibroblasts. I. Paracrine cells important in health and disease. The American journal of physiology. 1999;277:C1–C9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal immunology. 2009;2:103–121. doi: 10.1038/mi.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, et al. The myofibroblast: one function, multiple origins. The American journal of pathology. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Wang JH. Fibroblasts and myofibroblasts in wound healing: force generation and measurement. J Tissue Viability. 2011;20:108–120. doi: 10.1016/j.jtv.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JW, et al. Wound healing in oral mucosa results in reduced scar formation as compared with skin: evidence from the red Duroc pig model and humans. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2009;17:717–729. doi: 10.1111/j.1524-475X.2009.00531.x. [DOI] [PubMed] [Google Scholar]
- Szpaderska AM, Zuckerman JD, DiPietro LA. Differential injury responses in oral mucosal and cutaneous wounds. Journal of dental research. 2003;82:621–626. doi: 10.1177/154405910308200810. [DOI] [PubMed] [Google Scholar]
- Hantash BM, Zhao L, Knowles JA, Lorenz HP. Adult and fetal wound healing. Frontiers in bioscience : a journal and virtual library. 2008;13:51–61. doi: 10.2741/2559. [DOI] [PubMed] [Google Scholar]
- Buchanan EP, Longaker MT, Lorenz HP. Fetal skin wound healing. Advances in clinical chemistry. 2009;48:137–161. doi: 10.1016/s0065-2423(09)48006-5. [DOI] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. The New England journal of medicine. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- Dumont N, et al. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia. 2013;15:249–262. doi: 10.1593/neo.121950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servais C, Erez N. From sentinel cells to inflammatory culprits: cancer-associated fibroblasts in tumour-related inflammation. The Journal of pathology. 2013;229:198–207. doi: 10.1002/path.4103. [DOI] [PubMed] [Google Scholar]
- Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell cycle. 2006;5:1597–1601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- Li X, et al. Targeting the cancer-stroma interaction: a potential approach for pancreatic cancer treatment. Current pharmaceutical design. 2012;18:2404–2415. doi: 10.2174/13816128112092404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrell JM, Caplan AI. Fibroblast heterogeneity: more than skin deep. Journal of cell science. 2004;117:667–675. doi: 10.1242/jcs.01005. [DOI] [PubMed] [Google Scholar]
- Cormack DH. Ham's Histology. J.B. Lippincott Company; 1987. pp. 450–474. [Google Scholar]
- Jahoda CA, Reynolds AJ. Dermal-epidermal interactions. Adult follicle-derived cell populations and hair growth. Dermatologic clinics. 1996;14:573–583. doi: 10.1016/s0733-8635(05)70385-5. [DOI] [PubMed] [Google Scholar]
- Jahoda CA, Reynolds AJ. Hair follicle dermal sheath cells: unsung participants in wound healing. Lancet. 2001;358:1445–1448. doi: 10.1016/S0140-6736(01)06532-1. [DOI] [PubMed] [Google Scholar]
- Sorrell JM, Baber MA, Caplan AI. Construction of a bilayered dermal equivalent containing human papillary and reticular dermal fibroblasts: use of fluorescent vital dyes. Tissue engineering. 1996;2:39–49. doi: 10.1089/ten.1996.2.39. [DOI] [PubMed] [Google Scholar]
- Harper RA, Grove G. Human skin fibroblasts derived from papillary and reticular dermis: differences in growth potential in vitro. Science. 1979;204:526–527. doi: 10.1126/science.432659. [DOI] [PubMed] [Google Scholar]
- Schafer IA, Pandy M, Ferguson R, Davis BR. Comparative observation of fibroblasts derived from the papillary and reticular dermis of infants and adults: growth kinetics, packing density at confluence and surface morphology. Mechanisms of ageing and development. 1985;31:275–293. doi: 10.1016/0047-6374(85)90095-8. [DOI] [PubMed] [Google Scholar]
- Sorrell JM, Baber MA, Caplan AI. Site-matched papillary and reticular human dermal fibroblasts differ in their release of specific growth factors/cytokines and in their interaction with keratinocytes. Journal of cellular physiology. 2004;200:134–145. doi: 10.1002/jcp.10474. [DOI] [PubMed] [Google Scholar]
- Sharon Y, Alon L, Glanz S, Servais C, Erez N. Isolation of normal and cancer-associated fibroblasts from fresh tissues by Fluorescence Activated Cell Sorting (FACS) Journal of visualized experiments : JoVE. 2013. p. e4425. [DOI] [PMC free article] [PubMed]
- Walmsley GG, et al. Live Fibroblast Harvest Reveals Surface Marker Shift in vitro. Tissue engineering. Part C, Methods. 2014. [DOI] [PMC free article] [PubMed]
- Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, Januszyk M, Krampitz GW, Gurtner GC, Lorenz HP, Weissman IL, Longaker MT. Identification and Isolation of a Dermal Lineage with Intrinsic Fibrogenic Potential. Science. 2015. [DOI] [PMC free article] [PubMed]
- Seluanov A, Vaidya A, Gorbunova V. Establishing primary adult fibroblast cultures from rodents. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nature protocols. 2008;3:799–810. doi: 10.1038/nprot.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M, Fitzgerald LR. Enzymatic separation of intact epidermal sheets from mouse skin. The Journal of investigative dermatology. 1962;39:111–114. doi: 10.1038/jid.1962.90. [DOI] [PubMed] [Google Scholar]
- Biburger M, Trenkwald I, Nimmerjahn F. yThree blocks are not enough - Blocking of the murine IgG receptor FcgammaRIV is crucial for proper characterization of cells by FACS analysis. European journal of immunology. 2015. [DOI] [PubMed]